Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(5):1346-1359. doi:10.7150/thno.18804 This issue Cite
Research Paper
Downregulation of SIRT7 by 5-fluorouracil induces radiosensitivity in human colorectal cancer
Ming Tang1*, Xiaopeng Lu1*, Chaohua Zhang1, 2, Changzheng Du1, 3, Linlin Cao1, Tianyun Hou1, Zhiming Li1, 2, Bo Tu1, Ziyang Cao1, 2, Yinglu Li1, 2, Yongcan Chen1, 2, Lu Jiang1, Hui Wang1, Lina Wang1, Baohua Liu2, Xingzhi Xu2, Jianyuan Luo4, Jiadong Wang5, Jin Gu3, Haiying Wang1 ![]() , Wei-Guo Zhu1, 2, 6
, Wei-Guo Zhu1, 2, 6 ![]()
1. Key Laboratory of Carcinogenesis and Translational Research (Ministry of Education), Beijing Key Laboratory of Protein Posttranslational Modifications and Cell Function; Department of Biochemistry and Molecular Biology, School of Basic Medical Sciences, Peking University Health Science Center, Beijing 100191, China;
2. Department of Biochemistry and Molecular Biology, School of Medicine, Shenzhen University, Shenzhen 518060, China;
3. Department of Colorectal Surgery, Peking University Cancer Hospital & Institute, Beijing 100142, China;
4. Department of Medical Genetics, Peking University Health Science Center, Beijing 100191, China;
5. Institute of Systems Biomedicine, Department of Radiation Medicine, School of Basic Medical Sciences, Peking University, Beijing 100191, China;
6. Peking University-Tsinghua University Center for Life Sciences, Beijing 100871, China.
* These authors are co-first authors
Received 2016-12-18; Accepted 2017-2-23; Published 2017-3-22
Abstract

5-Fluorouracil (5-FU) combined with radiotherapy is a common treatment strategy to treat human cancers, but the underlying mechanisms of this combination treatment remain unclear. Here, we report that NAD+-dependent deacetylase sirtuin-7 (SIRT7) protein levels were decreased due to 5-FU exposure rendering colorectal cancer cells sensitive to radiation. We found that SIRT7 downregulation was mediated via a Tat-binding Protein 1 (TBP1) proteasome-dependent pathway. Specifically, TBP1 was dephosphorylated at tyrosine 381 upon 5-FU treatment, which enhanced its direct interaction with SIRT7 and targeted it for degradation. Depletion of SIRT7 in cultured colorectal cancer cells induced radiosensitivity triggering cell death. Interestingly, decreased levels of SIRT7 mediated by 5-FU correlated well with improved therapeutic effect in patients with rectal cancer and with inhibited tumor growth in immune-compromised mice post-irradiation. Taken together, these data suggest that 5-FU induces radiosensitivity via SIRT7 degradation to favor a cell death pathway in targeted cancer cells. Thus, downregulation of SIRT7 could be a promising pharmacologic strategy to increase the effectiveness of chemoradiation therapy in cancer patients.
Keywords: SIRT7, TBP1, 5-FU, phosphorylation, therapeutic effect.
Introduction
The combination of chemotherapy with radiation (chemoradiation) is one of the most effective strategies to treat human cancers (1-3). 5-Fluorouracil (5-FU) and radiation is a standard treatment regimen for colorectal cancers (4, 5). The synergistic effects of 5-FU and radiation observed in vitro have driven the development of successful regimens in the clinic for the treatment of several human tumors, such as colorectal, pancreatic and biliary cancers (6-8). Several studies have aimed to identify how 5-FU induces radiosensitivity in cancer cells (6, 9-11), but the underlying mechanisms are still not fully understood.
Sirtuins are mammalian homologs of the yeast protein Sir2 (12) and have NAD+-dependent class III histone deacetylase activity. Sirtuins are conserved from bacteria to humans and perform diverse cellular and biological functions based on their subcellular localization and catalytic activity (12-14). Mammals possess seven sirtuins (SIRT1 - SIRT7); SIRT1, SIRT6, and SIRT7 primarily localize to the nucleus and likely contribute to the modulation of chromatin and the DNA damage response pathway. SIRT1 regulates cellular resistance to genotoxic stress through its interactions with Ku70 protein (15) and FOXO3 (16). SIRT6 participates in DNA damage repair and maintenance of genome integrity by modulating the activity of poly [adenosine diphosphate (ADP)-ribose] polymerase 1 (PARP1) (17) and by mediating base excision repair (18). SIRT6 also participates in the homologous recombination (HR) repair pathway by deacetylating CtIP (19), and in the non-homologous end joining (NHEJ) repair pathway by recruiting DNA-dependent protein kinase catalytic subunits (DNA-PKcs) to sites of DNA damage (20). Recent data have shown SIRT7-deficient cells exhibit impaired NHEJ due to H3K18 acetylation, which disrupts p53-binding protein 1 (53BP1) recruitment to DNA double-strand breaks (DSBs) (21). Finally, SIRT7 is recruited to sites of DNA damage by PARP1 and promotes chromatin condensation and DNA damage repair in human cancer cell lines (22). These studies demonstrate the importance of nuclear sirtuins in mediating the cellular response to DNA damage.
The sensitivity of radiation-induced cells has been a research focus in the field of radiation biology and oncology. Accumulating evidence suggests that the radiosensitivity of the tumor cells is a major determinant of tumor response to radiation (23, 24). Decreased SIRT1 protein levels result in enhanced radiosensitivity of hypoxic cells compared to normoxic cells due to increased c-Myc acetylation and expression (25). Furthermore, the deficiency of SIRT6 sensitizes Mouse Embryo Fibroblasts (MEF) and embryonic stem cells to radiation and inhibits their proliferation (18). Recently, evidence has indicated that SIRT7 deficient cells show increased susceptibility to externally induced DNA damage (26).
In this study, we investigated whether 5-FU enhances cellular radiosensitivity by affecting the function of nuclear sirtuins. 5-FU is known to interfere with nucleotide metabolism through its incorporation into RNA or DNA (10, 27). Our data show that SIRT7, but not SIRT1 or SIRT6, is markedly degraded in response to 5-FU treatment suggesting a specific role for SIRT7 in 5-FU-induced radiosensitivity. The enhanced degradation of SIRT7 was modulated by the dephosphorylation of TBP1 at tyrosine 381 and SIRT7 deficiency resulted in increased radiosensitivity and cell death. In human patients with rectal cancer, we confirmed a negative correlation between SIRT7 levels and therapeutic effect with 5-FU and radiation before surgical operation. Our data implicate SIRT7 as a critical factor in facilitating cell survival upon radiation of cancer cells, thus elucidating the mechanism underlying the efficacy of combined 5-FU-based chemotherapy and radiation.
Materials and Methods
Human sample collection
Tumor biopsy samples were obtained from 50 patients with locally advanced rectal adenocarcinoma who underwent neoadjuvant chemoradiotherapy and curative surgical resection between June 2012 and June 2015 at Peking University Cancer Hospital. Eligible patients were included according to the following criteria: (a) primary adenocarcinoma of the rectum identified histologically before treatment; (b) resectable rectal cancer ≤12 cm from the anal verge; (c) locally advanced rectal cancer (cT3-4N0 or cTanyN+) identified by endorectal ultrasonography or magnetic resonance imaging before treatment; (d) had no clinical evidence of synchronous distant metastasis; and (e) had an R0 resection (defined as no microscopic residual tumor cells at the longitudinal and circumferential resection margins). Hereditary colorectal cancer was excluded. All included patients underwent neoadjuvant radiotherapy with a gross tumor volume/clinical target volume of 50.6/41.8 Gy in 22 fractions, with concurrent capecitabine treatment (825 mg/m2 twice daily) over a period of 30 days, after which the patients underwent surgery for rectal cancer with an interval period of 8-10 weeks.
In each case, the pathological assessment included histological differentiation, depth of tumor invasion, lymph node metastases and lymphovascular invasion (LVI). The classification of our study was carried out according to TNM Classification of Malignant Tumors, 6th edition. Chemoradiation-induced tumor regression was assessed using the modified four-point tumor regression grade (TRG): TRG 0, no viable cancer cells; TRG 1, single or small groups of cancer cells with marked fibrosis; TRG 2, residual cancer outgrown by fibrosis; and TRG 3, fibrosis outgrown by cancer or no fibrosis with extensive residual cancer. In this study, TRG 0 tumors were excluded as SIRT7 expression could not be verified in these specimens.
Cell Culture
HCT116 (+/+), LoVo, HT-29, and HEK293T cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA). These cell lines were cultured in McCoy's 5A Medium or Dulbecco's modified Eagle's medium (M&C GENE TECHNOLOGY, Beijing, China) supplemented with 10% fetal bovine serum (FBS) (Gibco, Tennessee, USA) and 1% antibiotics (M&C GENE TECHNOLOGY, Beijing, China). All cell lines were maintained in a humidified incubator at 37° C with 5% CO2.
In vitro chemoradiation treatment
Oxaliplatin and 5-fluorouracil (5-FU) were obtained from Sigma-Aldrich (Sigma-Aldrich, Missouri, USA). The biological X-ray irradiator RS2000pro Rad Source was purchased from Rad Source Technologies (Rad Source Technologies, Georgia, USA) with radiation output of 160 KV, 25 mA at a dose rate of 4.125 Gy/min.
Plasmids and small interfering RNA
The human SIRT7 expression constructs (SIRT7-WT and SIRT7-H187Y) were gifts from Professor Katrin F. Chua (Stanford University, USA). The TBP1 plasmid was purchased from GeneChem (Shanghai, China). The following siRNAs were used to silence target genes by transfecting with Lipofectamine 2000 (Life Technologies-Invitrogen, CA, USA) according to the manufacturer's instructions:
scrambled siRNA: UUCUCCGAACGUGUCACGU;
PSMC1 siRNA: GCACCGTCCATCGTGTTTATT;
PSMC2 siRNA: CCTAAGATTGACCCAACAGTT;
TBP1 siRNA 1: CCUGGAUGUUGAUCCUAAUTT;
TBP1 siRNA 2: CAGCCCAACACCCAAGUUATT.
Protein extraction from cultured cells
Equal numbers of harvested cells were washed with PBS by centrifugation at 10, 000 rpm (9, 391 x g) at 4° C for 30 sec and the cell pellet was re-suspended in 30 μl (per 106 cells) 2X protease inhibitor buffer, consisting of 1 cocktail protease inhibitor pellet (Roche Holding AG, Basel, Switzerland) in 3.5 ml PBS. An equal volume of 2X SDS loading Buffer (950 μl Laemmle buffer + 50 μl 2-mercaptoethanol) was added to the re-suspended cells. The samples were boiled for 10 min with a pulse vortex every 5 min, and then pelleted by centrifugation at 12, 000 rpm (13, 523 x g) at 4° C for 15 min.
Tumor sample protein extraction
Human tumor samples were obtained from pretreatment biopsies and surgical specimens; each sample was washed with PBS three times to remove blood and feces and was ground on ice using a tissue grinder before protein extraction. The ground tumor tissue (10 mg for each sample) was mixed with PBS (5 μl/mg) and an equal volume of 2X SDS loading buffer (950 μl Laemmle buffer + 50 μl 2-mercaptoethanol), and then boiled for 10 min. The final solution was centrifuged at 12, 000 rpm (13, 523 x g) at 4° C for 10 min, and the upper phase of the solution was extracted for Western blotting.
SDS PAGE and Western blotting
Western blotting was used to evaluate protein levels as previously described, with minor modifications (28). Equal amounts of proteins were sized fractionated on 6-15% SDS-PAGE gel. The antibodies used were anti-SIRT7 (sc-135055), anti-TBP1 (PW8770-0025), anti-Ub (ab19247), anti-PSMC1 (11196-1-AP), anti-PSMC2 (A1985), anti-HA (M180-3, sc-805), anti-His (PM032), anti-pan-Serine (ab9332), anti-pan-Threonine (9386S), anti-pan-Tyrosine (9416S), anti-actin (TA-09), anti-FLAG (F1804).
Co-immunoprecipitation
For nuclear protein extraction, cell pellets were re-suspended in buffer A (10 mM HEPES pH 7.9, 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM dithiothreitol, 0.15% Nonidet P40, 1% protease inhibitor cocktail), and incubated on ice for 10 min prior to centrifugation at 12, 000 rpm (13, 523 x g) at 4° C for 30 sec. The supernatant fraction contained the cytoplasmic extract. The remaining pellet was washed and re-suspended in buffer B (20 mM HEPES pH 7.9, 400 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM dithiothreitol, 0.5% Nonidet P40, 1% protease inhibitor cocktail) and rocked for 15 min at 4° C with a pulse vortex every 5 min to mix. The samples were then centrifuged at 12, 000 rpm (13, 523 x g) for 15 min. The supernatant fraction from the centrifugation contained the nuclear extract.
Total protein lysate was obtained from cells by incubating on ice for 30 min in Nonidet P-40 buffer (20 mM Tris·HCl pH 7.5, 137 mM NaCl, 10% Glycerol, 1% NP-40, 2 mM EDTA, 1% cocktail), followed by centrifugation at 12,000 rpm (13, 523 x g) at 4° C for 15 min. The supernatant fraction from the centrifugation was used for immunoprecipitation.
These isolated protein fractions were used for detecting the phosphorylation of TBP1: after washing with Nonidet P-40 wash buffer (20 mM Tris·HCl pH 7.5, 137 mM NaCl, 10% Glycerol, 0.1% NP-40, 2 mM EDTA) and centrifugation at 1, 000 rpm (94 x g) at 4° C for 1 min three times , immunoprecipitated proteins were eluted with 3XFLAG peptide (F4799, 0.125 mg/ml in TBS (50 mM Tris-HCl, pH 7.4, with 150 mM NaCl) ) at volume of 50 μl overnight at 4° C and the supernatant fraction with SDS loading buffer for Western blotting.
For λ phosphatase treatment, immunoprecipitated FLAG-TBP1 was washed with Nonidet P-40 wash buffer, eluted with 3X FLAG peptide and then incubated the supernatant fraction with 400 units of λ phosphatase (New England Biolabs, MA, USA) at 30° C for 30 min.
GST pull-down
GST or GST-fusion proteins were constructed and induced by IPTG 0.1 mM for overnight at 28° C in Escherichia coli and purified using glutathione-Sepharose 4B beads (GE Healthcare, NY, USA). Equal amounts of the individual fusion proteins were incubated with GST fusion proteins (from E. coli) in TEN buffer (10 mM Tris·HCl, pH 8.0, 1 mM EDTA, 100 mM NaCl) for 4 h at 4° C. After washing in TEN buffer by centrifugation at 1, 000 rpm (94 x g) at 4° C for 1 min three times, the precipitated components were analyzed by Western blotting.
Real-Time RT-PCR Assay
Total RNA from cells was extracted using TRIzol reagent (TianGen, Beijing, China), precipitated the upper liquid using isopropanol at 12, 000 rpm (13, 523 x g) at 4° C for 10 min and washed with 75% ethanol. RNA was treated with DNase I for 30 min at 37° C to exclude potential contamination of DNA, and then reverse transcribed into cDNA using Quantscript RT Kit (Promega, WI, USA) according to the manufacturer's instructions. The relative expression of targets was measured by ABI 7500 (Life Technologies, NY, USA) real-time PCR with SYBR-green dye.
The primers (synthesized in Aoke Biological Technology, Beijing, China) used for RT-PCR were as follows:
SIRT7: AGAAGCGTTAGTGCTGCCG (sense),
GAGCCCGTCACAGTTCTGAG (antisense);
TBP1: CGTACCTTGTCTCCAACGTC (sense),
AGGAAGTACGTCTGTCGTGT (antisense);
GAPDH: GAAGGTGAAGGTCGGAGTC (sense),
GAAGATGGTGATGGGATTTC (antisense).
Establishment of stable cell lines
CRISPR-Cas9 based gene-editing: SIRT7-deficient stable HCT116 cell lines were generated via Lipofectamine transfection with sgRNA constructs that targeted SIRT7 (sequence 1: GAGCCGCTCCGAGCGCAAAG, sequence 2: CGAGAGCGCGGACCTGGTAA) cloned into a px459/Puro vector (the vector was a gift from Professor Chunyan Zhou at Peking University). Detailed information about the protocol was previously reported (29).
shRNA gene silencing: TBP1 stable knockdown HCT116 cells were generated by lentiviral transduction using an shRNA construct targeting TBP1 (sequence 1: CCUGGAUGUUGAUCCUAAUTT, sequence 2: CAGCCCAACACCCAAGUUATT, cloned into LV2/Puro vector) or a scrambled shRNA control (UUCUCCGAACGUGUCACGU) (shRNA constructs were purchased from GeneChem, Shanghai, China). shRNAs were separately packaged into lentiviruses by co-transfecting them with packaging plasmids PMD and PSP (these two plasmids were gifts from Professor Zebin Mao at Peking University) into HEK293T cells. 48 h after transfection, the supernatant was collected and used to infect HCT116 cells. Infections were repeated twice with an interval of 24 h to achieve maximal efficiency. Cells were selected with puromycin (2.5 μg/ml) for 14 days, and after colony formation, colonies were selected for cell growth. The efficiency of TBP1 knockdown was verified by Western blotting.
Colony formation
HCT116 cells were seeded in cell culture medium containing 10% FBS and 1% antibiotics in 60 mm dishes and were irradiated by X-ray after 12 h. For 5-FU treatment, cells were exposed to 10 μM 5-FU for 24 h and then washed at room temperature 800 rpm (60 x g) for 3 min with cell culture medium eight times and seeded with the equal number of cells. All cells were cultured for 14 days at 37° C, 5% CO2 for 2 weeks and then stained with crystal violet.
Tumor growth in vivo
Three groups of 6-8-week-old male NOD-SCID mice were caged and fed a diet of animal chow and water which were sterilized by high pressure.
Group 1 included six mice that were injected in the right hind leg with 107 HCT116 Cas9 SIRT7-WT cells or Cas9 SIRT7-KO cells in a volume of 0.2 ml in medium separately. When a tumor reached a mean diameter of 1 cm in three individual mice per group, the tumors were locally irradiated with a single dose of 10 Gy. The tumors were measured with calipers every day after radiation, and tumor volumes were calculated as follows: volume [mm3] = width2 X length/2).
Group 2 included twelve mice that were injected in the right hind leg with HCT116 cells. When all mice developed a tumor ~1 cm in mean diameter, six mice were injected with PBS (untreated control) and the remainder were injected with 5-FU for 3 days (10 mg/kg intraperitoneal injection). After injection, three of the PBS mice and three of the 5-FU mice were locally irradiated with a single dose of 10 Gy. Tumors were measured with calipers as described for group 1.
Group 3 included six mice that were injected in the right hind leg with scrambled shRNA HCT116 cells and six mice that were injected with TBP1 shRNA cells in a volume of 0.2 ml in medium. When all mice developed a tumor ~1 cm in diameter, half of the mice (three injected with scrambled shRNA cells and three injected with TBP1 shRNA) were injected (intraperitoneal) with 10 mg/kg 5-FU for 3 days followed by a single dose of local radiation at 10 Gy. Tumors were measured with calipers every day as described above. The mice were sacrificed through cervical dislocation.
Statistics
Statistical comparisons were carried out using the Student's t-test. A p < 0.05 was considered statistically significant (NS, p > 0.05, *p < 0.05, **p < 0.01, ***p < 0.001). Three independent experiments were performed in all cases.
Study approval
This study was approved by the Peking University Cancer Hospital's ethics committee. Informed consent was obtained from each patient before the use of their tumor tissue. Each animal study was conducted in accordance with principles outlined in the NIH Guide for the Care and Use of Laboratory Animals.
Results
SIRT7 protein levels are specifically modulated by 5-FU and not by X-ray radiation or oxaliplatin alone
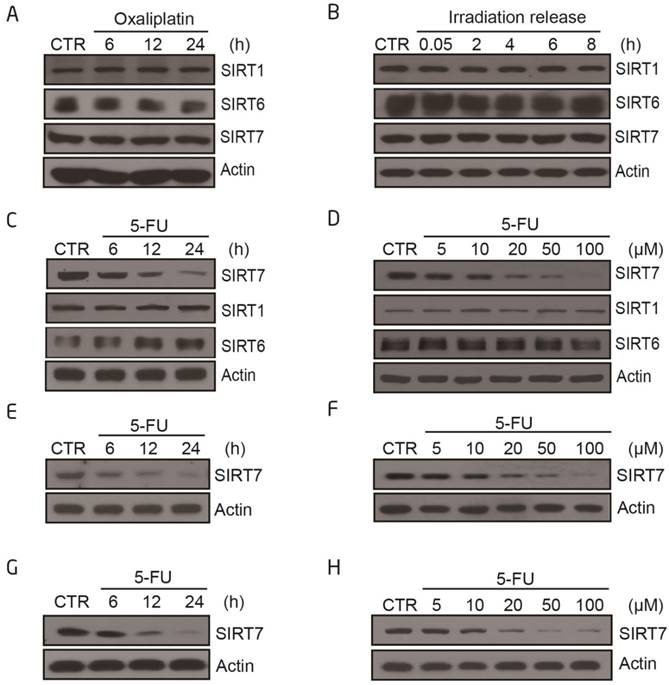
We first addressed whether chemotherapeutics sensitize cancer cells to radiation by altering the level and/or activity of class III HDACs sirtuins. We exposed the human colorectal cancer cell line, HCT116, either to a radiosensitizer (oxaliplatin or 5-FU) or X-ray radiation (IR) and analyzed the protein expression levels of nuclear sirtuins. SIRT1 and SIRT6 levels did not markedly change following any of the treatment modalities (Figure 1A-C), whereas SIRT7 levels decreased upon 5-FU treatment in a time-dependent and dose-dependent manner (Figure 1C-D). Treatment with oxaliplatin or radiation alone did not affect SIRT7 levels. These data were replicated in other human colorectal cancer cell lines, including LoVo (Figure 1E-F) and HT-29 cells (Figure 1G-H). These data showed that 5-FU specifically modulates SIRT7 expression and activity.
SIRT7 protein levels are specifically modulated by 5-FU and not by X-ray radiation or oxaliplatin alone A. Immunoblots for SIRT1, SIRT6 and SIRT7 levels in HCT116 cells following treatment with oxaliplatin (100 μM for 6, 12, 24 h). Actin was probed as a loading control and used to estimate protein quantification. B. Immunoblots for SIRT1, SIRT6 and SIRT7 levels in HCT116 cells following treatment with radiation (10 Gy, release for 0.05, 2, 4, 6, 8 h). C. Immunoblots for SIRT1, SIRT6 or SIRT7 levels in HCT116 cells following 100 μM 5-FU treatment for 6, 12, and 24 h. D. Immunoblots for SIRT1, SIRT6 or SIRT7 levels in HCT116 cells following 5-FU treatment at 5, 10, 20, 50, 100 μM for 24 h. E. Immunoblots for SIRT7 levels in LoVo cells following 100 μM 5-FU treatment for 6, 12, and 24 h. F. Immunoblots for SIRT7 levels in LoVo cells following 5-FU treatment at 5, 10, 20, 50, 100 μM for 24 h. G. Immunoblots for SIRT7 levels in HT-29 cells following 100 μM 5-FU treatment for 6, 12, and 24 h. H. Immunoblots for SIRT7 levels in HT-29 cells following 5-FU treatment at 5, 10, 20, 50, 100 μM for 24 h.
TBP1 specifically modulates SIRT7 degradation upon 5-FU treatment
We next sought to elucidate the molecular mechanisms by which SIRT7 protein levels are regulated by 5-FU. Real-time PCR analyses indicated that SIRT7 mRNA levels were not altered in 5-FU-treated cells (Supplementary Figure 1A-B), suggesting that decreased SIRT7 protein might be the result of altered protein stability.
To explore the pathways underlying the 5-FU-induced decrease in SIRT7 levels, we assayed the half-life of SIRT7 by using cycloheximide (CHX), which is an inhibitor of protein synthesis. Cultured HCT116 cells were pretreated with CHX and then exposed to either 5-FU or vehicle. The half-life of SIRT7 shortened from >4 h in untreated cells to 2 h in 5-FU-treated cells but not in oxaliplatin or radiation-treated cells (Figure 2A and Supplementary Figure 2A-B). We then pretreated HCT116 cells with either the proteasome inhibitor, MG132 or lysosome protease inhibitor, chloroquine (CHQ), to identify the mechanism involved in SIRT7 degradation. The increased clearance of SIRT7 protein was markedly inhibited by MG132 but not by CHQ indicating the involvement of the proteasome-dependent degradation pathway in mediating SIRT7 degradation in response to 5-FU (Figure 2B). When the levels of ubiquitination on immunoprecipitated SIRT7 were assessed, no obvious change in exogenous or endogenous ubiquitination of SIRT7 in response to 5-FU was detected (Figure 2C), suggesting that SIRT7 is degraded in an ubiquitin-independent and proteasome-dependent manner.
TBP1 specifically modulates SIRT7 degradation upon 5-FU treatment A. Immunoblots showing SIRT7 levels in HCT116 cells treated by 100 μg/ml cycloheximide (CHX) with or without 100 μM 5-FU for the indicated time. Line graph indicating the measured SIRT7 levels under each condition determined by scanning the SIRT7 bands in the left panel. B. Immunoblots showing SIRT7 levels in HCT116 cells treated with 20 μM MG132 for 12 h, or 50 μM chloroquine for 24 h, with or without 100 μM 5-FU for 24 h. C. Exogenous (left panel) and endogenous (right panel) ubiquitination of SIRT7 following treatment with 100 μM 5-FU for 24 h and 20 μM MG132 for 12 h. Cell nuclear lysates were immunoprecipitated with an anti-FLAG or anti-SIRT7 antibody and then blotted with an anti-HA or anti-Ub antibody. D. Mass spectrometry analysis of SIRT7 interacting proteins following treatment with 100 μM 5-FU for 24 h and 20 μM MG132 for 12 h. A plasmid expressing FLAG-SIRT7 was transfected into HCT116 cells, and cell lysates were extracted for immunoprecipitation followed by SDS-PAGE. The entire lane was excised, digested with trypsin, and analyzed by mass spectrometry. The proteins represent a selection of SIRT7 interacting proteins. E. For endogenous interaction between SIRT7 and TBP1 (upper panel), HCT116 cell nuclear lysates were extracted and precipitated with an anti-SIRT7 antibody. For exogenous interactions (middle and lower panel), HCT116 cells were co-transfected with GFP-tagged SIRT7 and FLAG-tagged TBP1 as indicated, anti-GFP or anti-FLAG immunoprecipitation was performed respectively. F. Changes of SIRT7 levels in HCT116 cells treated with 100 μM 5-FU for 24 h following TBP1 knockdown using two siRNAs. G. Interaction changes between SIRT7 and TBP1 upon treatment with oxaliplatin (100 μM, 24 h), irradiation (10 Gy, release for 6 h) and 5-FU (100 μM, 24 h). Cells were pretreated with MG132 to prevent protein degradation. H. Interaction changes between SIRT7 and TBP1 upon treatment with 0, 10, 50 or 100 μM 5-FU for 24 h. I. An in vitro GST-pull down assay showing the direct interaction between His-SIRT7 and GST-TBP1 purified from E. coli. J. Mapping the interacting fragments of TBP1 and SIRT7. Full-length GST-TBP1 or fragments of TBP1, including a coiled-coil-containing fragment (1-96 aa), an ATPase and helicase-containing fragment (97-439 aa), and a coiled-coil and helicase-containing fragment with 236-361 aa deleted, were incubated with His-SIRT7, and Western blotting was performed to detect interactions with an anti-His antibody (left panel). A reciprocal pull-down assay between full-length GST-SIRT7 or an N-terminus fragment (1-89 aa), a deacetylase core fragment (90-331 aa), a C-terminus fragment (332-400 aa), and FLAG-TBP1 (right panel). * Represents specific bands corresponding to the induced GST-tagged proteins stained by Coomassie brilliant blue.
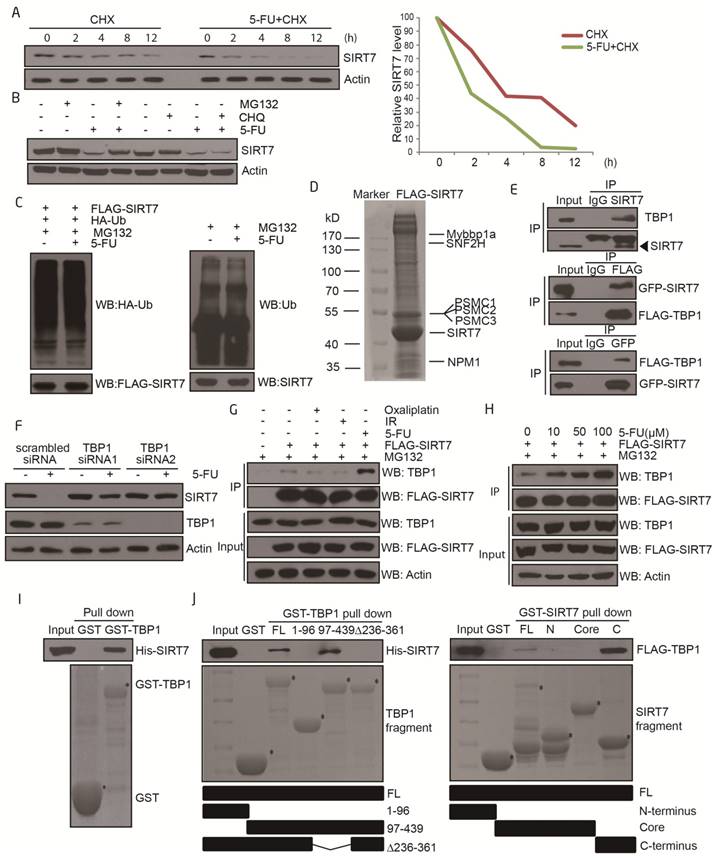
We performed SIRT7 co-immunopurification studies followed by mass spectrometry to help identify the modulators of SIRT7 degradation. SIRT7-interacting proteins were isolated and immunopurified from 5-FU-treated HCT116 cells in the presence of MG132 and then identified by mass spectrometry. SIRT7 co-purified with a few proteins, including SNF2H, NPM1, and Mybbp1a, as previously reported (30, 31). Also, we found the 26S protease regulatory proteins including PSMC1, PSMC2, and PSMC3 (also known as Tat-binding Protein 1, TBP1) to interact with SIRT7 (Figure 2D), which we then confirmed by co-immunoprecipitation (Figure 2E and Supplementary Figure 2C). These proteins have been previously reported to mediate ubiquitination-independent degradation (32, 33) which is consistent with our data. To identify which of these proteins were responsible for modulating SIRT7 degradation, we performed siRNA-mediated knockdown of TBP1, PSMC1, and PSMC2 and monitored the rate of degradation of SIRT7 after 5-FU incubation. Knockdown of TBP1 by siRNA markedly abrogated the 5-FU-induced reduction in SIRT7 levels in HCT116 (Figure 2F) and LoVo cells (Supplementary Figure 2D), whereas PSMC1 and PSMC2 had no effect on the rate of SIRT7 degradation (Supplementary Figure 2E). Interestingly, the levels of co-immunoprecipitation between SIRT7 and TBP1 seemed to increase only in a 5-FU dose-dependent manner but did not change with oxaliplatin or radiation (Figure 2G-H).
Furthermore, there was a direct interaction between SIRT7 and TBP1 as demonstrated by GST pull-down assay with either full-length or fragmented GST-TBP1 with His-SIRT7 and full-length or fragmented GST-SIRT7 with FLAG-TBP1 (Figure 2I-J). The interaction was mediated via the ATPase-containing fragment of TBP1 and the C-terminus (amino acid 332-400) of SIRT7. Based on these data, we can conclude that TBP1 mediates 5-FU-induced SIRT7 degradation through a direct protein interaction.
TBP1 dephosphorylation at Y381 is required for SIRT7 degradation
To understand how TBP1 participates in SIRT7 degradation, HCT116 cells were treated with 5-FU and the mRNA and protein contents were extracted for quantitative real time PCR and Western blotting, respectively. The mRNA and protein levels of TBP1 were not altered in 5-FU-treated cells (Supplementary Figure 3A-B), indicating the potential involvement of a post-translational modification that mediates the activation of TBP1. Immunoprecipitation experiments confirmed that TBP1 tyrosine dephosphorylation occurred in response to 5-FU treatment but not to radiation or oxaliplatin (Figure 3A-B). It was further confirmed in vitro using the protein lambda phosphatase (λ-PP), which markedly decreased tyrosine phosphorylation of TBP1 (Supplementary Figure 3C).
TBP1 dephosphorylation at Y381 is required for SIRT7 degradation A. Changes in TBP1 phosphorylation status upon 5-FU treatment. FLAG-TBP1 was immunoprecipitated from HCT116 cells treated with 100 μM 5-FU for 24 h and eluted for immuoblotting with an anti-pan-serine, an anti-pan-threonine or an anti-pan-tyrosine antibody, respectively. B. Changes in TBP1 phosphorylation status upon radiation (10 Gy, release for 6 h) or treatment with 100 μM 5-FU for 24 h, 100 μM oxaliplatin for 24 h. C. Mass spectrometry analysis of tyrosine phosphorylation at TBP1 Y381. FLAG-TBP1 was transfected into HCT116 cells, immunoprecipitated and eluted for electrophoresis. The TBP1 band was excised, digested with trypsin, and analyzed by mass spectrometry. D. Changes in the phosphorylation status of TBP1-WT and TBP1-Y381F with an anti-pan-tyrosine antibody. E. Immunoblots were performed with a specific anti-TBP1-Y381p antibody to detect changes in the phosphorylation status of TBP1-WT and TBP1-Y381F. F. Changes in TBP1-Y381p when cells treated with 100 μM 5-FU for 24 h. G. SIRT7 levels in FLAG-TBP1-WT or FLAG-TBP1-Y381F transfected HCT116 cells following treatment with 100 μg/ml cycloheximide (CHX) for the indicated time. H. Interaction changes between SIRT7 and TBP1-WT-FLAG or TBP1-Y381F-FLAG.
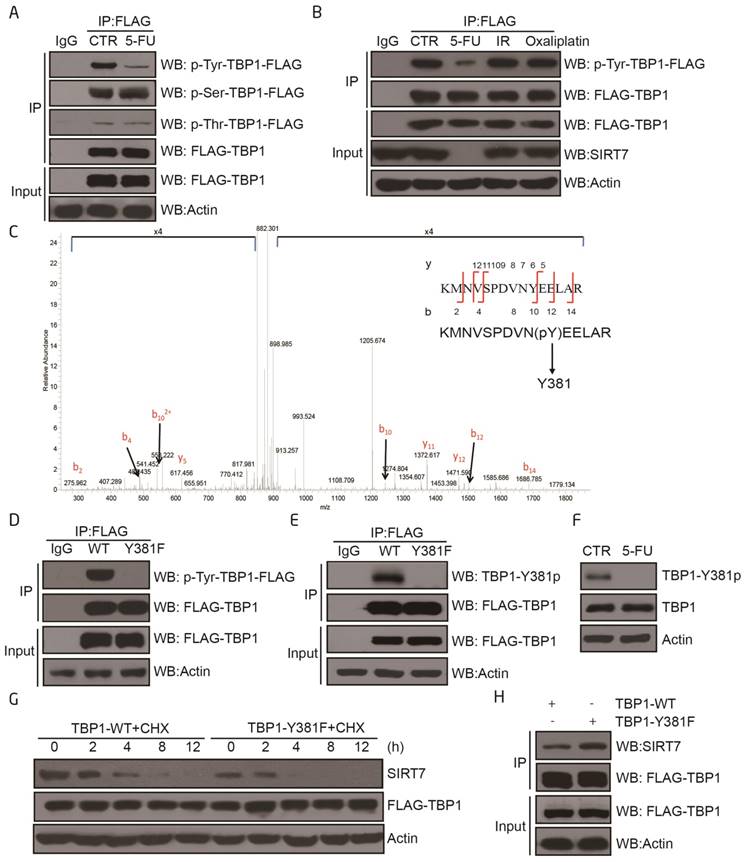
We next immunoprecipitated TBP1 from HCT116 cells and performed mass spectrometry to map the tyrosine phosphorylation sites on the protein. Tyrosine 381 (Y381) was identified as the only phosphorylated tyrosine residue under these experimental conditions (Figure 3C). Mutation of this residue from tyrosine (Y) to a phenylalanine (F) (TBP1-Y381F) abolished TBP1 tyrosine phosphorylation indicating that Y381 may be the predominant residue involved in the tyrosine phosphorylation of TBP1 (Figure 3D). We, therefore, generated a phospho-specific antibody for TBP1-Y381p. The efficiency and specificity of this antibody were demonstrated to be sufficient for subsequent dot blot experiments with non-specific and specific phospho peptides as indicated (Supplementary Figure 3D). We used it to confirm that TBP1 was phosphorylated at Y381 and dephosphorylated upon 5-FU treatment in both HCT116 and LoVo cells (Figure 3E-F and Supplementary Figure 3E-F). We next tested the functional impact of mutant TBP1 (Y381F) by over-expressing it in HCT116 cells. Over-expression of TBP1-Y381F resulted in enhanced degradation of SIRT7 which was mechanistically linked to an increased association between the mutant TBP1 and SIRT7 as determined by co-immunoprecipitation (Figure 3G-H). Overall, these data further support Y381 as a specific phosphorylation site that mediates TBP1 function to target SIRT7 when cells are exposed to 5-FU.
5-FU-induced SIRT7 degradation sensitizes NOD-SCID mice to IR and abrogates tumor growth
It has been well recognized that as a standard therapeutic strategy, 5-FU is often used to synergize radiation in killing human colorectal cancers clinically (34). Most recently, two groups reported that SIRT7 is involved in DNA double-strand break (DSB) repair pathway, and SIRT7 deficient cells show increased susceptibility to externally induced DNA damage (21, 22) providing evidence for SIRT7's role in radiosensitivity. Consistent with these observations, our data have suggested that 5-FU and IR synergistically induce cancer cell death via SIRT7 degradation.
To explore the role of SIRT7 in response to radiation, a stable SIRT7 deficient cell line (SIRT7-KO) was established using human colorectal cancer cell line HCT116 with Clustered Regularly Interspaced Short Palindromic Repeats (CRISPRs) RNA-guided Cas9 nucleases (CRISPR-Cas9) -based gene-editing technology (29). Both SIRT7 knockout (SIRT7-KO) and SIRT7 wild-type (SIRT7-WT) HCT116 cells were then exposed to ionizing radiation (IR) at different doses (0-5 Gy), and subsequently, cell survival was determined by colony formation assay. As shown in Figure 4A, deficiency of SIRT7 led to a significantly (p<0.001) enhanced cell sensitivity to IR treatment in an IR dose-dependent manner. To establish whether SIRT7 deacetylase activity is required to protect against IR, a plasmid expressing either SIRT7-WT or SIRT7-H187Y was introduced into SIRT7-KO cells and a colony formation assay was performed after cellular irradiation. SIRT7-WT, but not mutated SIRT7-H187Y, rescued cell viability following IR (Figure 4A). As expected, treatment with both 5-FU and radiation induced a significant (p<0.01) synergistic effect to inhibit the survival of cancer cells, but oxaliplatin with radiation did not show a similar synergistic effect (Figure 4B and Supplementary Figure 4A).
5-FU-induced SIRT7 degradation sensitizes NOD-SCID mice to IR and abrogates tumor growth A. A colony formation assay was performed with SIRT7-WT cells, SIRT7-KO cells, or SIRT7-KO cells transfected with plasmids expressing SIRT7-WT or SIRT7-H187Y. Cells were exposed to different doses of IR. Colonies containing >50 cells were counted. Error bars represent standard deviations. Western blots indicate the efficiencies of SIRT7 knockout and overexpression (right panel). *** represents p<0.001. B. A colony formation assay was performed with HCT116 cells treated with 10 μM 5-FU alone, 2 Gy IR alone or both treatments combined, as indicated. All samples were seeded with 20,000 cells. Colonies containing >50 cells were counted (lower graph). Error bars represent standard deviations. ** represents p<0.01. C. A colony formation assay was performed with scrambled shRNA or TBP1 shRNA cells exposed to different doses of ionizing radiation (IR). Cells were pretreated with 10 μM 5-FU for 24 h. Western blotting was performed to detect TBP1 knockdown efficiency (right panel). Error bars represent standard deviations. *** represents p<0.001. D. Tumor growth rate in different groups of treated mice. NOD-SCID mice were divided into the following four groups: group 1: WT group (injected with SIRT7-WT cells); group 2: KO group (injected with SIRT7-KO cells); group 3: WT with radiation group (injected with SIRT7-WT cells and then irradiated); group 4: KO with radiation group (injected with SIRT7-KO cells and then irradiated). Tumor size was measured on the indicated days following radiation. The initial mouse tumor size was designated as 1, and subsequent tumor sizes were quantified via comparison to the initial tumor size. SIRT7 KO efficiency was detected by immunoblotting tumor extracts (lower panel). E. The effects of combination treatment with 5-FU and radiation on tumor growth in mice. NOD-SCID mice were grouped into the following four groups: group 1: control group; group 2: radiation alone (10 Gy one time); group 3: 5-FU alone (10 mg/kg intraperitoneal injection for three days); group 4: radiation and 5-FU (one dose at 10 Gy and intraperitoneal injection of 10 mg/kg 5-FU for 3 days prior to radiation). All mice were injected with HCT116 cells. Tumor size was measured on the indicated days following IR. The analysis was performed as described above. SIRT7 was detected by immunoblotting protein samples obtained from tumor extracts (lower panel). F. The effects of TBP1 knockdown in combination with radiation and 5-FU treatment on tumor growth. NOD-SCID mice were grouped into the following four groups: group 1: control group (injected with scrambled shRNA cells); group 2: TBP1 shRNA group (injected with TBP1 shRNA cells); group 3: 5-FU and radiation combination group (injected with scrambled shRNA cells and intraperitoneal injection of 10 mg/kg 5-FU for 3 days prior to radiation); group 4: TBP1 shRNA and 5-FU and radiation combination group (injected with TBP1 shRNA cells and intraperitoneal injection of 10 mg/kg 5-FU for 3 days prior to radiation). Tumor size was measured on the indicated days following IR. The analysis was performed as described above. The efficiency of TBP1 knockdown was detected by immunoblotting protein samples obtained from tumor extracts (right panel).
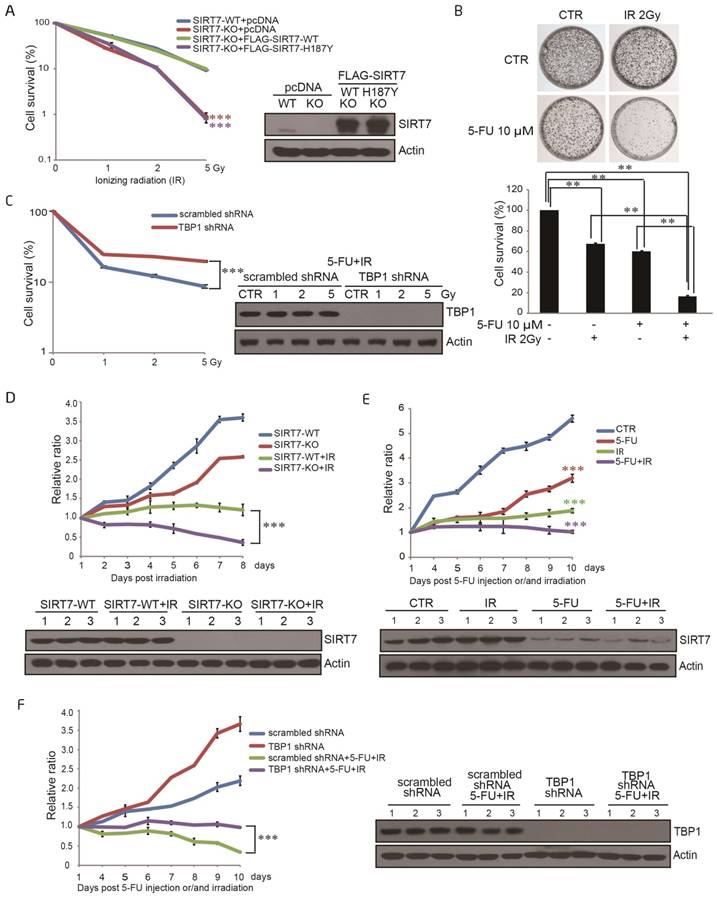
To determine whether the degradation of SIRT7 is the primary mechanism underlying 5-FU-induced radiosensitivity, HCT116 cells carrying scrambled shRNA or TBP1 shRNA were treated with combined 5-FU and IR across a range of doses. Cell survival, as evaluated by colony formation assay, was significantly (p<0.001) enhanced in TBP1 shRNA cells compared to that in scrambled shRNA cells indicating that SIRT7 degradation primarily results in 5-FU-induced radiosensitivity (Figure 4C).
Finally, we performed a tumor growth experiment in non-obese diabetic severe combined immunodeficiency (NOD-SCID) mice to confirm the synergistic role of IR and 5-FU-induced SIRT7 degradation on cancer cell death in vivo. SIRT7-WT or SIRT7-KO HCT116 cells were separately injected into NOD-SCID mice and tumor size was measured after the mice were exposed to IR. The tumor sizes in mice injected with SIRT7-WT cells were significantly (p<0.001) larger than those in mice injected with SIRT7-KO cells in response to radiation (Figure 4D). Also, tumor size was further decreased when NOD-SCID mice were treated with a combination of 5-FU and radiation compared to 5-FU or radiation alone (p<0.001) (Figure 4E). However, tumor sizes in mice injected with cells containing intact TBP1 (scrambled shRNA) were notably (p<0.001) smaller than those in mice injected with TBP1 shRNA in response to combined 5-FU and IR exposure (Figure 4F). These data are consistent with our cell-based findings and support that 5-FU-mediated radiosensitivity is attributable to the pharmacological effects resulting in SIRT7 degradation in vivo.
Reduced SIRT7 levels are associated with improved therapeutic effect in rectal cancer patients treated with 5-FU and radiation
The 5-FU analog, capecitabine, is catalyzed to 5-FU in living cells and is typically used in combination with radiation as a preoperative adjuvant therapy to reduce tumor size and stage (35). We were interested in determining if the synergistic effect of 5-FU with radiation is due to the 5-FU-induced degradation of SIRT7 in colorectal cancer. We analyzed the protein expression levels of SIRT7 in 50 paired human cancer biopsies collected from patients before and after chemoradiotherapy with a single dose of capecitabine (825 mg/m2) and radiation (50.6 Gy/22f).
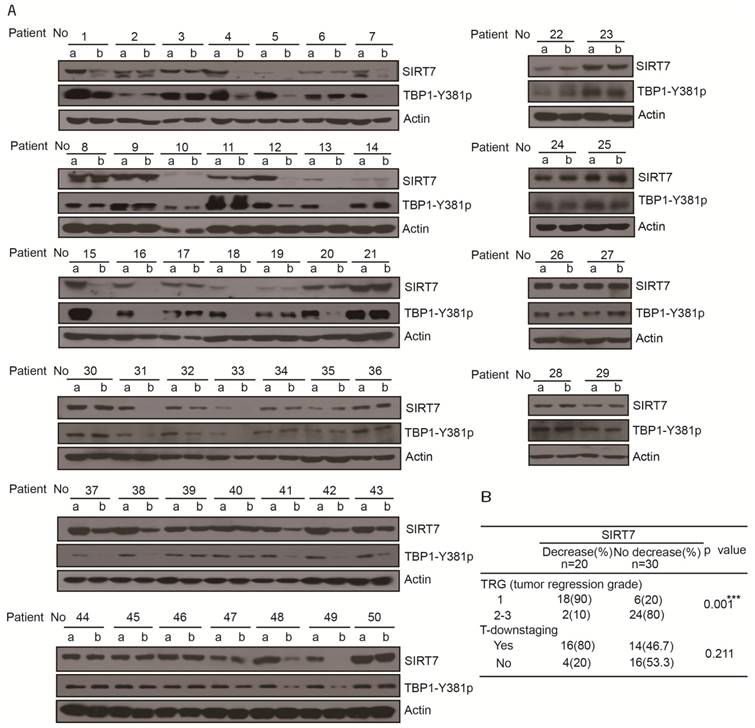
Samples were acquired from 30 male and 20 female patients (sample acquisition process is shown Supplementary Figure 5A) with a mean age of 57.6 years (median: 59; range: 23-85) (Table 1). Assessment of SIRT7 protein levels in the tumor samples collected before chemoradiotherapy showed no significant difference between patients in terms of gender, age, tumor location, serum carcinoembryonic antigen (CEA), tumor stage, or other clinicopathological variables (Table 1). After chemoradiotherapy, SIRT7 protein levels were significantly decreased in 20 patients (40%) (patient's number: 1, 4, 5, 7, 12, 13, 15, 16, 18, 20, 31, 32, 33, 37, 38, 41, 42, 43, 48, 49) compared to their corresponding protein levels prior to treatment. There was no change in the SITR7 protein levels in the remaining 30 (60%) paired tumor samples before and after treatment with 5-FU combined with radiation (Figure 5A). Tumor down-staging did not correspond with decreased SIRT7 levels but decreased SIRT7 levels were associated with improved therapeutic effect in patients after surgical intervention (Figure 5B). Among the patients whose SIRT7 levels significantly decreased following treatment, 90% achieved tumor regression grade 1 (TRG1) compared to 20% of those with no decreased SIRT7 levels (Figure 5B). To further confirm whether TBP1 tyrosine phosphorylation levels in treated tumor samples are consistent with the SIRT7 levels, we used anti-TBP1-Y381p antibody to perform Western blotting of tumor tissues before and after treatment with 5-FU combined with radiation therapy. As shown in Figure 5A, TBP1-Y381 phosphorylation levels clearly showed the same trend as the changes in SIRT7 levels following chemoradiotherapy. TBP1-Y381p levels in 20 tumor tissue pairs (patient's number: 1, 4, 5, 7, 12, 13, 15, 16, 18, 20, 31, 32, 33, 37, 38, 41, 42, 43, 48, 49) were significantly decreased in post-treatment tumors compared with those prior to treatment. These data indicated that 5-FU caused tyrosine dephosphorylation at tyrosine 381 of TBP1 and led to the degradation of SIRT7. The therapeutic effect was improved in cases of colorectal cancer where SIRT7 levels were decreased as a result of adjuvant therapy. Together with the in vitro results that 5-FU alone could cause a reduction in SIRT7, our data in rectal cancer patients suggest that a 5-FU-mediated reduction in SIRT7 renders colorectal cancer cells sensitive to radiotherapy.
Association between the patient clinical characteristics and the change in SIRT7 expression before and after chemoradiotherapy
| Characteristics | SIRT7 | p value | |
|---|---|---|---|
| Decrease (%) n=20 | No decrease (%) n=30 | ||
| Gender | |||
| Male | 8(40) | 22(73.3) | 0.122 |
| Female | 12(60) | 8(26.7) | |
| Age (yr) | |||
| <60 | 12(60) | 14(46.7) | 0.688 |
| ≥60 | 8(40) | 16(53.3) | |
| Histological differentiation | |||
| Moderate | 16(80) | 28(93.3) | 0.543 |
| Poor | 4(20) | 2(6.7) | |
| cT stage | |||
| T3 | 14(70) | 24(80) | 0.653 |
| T4 | 6(30) | 6(20) | |
| cN stage | |||
| N0 | 0(0) | 2(6.7) | 1 |
| N1-2 | 20(100) | 28(93.3) | |
| ypT stage | |||
| yT2 | 12(60) | 8(26.7) | 0.122 |
| yT3 | 8(40) | 22(73.3) | |
| ypN stage | |||
| N0 | 16(80) | 14(46.7) | 0.21 |
| N1-2 | 4(20) | 16(53.3) | |
| Lymphovascular invasion | |||
| Yes | 0(0) | 6(18.8) | 0.25 |
| No | 20(100) | 24(81.2) | |
| Serum CEA (ng/ml) | |||
| <5 | 12(60) | 12(40) | 0.428 |
| ≥5 | 8(40) | 18(60) | |
Independent χ2 test to determine the relationship between the change of SIRT7 before and after chemoradiotherapy and clinical characteristics of patients. p > 0.05 represents no significance correlation.
Reduced SIRT7 levels are associated with improved therapeutic effect in rectal cancer patients treated with 5-FU and radiation A. Immunoblots for SIRT7 levels in human rectal tumor samples (50 pairs) obtained before and after chemoradiotherapy (a, samples obtained from diagnostic biopsies; b, samples obtained from surgery). B. Independent χ2 test, separately to determine the relationship SIRT7 of decrease/no decrease with tumor response to chemoradiotherapy. ***p < = 0.001 represents very significant relationship, p > 0.05 represents no significance correlation.
Discussion
5-FU sensitizes cells to radiation by exerting effects on multiple biological processes. For example, 5-FU-induced thymidylate synthase expression is a key determinant of 5-FU radiosensitivity (10, 11). Also, 5-FU inhibits the mRNA expression of excision repair cross-complementing 1 (ERCC1), which is responsible for the nucleotide excision repair of radiation-induced or chemotherapy-induced DNA damage, accounting for the higher sensitivity of tumors to radiation (6). Moreover, 5-FU increases radiosensitivity by killing S-phase cells, which are typically radioresistant (6).
In this study, we present, for the first time, a novel mechanism in which 5-FU degrades SIRT7 through the TBP1-modulated 26S proteasome-dependent pathway resulting in increased radiosensitivity of cancer cells. More importantly, clinical evidence supports the association of SIRT7 levels with therapeutic effect to neoadjuvant chemoradiotherapy in human colon cancer.
TBP1, first identified as human immunodeficiency virus (HIV) Tat binding protein-1, suppresses Tat-mediated transactivation (36). It has been reported that TBP1-mediated protein degradation is stimulus-dependent. For example, TBP1 specifically interacts with von Hippel-Lindau (VHL) and Hif1α and promotes the VHL-dependent degradation of Hif1α in an ubiquitin-dependent manner under conditions of normoxia (37). TBP1 also mediates protein degradation via a ubiquitin-independent pathway. Fos-related antigen-1 (Fra-1) is a member of the activator protein-1 transcription factor superfamily, and its turnover does not require ubiquitination but is destabilized via a TBP1 ubiquitin-independent pathway (33).
Our study provides evidence that 5-FU-induced SIRT7 degradation is mediated by TBP1 through a ubiquitin-independent and proteasome-dependent pathway (Figure 2). We identified a novel pathway for TBP1-mediated protein degradation that involves dephosphorylation of TBP1 (Figure 3). However, it is not clear how 5-FU treatment activates TBP1 dephosphorylation to mediate SIRT7 degradation.
SIRT7 is implicated in a wide variety of biological and oncological processes. It has been reported that SIRT7 can serve as a prognostic factor (38) as there was an association between its protein levels and tumor stage (39). SIRT7 has a crucial function in maintaining the malignant features of cancer cells and is over-expressed in various cancer types (39, 40). SIRT7 is also an NAD+-dependent deacetylase. It localizes to chromatin to deacetylate lysine 18 of histone H3 and stabilizes the transformed state of cancer cells (40). This oncogenic feature of SIRT7 renders it a promising pharmacological target for cancer therapy. SIRT7 orchestrates the activity and function of several other molecules. It enables the association of the B-WICH chromatin remodeling complex with RNA Pol I machinery during transcription (31). SIRT7 also regulates the RNA and protein synthesis of polymerase III through mTOR and the TFIIIC2 complex (41) and Pol I-mediated transcription (42), respectively. In addition, SIRT7 deacetylates GABPβ1 (43) and NPM1 (30) thus regulating mitochondrial homeostasis and aging, respectively. SIRT7 also promotes the regenerative capacity of aged hematopoietic stem cells (HSCs) by driving the mitochondrial unfolded protein response (UPRmt) (44). In the context of DNA damage, SIRT7 is associated with the DNA damage repair process, and SIRT7 knockdown cells exhibit increased susceptibility to DNA damage and undergo high levels of apoptosis (45). The recent identification of a direct role for SIRT7 in the DSB repair of the NHEJ pathway established a functional link between SIRT7-mediated H3K18 deacetylation and 53BP1 recruitment to sites of DNA damage (26). Interestingly, SIRT7 overexpression increased both NHEJ and HR repair efficiency in reporter cell lines (17, 22). Although we did not determine whether SIRT7 affects the NHEJ or HR repair pathways, we provide clear evidence that 5-FU induced SIRT7 degradation is involved in increased radiosensitivity in colorectal cancer cell lines and immune-compromised mice.
In view of the involvement of SIRT7 in numerous cellular functions outlined above, we explored its clinical significance in colorectal cancer. By comparing the expression levels of SIRT7 before and after chemoradiotherapy, we discovered that the decrease in SIRT7 levels correlated well with better therapeutic effect following combined 5-FU and IR therapy (Figure 5B). These findings are of great clinical relevance as downregulation of SIRT7 during radiotherapy would be expected to result in an improved therapeutic outcome.
In summary, we have described a novel mechanism in which 5-FU-induced radiosensitivity derives from SIRT7 degradation resulting in the sensitization of cancer cells to radiation therapy. This study provides mechanistic insight into the biological relevance of cancer treatment with 5-FU and radiation and suggests a useful strategy of inducing degradation of SIRT7 to achieve better therapeutic effect with radiation therapy. This observation is related with the clinical translation of deficient SIRT7 as a radiation sensitizer combined with radiation therapy.
Abbreviations
5-FU: 5-Fluorouracil; TBP1: Tat-binding Protein 1; PARP1: poly [adenosine diphosphate (ADP)-ribose] polymerase 1; HR: homologous recombination; NHEJ: non-homologous end joining; DNA-PKcs: DNA-dependent protein kinase catalytic subunits; 53BP1: p53-binding protein 1; DSBs: DNA double-strand breaks; CHX: cycloheximide; CHQ: chloroquine; λ-PP: lambda phosphatase; CEA: carcinoembryonic antigen; ERCC1: excision repair cross-complementing 1; HIV: human immunodeficiency virus; VHL: von Hippel-Lindau; Fra-1: Fos-related antigen-1; HSCs: hematopoietic stem cells; UPRmt: mitochondrial unfolded protein response.
Supplementary Material
Supplementary figures.
Acknowledgements
The authors would like to thank Dr Katrin F. Chua at the University of Stanford for kindly providing the SIRT7-WT and SIRT7-H187Y FLAG-tagged plasmids. This study was supported by the National Key Basic Research Program of China (grant numbers 2011CB504200, 2013CB911000 and 2013CB911001) and the National Natural Science Foundation of China (grant numbers 31070691, 81321003, 81472627 and 91319302), Discipline Construction Funding of Shenzhen (2016), and Shenzhen Municipal Commission of Science and Technology Innovation (grant number JCYJ20160427104855100).
Author contributions
W. G. Z, H. Y. W, M. T, X. L conceived the project. W. G. Z, H. Y. W, M. T, X. L conceived, designed and performed the experiments. W. G. Z, M. T, wrote the manuscript. M. T, X. L, C. Z, C. D, L. C, T. H, Z. L, B. T, Z. C, Y. L, Y. C, L. J, H. W, L. W, analyzed the data and performed material preparation. B. L, X. X, J. L, J. W, discussed the results and commented on the manuscript. C. D, J. G provided the human tumor samples.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Glynne-Jones R, Dunst J, Sebag-Montefiore D. The integration of oral capecitabine into chemoradiation regimens for locally advanced rectal cancer: how successful have we been? Annals of oncology: official journal of the European Society for Medical Oncology / ESMO. 2006;17:361-71
2. Wilson GD, Bentzen SM, Harari PM. Biologic basis for combining drugs with radiation. Seminars in radiation oncology. 2006;16:2-9
3. Gerard JP, Conroy T, Bonnetain F, Bouche O, Chapet O, Closon-Dejardin MT. et al. Preoperative radiotherapy with or without concurrent fluorouracil and leucovorin in T3-4 rectal cancers: results of FFCD 9203. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2006;24:4620-5
4. Sauer R, Becker H, Hohenberger W, Rodel C, Wittekind C, Fietkau R. et al. Preoperative versus postoperative chemoradiotherapy for rectal cancer. The New England journal of medicine. 2004;351:1731-40
5. Sauer R, Liersch T, Merkel S, Fietkau R, Hohenberger W, Hess C. et al. Preoperative versus postoperative chemoradiotherapy for locally advanced rectal cancer: results of the German CAO/ARO/AIO-94 randomized phase III trial after a median follow-up of 11 years. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2012;30:1926-33
6. Ojima E, Inoue Y, Watanabe H, Hiro J, Toiyama Y, Miki C. et al. The optimal schedule for 5-fluorouracil radiosensitization in colon cancer cell lines. Oncology reports. 2006;16:1085-91
7. McGinn CJ, Zalupski MM. Combined-modality therapy in pancreatic cancer: current status and future directions. Cancer journal. 2001;7:338-48
8. Rich TA. Chemoradiation for pancreatic and biliary cancer: current status of RTOG studies. Annals of oncology: official journal of the European Society for Medical Oncology / ESMO. 1999;10(Suppl 4):231-3
9. Flanagan SA, Krokosky CM, Mannava S, Nikiforov MA, Shewach DS. MLH1 deficiency enhances radiosensitization with 5-fluorodeoxyuridine by increasing DNA mismatches. Molecular pharmacology. 2008;74:863-71
10. Longley DB, Harkin DP, Johnston PG. 5-fluorouracil: mechanisms of action and clinical strategies. Nature reviews Cancer. 2003;3:330-8
11. Johnston PG, Lenz HJ, Leichman CG, Danenberg KD, Allegra CJ, Danenberg PV. et al. Thymidylate synthase gene and protein expression correlate and are associated with response to 5-fluorouracil in human colorectal and gastric tumors. Cancer research. 1995;55:1407-12
12. Michishita E, Park JY, Burneskis JM, Barrett JC, Horikawa I. Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Molecular biology of the cell. 2005;16:4623-35
13. Finkel T, Deng CX, Mostoslavsky R. Recent progress in the biology and physiology of sirtuins. Nature. 2009;460:587-91
14. Zhang P, Tu B, Wang H, Cao Z, Tang M, Zhang C. et al. Tumor suppressor p53 cooperates with SIRT6 to regulate gluconeogenesis by promoting FoxO1 nuclear exclusion. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:10684-9
15. Cohen HY, Miller C, Bitterman KJ, Wall NR, Hekking B, Kessler B. et al. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science. 2004;305:390-2
16. Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y. et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011-5
17. Mao Z, Hine C, Tian X, Van Meter M, Au M, Vaidya A. et al. SIRT6 promotes DNA repair under stress by activating PARP1. Science. 2011;332:1443-6
18. Mostoslavsky R, Chua KF, Lombard DB, Pang WW, Fischer MR, Gellon L. et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell. 2006;124:315-29
19. Kaidi A, Weinert BT, Choudhary C, Jackson SP. Human SIRT6 promotes DNA end resection through CtIP deacetylation. Science. 2010;329:1348-53
20. McCord RA, Michishita E, Hong T, Berber E, Boxer LD, Kusumoto R. et al. SIRT6 stabilizes DNA-dependent protein kinase at chromatin for DNA double-strand break repair. Aging. 2009;1:109-21
21. Vazquez BN, Thackray JK, Simonet NG, Kane-Goldsmith N, Martinez-Redondo P, Nguyen T. et al. SIRT7 promotes genome integrity and modulates non-homologous end joining DNA repair. The EMBO journal. 2016;35:1488-503
22. Li L, Shi L, Yang S, Yan R, Zhang D, Yang J. et al. SIRT7 is a histone desuccinylase that functionally links to chromatin compaction and genome stability. Nature communications. 2016;7:12235
23. Dunne AL, Price ME, Mothersill C, McKeown SR, Robson T, Hirst DG. Relationship between clonogenic radiosensitivity, radiation-induced apoptosis and DNA damage/repair in human colon cancer cells. British journal of cancer. 2003;89:2277-83
24. Gerweck LE, Vijayappa S, Kurimasa A, Ogawa K, Chen DJ. Tumor cell radiosensitivity is a major determinant of tumor response to radiation. Cancer research. 2006;66:8352-5
25. Xie Y, Zhang J, Ye S, He M, Ren R, Yuan D. et al. SirT1 regulates radiosensitivity of hepatoma cells differently under normoxic and hypoxic conditions. Cancer science. 2012;103:1238-44
26. Vazquez BN, Thackray JK, Simonet NG, Kane-Goldsmith N, Martinez-Redondo P, Nguyen T. et al. SIRT7 promotes genome integrity and modulates non-homologous end joining DNA repair. The EMBO journal. 2016
27. Pinedo HM, Peters GF. Fluorouracil: biochemistry and pharmacology. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 1988;6:1653-64
28. Zhu WG, Hileman T, Ke Y, Wang P, Lu S, Duan W. et al. 5-aza-2'-deoxycytidine activates the p53/p21Waf1/Cip1 pathway to inhibit cell proliferation. The Journal of biological chemistry. 2004;279:15161-6
29. Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nature protocols. 2013;8:2281-308
30. Lee N, Kim DK, Kim ES, Park SJ, Kwon JH, Shin J. et al. Comparative interactomes of SIRT6 and SIRT7: Implication of functional links to aging. Proteomics. 2014;14:1610-22
31. Tsai YC, Greco TM, Boonmee A, Miteva Y, Cristea IM. Functional proteomics establishes the interaction of SIRT7 with chromatin remodeling complexes and expands its role in regulation of RNA polymerase I transcription. Mol Cell Proteomics. 2012;11:60-76
32. Craxton A, Butterworth M, Harper N, Fairall L, Schwabe J, Ciechanover A. et al. NOXA, a sensor of proteasome integrity, is degraded by 26S proteasomes by an ubiquitin-independent pathway that is blocked by MCL-1. Cell death and differentiation. 2012;19:1424-34
33. Pakay JL, Diesch J, Gilan O, Yip YY, Sayan E, Kolch W. et al. A 19S proteasomal subunit cooperates with an ERK MAPK-regulated degron to regulate accumulation of Fra-1 in tumour cells. Oncogene. 2012;31:1817-24
34. Crane CH, Janjan NA, Mason K, Milas L. Preoperative chemoradiation for locally advanced rectal cancer: emerging treatment strategies. Oncology. 2002;16:39-44
35. Salem ME, Hartley M, Unger K, Marshall JL. Neoadjuvant Combined-Modality Therapy for Locally Advanced Rectal Cancer and Its Future Direction. Oncology. 2016;30:546-62
36. Nelbock P, Dillon PJ, Perkins A, Rosen CA. A cDNA for a protein that interacts with the human immunodeficiency virus Tat transactivator. Science. 1990;248:1650-3
37. Corn PG, McDonald ER 3rd, Herman JG, El-Deiry WS. Tat-binding protein-1, a component of the 26S proteasome, contributes to the E3 ubiquitin ligase function of the von Hippel-Lindau protein. Nature genetics. 2003;35:229-37
38. Yu H, Ye W, Wu J, Meng X, Liu RY, Ying X. et al. Overexpression of sirt7 exhibits oncogenic property and serves as a prognostic factor in colorectal cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2014;20:3434-45
39. Zhang S, Chen P, Huang Z, Hu X, Chen M, Hu S. et al. Sirt7 promotes gastric cancer growth and inhibits apoptosis by epigenetically inhibiting miR-34a. Sci Rep. 2015;5:9787
40. Barber MF, Michishita-Kioi E, Xi Y, Tasselli L, Kioi M, Moqtaderi Z. et al. SIRT7 links H3K18 deacetylation to maintenance of oncogenic transformation. Nature. 2012;487:114-8
41. Tsai YC, Greco TM, Cristea IM. Sirtuin 7 plays a role in ribosome biogenesis and protein synthesis. Mol Cell Proteomics. 2014;13:73-83
42. Ford E, Voit R, Liszt G, Magin C, Grummt I, Guarente L. Mammalian Sir2 homolog SIRT7 is an activator of RNA polymerase I transcription. Genes & development. 2006;20:1075-80
43. Ryu D, Jo YS, Lo Sasso G, Stein S, Zhang H, Perino A. et al. A SIRT7-dependent acetylation switch of GABPbeta1 controls mitochondrial function. Cell metabolism. 2014;20:856-69
44. Mohrin M, Shin J, Liu Y, Brown K, Luo H, Xi Y. et al. Stem cell aging. A mitochondrial UPR-mediated metabolic checkpoint regulates hematopoietic stem cell aging. Science. 2015;347:1374-7
45. Kiran S, Oddi V, Ramakrishna G. Sirtuin 7 promotes cellular survival following genomic stress by attenuation of DNA damage, SAPK activation and p53 response. Experimental cell research. 2015;331:123-41
Author contact
![]() Corresponding authors: Wei-Guo Zhu, Tel: 0086-10-82802235; Fax: 86-10-82805079; Email: zhuweiguoedu.cn or zhuweiguoedu.cn or Haiying Wang at wendyedu.cn
Corresponding authors: Wei-Guo Zhu, Tel: 0086-10-82802235; Fax: 86-10-82805079; Email: zhuweiguoedu.cn or zhuweiguoedu.cn or Haiying Wang at wendyedu.cn