Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and methods
Results
Discussion
Summary and Conclusions
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(3):733-750. doi:10.7150/thno.17450 This issue Cite
Research Paper
Exosomes derived from human platelet-rich plasma prevent apoptosis induced by glucocorticoid-associated endoplasmic reticulum stress in rat osteonecrosis of the femoral head via the Akt/Bad/Bcl-2 signal pathway
Shi-Cong Tao1, Ting Yuan1, Bi-Yu Rui1, Zhen-Zhong Zhu1, Shang-Chun Guo2, ![]() , Chang-Qing Zhang1,2,
, Chang-Qing Zhang1,2, ![]()
1. Department of Orthopedic Surgery, Shanghai Jiao Tong University Affiliated Sixth People's Hospital, 600 Yishan Road, Shanghai 200233, China;
2. Institute of Microsurgery on Extremities, Shanghai Jiao Tong University Affiliated Sixth People's Hospital, 600 Yishan Road, Shanghai 200233, China.
Received 2016-9-3; Accepted 2016-11-22; Published 2017-1-15
Abstract

An excess of glucocorticoids (GCs) is reported to be one of the most common causes of osteonecrosis of the femoral head (ONFH). In addition, GCs can induce bone cell apoptosis through modulating endoplasmic reticulum (ER) stress. Among the three main signal pathways in ER stress, the PERK (protein kinase RNA-like ER kinase)/CHOP (CCAAT-enhancer-binding protein homologous protein) pathway has been considered to be closely associated with apoptosis. Platelet-rich plasma (PRP) has been referred to as a concentration of growth factors and the exosomes derived from PRP (PRP-Exos) have a similar effect to their parent material. The enriched growth factors can be encapsulated into PRP-Exos and activate Akt and Erk pathways to promote angiogenesis. Activation of the Akt pathway may promote the expression of anti-apoptotic proteins like Bcl-2, while CHOP can inhibit B-cell lymphoma 2 (Bcl-2) expression to increase the level of cleaved caspase-3 and lead to cell death. Consequently, we hypothesized that PRP-Exos prevent apoptosis induced by glucocorticoid-associated ER stress in rat ONFH via the Akt/Bad/Bcl-2 signal pathway. To verify this hypothesis, a dexamethasone (DEX)-treated in vitro cell model and methylprednisolone (MPS)-treated in vivo rat model were adopted. Characterization of PRP-Exos, and effects of PRP-Exos on proliferation, apoptosis, angiogenesis, and osteogenesis of cells treated with GCs in vitro and in vivo were examined. Furthermore, the mechanism by which PRP-Exos rescue the GC-induced apoptosis through the Akt/Bad/Bcl-2 pathway was also investigated. The results indicate that PRP-Exos have the capability to prevent GC-induced apoptosis in a rat model of ONFH by promoting Bcl-2 expression via the Akt/Bad/Bcl-2 signal pathway under ER stress.
Keywords: platelet-rich plasma, exosomes, glucocorticoids, osteonecrosis of the femoral head, endoplasmic reticulum stress, apoptosis.
Introduction
Osteonecrosis is defined as the cellular death of bone and occurs most commonly in the hip [1, 2]. Steroid-induced osteonecrosis of the femoral head (ONFH) is a serious side effect of glucocorticoids (GCs) use [3], and excessive use of GCs is one of the most common causes of ONFH [4, 5]. Although the exact mechanism of GC-induced ONFH remains unclear, it appears to involve cell death, vascular compromise, or deficient bone repair [6].
Firstly, high-dose GCs treatment increases cell apoptosis and GC-induced apoptosis is known to be due to direct hormonal effects on bone cells [7-9]. GC-induced apoptosis of bone cells could account for the loss of bone strength that occurs before the loss of bone mineral density. Recent evidence demonstrates that increased bone cell apoptosis is associated with endoplasmic reticulum (ER) stress, which results from the accumulation of misfolded/unfolded proteins [10, 11]. The accumulation of misfolded/unfolded proteins induces the phosphorylation of protein kinase-like endoplasmic reticulum kinase (PERK), activates the unfolded protein response (UPR) and helps the cell to adapt to ER stress in mild ER stress conditions [12].
GCs excess often elicits serious adverse effects on the vascular system [13]. The endothelium is a critical site for the control of apoptosis during processes such as inflammation, vascular remodeling, and angiogenesis [14]. Furthermore, the suppression of endothelial cell (EC) apoptosis is necessary for the maintenance of blood vessel integrity and for angiogenesis [15].
GCs treatment is known to inhibit the expression of VEGF-A and consequent vasculogenesis in ECs [16]. The vascular impairment is indicated by reduced circulating angiogenic cell function, with the migratory function and VEGF protein secretion weakened [17]. Previously, we blocked local angiogenesis by injecting VEGF receptor 2 (VEGFR2) antibody into the capsular attachment to the proximal femur, in order to establish an animal model of ONFH. This also showed that impairment of angiogenesis can induce ONFH [18]. In contrast, treatment to enhance angiogenesis can prevent progression of ONFH [19].
At the same time, osteogenesis is severely affected by GCs hormones [20]. GCs have complex stimulatory and inhibitory effects on skeletal metabolism. Endogenous GC signaling is required for normal bone formation in vivo, and synthetic GCs, such as low-dose dexamethasone (DEX), promote osteoblastic differentiation in several in vitro model systems [21]. Higher doses of GCs and prolonged treatment are associated with a greater risk, although osteonecrosis may also occur as a result of short-term exposure to high doses, suggesting that there are adverse effects of GCs on osteogenesis and that ONFH is somewhat compromised by high-dose GCs therapy [22]. The administration of high-dose GCs reduces bone mass and induces a decline in bone density, ultimately leading to bone loss [23].
As a common disease, osteonecrosis is attracting more and more attention. In order to improve the early diagnosis of GC-induced ONFH or guide clinical trials in GC-induced osteonecrosis, studies of circulating exosomes [24] and microvesicles [25] have aroused widespread interest. Within the past decade, extracellular vehicles (EVs) have emerged as key mediators involved in signal transduction between cells to regulate a diverse range of biological processes in both prokaryotes and eukaryotes [26]. One type of EVs, named microvesicles, which can be formed and released from the cell membrane and displays a varied range of sizes (100-1,000 nm in diameter), is composed of a lipid bilayer containing transmembrane proteins, enclosing cytosolic proteins and RNA. Another type of EVs, named exosomes, are vesicles smaller than 100 nm in diameter and enriched in endosome-derived components [27]. In addition to their key role as vehicles of intercellular communication, exosomes have the potential to be used as vehicles of gene and drug delivery for clinical application [28].
As is well known, platelet-rich plasma (PRP), an autologous derivative of whole blood that contains a supra-physiological concentration of platelets [29], promotes axon regeneration and wound healing, enhances bone regeneration [30] and cartilage repair [31], augments tendon and ligament repair [32, 33], cures chronic soft tissue lesions [34, 35], and treats chronic femoral osteomyelitis [36]. Furthermore, there have been studies reporting that ONFH could be treated completely with adipose tissue-derived stem cells and PRP in the early stage [37], with injected PRP as a nonsurgical approach for advanced stage disease [38], and that a single injection of PRP accelerates surgical angiogenesis in vascular-implanted necrotic bone [39].
All these functions were thought to be attributable to the many growth factors secreted by activated platelets, which can improve cellular chemotaxis, proliferation and differentiation, angiogenesis, and the production of extracellular matrix. Activated platelets can also promote capillary construction and bone formation by promoting the proliferation and migration of ECs and bone mesenchymal stem cells (BMSCs) [29].
In spite of these benefits, one problem restricting PRP application in the clinical setting is the requirement for autologous platelets.
Given that these effective factors can be encapsulated and enriched in PRP-derived exosomes (PRP-Exos) [40], that there is solid evidence that these nanoparticles have a similar biological action to the source cells, and that direct application of these exosomes has no obvious adverse effects such as immunogenicity or tumorigenicity [41, 42], this study aimed to adopt the method of injecting PRP-Exos to treat ONFH, to observe whether PRP-Exos could reduce GC-ER stress-induced apoptosis and exert a protective effect, and to study this molecular mechanism on the cells by inhibiting apoptosis, providing rational approaches to interventions that could prevent ONFH.
Materials and methods
Human PRP extraction
The extraction methods and characterization of PRP have been described in our previous reports [31, 35]. All of the blood specimens used in the present study were approved by the ethical committee of the Shanghai Sixth People's Hospital, Shanghai Jiao Tong University School of Medicine. Briefly, whole blood was taken from healthy donors and added to acid citrate dextrose solution A (ACD-A) anticoagulant (1 mL ACD-A/9 mL blood). To obtain platelet-containing plasma separate from erythrocytes and leukocytes, 40 mL of whole blood was centrifuged at 160 × g for 10 min in a 50 mL centrifuge tube. Then the separated plasma was added to another tube, and centrifuged again for 15 min at 250 × g. The supernatant plasma was discarded, before the platelet pellet was resuspended in the residual plasma to obtain 4 mL PRP.
PRP-Exos extraction
Based on the methods of Torreggiani et al. [40] and Thery et al. [43], exosomes were extracted carefully from PRP. Briefly, PRP samples were centrifuged at 250 × g for 15 min and the platelet pellet was washed with PBS (calcium-free, magnesium-free and phenol red-free; Gibco; Thermo Fisher Scientific, Waltham, MA, USA) three times. The resuspended platelet pellet was activated and centrifuged in series at low speeds (300 × g for 10 min, 2,000 × g for 10 min) to remove cell debris. Then, the supernatant was filtered through a 0.22 μm filter (Merck-Millipore, Darmstadt, Germany). The filtered supernatant was transferred to a 15 mL Amicon Ultra-15 Centrifugal Filter Unit (Merck-Millipore) and centrifuged at 4,000 × g. The ultrafiltered liquid was washed with PBS three times and re-ultrafiltered at 4,000 × g. To purify the exosomes, the ultrafiltered liquid was transferred onto a 30% sucrose-D2O cushion in an Ultra-Clear™ tube (Beckman Coulter, Brea, CA, USA) and ultra-centrifuged at 100,000 × g for 70 min to pellet exosomes, which were then washed in a large volume of PBS and ultra-centrifuged again at the same high speed for 70 min. All steps were performed at 4°C. The exosomes were carefully resuspended in sterile PBS and stored at -80°C for subsequent experiments.
Identification of PRP-Exos
First, NanosizerTM technology (Malvern Instruments, Malvern, UK) was applied carefully to estimate the size distribution of PRP-Exos. Secondly, after exosomes were coated onto a 2 nm copper grid and stained using 2% uranyl acetate, a Hitachi H-7650 transmission electron microscope (TEM) was employed to observe the morphology of exosomes derived from PRP. Finally, western blotting was used to examine the specific exosome biomarkers TSG101, CD9, CD63, and CD81, and the source marker CD41. Calnexin was examined using western blotting to confirm the purity of exosomes. Platelet lysate (PL) was prepared, to act as control, according to the methods of Torreggiani et al [40].
Cell culture and treatments
Murine osteoblastic MC3T3-E1 cells and the human microvascular endothelial cell line (HMEC-1) were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). MC3T3-E1 cells were maintained in α-MEM (Hyclone; GE Healthcare, Little Chalfont, UK) with 10% fetal bovine serum (FBS, Gibco), 100 U/mL penicillin, and 100 μg/mL streptomycin (Gibco). HMEC-1 were cultured in MCDB131 medium (Gibco), supplemented with 10% FBS (Gibco), 2 mM L-glutamine (Gibco), 10 ng/mL epidermal growth factor (EGF, Sigma-Aldrich, St. Louis, MO, USA) and 1 μg/mL hydrocortisone (Sigma-Aldrich).
BMSCs were isolated from healthy donors (aged 30-50 years) who underwent amputation because of severe trauma, with permission from the patients and the Institutional Review Board at Shanghai Sixth People's Hospital. Cancellous bone was collected under aseptic conditions, flushed with α-MEM (Hyclone) and briefly vibrated for 1 min, 10 times. After the vibrated mixture was filtered through a 70 μm cell strainer (BD Biosciences, Franklin Lakes, NJ, USA), the cell suspension was seeded into a T25 culture flask. BMSCs were cultured in α-MEM containing 10% FBS and purified through passaging. BMSCs from passage 4 to 6 were used in this study.
To investigate the effects of PRP-Exos, cells were treated with DEX (Sigma-Aldrich), PRP-Exos or both. At certain intervals, a series of in vitro assays were performed on the cells. The concentration of DEX used was 10 μM in the viability experiment, proliferation experiment and osteogenic differentiation experiment including analysis by alizarin red staining, western blotting and ELISA, and 100 μM in the apoptosis experiment including analysis by flow cytometry and western blotting. The concentration of PRP-Exos was 50 μg/mL.
Lentivirus infection and RNA interference
Lentiviral transduction particles for shBad (sc-29778-V) and Bcl-2 lentiviral activation particles (sc-400025-LAC) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Cell transfection was executed in accordance with the protocol provided by Santa Cruz. Briefly, cells were incubated in retroviral supernatant with 5 μg/mL polybrene (Sigma-Aldrich) for 48 h, at the end of which cells were selected with 2.5 μg/mL puromycin (Sigma-Aldrich) in culture medium.
Inhibitor and agonist
GSK2656157, MK-2206 2HCl and SC79 were purchased from Selleckchem (Houston, TX, USA).
GSK2656157 is a specific inhibitor of PERK [44]; MK-2206 2HCl is a selective inhibitor of AKT [45], and SC79 is a unique specific Akt activator [46]. BMSCs were cultured in conditioned medium containing the specified concentration of PRP-Exos (50 μg/mL), GSK2656157 (20 μM), MK-2206 2HCl (5 μM) or SC79 (8 μg/mL). After treatment, western blotting was performed to examine expression of signal molecules.
Cell viability assay
Cell counting kit-8 assay (CCK-8; Dojindo, Kumamoto, Japan) was used to estimate cell proliferation. A total of 5,000 cells per well were seeded into 96-well plates. One group without cells served as the blank. On days 0, 1, 3, 5 and 7, 10 μL CCK-8 solution was added to cells in each well and incubated at 37°C for one hour. The absorbance was measured at 450 nm on a microplate reader and the difference between the optical density (OD) of the cells in medium minus the absorbance of the blank medium-only wells represented the survival/proliferation of cells.
Cell proliferation assay
To confirm the change in the proliferation rate of the three cell types (HMEC-1, BMSCs and MC3T3-E1), an EdU-488 Cell Proliferation Kit (Ribobio, Guangzhou, China) was used and results were evaluated by flow cytometry following the manufacturer's instructions. Cells were seeded into a 48-well plate at an initial density of approximately 2 × 104 cells/well, and cultured in the specified conditions for 12 h. Then, the EdU working solution, mixed with 150 μL complete culture medium and 0.15 μL EdU, was added into each well and incubated at 37°C for 3 h. After incubation, cultured cells were harvested using trypsin-EDTA, washed twice with PBS and fixed in 4% paraformaldehyde (PFA) for 10 min. The fixed cells were neutralized using 2 mg/mL glycine and washed twice in PBS. After being permeabilized with 0.4% Triton X-100 for 3 min and washed twice with PBS, the labeled cells were resuspended in Apollo staining solution (Ribobio) and incubated for 10 min. After another two washes in 0.4% Triton X-100, the prepared cells were resuspended in PBS and analyzed using a Guava® easyCyte™ flow cytometer.
HMEC-1 tube formation assay
To investigate the vessel-like construction activity of the HMEC-1 cells, tube formation in ECM gel (Sigma-Aldrich) was assessed. First, 50 μL of cold ECM gel was added to each well in a 96-well plate. After pretreatment with medium in the presence or absence of PRP-Exos and DEX for 48 hours, 1 × 104 HMEC-1 cells in 100 μL medium were seeded onto the ECM gel, which had been allowed to gel for 15 min. After 12 h, the ability to form capillary-like structures was examined by viewing the polygonal construction after the cells were added to the ECM gel under a light microscope.
HMEC-1 migration assay
To observe the effect of the PRP-Exos on the migration of HMEC-1 cells, the transwell assay was adopted. Firstly, 10,000 cells/well were seeded into the upper chamber of a 24-well transwell plate (Corning; pore size = 8 μm). Secondly, 10 μM DEX together with or without 50 μg/mL PRP-Exos was added to the lower chamber at 700 μL/well. Twelve hours after incubation, the insert was transferred to another well, washed with PBS and stained with 0.5% crystal violet for one minute. After staining, the cells from the upper surface of the filter membranes were removed with a cotton swab. Stained cells and thus migration activity were evaluated by observing the stained cells under an optical microscope.
Cell apoptosis assay
The Annexin V-FITC early apoptosis detection kit (Cell Signaling Technology, Danvers, MA, USA) was used to estimate the effect of PRP-Exos on HMEC-1, MC3T3-E1, and BMSC cell apoptosis according to the manual. Briefly, after 96 hours of culture with serum-free medium in the presence or absence of PRP-Exos and DEX [4], cells were harvested, washed with DPBS, and resuspended in 96 μL of Annexin V binding buffer, before being incubated with 1 μL of Annexin V-FITC and 12.5 μL of propidium iodide (PI) for 10 min on ice in the dark. Next, the cell suspension was diluted to a final volume of 250 μL with Annexin V binding buffer and analyzed individually by flow cytometry.
Osteogenesis of cells exposed to DEX
Osteogenic differentiation was induced by osteogenic differentiation medium (Cyagen, Suzhou, China), in accordance with the manufacturer's instructions. BMSC and MC3T3-E1 cells were cultured in osteogenic differentiation medium in the presence or absence of DEX (10 μM) or PRP-Exos. After 21 days of induction, calcium mineral deposits were measured by Alizarin red (Cyagen) staining.
Enzyme-linked immunosorbent assay (ELISA)
The anti-osteocalcin (OCN) and anti-alkaline phosphatase (ALP) antibodies for ELISA were obtained from Santa Cruz Biotechnology. The primary antibodies (1:1000) were diluted with ELISA coating solution (SolarBio, Beijing, China), 0.1 mL was added into each well and the cells were incubated at 4°C. Next day, each well was washed three times with PBS before being lysed for assay.
The cell lysates were diluted with PBS and 0.1 mL diluted cell lysate was added into appropriate wells. After the plates were incubated at 37°C for 2 h, the lysate was removed and washed four times with 200 μL PBS/ well. Then, 100 μL of the appropriate primary antibodies (1:3000) were added to each well and the plate was incubated at 37°C for 1 h. After washing four times with PBS, 100 μL HRP-linked secondary antibody was added to each well and the plate was incubated at 37°C for 30 min. After washing four times with PBS, 100 μL of TMB Substrate (SolarBio) was added to each well and the plate was incubated at 37°C for 10 min, and at 25°C for 30 min. Then 100 μL of ELISA STOP Solution (SolarBio) was added to each well and the plate was shaken gently for a few seconds. Finally, the absorbance at 450 nm was read using a BioRad microplate reader.
Western blotting analysis
The protein of exosomes was extracted using a Total Exosome RNA & Protein Isolation Kit (Invitrogen, Carlsbad, CA, USA) following the attached instructions. Cell or exosome lysates were diluted at a ratio of 1:5 with protein loading buffer (6 ×) and heated at 95°C for 5 min. Protein extracts were separated on a 10% FastPAGE Plus premixed gel (New Cell & Molecular Biotech, Suzhou, China) at 120 V for 1 h and blotted onto a polyvinylidene difluoride (PVDF) membrane (Merck-Millipore) for 90 min at 200 mA. The membranes were then blocked for 120 min with 5% non-fat dried milk in TBST (Tris-buffered saline, 10 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.1% Tween-20). Subsequently, the membranes were incubated with primary antibodies at 4°C overnight, followed by incubation with horseradish peroxidase (HRP)-labeled secondary antibodies (Cell Signaling Technology) at 37°C for 1 h. The immunoreactive bands were visualized using enhanced chemiluminescence reagent (Thermo Fisher Scientific) and imaged by an Image Quant LAS 4000 mini bio-molecular imager (GE Healthcare).
Antibodies for western blotting
The primary antibodies used were anti-collagen I, anti-VEGF and anti-PDGFBB (Abcam, Cambridge, UK); anti-TSG101, anti-CD9, anti-CD63, and anti-CD81 (System Biosciences, Mountain View, CA, USA). The primary antibodies anti-CD41, anti-TGF-β, anti-bFGF, anti-cleaved-caspase-3, anti-Runx2, anti-β-catenin, anti-β-tubulin, anti-calnexin, anti-Bcl-2, anti-CHOP, anti-Bad and phosphorylated Bad (p-Bad), anti-Erk1/2 and phosphorylated Erk1/2 (p-Erk1/2), anti-Akt and phosphorylated Akt (p-Akt) antibody were all obtained from Cell Signaling Technology (Danvers, MA, USA). The anti-PERK and phosphorylated PERK (p-PERK) antibodies were obtained from Santa Cruz Biotechnology.
Animal model and grouping
All experimental and animal care procedures were approved by the Animal Research Ethics Committee of Shanghai Sixth People's Hospital and performed in accordance with the guidelines of the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals.
All the animal experiments were performed in an SPF grade animal laboratory. Sixty 8-week-old healthy female Sprague-Dawley (SD) rats weighing 300-350 g were used in this study. The rats were randomly and equally divided into three groups: (1) control group (rats served as controls treated with normal saline) (n = 20); (2) MPS group (methylprednisolone; rats with MPS-induced ONFH) (n = 20); (3) PRP-Exos group (rats with MPS-induced ONFH treated with PRP-Exos) (n = 20). Each group was divided into a long-term subgroup (n = 15) and a short-term subgroup (n = 5).
In the long-term subgroup, to induce ONFH in the rats, MPS (Pfizer Inc., New York, NY, USA) (20 mg/kg/d) was intramuscularly injected into rats once a day, on the first three days of every week, for three weeks. After each MPS injection, rats in the PRP-Exos group and the MPS group were administered 100 μg exosomes (dissolved in 200 μL of PBS) or an equal volume of PBS, via the tail vein. Six weeks later, the rats were sacrificed and the femoral heads were examined by micro-CT, immunohistochemical (IHC) and histomorphological analyses.
In the short-term subgroup, MPS was only injected once a day on the first three days. After each MPS injection, rats in the PRP-Exos group and the MPS group were administered 100 μg exosomes (dissolved in 200 μL of PBS) or an equal volume of PBS via the tail vein. After three days of injections, the five rats of each group were anesthetized by intraperitoneal injection of 400 mg/kg chloral hydrate and the femoral heads were obtained for TUNEL assay and Ki67 immunostaining.
Throughout the experiment, none of the rats in any of the groups died before these tests and no antibiotic agents were used.
Angiography and osteogenesis with micro-CT analysis
Six weeks after the first MPS injection, the rats were sacrificed, and the abdominal aorta and vein were dissected. The proximal aorta was ligated and the abdominal vein was cut off. Heparinized saline, 4% paraformaldehyde and Microfil (MV-112, Flow Tech, Inc., Carver, MA, USA) were injected in sequence through the distal abdominal aorta, until a constant outflow was observed exiting the abdominal vein. Then, the rats were stored at 4°C overnight to ensure complete polymerization, and the bilateral femoral heads were resected.
The femoral heads were fixed and decalcified with 10% EDTA solution and then analyzed by SkyScan1178 (Bruker MicroCT, Kontich, Belgium). Micro-CT scanning was set at a resolution of 9 μm per pixel. Two-dimensional images were captured with CTVol and three-dimensional images of the vasculature were reconstructed. The total vessel volume was calculated using CTAn (Bruker).
The scanner was set at the trabecular bone, segmented from bone marrow and analyzed to determine trabecular thickness (Tb.Th), trabecular separation (Tb.Sp), bone volume per tissue volume (BV/TV), and trabecular number (Tb.N). Three planes (coronal section, sagittal section and transverse section) of each sample were generated using DataViewer (Bruker MicroCT).
After micro-CT scanning, the femoral heads were excised and embedded in paraffin. Samples were cut into 5-µm sections, deparaffinized in xylene, rehydrated through a graded series of ethanol, and rinsed in distilled water. Hematoxylin and eosin (HE) staining was performed for histological observation. The extent of osteogenesis was assessed by IHC analysis of type I collagen. Images were acquired with a microscope (IX81; Olympus, Tokyo, Japan). Antibodies were obtained from Abcam.
TUNEL assay and Ki67 immunostaining
The femoral head samples were cut into 5-µm sections, deparaffinized in xylene, and rehydrated through a graded ethanol series. A TUNEL staining kit (Roche, Basel, Switzerland) was used to detect DNA strand breaks according to the manufacturer's instructions. Proliferating cells were immunostained with Ki67 antibody (Abcam), and nuclei were counter-stained with DAPI. Images were acquired with an LSM-880 confocal microscope (Carl Zeiss, Jena, Germany). The number of TUNEL-positive cells (apoptotic cells) and Ki67-positive cells (proliferative cells) per field was evaluated in five fields per section and five sections per femoral head.
Statistical analysis
All of these experiments were repeated three times. The data are shown as means ± standard deviation (SD). Means of multiple groups were compared by one-way analysis of variance (ANOVA). Independent-sample t tests were used to compare means between two different groups. Fisher's exact test was used to compare the incidence of disease between two different groups. Statistical analysis was conducted using SPSS 20.0 (IBM Corp., Armonk, NY, USA). P values < 0.05 were considered statistically significant.
Results
Characterization of PRP-Exos
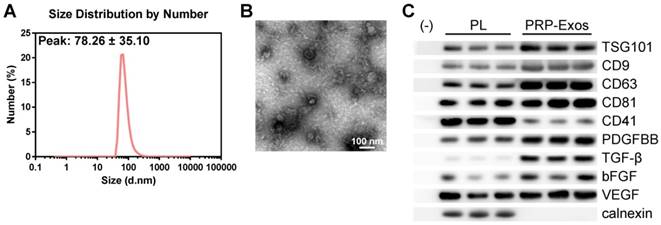
In order to comprehensively characterize the purified nanoparticles derived from PRP, transmission electron microscopy (TEM), dynamic light scattering (DLS) analysis, and western blotting were employed. The DLS measurements revealed that the approximate size of these particles varied between 30 and 100 nm (Fig. 1A), which was consistent with that previously reported [47]. TEM showed that PRP-Exos exhibited a round-shaped morphology, with a size ranging from 30 to 100 nm (Fig. 1B), indicating the presence of exosomes. Moreover, western blotting (Fig. 1C) showed that PRP-Exos were positive for the characteristic exosomal surface markers TSG101, CD9, CD63, and CD81 and the platelet marker CD41 (Fig. 1C), which further confirmed their exosome identity. These data suggested that the nanoparticles were exosomes. At the same time, analysis of the exosome cargo showed that PDGFBB, TGF-β, bFGF and VEGF were contained within PRP-Exos (Fig. 1C). Further, PRP-Exos were negative for a ER membrane protein calnexin (Fig. 1C), which is contained in platelets [48] and not contained in exosomes [49, 50].
Characterization of exosomes derived from PRP. (A) Particle size distribution of PRP-Exos measured by DLS. (B) Morphology of PRP-Exos examined by transmission electron microscopy. Scale bar: 100 nm. (C) Western blotting analysis of the surface biomarkers TSG101, CD9, CD63, and CD81 on PRP-Exos, the source marker CD41 and the encapsulated proteins calnexin, PDGFBB, TGF-β, bFGF, and VEGF, as compared with platelet lysate (PL).
Effects of PRP-Exos on proliferation of cells treated with GCs in vitro and in vivo
To explore the functional roles of PRP-Exos in cell proliferation under normal or high levels of GCs, HMEC-1, BMSCs and MC3T3-E1 cells were cultured in conditioned medium supplemented with DEX with or without PRP-Exos for a series of functional assays. The proliferation of cells was examined by CCK-8 analysis (Fig. 2A) and confirmed by EdU assay (Fig. 2B). The results revealed that DEX treatment markedly reduced the proliferative capability of cells, as previous studies showed that an excess of GCs could directly reduce proliferation, whereas incubation with PRP-Exos resulted in a remarkable increase in the proliferation of all three cell types. Moreover, the DEX-induced down-regulation of proliferation could be significantly rescued by PRP-Exos in all three cell lines.
Effects of PRP-Exos on cell proliferation in vitro and in vivo. (A) The cell viability of HMEC-1, BMSC and MC3T3-E1 cells treated with DEX together with or without PRP-Exos, examined by CCK-8 analysis. (B) The cell proliferation of HMEC-1, BMSC and MC3T3-E1 cells treated with DEX together with or without PRP-Exos was confirmed by EdU kit assay. (C) The effects of PRP-Exos on cellular proliferation in the femoral heads of rats in the short-term subgroup were evaluated by Ki67 immunostaining in vivo. Scale bar: 50 μm.
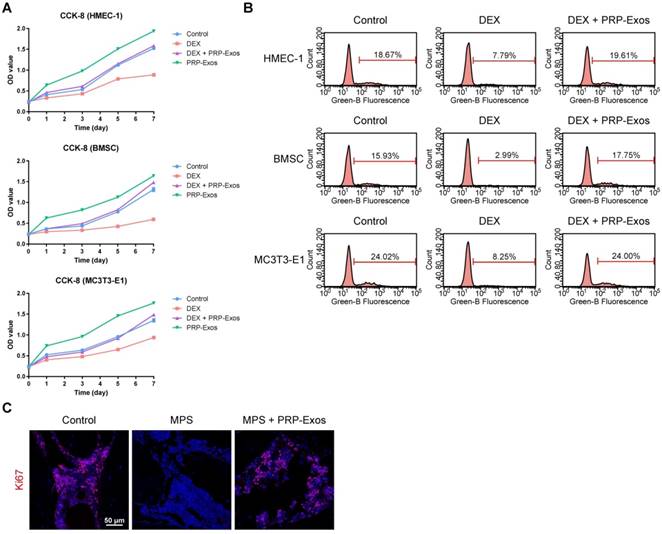
In vivo, the effects of PRP-Exos on cellular proliferation in the femoral heads of rats with GC-induced ONFH in the early stage were evaluated by Ki67 immunostaining. This indicated that the proliferation of cells in the MPS group was reduced compared to normal cells, but PRP-Exos treatment reversed the MPS-induced anti-proliferative effect (Fig. 2C).
These data indicated that PRP-Exos could promote bone tissue maintenance and regeneration, and enhance cellular proliferation.
Effects of PRP-Exos on cell apoptosis induced by GCs in vitro and in vivo
In vitro, cell apoptosis was assessed through Annexin V-FITC/PI double staining with flow cytometric analysis, while the early apoptosis rate of cells was measured. The results suggested that exposure to DEX dramatically increased apoptosis compared to the control group. In contrast, supplementation with PRP-Exos attenuated the apoptotic effect of DEX in HMEC-1, BMSC and MC3T3-E1 cells. Furthermore, HMEC-1, BMSC and MC3T3-E1 cells treated with PRP-Exos alone exhibited fewer apoptotic cells compared to the control group with serum starvation-induced apoptosis (Fig. 3A-C).
Effects of PRP-Exos on GC-induced apoptosis in vitro and in vivo. (A-C) In vitro, the apoptosis of HMEC-1, BMSC and MC3T3-E1 cells induced by DEX together with or without PRP-Exos were assessed through Annexin V-FITC/PI double staining with flow cytometric analysis. (D) Western blotting was used to examine the cleaved caspase-3 expression in HMEC-1, BMSC and MC3T3-E1 cells exposed to DEX and co-treated with or without PRP-Exos. (E) In vivo, the effects of PRP-Exos on the rat model in the short-term subgroup were evaluated by TUNEL assay. Scale bar: 50 μm.
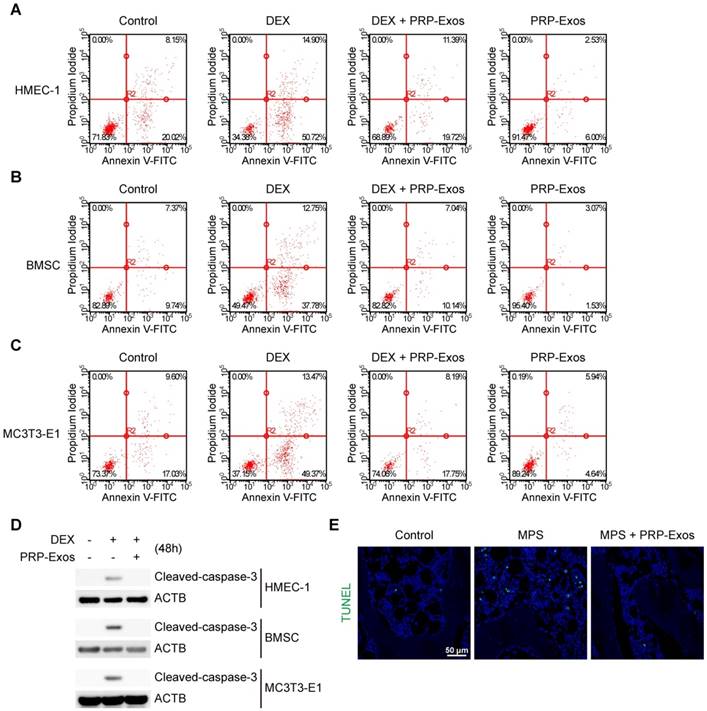
Western blotting results of HMEC-1, BMSC and MC3T3-E1 cells indicated that caspase-3 was activated (termed 'cleaved caspase-3') after 48 h of exposure to DEX, and rescued by co-treatment with PRP-Exos, compared to the control group (Fig. 3D).
In vivo, the anti-apoptosis-promoting effects of PRP-Exos on the rat model of GC-induced ONFH in the early stage were evaluated by TUNEL assay. The results showed that the number of apoptotic cells in the MPS group was notably enhanced compared to the control group, whereas the MPS-induced apoptotic effect was rescued as a result of treatment with PRP-Exos in rats (Fig. 3E).
These data indicated that PRP-Exos could promote bone tissue resistance to apoptosis, and thereby protect the femoral head against necrosis.
Effects of PRP-Exos on angiogenesis of cells or tissue treated with GCs in vitro and in vivo
From tube formation images, the DEX group showed strong anti-angiogenic activity in the angiogenesis assay model with no loop formation. There was not even the slightest indication of loop structure formation after 12 h (Fig. 4A).
Effects of PRP-Exos on angiogenesis of cells or tissue treated with GCs in vitro and in vivo. (A) The angiogenesis of HMEC-1 cells induced by DEX together with or without PPR-Exos was evaluated by tube formation assay. Scale bar: 50 μm. (B) After 3 h induction by DEX together with or without PRP-Exos, the protein expression of Erk, p-Erk, Akt and p-Akt was examined by western blotting. (C) After induction by DEX together with or without PRP-Exos for 24 h, the protein expression of VEGF-A was examined by western blotting. (D) In vivo, the blood supply of the rat model in the long-term subgroup treated with PRP-Exos was examined using MicroFil.
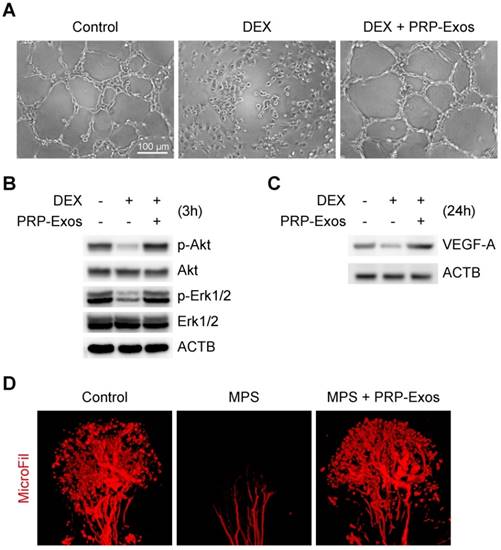
The western blotting results showed that VEGF, bFGF and PDGFBB were highly expressed in the PRP-Exos (Fig. 1C), suggesting that Akt and Erk pathways can be easily activated. To verify this hypothesis and confirm whether these effector proteins can respond to PRP-Exos stimulation, western blotting was carried out to assess the protein levels of Akt and p-Akt, Erk1/2 and p-Erk1/2 in HMEC-1 cells with or without DEX treatment with PRP-Exos for 3 h. Compared to DEX, incubation with PRP-Exos resulted in a significant increase in Akt and Erk phosphorylation level, whereas in the control group, the phosphorylation level was not significantly different, indicating that PRP-Exos can rescue angiogenesis during DEX exposure (Fig. 4B).
The VEGF-A expression level of HMEC-1 cells was also measured using western blotting analyses, and the results showed that the protein expression level of VEGF-A in cells treated with DEX was significantly lower than the control group. However, compared with DEX treatment alone, the VEGF-A protein content of cells co-treated with DEX and PRP-Exos was clearly higher (Fig. 4C).
In vivo, the blood supply of the rat model of GC-induced ONFH was examined by MicroFil. After treatment using the appropriate protocol for six weeks, the femoral heads of all the rats were harvested. Micro-CT analysis showed that GCs significantly damaged the blood supply of femoral heads but that co-administration with PRP-Exos had a notable protective effect on blood vessels in femoral heads, which was demonstrated by both reconstructed images and total vessel volume (Fig. 4D).
Effects of PRP-Exos on osteogenesis of BMSC and MC3T3-E1 cells treated with DEX in vitro
In order to observe the effects of the presence or absence of PRP-Exos with DEX on osteogenesis of BMSCs and MC3T3-E1 cells, the capacity of BMSCs and MC3T3-E1 cells to differentiate into osteogenic lineages was tested using specific differentiation medium (Cyagen), and the formation of calcium mineral deposits was measured by Alizarin red staining after 21 days of induction (Fig. 5A). The results indicated that PRP-Exos can prevent the inhibition of osteogenesis observed in BMSCs and MC3T3-E1 cells caused by DEX in vitro.
Effects of PRP-Exos on osteogenesis of BMSCs and MC3T3-E1 cells treated with DEX in vitro. Osteogenesis was induced together with or without DEX or PRP-Exos in BMSCs and MC3T3-E1 cells. (A) After 21 days, the formation of calcium mineral deposits was measured by Alizarin red staining. Scale bar: 50 μm. (B) After 7 days, the protein expression levels of Runx2, type I collagen and β-catenin were examined by western blotting. (C) After 7 days, the ALP expression level and (D), after 14 days, the OCN expression level, were examined by ELISA.
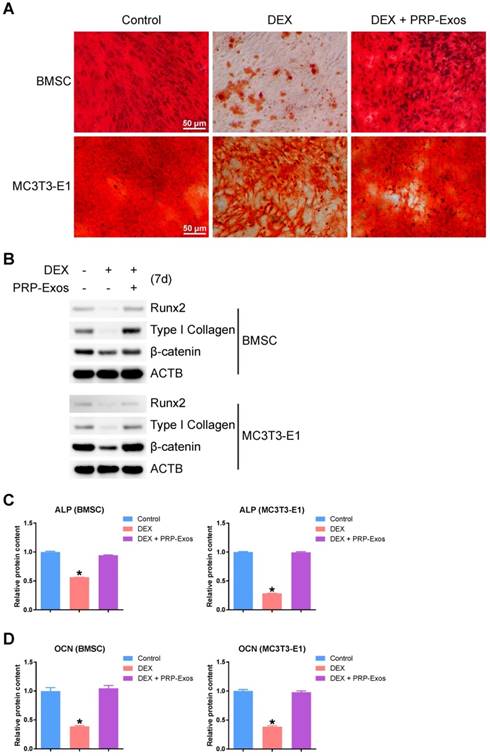
Using western blotting, analysis of the protein expression level of Runx2 (runt-related transcription factor 2), type I Collagen and β-catenin in BMSCs and MC3T3-E1 cells showed that PRP-Exos could rescue the expression of osteogenesis-related proteins and maintain the cells' osteogenic differentiation capacity (Fig. 5B).
Using ELISA, the ALP expression level on day 7 (Fig. 5C) and the OCN expression level on day 14 (Fig. 5D) were measured after osteogenic induction. The results indicated that the ALP and OCN expression levels were both rescued by PRP-Exos in BMSCs and MC3T3-E1 cells, compared to treatment with DEX alone.
Osteogenesis-promoting effects of PRP-Exos on the rat model of GC-induced ONFH
To investigate the effects of PRP-Exos on MPS-induced ONFH, the rat model of ONFH was created by intramuscular injection of MPS, followed by intravenous administration of PRP-Exos or an equal volume of PBS. Six weeks after treatment, micro-CT scanning was carried out to qualitatively evaluate the bone tissues within the femoral head (Fig. 6A). The results showed that 80% of rats treated with long-term high-dose MPS exhibited significant trabecular changes, such as bone mineral loss and cystic degeneration in the subchondral area of the femoral heads, compared with the normal rats. In contrast, the subchondral trabeculae of the femoral heads appeared intact and well distributed in rats co-treated with PRP-Exos, and only 13.3% of rats were found to have very mild osteonecrotic changes. Qualitative analyses of all the micro-CT parameters then further confirmed the preventive efficacy of PRP-Exos on GC-induced ONFH in rats.
The osteogenesis-promoting effects of PRP-Exos on the rat model of GC-induced ONFH. (A) Reconstructed coronal, transverse and sagittal images of bones within the normal, MPS and PRP-Exos groups. (B) Quantitative analyses of trabecular thickness (Tb.Th), trabecular separation (Tb.Sp), bone volume per tissue volume (BV/TV), and trabecular number (Tb.N) in the different treatment groups. (C) HE staining of the femoral heads in rats receiving different treatments. Scale bar: 100 μm. (D) Immunohistochemical staining of type I collagen in samples from the different treatment groups. Scale bar: 100 μm.
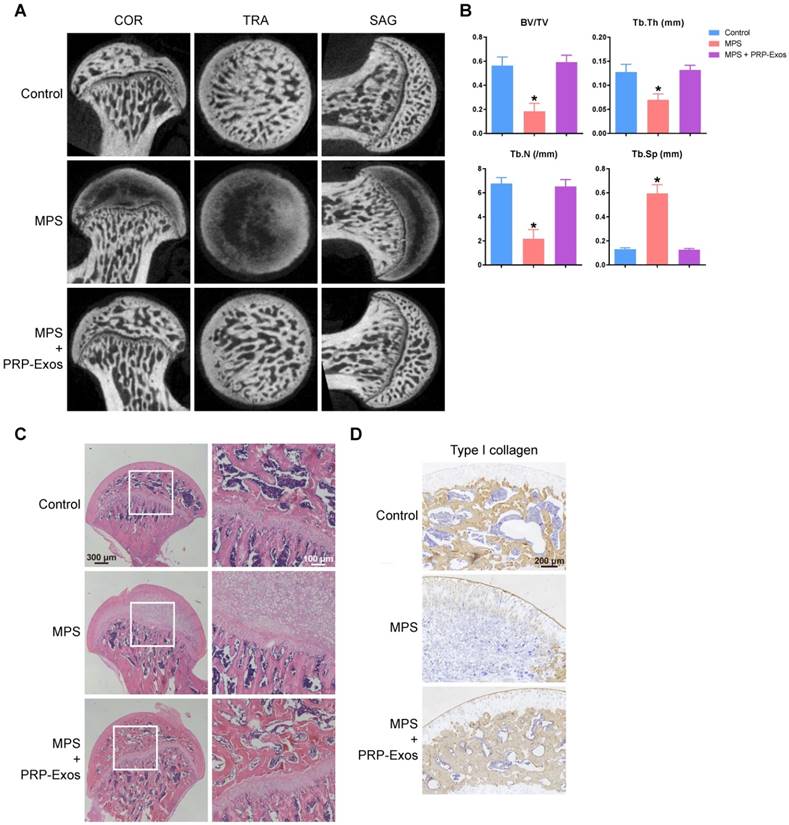
As shown in Fig. 6B, the microstructural parameters Tb.Th, BV/TV, and Tb.N in the MPS group were markedly reduced compared with the normal group, but additional treatment with PRP-Exos could remarkably reverse the MPS-induced reduction of these parameters. Moreover, the parameter Tb.Sp was significantly increased in the MPS group compared to the normal group, and this increase could be profoundly inhibited by PRP-Exos.
In keeping with the above findings, histological evidence based on HE staining (Fig. 6C) revealed that osteonecrosis was obvious in the MPS group. In these MPS-treated rats, the trabecular bone of the femoral head became sparser and even disappeared, replaced by abnormal morphology. In contrast, the trabecular bone in the rats additionally treated with PRP-Exos was well organized, and few trabecular structures or bone marrow were replaced by necrotic tissues. The incidence of histological ONFH in the PRP-Exos group was markedly lower than in the MPS group, and no osteonecrosis was observed in the normal group.
Type I collagen is the most abundant protein of bone matrix [51] and is closely related to various bone diseases [52]. IHC staining for type I collagen revealed that osteogenesis of femoral heads decreased in the MPS group, but this effect was markedly inhibited by PRP-Exos (Fig. 6D).
PRP-Exos rescued cells from GC-induced apoptosis through the Akt/Bad/Bcl-2 pathway
Twenty-four h after treating BMSCs with PRP-Exos or DEX, western blotting was performed to detect PERK phosphorylation. The results showed that high-dose GCs activated ER stress and then PERK was activated by phosphorylation to perform its protein-folding function. CHOP, one downstream effector protein of PERK [53], inhibits B-cell lymphoma 2 (Bcl-2) protein expression, and then caspase-3 is cleaved, leading to cell apoptosis [54]. The PERK pathway is the critical mechanism of ER stress-induced apoptosis.
When GSK2656157, a PERK inhibitor [55], was added to BMSCs together with DEX, the phosphorylation level of PERK and the protein expression level of CHOP decreased, and Bcl-2 protein expression levels increased, then cleaved-caspase-3 could not be detected (Fig. 7A).
PRP-Exos rescued cells from GC-induced apoptosis through the Akt/Bcl-2 pathway. (A) In the different treatment groups, the p-PERK, PERK, CHOP, Bcl-2 and cleaved-caspase-3 protein expression levels were examined by western blotting. (B) In the different treatment groups, the p-Akt, Akt, P-Bad, Bad, Bcl-2 and cleaved-caspase-3 protein expression levels were examined by western blotting. (C) A brief schematic diagram showing the underlying mechanism of PRP-Exos prevention of GC-induced ONFH.
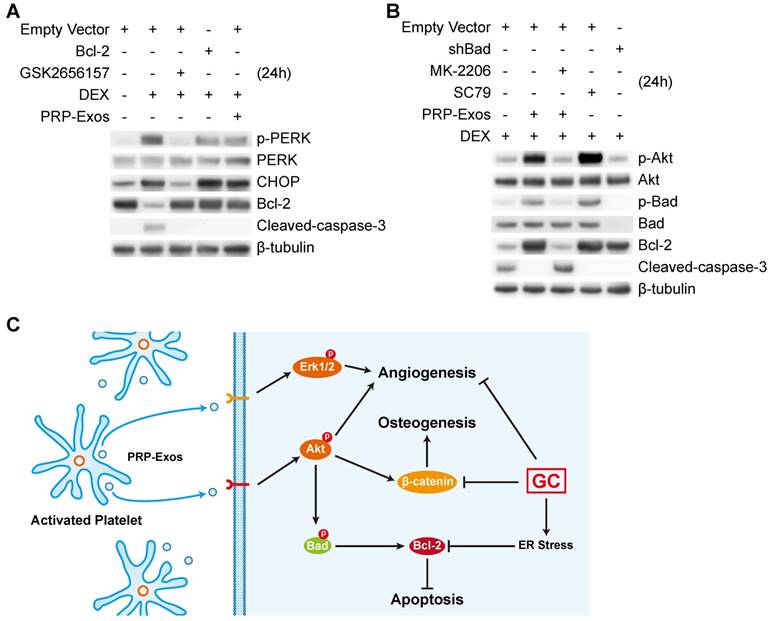
To investigate this further, we over-expressed Bcl-2, which is an anti-apoptotic protein [56], in BMSCs. With Bcl-2 over-expression, DEX still activated PERK and upregulated CHOP, but the inhibition of Bcl-2 by CHOP was blocked, and cleaved-caspase-3 could not be detected.
When PRP-Exos were added to BMSCs together with DEX, the Bcl-2 protein level was rescued by PRP-Exos and cleaved-caspase-3 could not be detected.
As shown in Figure 7A, the results showed that PRP-Exos could block ER stress-induced apoptosis without altering PERK activation and CHOP expression, effects which may be exerted through maintaining Bcl-2 expression.
Therefore, in order to study the molecular mechanism of the rescue effect of PRP-Exos on GC-induced apoptosis, PRP-Exos were added, together with DEX, to BMSCs, with the result that Akt and Bad were phosphorylated, Bcl-2 expression increased, and cleaved-caspase-3 could then not be detected. Akt is a potent inhibitory signal for apoptosis and apoptosis is activated by several members of the Bcl-2 and Caspase families. Following activation, Akt inhibits pro-apoptotic mediators such as Bad; Bcl-2 expression is enhanced and caspase-3 is then inhibited [57].
When Bad is inactivated by phosphorylation, Bcl-2 level is enhanced. MK-206, an Akt inhibitor, was added together with PRP-Exos. We found that the PRP-Exos promoted the phosphorylation level of Akt, Bad was blocked, and Bcl-2 decreased. Then cleavage of caspase-3 was activated and cell apoptosis returned. When SC79, a novel small molecular activator of Akt, was added with DEX to BMSCs, the effect was similar to PRP-Exos and the results indicated that Bcl-2 might be the key protein in the anti-apoptotic effect of PRP-Exos.
Finally, we used lentiviral shRNA to knock down Bad in BMSCs, which can enhance Bcl-2 expression, and block the apoptosis induced by the suppression of Bcl-2.
Here we demonstrated that the stimulatory effects of PRP-Exos on anti-apoptosis mainly resulted from the activation of the Akt/Bad/Bcl-2 signal pathway, as shown in Figure 7B.
Figure 7C is a brief schematic diagram showing the mechanism underlying the preventive effect of PRP-Exos on GC-induced ONFH.
Discussion
The key point of ONFH is cell death [19], which is induced mainly by GCs. The effect of PRP-Exos on the rescue of EC apoptosis is mediated by significant enhancement of the activation of the Akt and Erk signaling cascade under GCs treatment. Erk and Akt were phosphorylated to activate the angiogenesis signaling pathways. Akt conveys anti-apoptotic signals [58], thus the Akt pathway is considered the main survival pathway in ECs.
Among these anti-apoptotic factors acting on ECs, VEGF, which is highly expressed in PRP-Exos, is the key modulator of normal vessel generation, whose signaling cascades in ECs will provide molecular targets for the development of angiogenic therapies [59]. Among the three receptors of VEGF, VEGFR2 appears to be critical for the regulation of angiogenesis, as it conveys signals for the promotion of the proliferation, survival, and migration of ECs. Thus, upon VEGF-A binding, phosphorylation of VEGFR2 leads to the activation of PKC phosphorylating mitogen-activated protein kinase (MAPK/Erk1/2), which leads to the proliferation of ECs. Through these two important pathways [60], EC apoptosis was rescued and vascularization was greatly improved, with rapid promotion of the blood supply. In addition, the results of our in vivo study showed that the blood supply was significantly protected by PRP-Exos.
In bone cells, at the molecular level, GCs regulate various functions, including osteoblastogenesis and apoptosis [61]. In this study, the inhibited formation of mineralized nodules, and decreased protein expression of Runx2, type I collagen, β-catenin, ALP and OCN were all rescued by PRP-Exos, compared to treatment with DEX alone in the BMSCs and MC3T3-E1 cells.
GCs excess inhibits osteoblast differentiation and induces osteoblast apoptosis due to high levels of GC-enhanced β-catenin degeneration [62]. At physiological concentrations, GCs directly stimulate normal osteoblasts to produce and release Wnt proteins, which can act in a paracrine manner to activate the canonical Wnt signaling cascade. This results in accumulation of β-catenin and expression of the master regulator of osteoblast differentiation, Runx2, which drives the differentiation of these pluripotent cells towards the osteoblastic lineage [63]. The addition of GCs to culture medium is crucial for the formation of mineralized nodules. GCs must govern osteoblast differentiation through direct actions [63]. Akt triggers phosphorylation-mediated deactivation of glycogen synthase kinase 3 beta (GSK-3β) and GSK-3β controls the stability of β-catenin. [64] Thus, we concluded that the PRP-Exos stabilized β-catenin via Akt/GSK-3β/β-catenin and finally maintained the osteogenic ability of BMSCs and other preosteoblastic cells.
There are studies suggesting that part of the pro-apoptotic actions of GCs on bone cells are mediated through ER stress [65].
Normally, the vast majority of proteins secreted onto, or displayed on, the cell surface first enter the ER to fold and assemble. Only correctly-assembled proteins can advance from the ER to the cell surface. To ascertain fidelity in protein folding, cells regulate the protein-folding capacity in the ER according to need. The ER responds to the burden of unfolded proteins in its lumen (ER stress) by activating intracellular signal transduction pathways, collectively termed the UPR. There are at least three mechanistically-distinct branches of the UPR, which regulate the expression of numerous genes that maintain homeostasis in the ER, or induce apoptosis if ER stress remains overloaded [66]. Three UPR signal pathways have been characterized as the pathways that are initiated by the stress sensors protein kinase-like endoplasmic reticulum kinase (PERK), inositol-requiring protein 1α (IRE1α) and activating transcription factor 6 (ATF6).
ER stress is a “double-edged sword”. It results from accumulation of misfolded/unfolded proteins, which can trigger apoptosis, and can be alleviated by phosphorylation of eukaryotic translation initiation factor 2α (eIF2α). Phosphorylated eIF2α slows the global rate of protein translation to provide time for the ER to recover from the excessive protein load, allowing the cell to escape from apoptosis and thus promoting cell viability [12, 66].
The UPR helps the cells to adapt to ER stress and facilitates cell survival [67]. Under acute ER stress, the UPR is activated to maintain ER protein-folding homeostasis. This involves the transient attenuation of protein synthesis, an increase in protein folding and transport in the ER, and an increase in ER-associated protein degradation and autophagy. To reduce the protein-folding load on the ER, PERK is activated to phosphorylate eIF2α, which halts the mRNA translation. After translation, ATF4 (Activating Transcription Factor 4) enters the nucleus. ATF4 also activates the transcription of the pro-apoptotic protein CHOP. Once ER protein-folding homeostasis is restored, ATF4 and CHOP induce transcription to direct eIF2α de-phosphorylation and restart global mRNA translation, which is essential for cells to survive an acute injury.
However, prolonged UPR activation induces apoptosis, and PERK-mediated phosphorylation of eIF2α can contribute to cell death by inhibiting the synthesis of pro-survival proteins. One of the downstream targets of PERK, the transcription factor CHOP, may repress Bcl-2 expression [68]. The pro-apoptotic action occurs mainly through activation of the PERK-eIF2α-ATF4-CHOP-Bcl-2 pathway [65, 69]. Studies have demonstrated that CHOP suppresses transcription of the anti-apoptotic protein Bcl-2. Apoptotic cell death also suppresses the synthesis of anti-apoptotic Bcl-2 proteins [70].
Interestingly, our results suggested that PRP-Exos enhanced the activation of Akt under ER stress and the Akt inhibitor MK-2206 2HCl blocked this anti-apoptotic effect. PRP-Exos increased the expression of Bcl-2 and blocked the activation of caspase-3. Analysis of the expression of key molecular factors involved in cell apoptosis indicated that the protective effects of PRP-Exos depend on the Akt/ Bcl-2 signaling pathway.
Upon stimulation by a variety of extracellular signals such as by PRP-Exos in this study, Akt was phosphorylated and thereby activated. Activated Akt inactivates Bad (Bcl-2-associated death promoter), which promotes cell death by inhibiting Bcl-2 protein expression [71]. Our results showed that Bcl-2 protein expression was re-activated through increased phosphorylation of Bad, which was in agreement with previous research [72].
Bcl-2 is an anti-apoptotic protein that is localized to the outer mitochondrial membrane where it prevents the release of cytochrome c from mitochondria by inhibiting apoptosis-induced mitochondrial pore formation. Even under ER stress when CHOP protein expression was high, the results indicated that Bcl-2 protein expression was near normal compared to the control in the PRP-Exos treatment group. Translocation of cytochrome c from the mitochondria to the cytoplasm is a key event in the activation of caspase-3 and the induction of apoptosis [73].
The Akt signaling pathway has been implicated in a wide range of cellular functions involving cell survival, proliferation, angiogenesis, metabolism and cell migration. Activation of Akt led to an increase of an anti-apoptotic protein of the Bcl family, Bcl-2, in BMSCs. Our results showed that using Bad shRNA to knock down Bad in BMSCs rescued the expression of Bcl-2 in BMSCs, indicating that the effect of Bad knockdown on Bcl-2 protein expression was similar to that of Akt activation modulating Bad inactivation.
Moreover, when Akt was directly activated by its activator SC79 in DEX-induced BMSCs, the activated Akt up-regulated the Bcl-2 protein level, as well as inducing a synergistic effect of cleaved-caspase-3 down-regulation. When Akt was inhibited by MK-2206 2HCl in DEX-induced cells in the presence of PRP-Exos, the results demonstrated that treatment with PRP-Exos rescued Bcl-2 protein expression, which was blocked by the inhibition of Akt. In addition, the fact that the level of Bcl-2 protein stayed the same indicated that it may cause apoptosis resistance, which contributed to the overall mechanism of GC-induced apoptosis. This steady-state Bcl-2 expression level was regulated by the activation of both CHOP and Akt.
In summary, the present results first confirmed that the Akt/Bad signal pathway plays a crucial role in the protective effect of PRP-Exos on GC-ER stress-induced apoptosis. Findings from this study will help shed light on the mechanism of PRP-Exos' anti-apoptotic effects under GC-induced ER stress.
On top of these benefits, the prominent feature of PRP-Exos is their potential as mediators of intercellular communication, even across species. Cross-species communication involving EVs has been reported several times. For example, EVs derived from LX2, a human liver stellate cell line, have been used in a rat model [74], and human MSC-derived EVs have been shown to have therapeutic effects on a mouse model [75] and a rat model [76]. These studies focused on the communication from humans to other species. There was also a study which investigated the communication from other species to humans. Kusuma et al. [77] demonstrated that humans could absorb cows' milk exosomes and transport them to peripheral tissues across species boundaries. The immune evasion mechanisms of exosomes remain unclear and should be further studied. This hypothesis by our team assumed that the majority of protein was encapsulated in the lipid bilayers and exposed only during the exosome-cytomembrane interaction. The exposed protein produced the effect quickly and was then endocytosed and degraded by target cells. Therefore, without direct contact, the immune response was potentially overridden. Another advantage of exosomes is that the lipid bilayer could protect the proteins and RNAs from the actions of proteinases and RNAases. This characteristic means that exosomes could reach the whole body while directly-applied growth factors would mostly be degraded before reaching the damaged area.
Platelets are produced from megakaryocytes (MKs), predominantly to maintain the normal blood platelet count and express the most platelet proteins [78]. Platelets themselves have little or no ability to manufacture proteins. The exact criterion for the confirmation of MKs is described as high expression of a platelet biomarker such as CD41 or CD61. Analysis of protein expression levels showed that CD41 was present not only in platelets but also in exosomes derived from PRP. To confirm the purity of exosomes, calnexin was examined and the result showed that PRP-Exos were negative for calnexin, which is contained in platelets [48] and not contained in exosomes [49, 50]. This result further confirmed that the exosomes derived from PRP indeed contained CD41.
These results indicated that PRP-Exos might be produced in multivesicular bodies (MVBs) of MKs and carried by platelets. The relationship between exosomes derived from MKs and PRP-Exos needs further study to thoroughly elucidate the intricate molecular mechanism of PRP therapy.
Summary and Conclusions
In summary, our study demonstrates for the first time that PRP-Exos play an anti-apoptotic role against GC-ER stress-induced apoptosis in vitro and in vivo. In ECs, PRP-Exos activate the Akt and Erk pathways to promote cell proliferation and survival, thus promoting angiogenesis. In bone cells, PRP-Exos rescue the osteogenic protein expression level through the Wnt/β-catenin signaling pathway, thus maintaining osteogenic differentiation and osteogenesis. Under ER stress, PRP-Exos block the CHOP-mediated inhibition of Bcl-2 protein expression via the Akt/Bad/Bcl-2/Caspase-3 signal pathway, saving the cell from apoptosis.
Abbreviations
ONFH: osteonecrosis of the femoral head
GCs: glucocorticoids
ER: endoplasmic reticulum
ER stress: endoplasmic reticulum stress
ACD-A: acid citrate dextrose solution A
PRP: platelet-rich plasm
PRP-Exos: exosomes derived from PRP
Akt: protein kinase B
Erk: extracellular signal regulated kinase
PERK: Protein kinase-like endoplasmic reticulum kinase
eIF2α: eukaryotic translation initiation factor 2α
IRE1α: inositol-requiring protein 1α
ATF4: activating transcription factor 4
ATF6: activating transcription factor 6
CHOP: CCAAT-enhancer-binding protein homologous protein
UPR: unfolded protein response
ECs: endothelial cells
EVs: extracellular vesicles
Bcl-2: B-cell lymphoma 2
Bad: Bcl-2-associated death promoter
ROS: reactivate oxygen species
HE: Hematoxylin and eosin
IHC: immunohistochemistry
OCN: osteocalcin
ALP: alkaline phosphatase
Runx2: runt-related transcription factor 2
PVDF: polyvinylidene Fluoride
TBS: tris-buffered Saline
TUNEL: terminal deoxynucleotidyl transferase dUTP nick end labeling
BMSCs: bone mesenchymal stem cells
HMEC-1: the human microvascular endothelial cell line
MVBs: multivesicular bodies
PS: penicillin-streptomycin solution
PBS: phosphate buffered saline
DMEM: Dulbecco's modified Eagle's medium
FBS: fetal bovine serum
DLS: dynamic light scattering
TEM: transmission electron microscopy
Tb.Th: trabecular thickness
Tb.Sp: trabecular separation
BV/TV: bone volume per tissue volume
Tb.N: trabecular number
EDTA: ethylene diamine tetraacetic acid
IHC: immunohistochemistry
α-MEM: α-modified minimum essential medium
DEX: dexamethasone
CCK-8: cell counting kit-8
OD: optical density
PI: propidium iodide
HRP: horseradish peroxidase
MPS: methylprednisolone
CT: computed tomography
Acknowledgements
We thank the National Natural Science Foundation of China for their support of our work (Grant No. 81301589, No. 81572239 and No. 81472066). This work was also supported by Joint Project Funding for Major Diseases in Shanghai (Grant No.2014ZYJB0301).
The authors would also like to acknowledge Prof. Jun-Feng Yin from Tong Ji University for his assistance with the statistical analysis and thank Shanghai Institute of Microsurgery on Extremities for providing experimental equipment. We acknowledge all the scientific and technical support from the animal experimental center affiliated with Shanghai Sixth People's Hospital.
Ethics
This study was carried out in adherence to the Declaration of Helsinki. All experimental and animal care procedures were approved by the Animal Research Ethics Committee of Shanghai Sixth People's Hospital and performed in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. Written informed consent was obtained from each donor with the permission of the Institutional Review Board at Shanghai Sixth People's Hospital.
Author Contributions
S.C.T. performed the experiments and statistical analysis, and drafted the manuscript. B.Y.R. helped with the animal experiment. T.Y. and Z.Z.Z participated in the statistical analysis. S.C.G. participated in the design of the study and the statistical analysis, and helped to draft the manuscript. C.Q.Z. conceived of the study and participated in its design. All authors read and approved the final manuscript.
Competing interests
The authors have declared that there are no competing interests.
References
1. Bose VC, Baruah BD. Resurfacing arthroplasty of the hip for avascular necrosis of the femoral head: a minimum follow-up of four years. J Bone Joint Surg Br. 2010;92:922-8
2. Steffen RT, Athanasou NA, Gill HS, Murray DW. Avascular necrosis associated with fracture of the femoral neck after hip resurfacing: histological assessment of femoral bone from retrieval specimens. J Bone Joint Surg Br. 2010;92:787-93
3. Hao C, Yang S, Xu W, Shen JK, Ye S, Liu X. et al. MiR-708 promotes steroid-induced osteonecrosis of femoral head, suppresses osteogenic differentiation by targeting SMAD3. Sci Rep. 2016;6:22599
4. Zhang Y, Yin J, Ding H, Zhang C, Gao YS. Vitamin K2 Ameliorates Damage of Blood Vessels by Glucocorticoid: a Potential Mechanism for Its Protective Effects in Glucocorticoid-induced Osteonecrosis of the Femoral Head in a Rat Model. Int J Biol Sci. 2016;12:776-85
5. Zhang YL, Yin JH, Ding H, Zhang W, Zhang CQ, Gao YS. Vitamin K2 Prevents Glucocorticoid-induced Osteonecrosis of the Femoral Head in Rats. Int J Biol Sci. 2016;12:347-58
6. Liu YF, Chen WM, Lin YF, Yang RC, Lin MW, Li LH. et al. Type II collagen gene variants and inherited osteonecrosis of the femoral head. N Engl J Med. 2005;352:2294-301
7. Jilka RL, Weinstein RS, Bellido T, Roberson P, Parfitt AM, Manolagas SC. Increased bone formation by prevention of osteoblast apoptosis with parathyroid hormone. J Clin Invest. 1999;104:439-46
8. Plotkin LI, Manolagas SC, Bellido T. Glucocorticoids induce osteocyte apoptosis by blocking focal adhesion kinase-mediated survival. Evidence for inside-out signaling leading to anoikis. J Biol Chem. 2007;282:24120-30
9. Plotkin LI, Weinstein RS, Parfitt AM, Roberson PK, Manolagas SC, Bellido T. Prevention of osteocyte and osteoblast apoptosis by bisphosphonates and calcitonin. J Clin Invest. 1999;104:1363-74
10. Hamamura K, Yokota H. Stress to endoplasmic reticulum of mouse osteoblasts induces apoptosis and transcriptional activation for bone remodeling. FEBS Lett. 2007;581:1769-74
11. Saito A, Ochiai K, Kondo S, Tsumagari K, Murakami T, Cavener DR. et al. Endoplasmic reticulum stress response mediated by the PERK-eIF2(alpha)-ATF4 pathway is involved in osteoblast differentiation induced by BMP2. J Biol Chem. 2011;286:4809-18
12. Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M. et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell. 2003;11:619-33
13. Iuchi T, Akaike M, Mitsui T, Ohshima Y, Shintani Y, Azuma H. et al. Glucocorticoid excess induces superoxide production in vascular endothelial cells and elicits vascular endothelial dysfunction. Circ Res. 2003;92:81-7
14. Williams TA, Verhovez A, Milan A, Veglio F, Mulatero P. Protective effect of spironolactone on endothelial cell apoptosis. Endocrinology. 2006;147:2496-505
15. O'Connell BJ, Genest J Jr. High-density lipoproteins and endothelial function. Circulation. 2001;104:1978-83
16. Greenberger S, Boscolo E, Adini I, Mulliken JB, Bischoff J. Corticosteroid suppression of VEGF-A in infantile hemangioma-derived stem cells. N Engl J Med. 2010;362:1005-13
17. Aschbacher K, Derakhshandeh R, Flores AJ, Narayan S, Mendes WB, Springer ML. Circulating angiogenic cell function is inhibited by cortisol in vitro and associated with psychological stress and cortisol in vivo. Psychoneuroendocrinology. 2016;67:216-23
18. Gao YS, Wang HF, Ding H, Zhang CQ. A novel rat model of osteonecrosis of the femoral head induced by periarticular injection of vascular endothelial growth factor receptor 2 antibody. J Surg Res. 2013;183:e1-5
19. Yu Z, Fan L, Li J, Ge Z, Dang X, Wang K. Lithium prevents rat steroid-related osteonecrosis of the femoral head by beta-catenin activation. Endocrine. 2016;52:380-90
20. Hartmann K, Koenen M, Schauer S, Wittig-Blaich S, Ahmad M, Baschant U. et al. Molecular Actions of Glucocorticoids in Cartilage and Bone During Health, Disease, and Steroid Therapy. Physiol Rev. 2016;96:409-47
21. Phillips JE, Gersbach CA, Wojtowicz AM, Garcia AJ. Glucocorticoid-induced osteogenesis is negatively regulated by Runx2/Cbfa1 serine phosphorylation. J Cell Sci. 2006;119:581-91
22. Weinstein RS. Clinical practice. Glucocorticoid-induced bone disease. N Engl J Med. 2011;365:62-70
23. Israel E, Banerjee TR, Fitzmaurice GM, Kotlov TV, LaHive K, LeBoff MS. Effects of inhaled glucocorticoids on bone density in premenopausal women. N Engl J Med. 2001;345:941-7
24. Zhu HY, Gao YC, Wang Y, Zhang CQ. Circulating exosome levels in the diagnosis of steroid-induced osteonecrosis of the femoral head. Bone Joint Res. 2016;5:276-9
25. Wu Z, Ji C, Li H, Qiu G, Gao C, Weng X. Elevated level of membrane microparticles in the disease of steroid-induced vascular osteonecrosis. J Craniofac Surg. 2013;24:1252-6
26. S ELA, Mager I, Breakefield XO, Wood MJ. Extracellular vesicles: biology and emerging therapeutic opportunities. Nat Rev Drug Discov. 2013;12:347-57
27. Tkach M, Thery C. Communication by Extracellular Vesicles: Where We Are and Where We Need to Go. Cell. 2016;164:1226-32
28. Gyorgy B, Hung ME, Breakefield XO, Leonard JN. Therapeutic applications of extracellular vesicles: clinical promise and open questions. Annu Rev Pharmacol Toxicol. 2015;55:439-64
29. Yuan T, Guo SC, Han P, Zhang CQ, Zeng BF. Applications of leukocyte- and platelet-rich plasma (L-PRP) in trauma surgery. Curr Pharm Biotechnol. 2012;13:1173-84
30. Kim YH, Furuya H, Tabata Y. Enhancement of bone regeneration by dual release of a macrophage recruitment agent and platelet-rich plasma from gelatin hydrogels. Biomaterials. 2014;35:214-24
31. Yin WJ, Xu HT, Sheng JG, An ZQ, Guo SC, Xie XT. et al. Advantages of Pure Platelet-Rich Plasma Compared with Leukocyte- and Platelet-Rich Plasma in Treating Rabbit Knee Osteoarthritis. Med Sci Monit. 2016;22:1280-90
32. Yuan T, Zhang CQ, Wang JH. Augmenting tendon and ligament repair with platelet-rich plasma (PRP). Muscles Ligaments Tendons J. 2013;3:139-49
33. Yuan T, Zhang J, Zhao G, Zhou Y, Zhang CQ, Wang JH. Creating an Animal Model of Tendinopathy by Inducing Chondrogenic Differentiation with Kartogenin. PLoS One. 2016;11:e0148557
34. Liu J, Yuan T, Zhang C. Three cases using platelet-rich plasma to cure chronic soft tissue lesions. Transfus Apher Sci. 2011;45:151-5
35. Yuan T, Zhang CQ, Tang MJ, Guo SC, Zeng BF. Autologous Platelet-rich Plasma Enhances Healing of Chronic Wounds. Wounds. 2009;21:280-5
36. Yuan T, Zhang C, Zeng B. Treatment of chronic femoral osteomyelitis with platelet-rich plasma (PRP): a case report. Transfus Apher Sci. 2008;38:167-73
37. Pak J, Lee JH, Jeon JH, Lee SH. Complete resolution of avascular necrosis of the human femoral head treated with adipose tissue-derived stem cells and platelet-rich plasma. J Int Med Res. 2014;42:1353-62
38. Ibrahim V, Dowling H. Platelet-rich plasma as a nonsurgical treatment option for osteonecrosis. PM R. 2012;4:1015-9
39. Yokota K, Ishida O, Sunagawa T, Suzuki O, Nakamae A, Ochi M. Platelet-rich plasma accelerated surgical angio-genesis in vascular-implanted necrotic bone: an experimental study in rabbits. Acta Orthop. 2008;79:106-10
40. Torreggiani E, Perut F, Roncuzzi L, Zini N, Baglio SR, Baldini N. Exosomes: novel effectors of human platelet lysate activity. Eur Cell Mater. 2014;28:137-51 discussion 51
41. Burger D, Vinas JL, Akbari S, Dehak H, Knoll W, Gutsol A. et al. Human endothelial colony-forming cells protect against acute kidney injury: role of exosomes. Am J Pathol. 2015;185:2309-23
42. Xin H, Li Y, Chopp M. Exosomes/miRNAs as mediating cell-based therapy of stroke. Front Cell Neurosci. 2014;8:377
43. Thery C, Amigorena S, Raposo G, Clayton A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol. 2006 Chapter 3: Unit 3 22
44. Jia XE, Ma K, Xu T, Gao L, Wu S, Fu C. et al. Mutation of kri1l causes definitive hematopoiesis failure via PERK-dependent excessive autophagy induction. Cell Res. 2015;25:946-62
45. Yang J, Wu Z, Renier N, Simon DJ, Uryu K, Park DS. et al. Pathological axonal death through a MAPK cascade that triggers a local energy deficit. Cell. 2015;160:161-76
46. Jo H, Mondal S, Tan D, Nagata E, Takizawa S, Sharma AK. et al. Small molecule-induced cytosolic activation of protein kinase Akt rescues ischemia-elicited neuronal death. Proc Natl Acad Sci U S A. 2012;109:10581-6
47. De Jong OG, Van Balkom BW, Schiffelers RM, Bouten CV, Verhaar MC. Extracellular vesicles: potential roles in regenerative medicine. Frontiers in immunology. 2014;5:608
48. Crescente M, Pluthero FG, Li L, Lo RW, Walsh TG, Schenk MP. et al. Intracellular Trafficking, Localization, and Mobilization of Platelet-Borne Thiol Isomerases. Arterioscler Thromb Vasc Biol. 2016;36:1164-73
49. Gastpar R, Gehrmann M, Bausero MA, Asea A, Gross C, Schroeder JA. et al. Heat shock protein 70 surface-positive tumor exosomes stimulate migratory and cytolytic activity of natural killer cells. Cancer Res. 2005;65:5238-47
50. Lasser C, Alikhani VS, Ekstrom K, Eldh M, Paredes PT, Bossios A. et al. Human saliva, plasma and breast milk exosomes contain RNA: uptake by macrophages. J Transl Med. 2011;9:9
51. Garcia-Giralt N, Nogues X, Enjuanes A, Puig J, Mellibovsky L, Bay-Jensen A. et al. Two new single-nucleotide polymorphisms in the COL1A1 upstream regulatory region and their relationship to bone mineral density. J Bone Miner Res. 2002;17:384-93
52. Stover DA, Verrelli BC. Comparative vertebrate evolutionary analyses of type I collagen: potential of COL1a1 gene structure and intron variation for common bone-related diseases. Mol Biol Evol. 2011;28:533-42
53. Mihailidou C, Panagiotou C, Kiaris H, Kassi E, Moutsatsou P. Crosstalk between C/EBP homologous protein (CHOP) and glucocorticoid receptor in lung cancer. Mol Cell Endocrinol. 2016;436:211-23
54. Holmes A, Abraham DJ, Sa S, Shiwen X, Black CM, Leask A. CTGF and SMADs, maintenance of scleroderma phenotype is independent of SMAD signaling. J Biol Chem. 2001;276:10594-601
55. Sagi I, Chia G, Golan-Lev T, Peretz M, Weissbein U, Sui L. et al. Derivation and differentiation of haploid human embryonic stem cells. Nature. 2016;532:107-11
56. Llambi F, Wang YM, Victor B, Yang M, Schneider DM, Gingras S. et al. BOK Is a Non-canonical BCL-2 Family Effector of Apoptosis Regulated by ER-Associated Degradation. Cell. 2016;165:421-33
57. Zhu Y, Wu G, Zhu G, Ma C, Zhao H. Chronic sleep restriction induces changes in the mandibular condylar cartilage of rats: roles of Akt, Bad and Caspase-3. Int J Clin Exp Med. 2014;7:2585-92
58. Ho L, Tan SY, Wee S, Wu Y, Tan SJ, Ramakrishna NB. et al. ELABELA Is an Endogenous Growth Factor that Sustains hESC Self-Renewal via the PI3K/AKT Pathway. Cell Stem Cell. 2015;17:435-47
59. Karali E, Bellou S, Stellas D, Klinakis A, Murphy C, Fotsis T. VEGF Signals through ATF6 and PERK to promote endothelial cell survival and angiogenesis in the absence of ER stress. Mol Cell. 2014;54:559-72
60. She QB, Halilovic E, Ye Q, Zhen W, Shirasawa S, Sasazuki T. et al. 4E-BP1 is a key effector of the oncogenic activation of the AKT and ERK signaling pathways that integrates their function in tumors. Cancer Cell. 2010;18:39-51
61. Moutsatsou P, Kassi E, Papavassiliou AG. Glucocorticoid receptor signaling in bone cells. Trends Mol Med. 2012;18:348-59
62. Libro R, Giacoppo S, Bramanti P, Mazzon E. Is the Wnt/beta-catenin pathway involved in the anti-inflammatory activity of glucocorticoids in spinal cord injury?. Neuroreport. 2016
63. Zhou H, Mak W, Zheng Y, Dunstan CR, Seibel MJ. Osteoblasts directly control lineage commitment of mesenchymal progenitor cells through Wnt signaling. J Biol Chem. 2008;283:1936-45
64. Gupta P, Srivastav S, Saha S, Das PK, Ukil A. Leishmania donovani inhibits macrophage apoptosis and pro-inflammatory response through AKT-mediated regulation of beta-catenin and FOXO-1. Cell Death Differ. 2016
65. Rozpedek W, Pytel D, Mucha B, Leszczynska H, Diehl JA, Majsterek I. The Role of the PERK/eIF2alpha/ATF4/CHOP Signaling Pathway in Tumor Progression During Endoplasmic Reticulum Stress. Curr Mol Med. 2016;16:533-44
66. Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081-6
67. Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13:89-102
68. McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol. 2001;21:1249-59
69. Lin JH, Li H, Yasumura D, Cohen HR, Zhang C, Panning B. et al. IRE1 signaling affects cell fate during the unfolded protein response. Science. 2007;318:944-9
70. Wang M, Kaufman RJ. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature. 2016;529:326-35
71. Yu F, Sugawara T, Maier CM, Hsieh LB, Chan PH. Akt/Bad signaling and motor neuron survival after spinal cord injury. Neurobiol Dis. 2005;20:491-9
72. Hu Y, Sun H, Owens RT, Gu Z, Wu J, Chen YQ. et al. Syndecan-1-dependent suppression of PDK1/Akt/bad signaling by docosahexaenoic acid induces apoptosis in prostate cancer. Neoplasia. 2010;12:826-36
73. Qin B, Xiao B, Liang D, Xia J, Li Y, Yang H. MicroRNAs expression in ox-LDL treated HUVECs: MiR-365 modulates apoptosis and Bcl-2 expression. Biochem Biophys Res Commun. 2011;410:127-33
74. Li L, Piontek K, Ishida M, Fausther M, Dranoff JA, Fu R. et al. Extracellular vesicles carry miR-195 to intrahepatic cholangiocarcinoma and improve survival in a rat model. Hepatology. 2016
75. Monsel A, Zhu YG, Gennai S, Hao Q, Hu S, Rouby JJ. et al. Therapeutic Effects of Human Mesenchymal Stem Cell-derived Microvesicles in Severe Pneumonia in Mice. Am J Respir Crit Care Med. 2015;192:324-36
76. Zhang B, Wang M, Gong A, Zhang X, Wu X, Zhu Y. et al. HucMSC-Exosome Mediated-Wnt4 Signaling Is Required for Cutaneous Wound Healing. Stem Cells. 2015;33:2158-68
77. Kusuma RJ, Manca S, Friemel T, Sukreet S, Nguyen C, Zempleni J. Human vascular endothelial cells transport foreign exosomes from cow's milk by endocytosis. Am J Physiol Cell Physiol. 2016;310:C800-7
78. Geddis AE, Kaushansky K. Immunology. The root of platelet production. Science. 2007;317:1689-91
Author contact
![]() Corresponding authors: Prof. Chang-Qing Zhang, Department of Orthopedic Surgery, Shanghai Jiao Tong University Affiliated Sixth People's Hospital, Shanghai 200233, China. E-mail: zhangcqedu.cn or Shang-Chun Guo, Institute of Microsurgery on Extremities, Shanghai Jiao Tong University Affiliated Sixth People's Hospital, 600 Yishan Road, Shanghai 200233, China. E-mail: achunicom; Tel: +86-188-1727-6288.
Corresponding authors: Prof. Chang-Qing Zhang, Department of Orthopedic Surgery, Shanghai Jiao Tong University Affiliated Sixth People's Hospital, Shanghai 200233, China. E-mail: zhangcqedu.cn or Shang-Chun Guo, Institute of Microsurgery on Extremities, Shanghai Jiao Tong University Affiliated Sixth People's Hospital, 600 Yishan Road, Shanghai 200233, China. E-mail: achunicom; Tel: +86-188-1727-6288.