Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Conclusion
Materials and Methods
Supplementary Material
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(4):974-986. doi:10.7150/thno.17830 This issue Cite
Research Paper
Integration of Receptor Tyrosine Kinases Determines Sensitivity to PI3Kα-selective Inhibitors in Breast Cancer
Yi-chao Xu1,2, Xiang Wang1, Yi Chen1, Si-meng Chen1, Xin-ying Yang1, Yi-ming Sun1, Mei-yu Geng1, Jian Ding1,2 ![]() , Ling-hua Meng1,2
, Ling-hua Meng1,2 ![]()
1. Division of Anti-tumor Pharmacology, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, China.
2. University of Chinese Academy of Sciences, Beijing, China.
Received 2016-10-6; Accepted 2016-12-8; Published 2017-2-23
Abstract

PI3Kα-selective inhibitor BYL719 is currently in phase II/III clinical trial for the treatment of breast cancer, but highly variable response has been observed among patients. We sought to discover predictive biomarker for the efficacy of BYL719 by dissecting the proliferative signaling pathway mediated by PI3K in breast cancer. BYL719 concurrently inhibited the phosphorylation of AKT and ERK in PIK3CA-mutated human breast cancer cells. PI3K-regulated ERK phosphorylation was independent of canonical PDK1/AKT/mTOR pathway, while it was associated with RAF/MEK. Hyper-activation of EGFR or RAS abrogated inhibition of ERK phosphorylation by BYL719. Furthermore, hyper-activation of receptor tyrosine kinases (RTKs) including EGFR, c-MET, FGFR and HER3 but not IGF-1R restored ERK phosphorylation and cell viability suppressed by BYL719, suggesting the discriminative functions of RTKs in cell signaling and proliferation. By profiling 22 breast cancer cell lines, we found that BYL719 was more potent in cell lines where phosphorylation of both AKT and ERK was attenuated than those where only AKT phosphorylation was inhibited. The potency of BYL719 was further found to be significantly correlated with the expression profile of RTKs in breast cancer cells. Specifically, overexpression of EGFR, c-MET and/or FGFR1 forecasted resistance, while overexpression of IGF-1R and/or HER2 predicted sensitivity to BYL719 in breast cancer cells. Similar correlation between BYL719 efficacy and expression profile of RTKs was found in patient-derived xenograft models of breast cancer. Thus, inhibition of ERK phosphorylation by PI3Kα inhibitor BYL719 contributes to its antitumor efficacy and is determined by the converged signaling from RTKs. The expression profile of RTKs in breast cancer tissue could be potentially developed as a predictive biomarker for the efficacy of PI3Kα inhibitors.
Keywords: BYL719, PI3K, ERK, receptor tyrosine kinase, breast cancer, predictive biomarker.
Introduction
Phosphatidylinositol 3-kinases (PI3Ks) integrate signals from diversified environmental cues and play important roles in multiple cellular processes including survival, growth and metabolism. Aberrant activation of PI3K signaling pathway occurs frequently in breast cancer through different mechanisms such as hyper-activation of upstream receptor tyrosine kinases (RTKs), mutant RAS, mutations and/or amplifications of PI3K and downstream effector AKT, and functional loss of PTEN or INPP4B. Specifically, PIK3CA, which encodes the p110α isoform, is the most frequently mutant oncogene with a frequency of more than one third in breast cancer (1). Because PI3Kα plays an important role in tumorigenesis and development, and specific targeting PI3Kα may improve therapeutic outcomes and diminish the side effects caused by inhibiting other isoforms of class I PI3K. PI3Kα has been validated as a promising target for cancer therapy and several PI3Kα-selective inhibitors have entered clinical trials (2).
BYL719 is one of the most advanced PI3Kα-specific inhibitor, which is currently in phase II/III clinical trial for the treatment of various types of breast cancer (https://www.clinicaltrials.gov). BYL719 displays a favorable safety profile, and tumor regression and prolonged disease control are observed in heavily pretreated patients with various tumor types carrying PIK3CA mutation in a phase I study (3, 4). However, a phase Ib study with BYL719 and letrozole for therapy of ER-positive breast cancer demonstrates that the efficacy is not restricted to patients with PIK3CA mutant tumors (5).
Breast cancer contains a group of highly heterogeneous tumors with diversified molecular features. By deep sequencing 100 tumor samples from different type of breast cancers, driver mutations in at least 40 cancer genes and 73 different combinations of mutated cancer genes have been identified (1), highlighting the complexity of the driving force of tumor development. PI3K pathway is frequently hyper-activated not only in ER-positive breast cancers but also in other types of breast cancer, which validates PI3K as a promising target for breast cancer therapy. On the other hand, multiple cancer driver genes existing in breast cancer may affect the efficacy of PI3K inhibitors. It is of great importance to understand the genetic context, where PI3K inhibitors would execute their activity alone or in combination with other regimen. Since PI3K and MAPK pathway represent the most important pathways mediating cell survival and proliferation (6), we dissected the effect of PI3Kα inhibitor, specifically BYL719, on the PI3K and MAPK signaling in breast cancer cells and investigated the impact of multiple RTKs on the activity of PI3Kα inhibitor in cell signaling as well as cell proliferation. We proposed that converged activity of RTKs in breast cancer would determine the efficacy of PI3Kα inhibitors.
Results
PI3K inhibitors concurrently down-regulate phosphorylated AKT and ERK in T47D and MCF-7 cells
Feedback up-regulation of MAPK pathway has been reported to alleviate the antitumor potency of PI3K inhibitors (7, 8). In an effort to elucidate the mechanism of variable response to PI3K inhibitors in breast cancer patients, we investigated the PI3K and MAPK signaling pathway upon PI3K inhibition in human breast cancer cells. BYL719 treatment for 1 h concentration-dependently decreased the level of phosphorylated AKT at both Ser473 and Thr308 in T47D cells (Fig. 1A), which harbor PIK3CA H1047R mutation and had been reported to be highly sensitive to PI3K inhibition (9). In addition, BYL719 concurrently inhibited phosphorylation of ERK, though MAPK was not the classic downstream effector of PI3K. Similar results were obtained with GDC0941, a potent inhibitor of class I PI3Ks (Fig. 1A) (10). BYL719 and GDC0941 also inhibited phosphorylation of AKT and ERK in MCF-7 cells, which harbor PIK3CA E545K mutation (Fig. S1). It has been reported that durable inhibition of PI3K causes feedback including up-regulation of expression and phosphorylation of multiple RTKs, which leads to reactivation of downstream AKT and MAPK signaling pathway (8). We found that BYL719 or GDC0941 strongly inhibited phosphorylation of AKT and ERK upon 1-h treatment, and the inhibition sustained for 72 h (Fig. 1B). These results indicated that BYL719 and GDC0941 simultaneously inhibited phosphorylation of AKT and ERK in breast cancer cells harboring activating PIK3CA alteration.
PI3K inhibitors concurrently down-regulate phosphorylated AKT and ERK. T47D cells were treated with BYL719 or GDC0941 for 1 h (A) or at 1 μM for indicated times (B). (C) T47D cells were treated with PI103 (1 μM), GDC0941 (0.3 μM), BYL719 (0.3 μM), A66 (1 μM), AZD6482 (3.3 μM), GSK2636771 (10 μM) or TGX221 (10 μM) for 1 h. (D) T47D cells were transfected with indicated siRNA for 72 h. Representative immunoblots for indicated proteins from at least three independent experiments are presented.
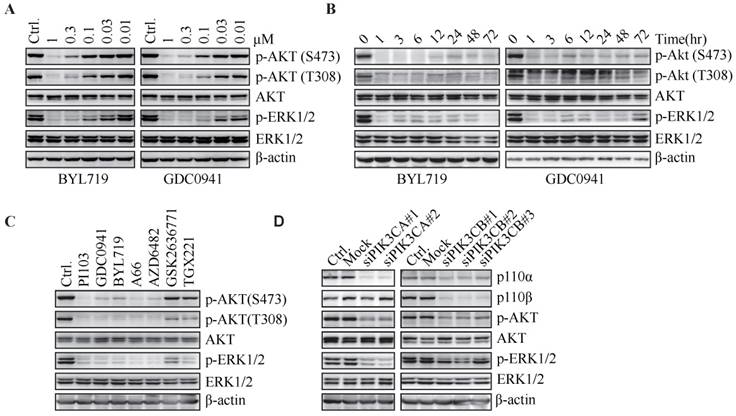
To investigate whether inhibition of ERK phosphorylation is a universal phenomenon upon blockade of PI3K, a series of PI3K inhibitors, including PI103 (a PI3K/mTOR dual inhibitor), A66 (a p110α-specific inhibitor), as well as three PI3Kβ-selective inhibitors (AZD6482, GSK2636771 and TGX221), were administrated to T47D cells. PI103, A66 and AZD6482 suppressed phosphorylation of AKT and ERK at the concentration to inhibit the proliferation of T47D cells by 50% (Fig. 1C). However, GSK2636771 and TGX221 had little effect on the level of phosphorylated AKT or ERK, which was consistent with the result that GSK2636771 and TGX221 failed to inhibit the proliferation of T47D cells at the same concentration (data not shown). The disparity in response of T47D cells to three PI3Kβ-specific inhibitors might reflect their different profiles in selectivity against PI3K isoforms. To further investigate which PI3K isoform was responsible for the phosphorylation of AKT and ERK in T47D cells, p110α or p110β was down-regulated by respective siRNAs. Decrease in p110α isoform significantly inhibited phosphorylation of AKT and ERK, whereas knockdown of p110β isoform had little effect on this process (Fig. 1D), indicating that PI3Kα played an important role in phosphorylation of AKT and ERK in T47D cells.
Hyper-activation of RAF/MEK overrides inhibition of ERK phosphorylation by PI3K blockade
To investigate the role of canonical PDK1/AKT/mTOR signaling cascade in PI3K-regulated ERK phosphorylation, T47D and MCF-7 cells were exposed to a series of inhibitors specifically targeting PDK1 (GSK2334470), AKT (MK2206 and GSK690693) or mTOR (AZD8055 and PP242). GSK2334470, MK2206 and GSK690693 inhibited their respective targets as described previously (11-13), while slightly enhanced the phosphorylation of ERK (Fig. 2A). Both AZD8055 and PP242 slightly elevated ERK phosphorylation as they abrogated phosphorylation of AKT and PRAS40 (Fig. 2A). Therefore, PI3K regulated ERK phosphorylation independent of PDK1/AKT/mTOR cascade.
Hyper-activation of RAF/MEK overrides inhibition of ERK phosphorylation by PI3K blockade. (A) Cells were treated with GSK2334470 (1 μM), MK2206 (1 μM), GSK690693 (1 μM), AZD8055 (0.1 μM) or PP242 (0.1 μM) for 1 h. (B) Cells were treated with SB590885 (10 μM), U0126 (1 μM) or AZD6244 (5 μM) for 1 h. (C) Cells stably expressing pBabe-BAF (V600E) were treated with BYL719 (1 μM), GDC0941 (1 μM) or SB590885 (10 μM) for 1 h. (D) Cells stably expressing MEK1 (S218D, S222D) were treated with BYL719 (1 μM), GDC0941 (1 μM) or U0126 (1 μM) for 1 h. Representative immunoblots for indicated proteins from at least three independent experiments are presented.
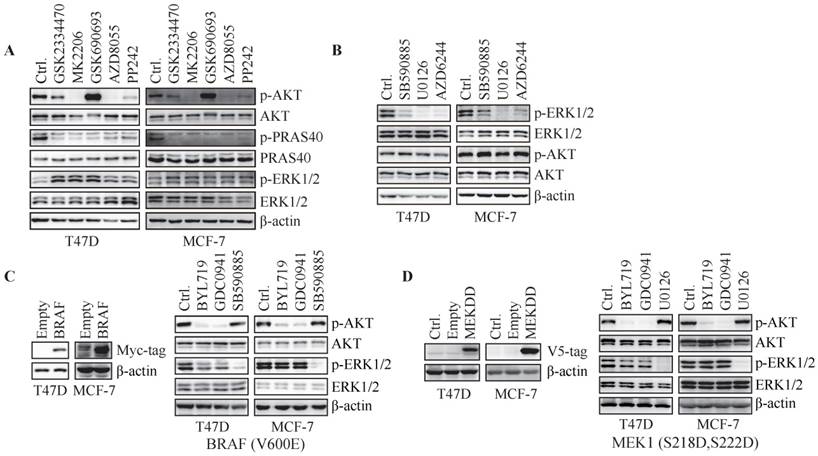
To investigate whether phosphorylation of ERK is regulated by RAF/MEK pathway, T47D and MCF-7 cells were treated with inhibitors specifically targeting RAF (SB590885) or MEK (U0126 and AZD6244) for 1 h. Inhibition of RAF or MEK resulted in reduced ERK phosphorylation in both cell lines (Fig. 2B). We next transfected T47D and MCF-7 cells with constitutively active BRAF(V600E). BYL719 and GDC0941 markedly inhibited phosphorylation of AKT but not ERK in cells over-expressing BRAF (Fig. 2C). SB590885 suppressed phosphorylation of ERK but not AKT under the same condition (Fig. 2C). Similarly, overexpression of constitutively active MEK1 (S218D, S222D) in T47D and MCF-7 cells abrogated inhibition of ERK phosphorylation by PI3K inhibitors (Fig. 2D). Thus, RAF/MEK regulated ERK phosphorylation downstream of PI3K in T47D and MCF-7 cells and hyper-activation of RAF/MEK overrode inhibition of ERK phosphorylation by PI3K blockade.
Hyper-activation of RAS or EGFR abrogates PI3K-regulated ERK phosphorylation in T47D and MCF-7 cells
As RAS regulates RAF/MEK/ERK signaling pathway, we further investigated the role of RAS in PI3K-regulated ERK phosphorylation. Both BYL719 and GDC0941 had no effect on the level of GTP-bound RAS that is the active form of RAS, while phosphorylation of AKT and ERK was blocked (Fig. 3A), suggesting PI3K-dependent ERK phosphorylation may not be mediated by RAS. We next over-expressed constitutively active HRAS (G12V) in T47D cells and found increased phosphorylated ERK compared to that in cells transfected with empty vector (Fig. 3B). BYL719 and GDC0941 failed to inhibit phosphorylation of ERK in T47D cells over-expressing HRAS (G12V) (Fig. 3B), indicating that PI3K and RAS may regulate ERK phosphorylation in parallel. Similar results were observed in MCF-7 cells (Fig. S2A and B).
Hyper-activation of RAS or EGFR abrogates PI3K-regulated ERK phosphorylation. (A) T47D cells were treated with BYL719 (1 μM) or GDC0941 (1 μM) for 1 h. RAS-GTP were pulled down using GST-RAF1-RBD agarose. (B) T47D cells stably expressing empty vector or HRAS (G12V) were treated with BYL719 (1 μM) or GDC0941 (1 μM) for 1 h. (C) Cell lysate of T47D cells was applied to phospho-RTK array. (D) T47D cells stably transfected with empty vector, wild type EGFR or EGFR (L858R) were treated with BYL719 (1 μM), GDC0941 (1 μM) or BIBW2992 (1 μM) for 1 h. (E) FBS-starved T47D cells were treated with BYL719 (1 μM) or BIBW2992 (1 μM) for 1 h and then stimulated with EGF (50 ng/ml) for 15 min. Representative immunoblots for indicated proteins from at least three independent experiments are presented.
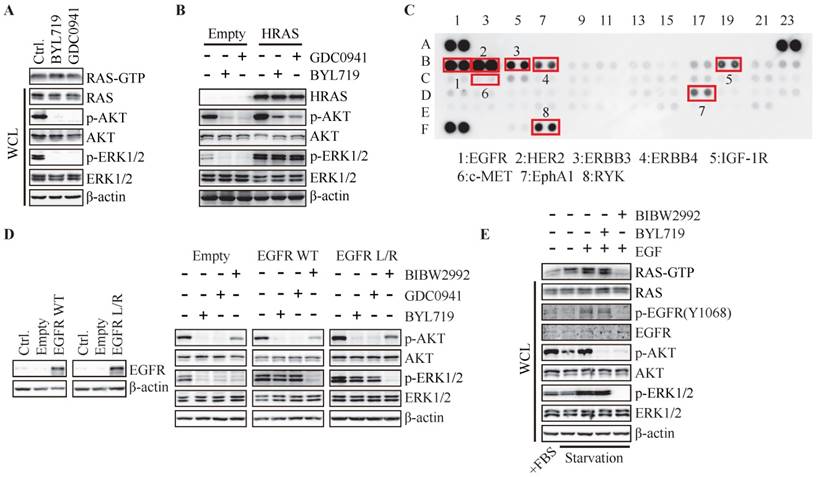
RAS and PI3K are well known as the key intracellular transducer of multiple RTKs, and signals from RTKs affect the crosstalk between RAS/MAPK and PI3K pathways (14, 15). We measured the basal phosphorylated level of RTKs in T47D cells via phospho-RTK array and found ERBB family, IGF-1R, RYK and EphA1 were highly phosphorylated in T47D cells (Fig. 3C). Considering the highly phosphorylated level of EGFR in breast cancer and its important function in RAS/MAPK pathway, we investigated the impact of EGFR on PI3K-dependent ERK phosphorylation by over-expressing wild-type or L858R-mutated EGFR in T47D cells. As shown in Fig. 3D, BYL719 and GDC0941 inhibited phosphorylation of AKT and ERK in T47D cells transfected with empty vector, while phosphorylation of AKT but not ERK was inhibited in cells over-expressing wild-type or mutated EGFR. EGFR inhibitor BIBW2992 was employed as a positive control to inhibit phosphorylation of both AKT and ERK (Fig. 3D). These results indicated that hyper-activation of EGFR abrogated dependence of ERK phosphorylation on PI3K. EGFR was further activated by EGF in T47D cells following serum-starvation for 24 h, which was accompanied with elevated GTP-bound RAS, phosphorylated AKT and ERK (Fig. 3E), demonstrating that EGFR transduced signaling to both PI3K/AKT and RAS/MAPK pathways. Consistently, BIBW2992 blocked both pathways induced by EGF (Fig. 3E). However, BYL719 had no effect on the level of Ras-GTP and phosphorylated ERK while inhibited AKT phosphorylation (Fig. 3E). Similar results were observed in MCF-7 cells (Fig. S2C). Therefore, hyper-activation of RAS or its upstream EGFR in T47D and MCF-7 cells may render the dependency of ERK activation to switch to RAS or EGFR.
The growth factors distinctly determine PI3K-dependent ERK phosphorylation in breast cancer cells
Given hyper-activation of EGFR abrogated inhibition of ERK phosphorylation by PI3K blockade, we attempted to investigate the effect of other RTKs on PI3K-regulated ERK phosphorylation. Except EGFR, IGF-1R, HER2, HER3, c-MET and FGFR are reported to be frequently amplified and/or overexpressed in breast cancer (Fig. S3). We examined the activation of PI3K and MAPK pathways after stimulating IGF-1R, EGFR, HER3, c-MET or FGFR with their respective ligands. IGF, EGF and NRG1 significantly elevated the phosphorylation of AKT and ERK, while HGF and FGF had little effect on this process, suggesting the abundance of IGF-1R, EGFR and HER3 in T47D cells (Fig. 4A). Inhibition of EGFR (BIBW2992) or HER3 (AZD8931) significantly reduced the phosphorylation of AKT and ERK, indicating the important role of EGFR and HER3 in PI3K and MAPK signaling (Fig. 4B). IGF-1R inhibitor OSI906 partially inhibited phosphorylation of AKT and ERK, while inhibition of c-MET (INCB28060) and FGFR (AZD4547) had no effect on AKT and ERK phosphorylation (Fig. 4B), suggesting minor effect of c-MET and FGFR on the signaling of PI3K and MAPK in T47D cells.
The growth factors distinctly determine PI3K-regulated ERK phosphorylation in breast cancer cells. T47D cells were stimulated with indicated growth factors (50 ng/mL) for 15 min (A) or treated with indicated inhibitors of receptor tyrosine kinases (1 μM) for 1 h (B). Phosphorylation of AKT and ERK was detected with immunoblot and quantified. Bars, mean ± SEM. ** P < 0.01; *** P < 0.001; **** P < 0.0001, compared with sample in control group using two way ANOVA analysis. (C) Starved T47D cells were treated with BYL719 (1 μM) or respective inhibitor of RTKs (1 μM) for 1 h and then stimulated with indicated growth factors (50 ng/mL) for 15 min. Phosphorylated AKT and ERK in the presence of BYL719 were quantified and plotted. Bars, mean ± SEM. **P < 0.01; ***P < 0.001, compared with respective control group using paired t test. (D) Cell viability of T47D cells treated with BYL719 alone or concurrently with indicated growth factors (50 ng/mL) for 72 h was measured. Bars, mean ± SEM. **P < 0.01; ***P < 0.001, compared with respective control group using the paired t test.
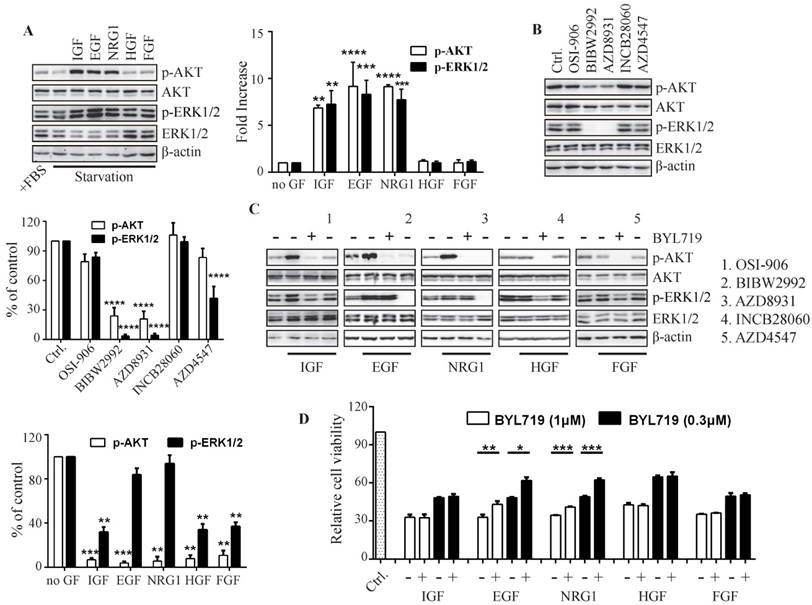
We next detected the effect of BYL719 on the growth factor-induced activation of PI3K and MAPK pathways (Fig. 4C). Pre-incubation of T47D cells with BYL719 significantly inhibited AKT and ERK phosphorylation induced by IGF, demonstrating that IGF-1R-transduced signaling was mostly mediated by PI3K. However, BYL719 had little effect on ERK phosphorylation while it reduced AKT phosphorylation in the presence of EGF or NRG1, suggesting that EGFR and HER3 may transduce signals to both PI3K and MAPK pathways. In consistency with the results in Fig. 4A, HGF and FGF failed to either activate PI3K and MAPK pathway or abrogate the inhibition of AKT and ERK phosphorylation by BYL719.
To further investigate the contribution of ERK activation to cell growth, T47D cells were exposed to BYL719 alone or concurrently with various growth factors (Fig. 4D). EGF or NRG1 significantly restored the growth of T47D cells impaired by BYL719, which was in agreement with the fact both EGF and NRG1 rescued ERK phosphorylation suppressed by BYL719. IGF, HGF or FGF had no effects on the activity of BYL719 against cell proliferation, which also failed to abrogate inhibition of ERK phosphorylation by BYL719 (Fig. 4C). Thus, ERK inhibition was necessary for the anti-proliferative activity of BYL719.
PI3K pathway is hyper-activated in MCF-7 and HCC1954 cells harboring PIK3CA E545K or H1047R mutation and we found that BYL719 inhibited phosphorylation of AKT and ERK in both cell lines (Fig. S1 & Fig. 5B). We next investigated whether RTKs were able to override PI3K-regulated ERK phosphorylation in different PIK3CA-mutated cell lines. Phosphorylated RTKs were detected with phospho-RTK assay in MCF-7 and HCC1954 cells. As shown in Fig. S4A, EGFR, HER3, IGF-1R and EphA1 were highly phosphorylated in MCF-7 cells, while EGFR, HER2, HER3, IGF-1R and c-MET were highly phosphorylated in HCC1954 cells. In MCF-7 cells, growth factors including EGF, NRG1 or FGF significantly up-regulated phosphorylation of AKT and ERK, while IGF enhanced the phosphorylation of AKT but not ERK (Fig. S4B). Meanwhile, supplement of EGF, NRG1 or HGF increased phosphorylation of AKT and ERK in HCC1954 cells (Fig. S4B). Accordingly, inhibition of EGFR, HER3 and FGFR in MCF-7 cells or inhibition of EGFR and HER3 in HCC1954 cells blocked phosphorylation of both AKT and ERK (Fig. S4C). Furthermore, supplement of EGF, NRG1 or FGF in MCF-7 cells, while supplement of EGF, NRG1 or HGF in HCC1954 cells abrogated inhibition of ERK phosphorylation as well as cell growth upon BYL719 treatment (Fig. S4D & E). These results suggested that differential expression of RTKs in different cell lines would affect the efficacy of PI3K inhibitors against cell signaling as well as cell proliferation.
The expression profile of RTKs determines PI3K-mediated signaling in breast cancer cells. (A) Immunoblots showing expression levels of indicated RTKs in 22 breast cancer cell lines. (B) Cells were treated with BYL719 for 1 h followed by Western blot analysis. (C) Scatterplot of expression level of indicated RTKs in breast cancer cells. Bars, mean ± SEM. * p < 0.05; ** p < 0.01, unpaired t test was performed.
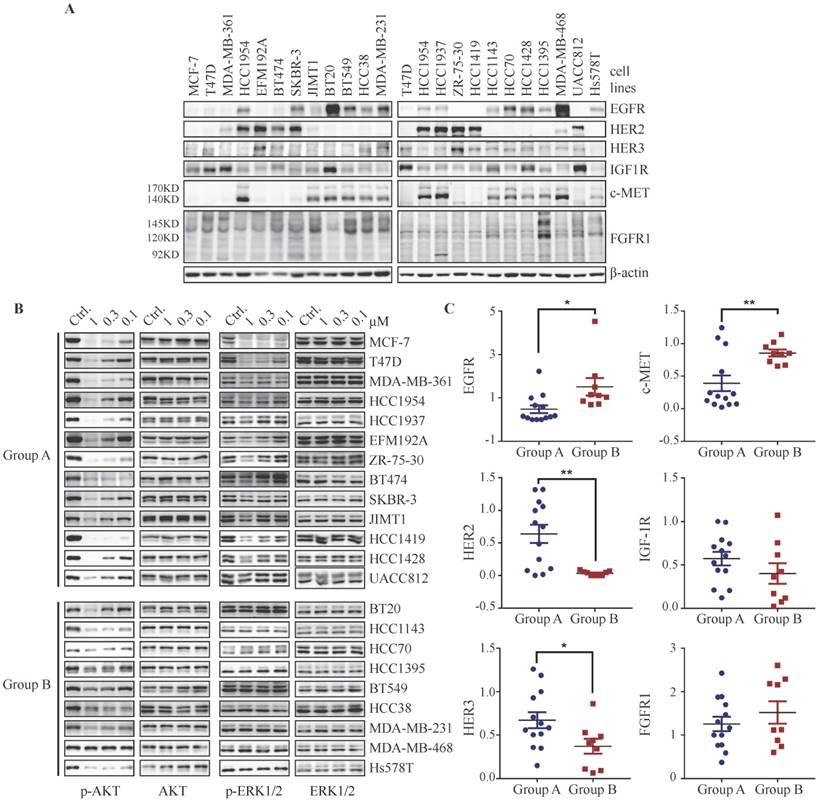
The disparity of RTKs expression profile determines PI3K-mediated signaling in breast cancer cells
Considering that PI3K-regulated ERK phosphorylation could be overrode by activation of RTKs such as EGFR, FGFR or c-MET in T47D, MCF-7 and HCC1954 cells, we attempted to investigate whether basal expression level of RTKs in breast cancer cells would affect the regulation of PI3K on ERK phosphorylation. We measured the basal protein level of EGFR, HER2, HER3, IGF-1R, c-MET and FGFR1 in a panel of 22 breast cancer cell lines. Each breast cancer cell line displays a distinct expression profile of RTKs (Fig. 5A). We next investigated the effect of BYL719 on the phosphorylation of AKT and ERK in these cells. As shown in Fig. 5B, this panel of breast cancer cells could be divided into two groups, i.e. Group A and Group B, according to their response to BYL719. In Group A, phosphorylation of both AKT and ERK was attenuated by BYL719, while phosphorylation of AKT but not ERK was blocked in Group B. To explore the association between expression profile of RTKs and PI3K-regulated ERK phosphorylation in breast cancer cells, we quantified the expression level of RTKs and found that the expression levels of EGFR and c-MET were significantly higher in Group B cells, where ERK phosphorylation was resistant to BYL719 (Fig. 5C). On the other hand, the expression level of HER2 was significantly higher in Group A cells, where phosphorylation of both AKT and ERK was inhibited by BYL719. There was no significant difference in IGF-1R expression between Group A and B cells. It is note-worthy that high expression levels of EGFR or c-MET may override PI3K-regulated ERK phosphorylation in BT20 cells (Fig. 5A). The expression level of IGF-1R was significantly higher in Group A cells if cell line BT20 was omitted (Fig. S5A). Though NRG1 abrogated suppression of ERK phosphorylation by PI3K inhibitor, the expression level of HER3 was significantly higher in Group A cells than that in Group B cells. There was no significant difference of FGFR1 expression between Group A and Group B cells, though expression level of FGFR1 was relatively higher in Group B cells. Altogether, signals from multiple RTKs convergently determined the phosphorylation of ERK in breast cancer cells, where high expression of EGFR or c-MET indicated independence of PI3K, while high expression of IGF-1R or HER2 indicated dependence of PI3K.
Down-regulation of ERK phosphorylation by PI3K blockade indicates sensitivity to PI3K inhibitor in breast cancer cells
As both PI3K and MAPK pathways play important roles in cell proliferation in breast cancer, we investigated the association between down-regulation of ERK signaling by BYL719 and its anti-proliferative activity in 22 breast cancer cell lines. The cell lines tested displayed a diversified sensitivity to BYL719 with IC50s ranging from 0.41 µmol/L to over 10 µmol/L (Fig. 6A). PIK3CA-mutated cells were significantly more sensitive to BYL719 than those with wild-type PIK3CA, while cells with PTEN alteration tend to be resistant to BYL719 (Fig. S6A & B). However, we noticed that PTEN-null HCC1937 cells harboring wild type PIK3CA were sensitive to BYL719, while HCC38 cells with PIK3CA mutation were resistant to BYL719, indicating that PIK3CA mutation or PTEN alteration alone failed to predict the sensitivity of BYL719 in breast cancer. Given inhibition of ERK phosphorylation was necessary for the maximal anti-proliferative activity of BYL719 in T47D, MCF-7 and HCC1954 cells, we plotted the IC50s of the tested cell lines and grouped them as shown in Figure 5B. As shown in Fig. 6B, IC50s obtained in Group A cells were significantly lower than those obtained in Group B cells, indicating that simultaneously inhibition of AKT and ERK sensitized cells to PI3Kα inhibitors. As we had shown that ERK signaling was determined by multiple RTKs, we attempted to investigate the correlation between expression level of RTKs and sensitivity to BYL719 in breast cancer cells. Bivariate correlation analysis between single RTK and IC50 values was performed due to the limited number of samples (Fig. S6C). Expression of EGFR or c-MET was positively correlated with IC50 values, suggesting that high expression of EGFR or c-MET would render breast cancer cells resistant to BYL719. On the other hand, expression of HER2 or IGF-1R was significantly negatively correlated with IC50 values in the same panel of breast cancer cells, indicating high expression of HER2 or IGF-1R would predict sensitivity to BYL719. There was no significant correlation between HER3 or FGFR1 and IC50 values. These results demonstrated that converged RTKs determined whether ERK phosphorylation was dependent on PI3K, which then determined the anti-proliferative activity of PI3Kα inhibitor in breast cancer cells.
Down-regulation of ERK phosphorylation indicates sensitivity to PI3K inhibitor in breast cancer cells. (A) IC50s of BYL719 in 22 breast cancer cell lines were determined with SRB assay. Bars, mean ± SEM.(B) Scatterplot of IC50s in breast cancer cells of Group A and B. Bars, mean ± SEM. Difference between Group A and B was analyzed with unpaired t test. **** p < 0.0001. (C) Expression of indicated RTKs in tumor tissues from PDX models was detected with IHC. Original magnification: ×200. (D) Quantitative heatmap showing expression level of RTKs in the PDX models. (E) Mice bearing PDX6717, PDX6254 or PDX6648 xenografts were treated with vehicle control or 50 mg/kg BYL719 once a day for 14 days. Relative tumor volume was measured and plotted. Data shown are mean ± SEM. (F) A proposed scheme representing that differential expression profile of RTKs determines the signaling pathway and cell proliferation.
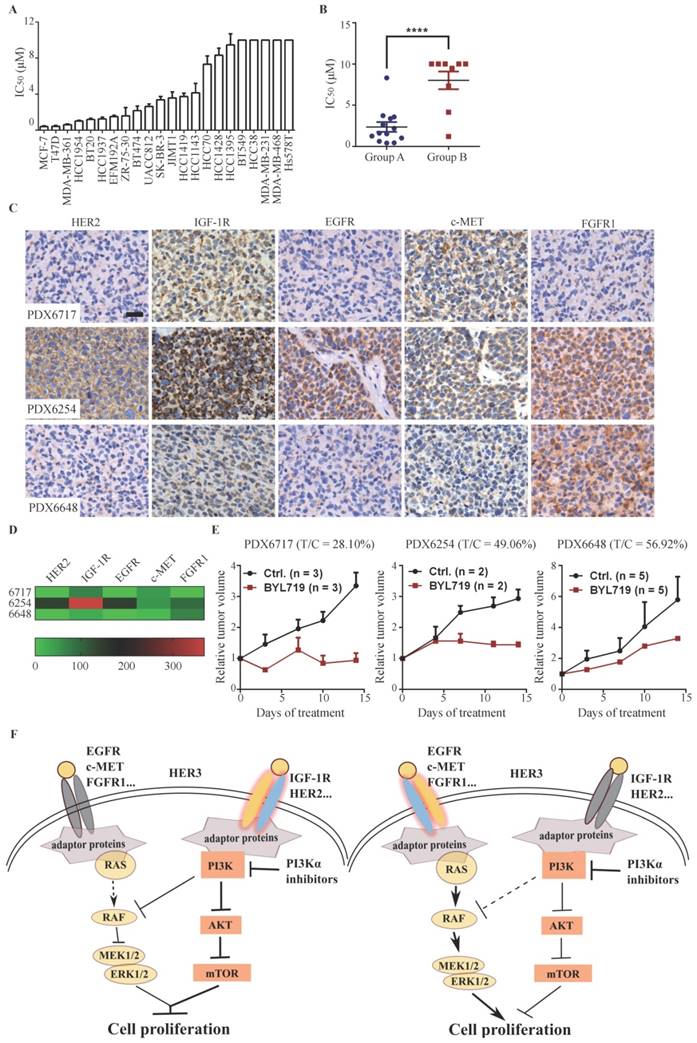
To examine the relationship between the expression profile of RTKs in patient breast cancer tissues and BYL719 efficacy, we examined the basal level expression of EGFR, HER2, IGF-1R, FGFR1 and c-MET by immunohistochemistry in a panel of 28 patient-derived xenograft (PDX) tissues. In consistency with the observation in breast cancer cells, each PDX tissue displayed a distinct profile of RTKs expression, indicating the heterogeneity of breast cancer (Fig. S6D). We randomly selected three PDX models and investigated the anti-tumor activity of BYL719. As shown in Fig. 6D&E, BYL719 (50 mg/kg) significantly inhibited the growth of PDX6717 with a T/C value of 28.10%, which expressed high level of IGF-1R but low levels of other detected RTKs. PDX6254, which expressed high level of EGFR, HER2 and IGF-1R, was less sensitive to BYL719 (T/C: 49.06%). Conversely, PDX6648 with high expression of FGFR1 was resistant to BYL719 treatment with a T/C value of 56.92%. Though the number of tested PDX models is limited, these results are consistent with those obtained in breast cancer cells, suggesting that high expression of HER2 and IGF-1R in breast cancer may predict sensitivity, while high expression of FGFR may predict resistance to PI3K inhibition.
Discussion
PI3Kα inhibitor BYL719 displays promising efficacy in advanced breast cancers, variable response to PI3Kα inhibitors provokes the demand to identify the predictive biomarker(s) (4, 5). In this study, we discovered that PI3K regulated ERK phosphorylation via RAF/MEK pathway independent of the canonical PI3K/AKT/mTOR signaling in PIK3CA-mutated breast cancer cells. However, dependence of ERK phosphorylation on PI3K switched to RAS or EGFR if hyper-activated RAS or EGFR was introduced into the same cell lines. By profiling expression of RTKs and cellular response to BYL719 in a panel of breast cancer cells and three PDX models of breast cancer, we found that high expression of HER2 and/or IGF-1R in breast cancer cells rendered dependence of ERK activation on PI3K and cells tended to be sensitive to PI3Kα inhibitors. On the other hand, high expression of EGFR, c-MET and/or FGFR1 conferred independence of ERK activation on PI3K and cells tended to be resistant to PI3Κα inhibitors (Fig. 6F).
We observed that blockade of PI3K, especially PI3Kα, resulted in decrease of phosphorylated AKT and ERK in PIK3CA mutant cells, which was in accordance with previous studies (16-19). It has been reported that PI3K but not AKT inhibition causes rapid inhibition of wild-type RAS followed by RAF/MEK/ERK signaling (19). However, PI3K blockade had no effect on RAS activity in our study. We found that PI3K-regulated ERK phosphorylation was independent of AKT and mTOR, which was consistent with the previous report (18). Ebi et al reported that PI3K inhibition led to a rapid suppression of Rac1/p21-activated kinase (PAK)/c-RAF/MEK/ERK signaling independent of RAS (18). Though we failed to observe inactivation of RAC upon PI3K inhibition (Fig. S2D), we did prove that PI3K regulated ERK phosphorylation via RAF/MEK signaling. These appearing conflicting results may reflect different cellular and experimental contexts. We found that over-expression of RAS abrogated PI3K-regulated ERK phosphorylation, revealing the converged role of PI3K and RAS in regulating ERK phosphorylation. Furthermore, activation of EGFR, HER3, c-MET or FGFR but not IGF-1R abrogated inhibition of ERK phosphorylation upon PI3K blockade, suggesting that integrated role of RTKs in regulating ERK phosphorylation. By profiling the basal level expression of RTKs and ERK phosphorylation upon PI3K inhibition in 22 breast cancer cell lines, we found that over-expression of EGFR, FGFR1 and/or c-MET but not IGF-1R predicted independence of ERK phosphorylation on PI3K, further revealing the complexity in regulating PI3K-dependent ERK signaling in breast cancers.
AKT and MAPK are important pathways to regulate cell survival and proliferation. Lack of efficacy of PI3K inhibitors in some types of cancer has been attributed to failure to block MAPK pathway and combination of PI3K and MEK inhibitor displays synergistic activity against mutant KRAS and PI3K murine lung cancers (20). We found that PI3Kα inhibitor was able to simultaneously inhibit phosphorylation of AKT and ERK in some types of breast cancer cells, which was consistent with the fact that these cells were sensitive to BYL719. Thus, simultaneous inactivation of AKT and MAPK could serve as a potential pharmacodynamic marker to monitor the efficacy of PI3Kα inhibitor in the breast cancer therapy. Interestingly, concurrent inhibition of AKT and ERK also indicated sensitivity of human primary leukemia cells to PI3Kδ inhibitors (21). Since integrated signals from multiple RTKs determined the dependence of ERK phosphorylation on PI3K, we proposed that the expression profile of RTKs may predict the efficacy of PI3Kα inhibitor in breast cancer therapy. Indeed, the expression of EGFR or c-MET was negatively correlated with the activity of BYL719 against a panel of 22 breast cancer cells, while expression of HER2 or IGF-1R was positively correlated with the activity of BYL719. Similar correlation was also observed in three PDX models of breast cancer. We failed to observe significant correlation between the expression of FGFR1 and BYL719 activity, which might be due to limited number of cell lines. Still, FGFR1 expression was slightly higher in cells where phosphorylation of ERK was resistant to BYL719, which was consistent with a recent report that over-expression of FGFR1 in breast cancer exhibited resistance to BYL719 (5). In this scenario, combination of FGFR inhibitor sensitizes FGFR1 over-expressing cells to BYL719 (5).
Conclusion
In this study, we revealed that integrated signals from RTKs determined the regulation of ERK phosphorylation by PI3K, which determined the efficacy of BYL719 against breast cancer. This observation suggests that expression profile of RTKs would potentially be developed as an integrative biomarker to predict the efficacy of PI3Kα inhibitor in the therapy of breast cancer. However, multivariate analysis of the correlation of RTK signature and drug efficacy based on the results from expanded samples in vivo and in clinical settings is warranted to achieve precise prediction.
Materials and Methods
Regents
U0126 was purchased from Beyotime (Shanghai, China). All other compounds used in this study were purchased from Selleckchem (Houston, USA). All the compounds were dissolved at 10 mM in DMSO and stored at -20°C. IGF, FGFα and HGF were purchased from Prepro Tech (New Jersey Rocky Hill, USA); NRG1 and EGF were purchased from R&D System (Minneapolis, MN, USA) and Sigma-Aldrich (St. Louis, MO, USA), respectively. All growth factors were dissolved in 0.1% BAS as stock solution and the aliquots were stored at -20°C.
Cell lines and proliferation assay
Human breast cancer cell lines JIMT1, EFM192A were obtained from Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH (Braunschweig, Germany). HCC38 was purchased from China Infrastructure of Cell Line Resources (Beijing, China). BT549 (TCHu 93) was obtained from Chinese Academy of Sciences Cell Bank (Shanghai, China). SK-BR-3 was provided by Dr. Li-guang Lou (Shanghai Institute of Materia Medica, China). BT20 was gift of Dr. Jing-yu Lang (Shanghai Institutes for Biological Sciences, China). MDA-MB-231 was provided by Fudan University Shanghai Cancer Center (Shanghai, China). All other cancer cell lines were obtained from American Type Culture Collection (Manassas, VA, USA). All the cell lines were authenticated by analyzing short-tandem repeats (STR) by Genesky Biotechnologies Inc. (Shanghai, China). Cell proliferation was evaluated by Sulforhodamine B (SRB; Sigma-Aldrich, St. Louis, MO, USA) assay as reported previously (22). Cells seeded in 96-well plate were treated in triplicate with BYL719 alone or with growth factors at 37°C for 72 h. Optic density for SRB assay was measured at 560 nm with microplate reader (Molecular Devices, Sunnyvale, CA, USA). The IC50 values were determined by four-parameter logit method with SoftMax Pro Software (23).
Western blotting
Cells were seeded in six-well plates and exposed to compounds at indicated concentrations for various times. The cells were collected and subjected to standard Western blot analysis as described previously (24), with the antibodies against phosphor-AKT (Ser473), phosphor-AKT (Thr308), AKT, phosphor-ERK (Thr202/Tyr204), ERK, p110α, p110β, phosphor-PRAS40 (Thr246), PRAS40, Myc-tag, HRAS, phosphor-EGFR (Tyr1068), EGFR, IGF-1R, FGFR1, HER3, c-MET, HER2 (Cell Signaling Technology, Danvers, MA, USA), RAS (Merck Millipore, Billerica, MA, USA), V5-tag (Invitrogen, Carisbad, CA, USA). The β-actin antibody was purchased from Sigma-Aldrich (St. Louis, MO, USA).
RNA interference
SiRNA duplexes targeting PI3Kα and β were synthesized by Ribobio (Guangzhou, China). The targeted genes sense sequences were as follows respectively, siPIK3CA#1: 5'-GAAUGAUAGUGACUUUAGAdTdT-3'; siPIK3CA#2: 5'-UCUUCACCAGAAUUGCCAAdTdT-3'; siPIK3CB#1: 5'-GGAUAUGUUGACACUCCAAdTdT-3'; siPIK3CB#2: 5'-GGACGUGUUUAGCUUUCAAdTdT-3'; siPIK3CB#3: 5'- GGUCUGCAUGAAUUUGAUUdTdT-3'. The negative control was provided by Ribobio (Guangzhou, China). The siRNA transfection was conducted using Lipofectamine RNAiMax (Invitrogen, Carisbad, CA, USA) according to the manufacturer's instruction.
Plasmids and transfection
Plasmids including pCMV-VSV-G (#8454), pCMV-dR8.2 dvpr (#8455), pDONR223-MEKDD (#31202), pDONR223-H-RAS V12 (#31201), pLX302 (#25896), pBabe-Puro-BRAF-V600E (#15269), EGFR L858R (#11012), EGFR WT (#11011) and pBabe-puro (#1764) were obtained from Addgene (Cambridge, MA, USA). Lentiviral donor plasmids were removed into the destination vector pLX302 using LR Clonase (Invitrogen, Carisbad, CA, USA). Recombined plasmids were verified by sequencing (Sangon Biotech, Shanghai, China). The infectious lentiviral particles were generated by co-transfecting pLX302 containing gene of interest, pCMV-VSV-G and pCMV-dR8.2 dvpr (at a 5:1:4 ratio) into the 293FT cells (Invitrogen, Carisbad, CA, USA). Retroviral particles were produced by transfection of Phenix293 cells (ATCC, Manassas, VA, USA) with pBabe containing gene of interest. Transfections were carried out using Lipofectamine 2000 according to the manufacture's instruction (Invitrogen, Carisbad, CA, USA). Breast cancer cells were infected with viruses in the presence of 8 μg/mL polybrene (Sigma-Aldrich, St. Louis, MO, USA). After 48 h, stable transfected cells were selected with puromycin (2 μg/mL for T47D cells, 1 μg/mL for MCF-7 cells) until control plates became cleared at 3 days post-treatment.
RAS-GTP Assay
RAS-GTP assay was performed using RAS Activation Assay Kit (Merck Millipore, Billerica, MA, USA) according to manufacturer's instruction. Briefly, cells grown to 70% confluence in 100-mm culture dish were treated with tested compounds for 1 h and then collected with ice-cold MLB. An equal amount of protein (500-800 μg) from each sample was incubated with GST-RAF1-RBD agarose for 45 minutes at 4°C with gentle agitation. GTPγS and GDP protein loading were used for positive and negative controls, respectively. Samples were then analyzed with Western blotting.
Animal studies
Patient-derived xenograft models of breast cancer were established and preserved by our lab. All studies were performed in compliance with Institutional Animal Care and Use Committee guidelines (Shanghai Institute of Materia Medica, Shanghai, China). Tumor section was cut into 1.5 mm3 small pieces which were then implanted into the right flank of female SCID (severe combined immunodeficiency) mice aged 3~4 weeks (Beijing HFK Bioscience Company, Beijing, China). Treatment began when the tumor volume reached a size of 100-200 mm3. Mice were randomized to receive vehicle control or tested compounds. BYL719 was administrated p.o. at the dose of 50 mg/kg every day. The average tumor volume (two perpendicular axes of the tumor were measured) was measured with vernier calipers twice a week, and animal weight was recorded every 3 to 4 days. The tumor volume (V) was calculated using the formula: V = a2b/2, and a and b represented the tumor's width and length respectively. Then relative tumor volume (RTV) was calculated as follows: RTV = Vt/V0. Vt was the tumor volume after treatments, and V0 was the tumor volume at the beginning of treatment. The treatment to control ration (T/C) was calculated using the formula: T/C (100%) = (TRTV/CRTV) × 100%, TRTV and CRTV represented the RTV of treatment and control group, respectively.
Immunohistochemistry
Tumors were harvested and fixed in 4% paraformaldehyde after the PDX models were established. The paraformaldehyde-fixed tumor tissues were stored at 4°C for 6-12 months before immunohistochemical analysis. Paraffin embedding, hematoxylin and eosin (H&E) staining as well as immunohistochemistry were conducted by Shanghai ZuoCheng Bio Company (Shanghai, China). All slides were observed under a Leica DM6 B microscope equipped with sCMOS camera with the same parameter setting for each RTK examined. Expression of tested RTKs was blindly quantified using Image-Pro Plus Software (Media Cybernetics Inc., Silver Springs, MD, USA).
Statistical analysis
Data were presented as mean ± SEM from at least three independent experiments. Statistically significant differences were determined using indicated analyses. Differences were considered statistically significant when P was less than 0.05.
Supplementary Material
Supplementary figures.
Abbreviations
PI3K: phosphatidylinositol 3-kinase; RTK: receptor tyrosine kinase; PTEN: phosphatase and tensin homolog; INPP4B: inositol polyphosphate-4-phosphatase; ER: estrogen receptor; MAPK: mitogen-activated protein kinase; MEK: mitogen-activated protein kinase kinase; PDK1: phosphoinositide-dependent kinase-1; mTOR: the mechanistic target of rapamycin; ERK: extracellular signal-regulated kinase; EGFR: epidermal growth factor receptor; IGF-1R: insulin-like growth factor 1 receptor; RYK: receptor-like tyrosine kinase; EphA1: ephrin type-A receptor 1; FGFR: fibroblast growth factor receptor; EGF: epidermal growth factor; FGF: fibroblast growth factor; HGF: hepatocyte growth factor; NRG1: neuregulin 1; HER: human epidermal growth factor receptor; IC50: half maximal inhibitory concentration.
Acknowledgements
This work was supported by National Science and Technology Major Project “Key New Drug Creation and Manufacturing Program” (2012ZX09301-001), National Natural Science Foundation of China (81321092, 81373445, 81402972) and "Personalized Medicines-Molecular Signature-based Drug Discovery and Development", Strategic Priority Research Program of the Chinese Academy of Sciences (XDA12020202).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Stephens PJ, Tarpey PS, Davies H. et al. The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012;486:400-4
2. Wang X, Ding J, Meng LH. PI3K isoform-selective inhibitors: next-generation targeted cancer therapies. Acta Pharmacol Sin. 2015;36:1170-6
3. Juric D, Argiles G, Burns HA. et al. Phase I study of BYL719, an alpha-specific PI3K inhibitor, in patients with PIK3CA mutant advanced solid tumors: preliminary efficacy and safety in patients with PIK3CA mutant ER -positive (ER plus ) metastatic breast cancer (MBC). Cancer Res. 2012:72
4. Juric D, Rodon J, Gonzalez-Angulo AM. et al. BYL719, a next generation PI3K alpha specific inhibitor: Preliminary safety, PK, and efficacy results from the first-in-human study. Cancer Res. 2012:72
5. Mayer IA, Abramson V, Formisano L. et al. A Phase Ib Study of Alpelisib (BYL719), a PI3Kalpha-specific Inhibitor, with Letrozole in ER+/HER2- Metastatic Breast Cancer. Clin Cancer Res. 2016 [Epub ahead of print]
6. De Luca A, Maiello MR, D'Alessio A. et al. The RAS/RAF/MEK/ERK and the PI3K/AKT signalling pathways: role in cancer pathogenesis and implications for therapeutic approaches. Expert Opin Ther Targets. 2012;16:S17-S27
7. Serra V, Scaltriti M, Prudkin L. et al. PI3K inhibition results in enhanced HER signaling and acquired ERK dependency in HER2-overexpressing breast cancer. Oncogene. 2011;30:2547-57
8. Chakrabarty A, Sanchez V, Kuba MG. et al. Feedback upregulation of HER3 (ErbB3) expression and activity attenuates antitumor effect of PI3K inhibitors. Proc Natl Acad Sci U S A. 2012;109:2718-23
9. Fritsch C, Huang A, Chatenay-Rivauday C. et al. Characterization of the novel and specific PI3Kalpha inhibitor NVP-BYL719 and development of the patient stratification strategy for clinical trials. Mol Cancer Ther. 2014;13:1117-29
10. Folkes AJ, Ahmadi K, Alderton WK. et al. The identification of 2-(1H-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-thieno[3,2-d]pyrimidine (GDC-0941) as a potent, selective, orally bioavailable inhibitor of class I PI3 kinase for the treatment of cancer. J Med Chem. 2008;51:5522-32
11. Najafov A, Sommer EM, Axten JM. et al. Characterization of GSK2334470, a novel and highly specific inhibitor of PDK1. Biochem J. 2011;433:357-69
12. Yap TA, Yan L, Patnaik A. et al. First-in-Man Clinical Trial of the Oral Pan-AKT Inhibitor MK-2206 in Patients With Advanced Solid Tumors. J Clin Oncol. 2011;29:4688-95
13. Levy DS, Kahana JA, Kumar R. AKT inhibitor, GSK690693, induces growth inhibition and apoptosis in acute lymphoblastic leukemia cell lines. Blood. 2009;113:1723-9
14. Sampaio C, Dance M, Montagner A. et al. Signal strength dictates phosphoinositide 3-kinase contribution to Ras/extracellular signal-regulated kinase 1 and 2 activation via differential Gab1/Shp2 recruitment: consequences for resistance to epidermal growth factor receptor inhibition. Mol Cell Biol. 2008;28:587-600
15. Borisov N, Aksamitiene E, Kiyatkin A. et al. Systems-level interactions between insulin-EGF networks amplify mitogenic signaling. Mol Syst Biol. 2009;5:256
16. Aksamitiene E, Kholodenko BN, Kolch W. et al. PI3K/Akt-sensitive MEK-independent compensatory circuit of ERK activation in ER-positive PI3K-mutant T47D breast cancer cells. Cell Signal. 2010;22:1369-78
17. Aksamitiene E, Achanta S, Kolch W. et al. Prolactin-stimulated activation of ERK1/2 mitogen-activated protein kinases is controlled by PI3-kinase/Rac/PAK signaling pathway in breast cancer cells. Cell Signal. 2011;23:1794-805
18. Ebi H, Costa C, Faber AC. et al. PI3K regulates MEK/ERK signaling in breast cancer via the Rac-GEF, P-Rex1. Proc Natl Acad Sci U S A. 2013;110:21124-9
19. Will M, Qin ACR, Toy W. et al. Rapid Induction of Apoptosis by PI3K Inhibitors Is Dependent upon Their Transient Inhibition of RAS-ERK Signaling. Cancer Discov. 2014;4:334-47
20. Engelman JA, Chen L, Tan X. et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14:1351-6
21. Wang X, Zhang X, Li BS. et al. Simultaneous targeting of PI3Kdelta and a PI3Kdelta-dependent MEK1/2-Erk1/2 pathway for therapy in pediatric B-cell acute lymphoblastic leukemia. Oncotarget. 2014;5:10732-44
22. Li T, Wang J, Wang X. et al. WJD008, a Dual Phosphatidylinositol 3-Kinase (PI3K)/Mammalian Target of Rapamycin Inhibitor, Prevents PI3K Signaling and Inhibits the Proliferation of Transformed Cells with Oncogenic PI3K Mutant. J Pharmacol Exp Ther. 2010;334:830-8
23. Li X, Tong LJ, Ding J. et al. Systematic combination screening reveals synergism between rapamycin and sunitinib against human lung cancer. Cancer Lett. 2014;342:159-66
24. Chen SM, Guo CL, Shi JJ. et al. HSP90 inhibitor AUY922 abrogates up-regulation of RTKs by mTOR inhibitor AZD8055 and potentiates its antiproliferative activity in human breast cancer. Int J Cancer. 2014;135:2462-74
Author contact
![]() Corresponding authors: Ling-hua Meng or Jian Ding, Division of Anti-tumor Pharmacology, State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, 555 Zu-chong-zhi Road, Shanghai 201203, P. R. China. E-mail: lhmengac.cn or jdingac.cn.
Corresponding authors: Ling-hua Meng or Jian Ding, Division of Anti-tumor Pharmacology, State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, 555 Zu-chong-zhi Road, Shanghai 201203, P. R. China. E-mail: lhmengac.cn or jdingac.cn.