Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Materials and Methods
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(5):1373-1388. doi:10.7150/thno.17826 This issue Cite
Research Paper
Simultaneous targeting of CD44 and EpCAM with a bispecific aptamer effectively inhibits intraperitoneal ovarian cancer growth
Jingying Zheng1,3 *, Shuhua Zhao3 *, Xiaolin Yu1, Shuang Huang2, Hong Yan Liu1 ![]()
1. Center for Biotechnology and Genomic Medicine, Medical College of Georgia, Augusta University, Augusta, GA 30912, USA;
2. Department of Anatomy and Cell Biology, University of Florida College of Medicine, Gainesville, FL 32610, USA.
3. Department of Gynecology and Obstetrics, The second hospital of Jilin University, Jinlin University, Changchun, 130041, China.
* These authors contributed equally to this work.
Received 2016-10-5; Accepted 2017-1-23; Published 2017-3-23
Abstract

CD44 and EpCAM play crucial roles in intraperitoneal ovarian cancer development. In this study, we developed an RNA-based bispecific CD44-EpCAM aptamer that is capable of blocking CD44 and EpCAM simultaneously by fusing single CD44 and EpCAM aptamers with a double stranded RNA adaptor. With the aid of a panel of ovarian cancer cell lines, we found that bispecific CD44-EpCAM aptamer was much more effective than either single CD44 or EpCAM aptamer in the ability to inhibit cell growth and to induce apoptosis. When these aptamers were tested in intraperitoneal ovarian cancer xenograft model, bispecific CD44-EpCAM aptamer suppressed intraperitoneal tumor outgrowth much more significantly than single CD44 and EpCAM aptamer either alone or in combination. The enhanced efficacy of bispecific CD44-EpCAM aptamer is most likely to be attributed to its increased circulation time over the single aptamers. Moreover, we showed that bispecific CD44-EpCAM aptamer exhibited no toxicity to the host and was unable to trigger innate immunogenicity. Our study suggests that bispecific CD44-EpCAM aptamer may represent a promising therapeutic agent against advanced ovarian cancer.
Keywords: CD44, EpCAM, aptamer, intraperitoneal tumor growth, bispecific molecule.
Introduction
Ovarian cancer (OC) is the most deadly cancer among all gynecologic malignancies. OC is often detected at an advanced stage with wide peritoneal metastasis, and 5-year survival rate of advanced OC is around 20-30%[1]. The combination of debulking and platinum-based chemotherapy is the standard treatment for advanced OC. Although patients initially respond favorably, most of them will develop chemoresistance and eventually demise with peritoneal metastasis [2, 3]. Therefore, it is urgently needed to develop effective therapeutics to overcome chemoresistance and to inhibit peritoneal metastasis.
Epithelial cell adhesion molecules (EpCAM), a glycosylated membrane protein, is overexpressed in over 70% OC and its level is closely associated with malignant ascites, chemoresistance, and decreased survival rate in OC patients [4]. Although normal epithelial tissues usually express EpCAM, its expression in the peritoneal cavity appears to be tumor specific because mesothelial cells in the abdominal cavity are negative for EpCAM expression [5, 6]. Since downregulation of EpCAM inhibits cell-cell adhesion and epithelial-mesenchymal transition, it has been postulated that EpCAM participates in ovarian cancer progression by regulating the process of EMT [7]. In addition, EpCAM-associated oncogenic features have recently also been connected to the enhanced transcription of c-Myc and the cyclin A/E [6]. Moreover, EpCAM positive cells are well recognized to possess tumor-initiating potential and EpCAM has thus been used as a key marker of ovarian cancer stem cells [8]. These findings strongly support the notion that EpCAM is an ideal therapeutic target for OC. Various EpCAM antagonists have been developed [9]; in fact, EpCAM-targeted antibodies have demonstrated the treatment efficacy in both experimental models and clinical trials. For instance, EpCAM antibody MT20 was able to effectively eliminate ovarian cancer cells in preclinical model [10] while treatment of EpCAM/CD3-bispecific antibody (Catumaxomab) led to substantial decrease of malignant ascites in OC patients [11].
In addition to EpCAM, CD44 has been identified as another important molecule in OC progression [12]. CD44 can organize a signaling platform which facilitates ovarian cancer cell growth, survival and metastasis [13]. CD44 can also serve as a receptor to mediate the attachment of ovarian cancer cells onto the peritoneal mesothelium by binding to mesothelium-associated hyaluronic acid (HA)[14]. A recent study revealed that population of CD44+/CD24- ovarian cancer cells were chemoresistant[12]. Analyses of ovarian cancer patient specimens further show that CD44 expression is associated with high-grade and advanced stage ovarian carcinoma [15]. These evidences clearly pinpoint a critical role of CD44 in ovarian cancer progression and metastasis [16]. This notion is buttressed by the observations that CD44-targeted antibodies or short hairpin RNAs inhibit ovarian cancer cell adhesion to mesothelial cells and peritoneal implantation [17, 18].
Combination therapy by simultaneously blocking dual or multiple signaling pathways [19] has demonstrated the promise in suppressing tumor progression and metastasis due to the capacity to overcome the function redundancy or synergistic action of targeted molecules [20]. Bispecific antibodies that can simultaneously target two different molecules have outshined conventional monoclonal antibody on the aspects of exhibiting better therapeutic efficacy and inciting less drug resistance [21]. With two-in-one format, bispecific molecules are less complicated in drug administration and have received more favorable regulatory approval than the proposed use of two single molecules in combination. However, antibody-based bispecific molecules are also suffered by high immunogenicity and difficult production involved in complicated technologies such as hybrid-hybridoma [22] and genetic engineering [23].
Aptamers are ssDNA or ssRNA that can bind target with high affinity and specificity [24]. Potent aptamers can be generated through in vitro enrichment process [25, 26] and are usually produced with little batch-to-batch variation [27, 28]. The nature of aptamer as a small oligonucleotide implicates that it offers many advantages over the antibody such as cell-free chemically synthesis, non-immunogenicity, high tissue penetration, thermostable and low cost [27, 29-32]. Pegaptanib, a vascular endothelial growth factor-targeted aptamer, is now approved for treating age-related macular degeneration [33]. Single EpCAM aptamer consisting of 19-nt RNA has been generated, which possesses the similar binding affinity as antibodies and is efficiently internalized through receptor-mediated endocytosis [34, 35]. Single CD44 aptamer with 90-nt has also been produced and can target cells with high CD44 expression effectively [36]. Importantly, single CD44 aptamer has been shown the capacity for drug delivery [37]. However, the therapeutic effect of these two aptamers against tumorigenesis has not been reported. In this study, we constructed an RNA-based bispecific molecule by fusing single CD44 and EpCAM aptamers into one unit. This fused aptamer displayed significantly improved circulation half-life and reduced renal filtration compared to the single aptamers. With the aid of both in vitro and in vivo experimental models, we showed that bispecific CD44-EpCAM aptamer inhibited ovarian cancer cell growth and suppressed intraperitoneal tumor progression much more efficiently than single CD44 and EpCAM aptamers used either alone or in combination. Our study not only offers a promising therapeutic agent against advanced ovarian cancer but also provides a methodology to develop bispecific aptamers from individual single aptamers.
Results
Construction and characterization of a bispecific CD44-EpCAM aptamer
Single CD44 aptamer[36] is 90 nucleotides long while single EpCAM aptamer[34] is much smaller and contains only 19 nucleotides (Figure 1A). The utilization of EpCAM aptamer is expected to confront with the problem of short circulation life time because molecular weight of 6.5 Kd of this aptamer is much smaller than the molecular mass cutoff of 30-50 Kd for renal glomerulus [38]. To overcome this problem, we fused single CD44 and EpCAM aptamers together with a 23bp double stranded RNA adaptor and purposely left 2-3 bases unpaired between adaptor and aptamer in order to give each single aptamer spatial space to form a 3-D structure shown in Figure 1A. The molecular weight of fused aptamer is 54.4Kd, which is large enough to avoid rapid renal depletion while is much smaller than antibody, and thus expected to possess an ideal bioavailability to many biological compartments. To increase nuclease resistance, we also incorporated 2'-fluoro-pyrimidines into entire RNA during the process of in vitro transcription.
Design and characterization of bispecific CD44-EpCAM aptamer. (A) Schematic illustration of the structure of CD44-EpCAM aptamer. EpCAM aptamer was conjugated with CD44 aptamer through 23bp adaptor and 2-3 unpaired base linkers. (B) Schematic of ELISA for evaluation of dual specificity of CD44-EpCAM aptamer. (C) Evaluation of dual specificity by ELISA. Recombinant human CD44 protein were coated on 96-well plates, and incubated with CD44-EpCAM mixed with EpCAM protein or control with a mixture of CD44 aptamer plus EpCAM aptamer and EpCAM protein. (D) Control experiment by coating plate with human serum albumin.
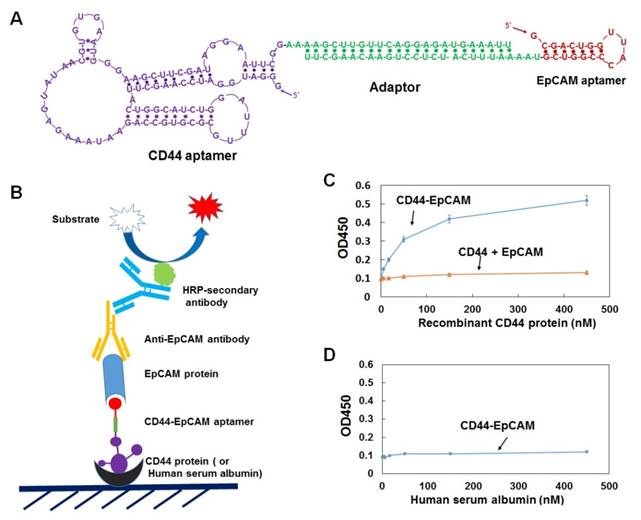
We next examined the binding of bispecific CD44-EpCAM aptamer by ELISA. Serially diluted human recombinant CD44 protein was immobilized on a 96-well plate. Following CD44-EpCAM incubated with 1µM EpCAM recombinant protein for 2h, the incubation mixture containing CD44-EpCAM aptamer and EpCAM protein were added into CD44 protein-immobilized plate. Control is the simply mixed CD44 aptamer and EpCAM aptamer. After washing, anti-EpCAM antibody was added to the plates followed by HRP-secondary antibody. As expected ( Figure 1C), CD44-EpCAM bispecific aptamers successfully linked to plate-immobilized CD44 protein and free EpCAM proteins, yielding CD44 protein concentration-dependent positive signals, whereas only the background signal was detected in a control experiment with a simple mixture of CD44 aptamer and EpCAM aptamer. In another control experiment, human serum albumin (HSA) has been immobilized to the plates and detected with CD44-EpCAM, however, there is no positive signal detectable (Figure 1D). These results suggest that bispecific CD44-EpCAM aptamer can efficiently bind both CD44 and EpCAM.
To verify if the 2-3 unpaired bases (linkers) between double-stranded adaptor and aptamer are important in keeping functionality of two aptamers, we have constructed two controls which contain no or only one unpaired linker. OVCAR8 cells were treated with linker-containing CD44-EpCAM and no linker controls. As shown in Figure S1, the control CD44-EpCAM No linker-1 which lacks the linker (unpaired two “A”) between CD44 aptamer and adaptor showed 25% of cytotoxicity at 2µM and 53% at 4µM, in contrast, cytotoxicity of linker-containing CD44-EpCAM is 77% at 2µM and 95 % at 4µM. Moreover, the removal of all unpaired base linkers (CD44-EpCAM No linker-2) has resulted about 95% activity loss (Figure S1). The results clearly demonstrate that 2-3 unpaired bases between aptamer and double stranded adaptor in this construct is crucial to render each aptamer function.
The expression of CD44 and EpCAM in ovarian cancer cell lines and cytotoxicity of bispecific CD44-EpCAM aptamer
To investigate the effect of bispecific CD44-EpCAM aptamer on ovarian cancer cell growth, we first analyzed the abundance of CD44 and EpCAM in a panel of ovarian cancer cell lines and human embryonic kidney HEK293T cell line. Western blotting with the specific antibodies showed that CD44 was highly expressed in all ovarian cancer cell lines (OVCAR8, SKOV3, OCC1 and ES2) while little was seen in HEK293T cells (Figure 2A). EpCAM was also expressed in all ovarian cancer cell lines and HEK293T cell line with highest expression detected in OVCAR8 cells (Figure 2A).
Evaluation of CD44 and EpCAM expression and cytotoxicity of CD44-EpCAM. (A) Western blot analysis of protein expression of CD44 and EpCAM in ovarian cancer cell lines and normal HEK293T cells. (B) Evaluation of cytotoxicity of CD44-EpCAM by CCK-8 assay. Ovarian cancer cell lines including OVCAR8, SKOV3, OCC1, ES2 and normal HEK293 cells were treated with varying concentration of CD44-EpCAM for 72 h, and cell viability was detected with CCK-8 agent. The results are the mean± SEM (N=3).
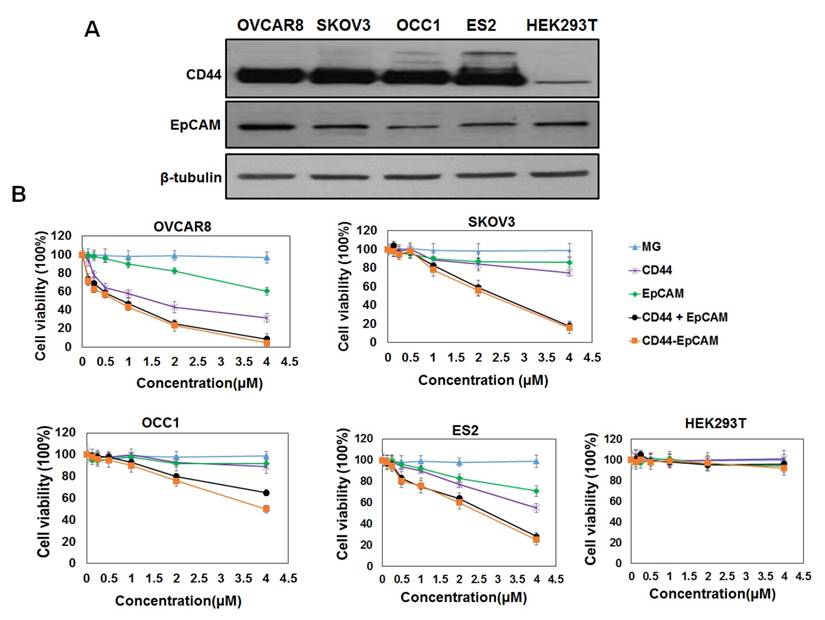
We next assessed the cytotoxicity of both single and bispecific CD44 and EpCAM aptamers on ovarian cancer cells by CCK-8 assay. Single EpCAM aptamer displayed little inhibitory effect at concentration <2µM but was able to reduce 10-30% of cell viability at 4µM in tested ovarian cancer cell lines (Figure 2B). In contrast, single CD44 aptamer dose-dependently reduced viability of OVCAR8, ES2 and SKOV3 but not OCC1 cells (Figure 2B). Interestingly, bispecific CD44-EpCAM at 4µM reduced viability of all four ovarian cancer cell lines with 95% reduction in OVCAR8, 92% in ES2, 84% in SKOV3 and 50% in OCC1 cells (Figure 2B). Effect of CD44-EpCAM aptamer was clearly specific because MG aptamer (specific to Malachite Green) displayed no any cytotoxicity up to 4µM in any of these cell lines. These results clearly demonstrate that bispecific CD44-EpCAM aptamer exhibits much greater potency in reducing ovarian cancer cell viability. Since none of the aptamer affected growth of HEK293T cells, these results indicate that bispecific CD44-EpCAM aptamer may specifically suppress ovarian cancer cell growth.
Serum stability of bispecific CD44-EpCAM aptamer
2'-fluoro-modified and unmodified CD44-EpCAM were incubated with final 50% human serum at 37°C for 2-24h followed by agarose gel electrophoresis to reveal RNA integrity. As shown in Figure 3A & 3B, unmodified CD44-EpCAM has been degraded as early as 2-h start test point, and no aptamer bands appear in 24-h incubation time range. On the contrary, 2'-fluoro-modified CD44-EpCAM kept its integrity (tight band) without degradation for 6h, and almost 45% of aptamer still remained at 24h.
Evaluation of serum stability and binding specificity. (A) Detection of serum stability by agarose gel electrophoresis. Unmodified and 2'-fluoro-modified CD44-EpCAM was incubated with 50% human serum for 2-24 h and subjected to analysis by 1% agarose gel electrophoresis. (B) Quantification of CD44-EpCAM from (A) by ImageJ. (C) Evaluation of binding pattern of different cell lines with CD44-EpCAM by flow cytometry. Cell lines were incubated with Cy5-labeled CD44-EpCAM and Cy5-labeled control MG aptamer, and detected by flow cytometry. Unstained cells are shown in solid grey, MG stained cells are in light blue, and CD44-EpCAM stained cells are in purple line. (D) Western blot analysis of CD44 and/or EpCAM gene knockdown. (E) Cell binding assay with flow cytometry. OVCAR8 cells were treated with CD44 and /or EpCAM siRNAs and stained with CD44-EpCAM-Cy5. Untreated: solid grey; MG-Cy5: light blue; normal cells stained with CD44-EpCAM-Cy5: red line; CD44 silenced cells stained with CD44-EpCAM-Cy5: green line; EpCAM silenced cells stained with CD44-EpCAM-Cy5: dark blue; both CD44 and EpCAM silenced cells stained with CD44-EpCAM-Cy5: purple line.
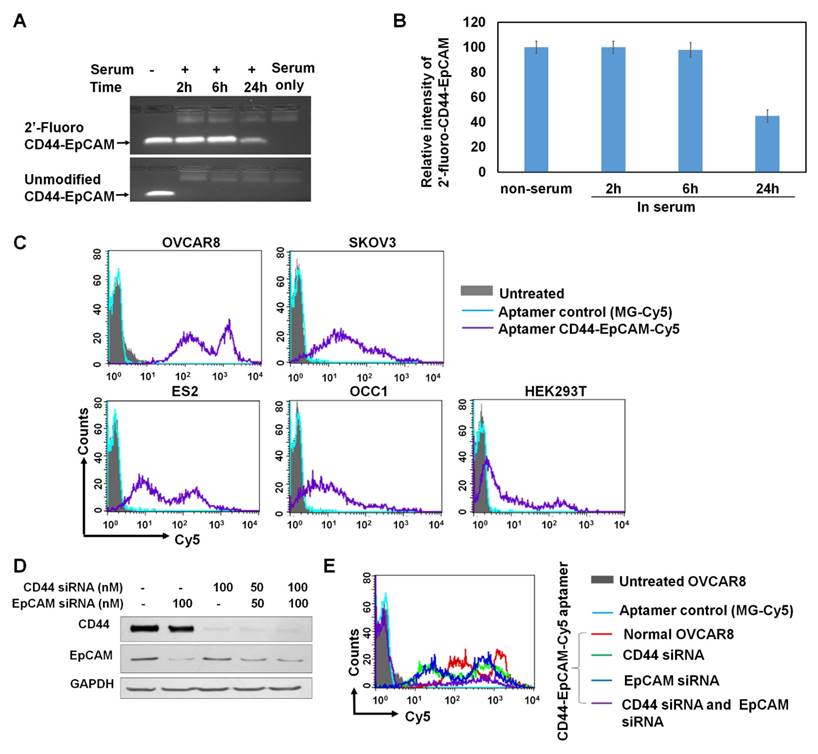
Binding specificity of bispecific CD44-EpCAM aptamer
To confirm target specificity of CD44-EpCAM aptamer, the binding patterns of different cell lines stained with CD44-EpCAM was assessed with flow cytometry. EpCAM aptamer was labeled with Cy5 at its 3' end (TriLink), and CD44-EpCAM-Cy5 was generated by annealing equal moles of EpCAM-Cy5 with CD44 aptamer. As shown in Figure 3C, OVCAR8, SKOV3 and ES2 show strong positive fluorescent intensity, and the intensity of OCC1 is weaker than OVCAR8, SKOV3 and ES2, but stronger than HEK293T. Furthermore, we identified if bispecific aptamer indeed binds to CD44 and EpCAM molecules. CD44 and EpCAM on OVCAR8 cells were subjected to knockdown by siRNAs. CD44+ (EpCAM silenced), EpCAM+ (CD44 silenced), and CD44-EpCAM- (both CD44 and EpCAM silenced) cells were used for evaluation of target specificity. First, Western blot was performed to detect gene knockdown. As shown in Figure 3D, by treatment with 100nM of CD44 siRNA and/ or EpCAM siRNA, OVCAR8 cells have significantly reduced protein levels of CD44 and/or EpCAM. Next, CD44+ EpCAM+ ( no siRNA treatment ), CD44+ ( EpCAM silenced), EpCAM+ ( CD44 silenced ), and CD44- EpCAM- ( both CD44 and EpCAM silenced) OVCAR 8 cells were stained with CD44-EpCAM-Cy5. As shown in Figure 3E, CD44+EpCAM+ OVCAR 8 cells (red lines) showed two strong fluorescence peaks and high fluorescence intensity, but after knockdown of CD44 or EpCAM, two fluorescence peaks of single positive CD44+ cells (blue line) or EpCAM+ cells (green line) significantly shift to left ( lower intensity areas), which indicate reduced the binding. Upon knockdown of both CD44 and EpCAM, CD44-EpCAM-cells (purple line) lose the binding with CD44-EpCAM by exhibiting significantly reduced fluorescence intensity with near disappearance of two strong fluorescence peaks (Figure 3E). The results demonstrated that the binding of CD44-EpCAM aptamer to cells is indeed through CD44 and EpCAM molecules.
Effect of bispecific CD44-EpCAM aptamer on ovarian cancer cell growth/survival
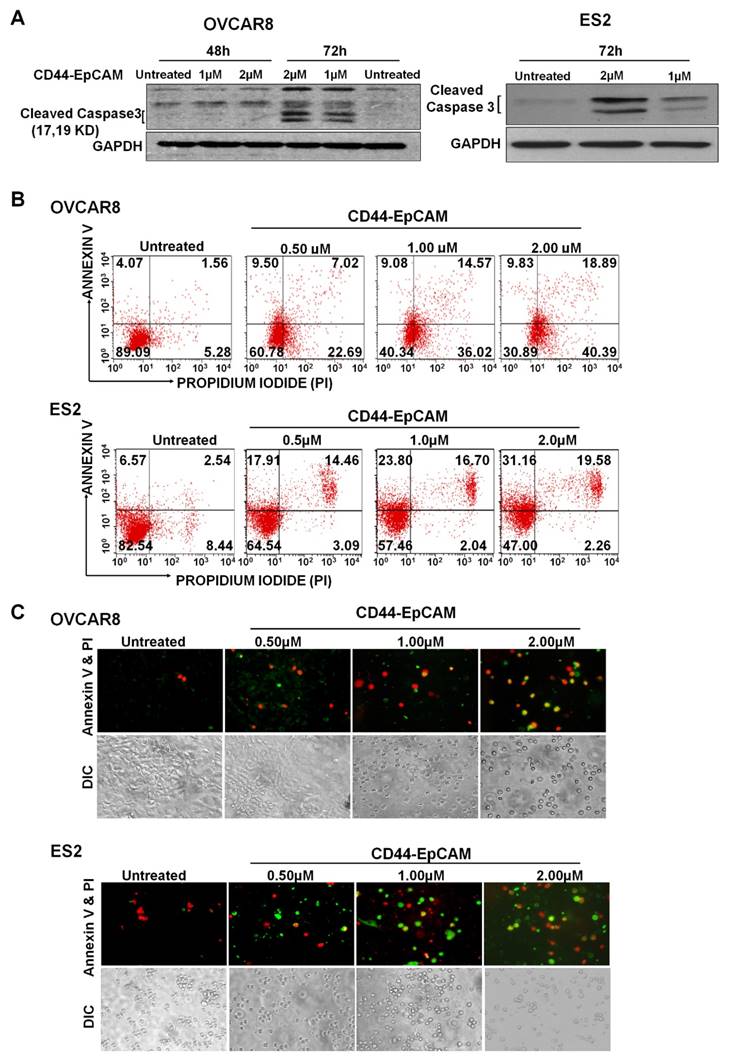
To explore if reduced cell viability by bispecific CD44-EpCAM aptamer was caused by apoptosis, we examined the level of cleaved caspase-3, an indicator of apoptosis, in cells treated with this aptamer for varying concentrations. Western blotting showed that treatment of bispecific CD44-EpCAM aptamer led to the appearance of cleaved caspase 3 (molecular weight 17Kd and 19Kd) in OVCAR8 and ES2 cells at 72h (Figure 4A). To further confirm the occurrence of apoptosis, OVCAR8 and ES2 cells treated with varying concentrations of bispecific CD44-EpCAM aptamer were subjected to Annexin V/Propidium Iodide (PI) staining-based flow cytometry. While the population of late stage apoptotic cell population (Annexin V+/PI+) was 1.56 % in control OVCAR8 cells, this population was increased to 18.89% in cells treated with 2µM bispecific CD44-EpCAM aptamer (Figure 4B). Similarly, population of late apoptotic cells was increased from 2.54% in control to 19.58% in ES2 cells treated with bispecific CD44-EpCAM aptamer. The apoptotic pattern was also demonstrated from fluorescence microscope imaging (Figure 4C). Compared with untreated controls, CD44-EpCAM treated OVCAR8 and ES2 cells have increased apoptosis signals of green (Annexin V) and red (PI) in a dose-dependent manner. These results suggest that bispecific CD44-EpCAM aptamer can effectively induce apoptosis in ovarian cancer cells.
Evaluation of Apoptosis. (A) Western blot analysis of Caspase-3 expression in OVCAR8 and ES2 cells after treatment with the varying concentrations of CD44-EpCAM. (B) Flow cytometry analysis of apoptosis after CD44-EpCAM treatment. OVCAR8 and ES2 cells were treated with different concentrations of CD44-EpCAM for 72h, and cells were stained with Alexa Fluor 488 Annexin V-Propidium Iodide and analyzed by flow cytometry. The apoptotic and dead cell population has increased after CD44-EpCAM treatment in a dose-dependent manner. Consistently (C) Fluorescence microscopy confirmed the increase of apoptotic and dead cells. The increased number of red (PI+) and green (Annexin V +) cells are visualized upon CD44-EpCAM treatment.
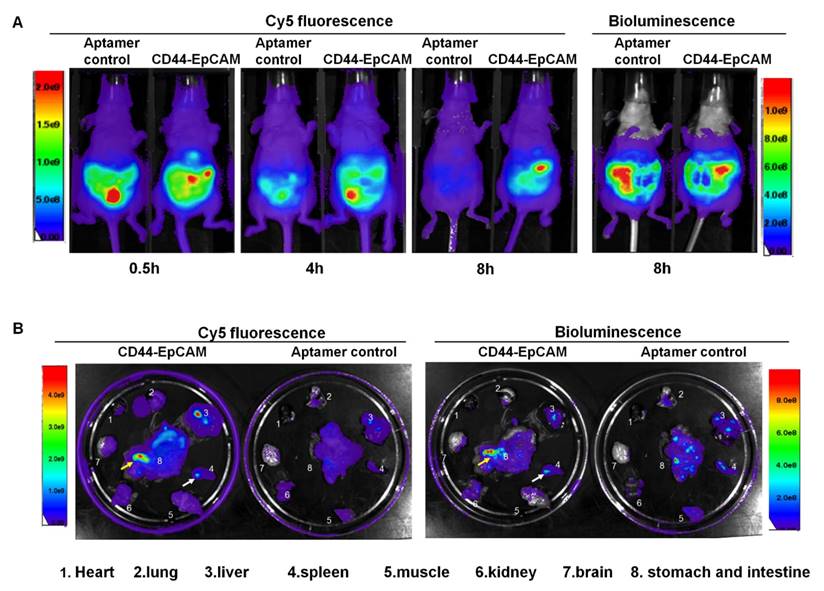
Biodistribution and tumor targeting capability
Female athymic nude mice were intraperitoneally injected with OVCAR8-Luc cells (5x106/mouse). After 8-day injection, Cy5-labeled CD44-EpCAM aptamer and Cy5-labeled non-targeting control MG aptamer were intraperitoneally injected to tumor bearing mice. At time points of 0.5h, 4h and 8h, Cy5 fluorescence of mice were detected with Xenogen IVIS100 imaging system to monitor aptamer distribution in whole body. After 8-h injection of aptamers, mice were injected with luciferin for bioluminescence imaging. Bioluminescence images were captured following Cy5 fluorescence imaging with Xenogen IVIS 100. Bioluminescence shows the distribution profile of OVCAR8 tumor cells. As shown in Figure 5A, after 4-h aptamer injection, non-targeting aptamer has shown the decreased Cy5 fluorescence compared with CD44-EpCAM, and was almost invisible post 8-h injection, while CD44-EpCAM still kept strong Cy5 fluorescence at 8h. Bioluminescence imaging of whole body demonstrated the distribution of tumor cells and confirmed that tumor cells have spread on entire peritoneum after 8-day implantation of OVCR8-luc tumor cells (Figure 5A, right). Following whole body imaging, organs were removed and ex vivo images were captured. As shown in Figure 5B, CD44-EpCAM aptamer (Cy5), but not non-targeting control aptamer, has greatly co-localized with spread tumors (bioluminescence). CD44-EpCAM aptamer was not observed in organs without tumors (brain, lung, heart). Notably, both CD44-EpCAM aptamer and control aptamer did not stagnate in kidney, which is different from nanoparticle-based materials [39, 40]. The results indicate CD44-EpCAM aptamer does not bind to tumor- free organs and has strong tumor targeting capability.
Biodistribution assay. (A) Athymic nu/nu female mice were intraperitoneally injected with luciferase-expressing OVCAR8 cells. 8-day post tumor cell implantation, mice were intraperitoneally injected with Cy5-CD44-EpCAM aptamer or non-targeting control aptamers. Cy5 fluorescence of whole body was captured at the time points of 0.5h, 4h and 8h. Bioluminescence was captured at 8h post aptamer injection. (B) Ex vivo organ imaging. Following whole-body imaging, major organs were removed to be visualized the locations of aptamers (Cy5) and tumors (bioluminescence). The arrows show the co-localization of aptamer and tumors.
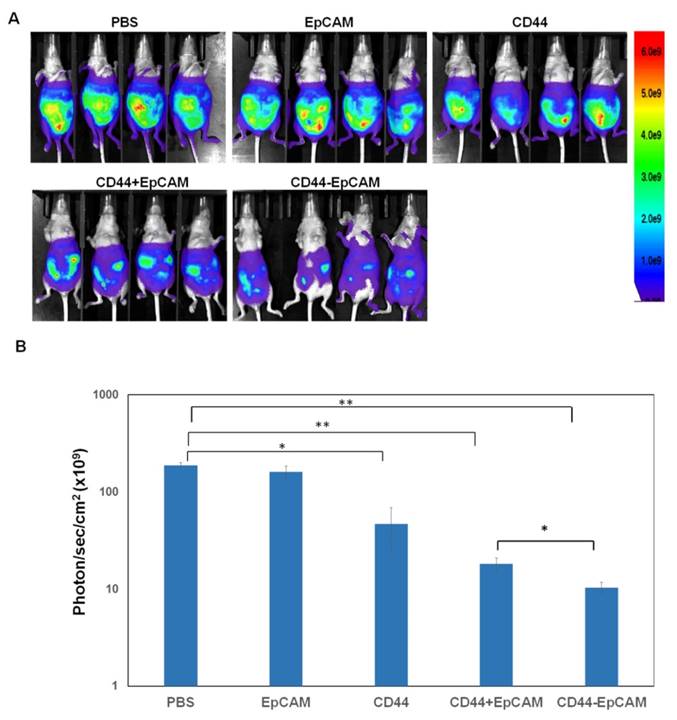
Effect of bispecific CD44-EpCAM aptamer on intraperitoneal xenograft development
The effectiveness of bispecific CD44-EpCAM aptamer to induce apoptosis led us to investigate whether this aptamer could suppress ovary tumorigenesis. Female athymic nude mice were intraperitoneally injected with OVCAR8-Luc cells (5x106/mouse). After 5 days, mice were intraperitoneally administered with PBS, single EpCAM aptamer, single CD44 aptamer, combination of single EpCAM and CD44 aptamer, and bispecific CD44-EpCAM aptamer at 2 nmoles per mouse every other day for first two weeks and every day for another two weeks. Visualized by bioluminescence imaging, we observed robust tumor grow in mice receiving PBS. Administering single EpCAM aptamer did not display treatment efficacy (Figure 6A and 6B). In contrast, single CD44 aptamer reduced tumor burden while the combined use of single CD44 and EpCAM aptamers retarded tumor growth even greater (Figure 6A and 6B). Notably, bispecific CD44-EpCAM aptamer decreased tumor growth much more significantly than either single aptamer alone or together (Figure 6A and 6B). The increased efficacy is likely due to the increased circulation life time of the larger molecular weight of the fused aptamer than the lower molecular weight of single aptamers.
CD44-EpCAM suppresses tumor peritoneal metastasis in xenograft mouse model. (A) Athymic nu/nu female mice were intraperitoneally injected with luciferase-expressing OVCAR8 cells. Treatment started 5-day post tumor cell implantation. Images by IVIS system were taken at the end of experiments. All exposure time and imaging parameter were set equally to keep results comparable. (B) Calculation of values of photo flux. The photo counts were calculated by the software installed with the instrument. (n=4, *P<0.05, **P<0.005).
To elucidate the molecular mechanism underlying bispecific CD44-EpCAM aptamer-led inhibition in intraperitoneal tumor growth, we performed H&E staining on tumor implants collected from sacrificed mice. Compared with control, bispecific CD44-EpCAM aptamer-treated tumors were highly vacuolated and contained highly condensed nucleus and cytoplasm (Figure 7A). Moreover, there were increased number of cells with body shrinkage and cells in specimens were loosely contacted with the neighboring cells in bispecific CD44-EpCAM aptamer-treated tumors. To link the observed histological alteration to the occurrence of apoptosis, we performed immunohistochemistry staining of cleaved caspase 3 and TUNEL (Terminal deoxynucleotidyl transferase dUTP nick end labeling). The staining intensity of cleaved caspase-3 and TUNEL was much greater in bispecific CD44-EpCAM aptamer-treated tumors than all other treatment groups (Figure 7A & 7B). This is apparently consistent with the observation that bispecific CD44-EpCAM aptamer was able to induce apoptosis in ovarian cancer cells (Figure 4). Moreover, we also observed that the staining intensity of proliferation marker Ki67 was greatly decreased in bispecific CD44-EpCAM aptamer-treated tumors (Figure 7A & 7B). These results suggest that bispecific CD44-EpCAM aptamer-led inhibition of intraperitoneal tumor progression results from triggering tumor cell apoptosis.
Histology analysis of tumor and detection of biomarkers associated with apoptosis and metastasis by immunohistochemistry. (A) Formalin-fixed paraffin-embedded sections of xenograft tumors were analyzed with H&E staining, IHC and TUNEL assay. Scale bar, 50µm. (B) Quantification of IHC staining with ImageJ and ImageJ plugin IHC profiler. P* <0.05, P** <0.01.
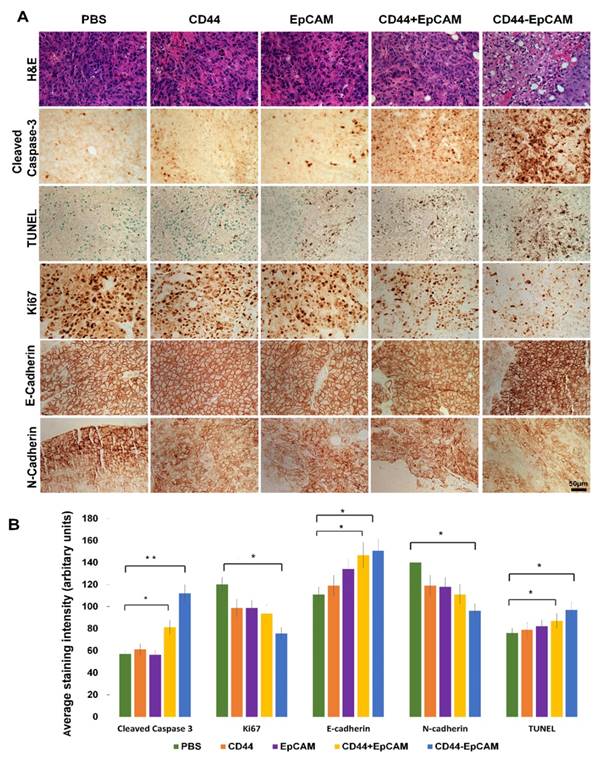
To study whether CD44-EpCAM aptamer inhibition of ovarian cancer peritoneal metastasis is also through interfering epithelial-mesenchymal transition (EMT), which contributes to early-stage migration of cancer cells and is indispensable for invasion and metastasis of cancer cells[41]. EMT is characterized by reduced E-cadherin and increased N-cadherin expression. Through IHC staining (Figure 7A & 7B), the results showed that, upon treated with CD44-EpCAM aptamer, the expression of E-Cadherin are increased and N-Cadherin are reduced. That indicates that the inhibition of metastasis of CD44-EpCAM aptamer is also though reversal of EMT in addition to inducing apoptosis.
Assessment of bispecific CD44-EpCAM aptamer toxicity
The promise of using bispecific CD44-EpCAM aptamer to suppress ovarian tumor growth led us to further assess the potential toxicity of this aptamer to the host. We collected all major organs including spleen, liver, kidney, heart, intestine and muscle from sacrificed mice and carried out histology examinations on these organs. H&E staining showed that there was no obvious histological difference among untreated, PBS- and bispecific CD44-EpCAM aptamer-treated mice (Figure 8A), suggesting that bispecific CD44-EpCAM aptamer is well tolerated by the host and can be safely used.
Assessment of toxicity. (A) Histology examination of CD44-EpCAM effect on major tissues. After treatment with CD44-EpCAM, the major tissues including spleen, lung, liver, kidney, heart, intestine and muscle were stained with H&E, and compared with PBS-treated and naïve mouse tissues. (B) Detection of potential immune response upon CD44-EpCAM treatment. Human peripheral blood mononuclear cells were treated with different concentrations of CD44-EpCAM for 24h, TNFα and IFNα in cell culture supernatants were quantified with ELISA kits. The results are the mean± SEM (N=3).
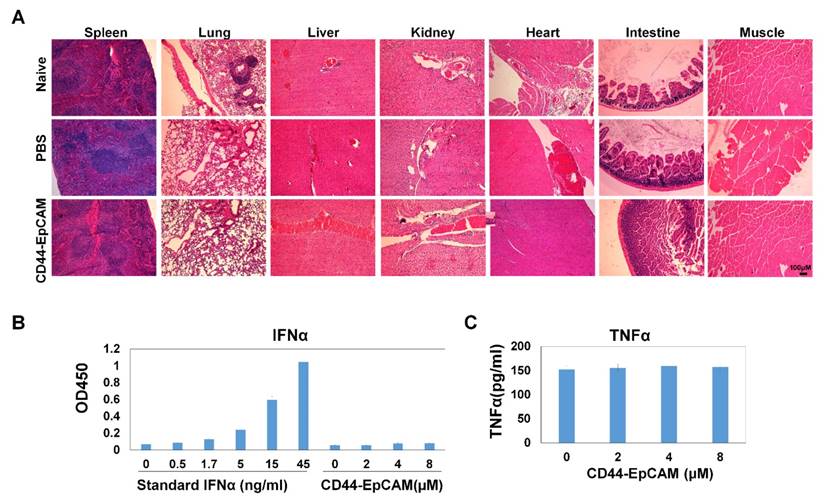
Exogenous RNA has been shown to induce innate immunogenicity-associated interferon upregulation [42]. To determine the effect of bispecific CD44-EpCAM aptamer on innate immunogenicity, we treated human peripheral mononuclear cells with bispecific CD44-EpCAM aptamer for 24 h followed by evaluating the amount of IFNα and TNFα released from these cells. ELISA showed that, at the concentration up to 8µM, there was no detectable elevation in the level of IFNα or TNFα over the untreated control (Figure 8B). Taken together, these results suggest that nucleic acid-based bispecific CD44-EpCAM aptamer has no toxicity to the host and does not trigger innate immune response.
Discussion
Intraperitoneal tumor growth represents an advanced late stage of ovarian cancer. As five-year survival rate of advanced patients remains at only 20-30%, developing effective strategy to inhibit ovary tumor development in peritoneal cavity is expected to improve survival of patients with advanced ovarian cancer. There are extensive evidences demonstrating CD44 as a critical molecule involved in ovary tumor development in peritoneal cavity. The observations that CD44-targeted antibodies and siRNAs were able to inhibit the formation of peritoneal implantation of ovarian cancer [18, 43] implicate that CD44 may be a promising ovarian cancer therapeutic target. Meanwhile, EpCAM, a molecule that mediates cell-to-cell contact, has also been shown to actively participate in ovary tumor progression and metastasis as targeting EpCAM was found to diminish the number of ovarian cancer cells in the peritoneal cavity[8, 10]. Interestingly, both CD44 and EpCAM are key cell surface markers of ovarian cancer stem cells which are recognized to be responsible for peritoneal metastatic dissemination and chemoresistance [44]. Co-expression of CD44 and EpCAM on ovarian cancer cells highlights the potential of co-targeting both molecules with a bispecific molecule to better fight ovarian cancer.
Most commonly used bispecific molecules are bivalent antibodies which has the capability to target two molecules simultaneously. However, high cost and immunogenicity of such antibodies have significantly impeded their broad application in disease treatment. Recent advance on aptamer development demonstrate clear advantages of aptamers over antibodies on the aspects of low immunogenicity, ease of production and modification [31]. Unfortunately, wide use of nucleic acid-based aptamers in clinical setting is limited by their short half-life in circulation due to small sizes and being susceptible to nuclease attack. Several attempts have been made to overcome these limitations. For example, PEG is conjugated to aptamer to increase the size of aptamer [45]. Aptamers are chemically modified with 2'-amino pyrimidines [46], 2-fluoro pyrimidines [47] or 2'-O-methyl ribose [45] to resist nuclease-mediated degradation. We previously showed that chemically modifying RNA-based aptamer with 2' F-pyrimidine rendered aptamers nuclease resistant [32, 48]. In this study, we adopted a new means to increase the size of aptamer by fusing two single aptamers via a double stranded complementary RNA adaptor and 2-3 unpaired bases between aptamer and adaptor, which render each single aptamer sufficient flexibility to bind its own target. It is clear that bispecific CD44-EpCAM aptamer effectively binds CD44 and EpCAM at the same time (Figure 1 and Figure 3E). A recent study reported that EpCAM aptamer-survivin siRNA chimera (about 20Kd) is ineffective for tumor-suppressing due to short blood circulation time. By conjugating a 20Kd PEG molecule onto the chimera, modified chimera was found to have significantly increased serum retention time and exhibited potent anti-tumor capability [49]. As our generated bispecific CD44-EpCAM aptamer has a molecular weight of 54.4Kd which is well above the molecular mass cutoff of 30-50 Kd for renal glomerulus, we expect that this aptamer possesses much improved half-life in circulation than either single CD44 or EpCAM aptamer. Meanwhile, significantly smaller size of CD44-EpCAM bispecific aptamer than antibody (55 vs 150Kd) also indicate that this aptamer will have much better tissue penetration than antibody.
Both CD44 and EpCAM are well recognized to contribute to ovarian cancer chemoresistance [50, 51]. They both are thus expected to be potential therapeutic targets. We envision that targeting both simultaneously will offer enhanced therapeutic efficacy. Our idea is apparently supported by our findings that bispecific CD44-EpCAM aptamer exhibits much better tumor-suppressing effect than either single aptamer (Figure 2 and 4). As expected that aptamers are generally not immunogenic and less toxic, we found that bispecific CD44-EpCAM aptamer was well tolerated by the host and triggered no innate immune responses (Figure 8). In conclusion, our study suggests the CD44-EpCAM bispecific aptamer represents a potent therapeutic agent against advanced ovarian cancer.
Materials and Methods
Materials
Cell culture media were purchased from Life technologies Corporation (Carlsbad, CA). Fetal bovine sera and Taq RNA polymerase were from Sigma-Aldrich (St Louis, MO). Antibodies were from Cell Signaling Technology (Danvers, MA). DuraScribe T7 Transcription kits were from Epicentre (Madison, WI). Recombinant human CD44 and EpCAM proteins were from Thermo Fisher Scientific. Alexa Fluor 488 Annexin V/Dead Cell apoptosis kits were from Invitrogen. ELISA Kits for detection of IFNα and TNFα were obtained from RayBiotech (Norcross, GA). TUNEL assay kits were purchased from R&D systems (Minneapolis, MN). CD44 siRNA and EpCAM siRNA were purchased from Life Technologies Corporation.
Cell lines
Cell lines including OVCAR8, SKOV3, OCC1, ES2 and HEK293 cells were purchased from the American Type Culture Collection (ATCC, Manassas, VA). Luciferase stable OVCAR8 cell line was developed by our laboratory. OVCAR8 cells were transfected with PGL4.51[luc2/CMV/neo] plasmid (Promega) by Lipofectamine 3000 reagent (Invitrogen) following the manufacture procedures. After 72 h transfection, cells were selected by adding 400µg/ml G418 antibiotic in DMEM containing 10% fetal bovine serum. After 1-month culture, luciferase activity in transfected cells was confirmed with One-Step Glow Assay kit (Fisher Scientific).
CD44-EpCAM construction
CD44 and EpCAM aptamers were individually synthesized by in vitro transcription with PCR products as templates. The CD44 aptamer ssDNA (5'-GGGATGGATCCAAGCTTACTGGCATCTGGATTTGCGCGTGCCAGAATAAAGAGTATAACGTGTGAATGGGAAGCTTCGATAGGAATTCGG) was synthesized from IDT as a PCR template. PCR was performed with forward primer (5'-TAATACGACTCACTATAGGGATGGATCCAAGCTTACT-3') and reverse primer (5'-AATTTCATCTCC TGAACAAGC TTTTCCGAAT-3'). Forward primer contains T7 RNA polymerase promoter binding sequence which is underlined. Reverse primer contains adaptor sequence which is bolded. The ssDNA of EpCAM aptamer containing T7 RNA polymerase promoter site (underlined) and adaptor sequence (bolded) (5'- TAATACGACTCACTATGCGACTGGTTACCCGGTCGTAAAA TTTCATCTCCTGAACAAGCTT) was synthesized from IDT as a PCR template. PCR was performed with forward primer (5'-TAATACGACT CACTATAGCGACTGGTTA-3) and reverse primer (5'-AAGCTTGTTCAGGAGATGAAATTTTACGA-3'). The PCR products were put into T-A cloning pCR 2.1 vector (Invitrogen) and sequenced. Transcription was performed with PCR product as templates using DuraScript transcription kits following manufacture's instruction. Two RNAs were mixed at molar ratio 1:1 and annealed to form one bispecific CD44-EpCAM molecule by heated at 94°C for 3 min followed by slowly cooling to room temperature within 1h.
Construction of controls without unpaired base linkers between aptamer and adaptor
For construction of CD44-EpCAM No linker control-1, the ssDNA of EpCAM aptamer is synthesized by IDT as PCR template with sequence of TAATACGACTCACTATGCGACTGGTTACCCGGTCGTAAAATTTCA TCTCCTGA ACAA G CTTTT, which has two more “T” (underlined) at the 5'-termini compared with linker-containing EpCAM aptamer. For construction of CD44-EpCAM No linker control-2, the sequence of the ssDNA of EpCAM aptamer is TAATACGACTCACTATGCGACTGGTTACCCGGTCG (TAA) AATTTCATCTCCTGAACAAGCTTTT, which has been removed three base TAA (bolded) and added two more “T”s at the 5-termini of EpCAM aptamer compared with above linker-containing EpCAM aptamer. PCRs of two constructs were performed with forward primer (5'-TAATACGACT CACTATAGCGACTGGTTA-3) and reverse primer (5'-AAAAGCTTGTTCAGGAGATGAAATT-3') with each ssDNA template. Transcription was performed with PCR products as templates. EpCAM aptamers without linkers were individually annealed with CD44 aptamer to generate no linker controls: CD44-EpCAM No linker-1 without unpaired bases between CD44 aptamer and double stranded adaptor, and CD44-EpCAM No linker-2 without any unpaired bases between aptamers (CD44 or EpCAM) and double stranded adaptor.
Characterization of dual binding functionality of CD44-EpCAM
Serially diluted recombinant human CD44 protein (0-500 nM) was loaded into 96-well microtiter plates at 50μg/well in triplicates and incubated overnight at 4°C. Plates were further blocked with 5% BSA/ sperm DNA (500 µg/ml)/ yeast tRNA (500µg/ml)/0.05% Tween-20 in PBS/T buffer overnight. CD44-EpCAM conjugates or simply mixed CD44 and EpCAM aptamers were incubated with 1μM recombinant human EpCAM for 4h at room temperature. For simply mixed CD44 and EpCAM, prior putting CD44 and EpCAM aptamers together, CD44 aptamer was blocked with ssDNA (5'-AATTTCATCTCCTGAACAAGCTT-3'). After incubation, the mixtures of CD44-EpCAM aptamer and EpCAM protein, simply mixed CD44 aptamer plus EpCAM aptamer and EpCAM protein were added into CD44-immobilized plates and incubated at 4 °C for 24h. After washing with 1xPBS/0.05% Tween-20 for 4 times, 100μl anti-EpCAM antibody at 1:1000 dilution was added and incubated for 4 h at room temperature. After washing 4 times, 100μl HRP-secondary antibody (1:5000) was added and incubated for 2h at room temperature. Detection of HRP was performed by incubating with 100μL/well soluble Turbo TMB‐ELISA substrate for 5 min, then reaction was quenched with 100μl/well Stop solution and the absorbance at 450nm was measured using TECAN Infinite M 200 plate reader.
Serum stability assay
2'-fluoro-modified and unmodified CD44-EpCAM (1nmol) were incubated with final 50% human serum at 37 °C for 2h, 6h, and 24h. RNA integrity was detected with 1% agarose gel electrophoresis in 1xTBE buffer. Aptamer intensity was measured with ImageJ (NIH).
Knockdown of CD44 and /or EpCAM
OVCAR8 cells were plated in 6-well plates at a density of 5 × 10 5 cells/well for 24 h. CD44 siRNA and / or EpCAM siRNA were transfected into cells using Lipofectamine.
RNAi MAX (Life Technologies) according to the manufacturer's instruction. Cells were harvested 72 h post-transfection and Western blot was performed to confirm gene knockdown. CD44 and /or EpCAM silenced cells were subjected to aptamer binding assay by BD FACSCalibur flow cytometry.
CD44-EpCAM aptamer binding specificity
OVCAR8, SKOV3, ES2, OCC1 and HEK293T cells were collected and washed with DPBS. OVCAR8 cells with CD44+ (EpCAM silenced), EpCAM+ (CD44 silenced) and CD44- EpCAM- (both CD44 and EpCAM silenced) were collected after 72h siRNA transfection. 3'-Cy5 labeled and 2'-fluoro-modified EpCAM (with adaptor) and MG aptamer were synthesized from TriLink. Cy5-labeled CD44-EpCAM was generated by annealing Cy5-EpCAM and CD44 aptamer through adaptor complementing. After washing, cells were incubated with Cy5-labeled CD44-EpCAM (1 µM) or Cy5-labeled MG aptamer (1µM) in 1xTBS buffer containing 5% BSA, yeast tRNA (500 μ g/ml), sperm DNA (500µg/ml), and 0.05% Tween-20 for 1 h at 37 °C incubator. Cell binding was detected using BD FACSCalibur flow cytometry.
Western blot
Cells were lysed in lysis buffer (M-PER Mammalian Protein Extraction Reagent, Thermo Fisher Scientific) containing 1x Halt Protease Inhibitor Cocktails. After 2-h lysis and centrifuged at 12,000 × g for 10 min at 4 °C, cell supernatant was collected and the protein concentration was determined with Bio-Rad Protein Assay (Bio-Rad, Hercules, CA). Protein (100 μg) was mixed with 2x Laemmli sample buffer containing 5% β-mercaptoethanol and heated at 95 °C for 10 min. Denatured samples were separated on 10% SDS-polyacrylamide gel and transferred to PVDF membrane. The membranes were blocked with 5% non-fat milk overnight at 4 °C, and then incubated with primary antibodies for 2 h at 4 ºC overnight, followed by incubation with horseradish peroxidase-conjugated secondary antibodies for 2 h at room temperature. After ECL Western Blotting Substrate (Pierce) was added onto membrane, the signals were captured by the exposure to X-ray film.
Cell viability assay
Proliferation and cytotoxicity of CD44-EpCAM was quantified by measuring WST-8 formazan using Cell Counting Kit-8 (CCK-8) (Dojindo, Japan). Cells in Dulbecco's Modified Eagle Medium containing 10% fetal bovine serum were seeded in 96-well plate at a density of 1-5 x103 in 5% CO2 incubator for 24h at 37°C. Cell lines of OVCAR8, SKOV3, OCC1, ES2 and HEK293T were incubated with the varying concentrations of aptamer constructs (CD44, EpCAM, CD44 plus EpCAM and CD44-EpCAM conjugate) for 72 hours. MG aptamer (specific to Malachite Green) (5'-GGAUCCCGACUGGCGAGAGCCAGGUAACGAAU GGAUCC-3') was used as an aptamer control. Absorbance was measured at 450nm using a spectrophotometer.
Assessment of apoptosis and cell death by flow cytometry and fluorescent microscopy
OVCAR8 cells were treated with different concentrations of CD44-EpCAM for 48h. The cells were harvested and stained with Alexa Fluor 488 Annexin V-Propidium Iodide (PI) solution for 15min at room temperature. Apoptosis was detected by flow cytometry and fluorescent microscope. A portion of collected cells (1x104/sample) were acquired by BD FACSCalibur and analyzed using BD FACStation software, and another portion of collected cells were imaged with fluorescence microscopy (Nikon Eclipse TE2000-S). Each channel signals were separately captured and merged with ImageJ Plugin for co-localization.
Peritoneal metastatic assay
All animal studies were approved by the Institutional Animal Care and Use Committee at Augusta University. Athymic nu/nu female mice were purchased from Harlan Laboratories. The methods were carried out in accordance with the approved guidelines. Peritoneal metastatic colonization assays were performed as previously described [52, 53]. OVCAR8-luc cells (5x106) in the log phase were intraperitoneally injected into the mice. 5-day after implantation, mice were treated with PBS, CD44 aptamer, EpCAM aptamer, mixed CD44 and EpCAM aptamers, and CD44-EpCAM conjugate at 2 nmole per mouse every other day in the start two weeks and every day in the last two weeks. After 1-month treatment, luciferin was intraperitoneally injected to mice at 150mg/kg. Before imaging, mice were anesthetized with isoflurane, bioluminescence imaging was performed with a Xenogen IVIS100 imaging system (Xenogen). Overall peritoneal metastasis was measured by selecting a region of interest (ROI) around the tumor sites of each mouse and quantified total flux using Living Image Software AMIVIEW 1.7.02 (Xenogen) with the units of photons/s/cm2/sr.
Biodistribution assay
OVCAR8-luc cells (5x106) in the log phase were intraperitoneally injected into the mice. After 8-day implantation, mice were intraperitoneally injected with Cy5-labeled CD44-EpCAM aptamer (15nmles) or equal mole amount of Cy5-labeled MG aptamer ( non-targeting control).The whole-body images were obtained at 0.5h, 4h and 8h using Xenogen IVIS100 imaging system by setting wavelength at excitation 640nm and emission 710nm. After 8-h injection of Cy5-labeled aptamers, mice were intraperitoneally treated with luciferin at 150mg/kg. Bioluminescence imaging was followed after Cy5 fluorescence imaging using Xenogen IVIS100. Mice were euthanized with CO2 after whole-body imaging and organs (heart, lung, liver, spleen, kidney, muscle, brain, stomach, and intestine) were removed. The ex vivo images of Cy5 (aptamer) and bioluminescence (tumor cells) were captured at the same time using Xenogen IVIS100.
Histology assay
Mice were euthanized with CO2, and tumors and organs (spleen, lung, kidney, intestine, heart, liver and muscle) were removed and fixed with 4% paraformaldehyde. Sections (6µm) were cut and mounted on the slides, and deparaffinized in xylene and ethyl alcohol. Each block has a section for H&E staining. For immunohistochemistry assay, sections were incubated in 3% normal goat serum for 2 h and incubated with primary antibodies: caspase-3 (1:20), Ki67 (1:100), E-cadherin (1:100) and N-cadherin (1:100). After washing, the sections were incubated with biotinylated secondary antibody (1:200, VECTOR, Burlingame, CA) for 1 hour. Following washing, the sections were incubated with VECTASTAIN ABC reagents for 30 min. The images were captured with Nuance fluorescence microscope with bright field imaging system. TUNEL assay was performed according to the manufacturer's instruction. Paraffin embedded tissues were sectioned, dewaxed, hydrated and digested with Proteinase K. After washing, slides were immersed into quenching solution for 5 min, and then incubated with TdT labeling buffer. After incubated with streptavidin-HRP for 10 min, following washing, DAB work solution was added into the slides. Then slides were counterstained with Methyl Green. The images were captured with Nuance fluorescence microscope.
Quantification of IHC staining
ImageJ and ImageJ plugin IHC profiler were applied for measurement. Images were changed to 8-bit grayscale type and inverted under “Edit” menu of ImageJ. After invert, the DAB stained areas are bright, and unstained areas are dark. The mean intensity was measured using “Measure” function under the “Analyze” menu of ImageJ. 8-10 fields of each treatment group were assessed. For TUNEL staining, before images were changed to greyscale, IHC profiler was used to do color deconvolution, by which DAB brown stain was separated from Methyl Green counterstain and then followed the same analysis as the above.
ELISA
Normal human peripheral blood mononuclear cells (PBMCs) were separated with BD Vacutainer cell preparation tubes with sodium citrate. Cells were seeded into 24-well plates at 106/well for 24 h in RPMI medium containing 10% fetal bovine serum. CD44-EpCAM with the varying concentrations was added into cells, and cells were incubated for 24 h in 5% CO2 incubator at 37 °C. The cell culture supernatant was detected with human IFNα and TNFα ELISA kits following the manufacture's instruction.
Statistical analysis
The Data were analyzed using two-tailed Student's t-test (Graph Pad Prism) by comparing with the control group, and expressed as a mean ± SEM. The differences of P < 0.05 was considered statistically significant.
Supplementary Material
Supplementary figures.
Acknowledgements
This work is supported by start-up funding of Augusta University and Department of Defense W81XWH-15-1-0333 (to H.Y.L.). H.Y.L. thanks the NIH for an F32 fellowship (CA150301)
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bowtell DD, Bohm S, Ahmed AA. et al. Rethinking ovarian cancer II: reducing mortality from high-grade serous ovarian cancer. Nat Rev Cancer. 2015;15:668-79
2. Jayson GC, Kohn EC, Kitchener HC. et al. Ovarian cancer. Lancet. 2014;384:1376-88
3. Kim A, Ueda Y, Naka T. et al. Therapeutic strategies in epithelial ovarian cancer. J Exp Clin Cancer Res. 2012;31:14
4. Tsikouras P, Tsagias N, Pinidis P, et al.The contribution of catumaxomab in the treatment of malignant ascites in patients with ovarian cancer. a review of the literature. Arch Gynecol Obstet. 2013;288:581-5
5. Balzar M, Winter MJ, de Boer CJ. et al. The biology of the 17-1A antigen (Ep-CAM). J Mol Med (Berl). 1999;77:699-712
6. Reichert JM, Valge-Archer VE. Development trends for monoclonal antibody cancer therapeutics. Nat Rev Drug Discov. 2007;6:349-56
7. Baeuerle PA, Gires O. EpCAM (CD326) finding its role in cancer. Br J Cancer. 2007;96:417-23
8. Imrich S, Hachmeister M, Gires O. EpCAM and its potential role in tumor-initiating cells. Cell Adh Migr. 2012;6:30-8
9. Nunna S, Reinhardt R, Ragozin S. et al. Targeted methylation of the epithelial cell adhesion molecule (EpCAM) promoter to silence its expression in ovarian cancer cells. PLoS One. 2014;9:e87703
10. Xiang W, Wimberger P, Dreier T. et al. Cytotoxic activity of novel human monoclonal antibody MT201 against primary ovarian tumor cells. J Cancer Res Clin Oncol. 2003;129:341-8
11. Burges A, Wimberger P, Kumper C. et al. Effective relief of malignant ascites in patients with advanced ovarian cancer by a trifunctional anti-EpCAM x anti-CD3 antibody: a phase I/II study. Clin Cancer Res. 2007;13:3899-905
12. Wang H, Tan M, Zhang S. et al. Expression and significance of CD44, CD47 and c-met in ovarian clear cell carcinoma. Int J Mol Sci. 2015;16:3391-404
13. Orian-Rousseau V. CD44 acts as a signaling platform controlling tumor progression and metastasis. Frontiers in Immunology. 2015:6
14. Gardner MJ, Catterall JB, Jones LM. et al. Human ovarian tumour cells can bind hyaluronic acid via membrane CD44: a possible step in peritoneal metastasis. Clin Exp Metastasis. 1996;14:325-34
15. Zhang J, Chang B, Liu JS. CD44 standard form expression is correlated with high-grade and advanced-stage ovarian carcinoma but not prognosis. Human Pathology. 2013;44:1882-1889
16. Sacks JD, Barbolina MV. Expression and Function of CD44 in Epithelial Ovarian Carcinoma. Biomolecules. 2015;5:3051-66
17. Strobel T, Swanson L, Cannistra SA. In vivo inhibition of CD44 limits intra-abdominal spread of a human ovarian cancer xenograft in nude mice: a novel role for CD44 in the process of peritoneal implantation. Cancer Res. 1997;57:1228-32
18. Zou L, Yi T, Song X. et al. Efficient inhibition of intraperitoneal human ovarian cancer growth by short hairpin RNA targeting CD44. Neoplasma. 2014;61:274-82
19. Wu C, Ying H, Grinnell C. et al. Simultaneous targeting of multiple disease mediators by a dual-variable-domain immunoglobulin. Nat Biotechnol. 2007;25:1290-7
20. van der Veeken J, Oliveira S, Schiffelers RM. et al. Crosstalk Between Epidermal Growth Factor Receptor- and Insulin-Like Growth Factor-1 Receptor Signaling: Implications for Cancer Therapy. Current Cancer Drug Targets. 2009;9:748-760
21. Kontermann RE. Dual targeting strategies with bispecific antibodies. Mabs. 2012;4:182-197
22. Lindhofer H, Mocikat R, Steipe B. et al. Preferential species-restricted heavy/light chain pairing in rat/mouse quadromas. Implications for a single-step purification of bispecific antibodies. J Immunol. 1995;155:219-25
23. Mack M, Riethmuller G, Kufer P. A small bispecific antibody construct expressed as a functional single-chain molecule with high tumor cell cytotoxicity. Proc Natl Acad Sci U S A. 1995;92:7021-5
24. Sun H, Zhu X, Lu PY. et al. Oligonucleotide aptamers: new tools for targeted cancer therapy. Mol Ther Nucleic Acids. 2014;3:e182
25. Ellington AD, Szostak JW. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990;346:818-22
26. Bock LC, Griffin LC, Latham JA. et al. Selection of single-stranded DNA molecules that bind and inhibit human thrombin. Nature. 1992;355:564-6
27. Dassie JP, Giangrande PH. Current progress on aptamer-targeted oligonucleotide therapeutics. Ther Deliv. 2013;4:1527-46
28. Zhou J, Bobbin ML, Burnett JC. Current progress of RNA aptamer-based therapeutics. Front Genet. 2012;3:234
29. Dassie JP, Liu XY, Thomas GS. et al. Systemic administration of optimized aptamer-siRNA chimeras promotes regression of PSMA-expressing tumors. Nat Biotechnol. 2009;27:839-49
30. Berezhnoy A, Castro I, Levay A. et al. Aptamer-targeted inhibition of mTOR in T cells enhances antitumor immunity. J Clin Invest. 2014;124:188-97
31. Keefe AD, Pai S, Ellington A. Aptamers as therapeutics. Nat Rev Drug Discov. 2010;9:537-50
32. Liu HY, Yu X, Liu H. et al. Co-targeting EGFR and survivin with a bivalent aptamer-dual siRNA chimera effectively suppresses prostate cancer. Sci Rep. 2016;6:30346. doi: 10.1038/srep30346
33. Wong TY, Liew G, Mitchell P. Clinical update: new treatments for age-related macular degeneration. Lancet. 2007;370:204-6
34. Shigdar S, Lin J, Yu Y. et al. RNA aptamer against a cancer stem cell marker epithelial cell adhesion molecule. Cancer Sci. 2011;102:991-8
35. Wang T, Gantier MP, Xiang D. et al. EpCAM Aptamer-mediated Survivin Silencing Sensitized Cancer Stem Cells to Doxorubicin in a Breast Cancer Model. Theranostics. 2015;5:1456-72
36. Ababneh N, Alshaer W, Allozi O. et al. In vitro selection of modified RNA aptamers against CD44 cancer stem cell marker. Nucleic Acid Ther. 2013;23:401-7
37. Alshaer W, Hillaireau H, Vergnaud J. et al. Functionalizing Liposomes with anti-CD44 Aptamer for Selective Targeting of Cancer Cells. Bioconjug Chem. 2015;26:1307-13
38. Healy JM, Lewis SD, Kurz M. et al. Pharmacokinetics and biodistribution of novel aptamer compositions. Pharm Res. 2004;21:2234-46
39. Liu L, Liu X, Xu Q. et al. Self-assembled nanoparticles based on the c(RGDfk) peptide for the delivery of siRNA targeting the VEGFR2 gene for tumor therapy. Int J Nanomedicine. 2014;9:3509-26
40. Huang Y, Wang X, Huang W. et al. Systemic Administration of siRNA via cRGD-containing Peptide. Sci Rep. 2015;5:12458. doi: 10.1038/srep12458
41. Zheng XF, Carstens JL, Kim J. et al. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature. 2015;527:525-30
42. Kim DH, Longo M, Han Y. et al. Interferon induction by siRNAs and ssRNAs synthesized by phage polymerase. Nat Biotechnol. 2004;22:321-5
43. Lessan K, Aguiar DJ, Oegema T. et al. CD44 and beta1 integrin mediate ovarian carcinoma cell adhesion to peritoneal mesothelial cells. Am J Pathol. 1999;154:1525-37
44. Zhan QL, Wang CM, Ngai S. Ovarian Cancer Stem Cells: A New Target for Cancer Therapy. Biomed Res Int. 2013 ArticleID916819
45. Burmeister PE, Lewis SD, Silva RF. et al. Direct in vitro selection of a 2'-O-methyl aptamer to VEGF. Chem Biol. 2005;12:25-33
46. Lin Y, Nieuwlandt D, Magallanez A. et al. High-affinity and specific recognition of human thyroid stimulating hormone (hTSH) by in vitro-selected 2'-amino-modified RNA. Nucleic Acids Res. 1996;24:3407-14
47. Rusconi CP, Scardino E, Layzer J. et al. RNA aptamers as reversible antagonists of coagulation factor IXa. Nature. 2002;419:90-94
48. Liu HY, Gao X. A Universal Protein Tag for Delivery of SiRNA-Aptamer Chimeras. Sci. Rep. 2013:3 doi: 10.1038/srep03129
49. Wang T, Gantier MP, Xiang DX. et al. EpCAM Aptamer-mediated Survivin Silencing Sensitized Cancer Stem Cells to Doxorubicin in a Breast Cancer Model. Theranostics. 2015;5:1456-1472
50. Gao Y, Foster R, Yang X. et al. Up-regulation of CD44 in the development of metastasis, recurrence and drug resistance of ovarian cancer. Oncotarget. 2015;6:9313-26
51. Chen Y, Yu DK, Zhang H. et al. CD133(+)EpCAM(+) Phenotype Possesses More Characteristics of Tumor Initiating Cells in Hepatocellular Carcinoma Huh7 Cells. Int J Biol Sci. 2012;8:992-1004
52. Yamada SD, Hickson JA, Hrobowski Y. et al. Mitogen-activated protein kinase kinase 4 (MKK4) acts as a metastasis suppressor gene in human ovarian carcinoma. Cancer Res. 2002;62:6717-23
53. Yang L, Fang D, Chen H. et al. Cyclin-dependent kinase 2 is an ideal target for ovary tumors with elevated cyclin E1 expression. Oncotarget. 2015;6:20801-12
Author contact
![]() Corresponding author: H.Y.L (Phone: 706-721-7149; Fax: 706-721-3688; email: HOLIUedu)
Corresponding author: H.Y.L (Phone: 706-721-7149; Fax: 706-721-3688; email: HOLIUedu)