Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Conclusion
Materials and Methods
Supplementary Material
Abbreviations
Acknowledgements
Conflicts of Interest
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(2):270-284. doi:10.7150/thno.16154 This issue Cite
Research Paper
Nanosized UCMSC-derived extracellular vesicles but not conditioned medium exclusively inhibit the inflammatory response of stimulated T cells: implications for nanomedicine
Marta Monguió-Tortajada1,*, Santiago Roura2,3,*, Carolina Gálvez-Montón2, Josep Maria Pujal4, Gemma Aran5, Lucía Sanjurjo5, Marcel·la Franquesa1,6, Maria-Rosa Sarrias5, Antoni Bayes-Genis2,7,8, Francesc E. Borràs1,6, ![]()
1. REMAR-IVECAT Group, Health Science Research Institute Germans Trias i Pujol, Can Ruti Campus, Badalona, Spain
2. ICREC Research Program, Health Science Research Institute Germans Trias i Pujol, Can Ruti Campus, Badalona, Spain
3. Center of Regenerative Medicine in Barcelona, Barcelona, Spain
4. Cell Processing Laboratory, Parc Científic i Tecnològic Universitat de Girona, Girona, Spain
5. Innate Immunity Group, Health Sciences Research Institute Germans Trias i Pujol, Badalona, Spain
6. Nephrology Service, Germans Trias i Pujol University Hospital, Badalona, Spain
7. Cardiology Service, Germans Trias i Pujol University Hospital, Badalona, Spain
8. Department of Medicine, UAB, Barcelona, Spain
*Both authors contributed equally to this work
Received 2016-5-13; Accepted 2016-9-18; Published 2017-1-1
Abstract

Undesired immune responses have drastically hampered outcomes after allogeneic organ transplantation and cell therapy, and also lead to inflammatory diseases and autoimmunity. Umbilical cord mesenchymal stem cells (UCMSCs) have powerful regenerative and immunomodulatory potential, and their secreted extracellular vesicles (EVs) are envisaged as a promising natural source of nanoparticles to increase outcomes in organ transplantation and control inflammatory diseases. However, poor EV preparations containing highly-abundant soluble proteins may mask genuine vesicular-associated functions and provide misleading data. Here, we used Size-Exclusion Chromatography (SEC) to successfully isolate EVs from UCMSCs-conditioned medium. These vesicles were defined as positive for CD9, CD63, CD73 and CD90, and their size and morphology characterized by NTA and cryo-EM. Their immunomodulatory potential was determined in polyclonal T cell proliferation assays, analysis of cytokine profiles and in the skewing of monocyte polarization. In sharp contrast to the non-EV containing fractions, to the complete conditioned medium and to ultracentrifuged pellet, SEC-purified EVs from UCMSCs inhibited T cell proliferation, resembling the effect of parental UCMSCs. Moreover, while SEC-EVs did not induce cytokine response, the non-EV fractions, conditioned medium and ultracentrifuged pellet promoted the secretion of pro-inflammatory cytokines by polyclonally stimulated T cells and supported Th17 polarization. In contrast, EVs did not induce monocyte polarization, but the non-EV fraction induced CD163 and CD206 expression and TNF-α production in monocytes. These findings increase the growing evidence confirming that EVs are an active component of MSC's paracrine immunosuppressive function and affirm their potential for therapeutics in nanomedicine. In addition, our results highlight the importance of well-purified and defined preparations of MSC-derived EVs to achieve the immunosuppressive effect.
Keywords: nanosized extracellular vesicles, exosomes, inflammation, umbilical cord mesenchymal stem cell, immunomodulation, size exclusion chromatography.
Introduction
Exacerbated immune responses drastically hamper outcomes in allogeneic cell and organ transplantation, and lead to autoimmune disorders causing morbidity and mortality. In this context, due to their regenerative and immunomodulatory capacities, mesenchymal stem cells (MSCs) have been proposed as powerful inhibitors to counteract such unwanted immune responses. Thus, MSCs represent a promising strategy for a variety of medical conditions, including treatment of damaged tissue, inflammatory diseases and transplantation [1,2].
MSCs comprise a population of multipotent progenitor cells that have been obtained from distinct human tissues [3]. In particular, MSCs can be isolated from several compartments of the umbilical cord such as umbilical cord blood, umbilical vein subendothelium and the Wharton's jelly [4]. Our study is confined to the MSCs from the subamnion connective tissue surrounding the umbilical vessels or Wharton's jelly (here namely UCMSC), which includes a primitive self-renewing cell population with the characteristics of MSCs defined by the International Society for Cellular Therapy [5]. While the immune capabilities of UCMSC have been poorly described yet, MSCs from bone marrow and adipose tissue have been suggested to acquire better immunosuppressive functions after encountering an inflammatory stimulus, such as in vitro priming by IFNγ [6-11]. Moreover, increasing evidence has shown that the restorative and immunosuppressive functions exerted by MSCs are both cell-contact dependent and also mediated through a variety of secreted soluble factors in a paracrine fashion, including tryptophan depletion by IDO, production of immunosuppressive molecules and cytokines such as adenosine, NO, PGE2, IL-10 and TGFβ [12-15] and also released extracellular vesicles (EVs) [16,17].
EVs are membrane nanovesicles, which range approximately from 30 to 200 nm, carrying molecules that reflect the phenotype and functions of the cells of origin [18]. To date, therapeutic applications of MSC-EVs include treatment of experimental acute and chronic kidney injury [19], reduction of ischemia/reperfusion injury [20] and Graft-versus-Host Disease suppression [21]. Taken together, these studies point to MSC-EVs as promising candidates for novel cell-free therapies [17,22]. In the context of bionanotechnology, however, there are still open questions regarding the best method of EV preparation and concentration, characterization in terms of biological activity [23,24], and definition of the underlying mechanisms of action for the standardization of EV preparations that can be used in the clinical setting [25]. These points, along with the reported non-beneficial effect of non-purified MSC conditioned medium (CM) itself [26,27], stress the need for refining more efficient MSC-EV preparations and characterize them in terms of immunomodulatory potential.
Thus, in the present study, we examined the suppressive potential of Size-Exclusion Chromatography (SEC)-enriched EVs derived from UCMSCs and compared the data with the non-EV containing fractions, non-purified CM and its ultracentrifuged pellet (UC pellet). The presented results demonstrate that nanosized EVs retain the immunosuppressive effect of MSCs mainly by inhibiting T cell proliferation and preventing the secretion of pro-inflammatory cytokines by polyclonally stimulated T cells.
Results
UCMSCs characterization
Primary cultures of elongated fibroblast-like cells established from UC were recognized as bona fide MSCs when evaluated by flow cytometry and in differentiation assays. In particular, over 95% of cells expressed a MSC-like profile, being positive for CD105, CD44, CD166, CD10, CD73, CD90, CD49c, CD49d, CD49e, and HLA-ABC, and negative for CD117, CD106, CD34, CD45, CD29, CD14, CD133, CD31, VEGFR2 and HLA-DR. Moreover, commitment of cells to the adipogenic, osteogenic and chondrogenic pathways resulted in accumulation of intracellular lipid droplets, in high extracellular deposition of calcium and in active synthesis of proteoglycans, respectively (Figure S1).
We subsequently explored the influence of IFNγ priming on the MSC's phenotype. As shown in Figure 1A, cultured cells had a typical spindle-shaped morphology regardless of IFNγ priming. In terms of MSC markers, 48h-IFNγ conditioning lead to unchanged CD73 and decreased CD90 levels, while MHC class II expression (HLA-DR) was significantly increased (Figure 1B,1C).
A: Bright-field images of UCMSC in culture media without (left) or with IFNγ (right). Scale bars = 100 µm. B-C: UCMSCs primed with IFNγ expressed unchanged levels of CD73, lower levels of CD90 and higher levels of HLA-DR (class II MHC) on surface. B: Representative histograms of unconditioned (blue line) and IFNγ-primed UCMSCs (red line) labeled for CD73, CD90 and HLA-DR. Isotype controls are depicted as shaded areas. C: MFI values for CD73, CD90 and HLA-DR of UCMSCs after culture without (white circles) or with IFNγ (black circles). Data is shown for nine independent experiments. ***p<0.001 by Paired T test; **p<0.01 by Wilcoxon matched-pairs signed rank test. D-E: Proliferation and viability of T cells stimulated with anti-CD2/CD3/CD28 coated microbeads in the absence or presence of unconditioned or IFNγ-primed UCMSCs at 1:15, 1:60, 1:240, 1:480, 1:960 or 1:1,920 cell ratios were analyzed by CFSE loss and FSC/SSC gating, respectively. D: Bars represent means ±SD of proliferation relative to their PBS control. E: Bars represent means ± SD of the percentage of viable cells. Data accounts for seven independent experiments from different donor samples, performed in triplicates. Statistical differences are indicated for groups with *p<0.05 and **p<0.01 by One sample T test to the 100%.
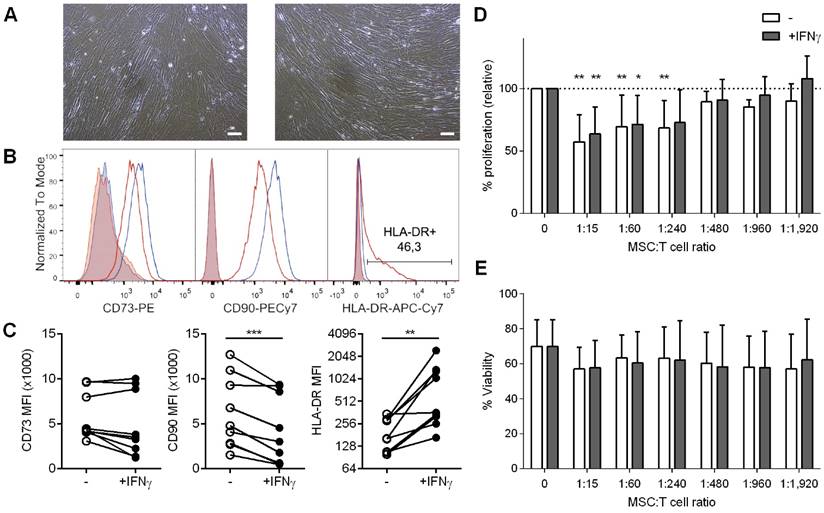
UCMSCs suppress T cells proliferation
It has been widely shown that MSCs from different origins inhibit T cell proliferation. To determine whether human UCMSC possess this capability and to analyze the influence of IFNγ priming on these cells, T cells were stimulated in the presence of increasing numbers of unconditioned or IFNγ-conditioned UCMSCs. The results confirmed that UCMSCs were able to inhibit polyclonal T cell proliferation and only at extremely low UCMSC:T cell ratios this effect was lost (Figure 1D). Our experiments showed that IFNγ-conditioning did not change their immune regulatory capabilities, as the reduced T cell proliferation was similar to the unconditioned UCMSCs. In these experiments T cell viability was not affected (Figure 1E).
Isolation and characterization of EVs from UCMSCs
After confirming the immunosuppressive potential of UCMSCs, we aimed to investigate whether EVs could be a mediator of the paracrine immunosuppressive action. First, EVs produced by UCMSCs were enriched from concentrated 48h-Conditioned Medium (CCM) by SEC (Figure 2A). EVs were found in fractions 5 to 7 according to their positivity for the EV-associated tetraspanins CD9 and CD63 (Figure 2B, C). The MSC markers CD73 (70 kDa) and CD90 (25-37 kDa) co-eluted in the EV fractions (F5-7), suggesting their presence in EVs as recently published [28]. Bulk protein content of CCM was detected from fraction 9 onwards. Of note, CCM from both IFNγ-conditioned and unconditioned UCMSCs showed the same SEC elution pattern, in which EVs were successfully separated from the bulk of protein. In this scenario, three distinct pools of SEC fractions were collected for further studies: i) the EV fraction, a pool of the tetraspanin-peak fractions (“EV”; F5-7); ii) the early tetraspanin-negative fractions, pooled as the proximal non-EV fractions (“non-EV prox”; F10-11); and iii) the late tetraspanin-negative fractions, pooled as the distal non-EV fractions (“non-EV dist”; F13-14).
EVs were successfully isolated by SEC. A: Scheme of the methodological procedure followed for the generation of the different study fractions obtained from UCMSC 48h-culture: supernatant was cleared of debris by centrifugation to obtain conditioned media (CM); concentrated CM (CCM) and eluted CM (ECM) were collected after ultrafiltration; CCM was loaded to the SEC column and fractions collected. B-C: UCMSC-EVs were found on fractions 5-7 while protein eluted after fraction 8 on both samples coming from unconditioned (B) and IFNγ-conditioned UCMSCs (C). SEC eluted fractions were checked for EV markers (CD9 and CD63), MSC markers (CD73 and CD90) by bead-based flow cytometry (left axis). Protein elution was monitored by absorption at 280nm (right axis). D-E: Cryo-EM images confirmed UCMSC-EVs presence in pooled EV fractions (F5-7) of unconditioned (D) and IFNγ-conditioned SEC preparations (E). Images of 20,000x and 30,000x magnifications are shown, with 200nm and 100nm scale bars, respectively. F: Box plot of the fold increase in protein content, CD9 and CD63 MFI of pooled EV fractions obtained from IFNγ-primed UCMSCs relative to unconditioned UCMSCs. Medians of ten independent experiments are depicted as horizontal bars, outliers as points. G: CD73, CD90, HLA-ABC (MHC-I) and HLA-DR (MHC-II) expression on EVs from unconditioned (white dots) and corresponding IFNγ-primed MSCs (black dots) are shown normalized to their CD9 MFI. Each dot corresponds to an independent experiment (n=10 and 6).
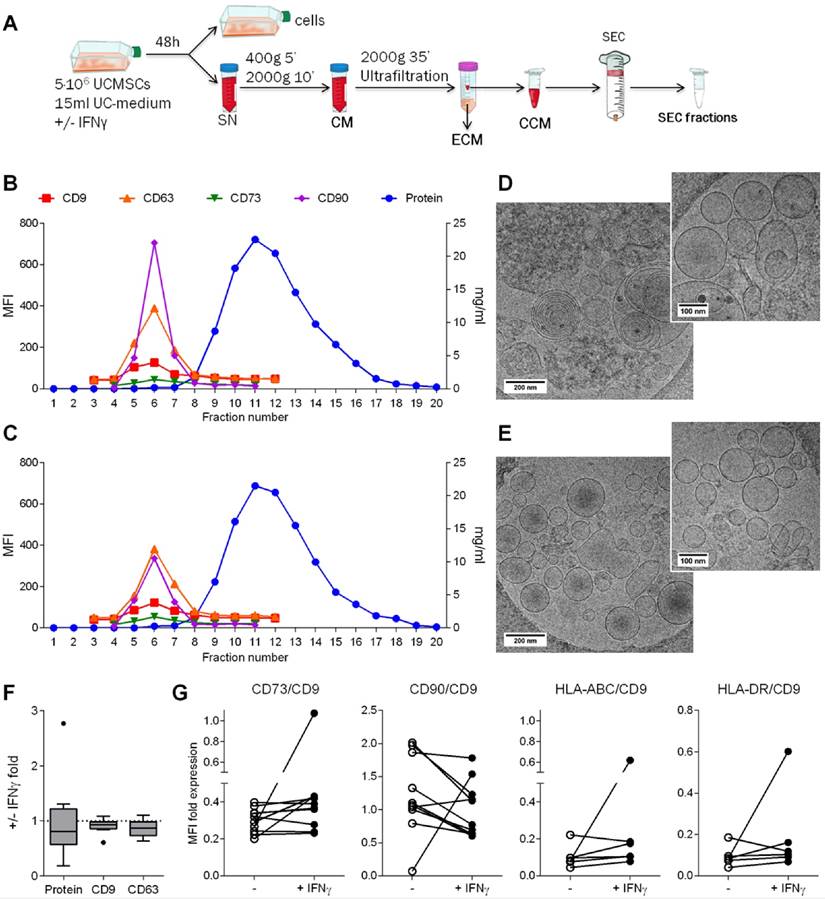
Subsequently, in order to confirm the presence of EVs, SEC fractions were processed for cryo-electron microscopy (cryo-EM) and analyzed by nanoparticle tracking analysis (NTA). EV fractions were confirmed to contain round-shaped nanovesicles free of contaminating clumps or protein aggregates by cryo-EM (Figure 2D, E and Figure S2A). NTA analysis of the tetraspanin-peak fractions showed the presence of particles with a mode diameter of 169.6 and 157.2 nm for unconditioned and IFNγ-primed UCMSC-EVs, respectively (Figure S2B). On the other hand, NTA indicated the presence of particles with a modal size of 118.1 nm in the proximal and 155.1 nm in the distal non-EV fractions, but cryo-EM images confirmed the lack of EVs in those fractions (Figure S2B). Overall, the concentration of particles calculated by NTA (regardless the presence or not of EVs) was very similar among all the experiments, ranging 2-6x1010 particles/ml (Figure S2A).
Analyses of the effect of IFNγ priming on EV production showed similar protein content, CD9 and CD63 presence and particle concentration in EV-containing SEC fractions (Figure 2F and Figure S2A). MFI values were then normalized to the expression of CD9 and, unlike the results observed in cells, HLA molecules remained absent in the majority of EV batches after IFNγ priming. Regarding MSC markers, CD73 was unchanged and CD90 decreased, resembling parental cells' behavior (Figure 2G).
Due to the apparent insubstantial benefits of IFNγ priming on the immunomodulatory capacities of UCMSCs, these would not recommend using IFNγ-priming on UCMSCs, especially in the allogeneic delivery setting given the HLA-DR overexpression in UCMSCs, also observed in few MSC-EV batches.
UCMSC-EVs reduce T cell proliferation and inflammatory cytokine production
Next, we aimed to delineate whether the EVs hold the immunosuppressive potential described for MSCs paracrine secretion. Thus, the three distinct pools of SEC fractions were compared by their ability to modify the T cell response. Additional controls included CM and its concentrated (CCM) and eluted (ECM) products after ultrafiltration (Figure 3A). Also, the UC pellet from CM was compared to SEC-EVs.
Only the pooled EV fraction reduced T cell polyclonal proliferation. A: The three different pooled SEC fractions: EV, proximal non-EV (“non-EV prox”) and distal non-EV fractions (“non-EV dist”) were analyzed for T cell proliferation suppression capacity compared to the CCM, ECM, full CM, and ultracentrifuged pellet (UC Pellet). B, C: Proliferation of T cells stimulated with anti-CD2/CD3/CD28 beads (10:1 ratio) was analyzed by CFSE loss in the presence of pooled UCMSC-EVs, proximal and distal non-EV fractions, CCM, ECM and CM (B) or the UC Pellet (C). Quantities were adjusted to 2.5x105 initial UCMSC. Bars represent proliferation relative to their PBS control. C, D: Viability of stimulated T cells assessed by FSC-A/SSC-A. Data represent means ± SD for seven (B, D) and four (C, E) independent experiments. Statistical differences are indicated for groups with p<0.05 by Kruskall-Wallis; #p<0.05 by Wilcoxon Signed Rank test (to the 100%); *p<0.05 and **p<0.01 by Mann-Whitney test.
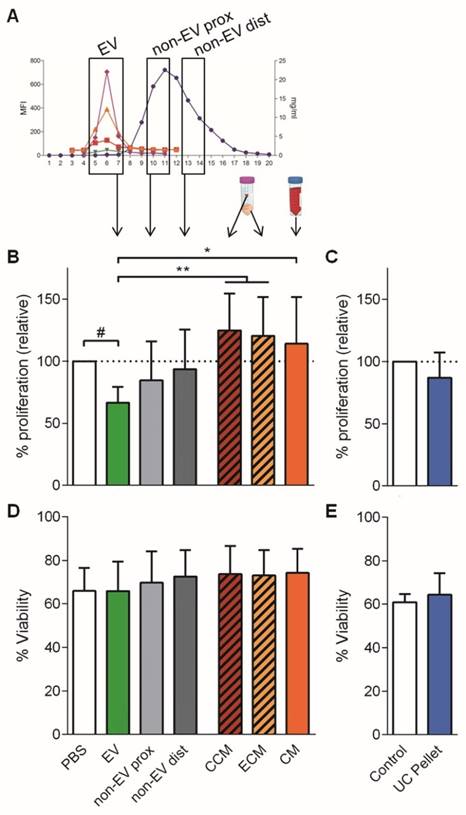
Only the SEC fractions containing EVs inhibited proliferation of stimulated T cells when added to the culture (34% reduction) (Figure 3B). This reduction of T cell proliferation showed a dose-dependent profile (Figure S3A, S3B). Importantly, when either proximal or distal non-EV fractions were added to the culture, T cell proliferation remained unaltered (Figure 3B), similar to the addition of CCM, ECM and CM. Moreover, the UC pellet did not impair T cell proliferation (Figure 3C). Noticeably, neither did EVs obtained from IFNγ-primed UCMSCs (Figure S3A). Viability of T cells was not altered in any of the conditions, thus discarding a principal apoptosis-mediated effect on T cell inhibited proliferation (Figure 3D, 3E). Finally, as an additional control to ratify the action by UCMSC-EV, complete medium alone was processed following the same SEC workflow, and the derived fractions did not suppress T cell proliferation (data not shown).
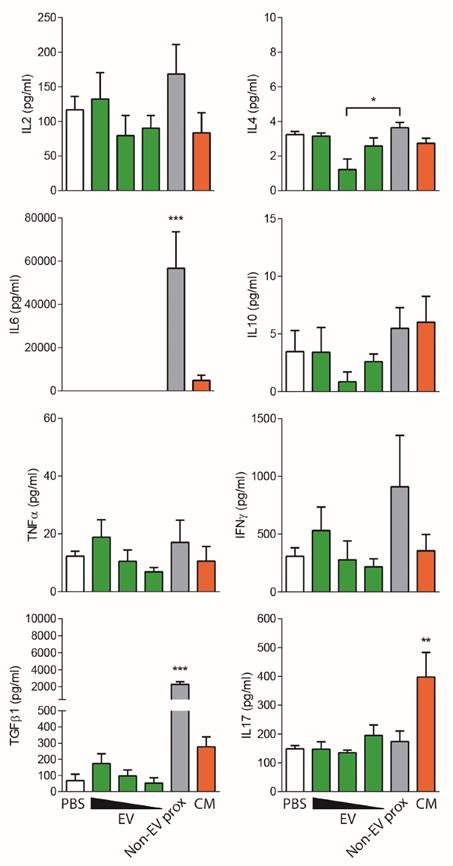
To further study the immunomodulatory effect of EVs, the cytokine profile of stimulated T cells was determined. Consistent with the reduced proliferation, no production of the pro-inflammatory cytokines IL-6, TNF-α or IFNγ nor IL-2, was observed in T cell cultures to which EVs were added. In sharp contrast, IL-6, TGFβ1, IL-17A and, to a lesser extent IFNγ, were highly produced in the presence of both proximal and distal non-EV fractions and CM (Figure 4).
Cytokines found in the supernatants of T cell proliferation assays corresponding to Figure 3 were analyzed by CBA (IL-2, IL-4, IL-6, IL-10, TNF-α, IFNγ, and IL-17A) and TGF-β1 ELISA. Bars represent means ± SD of seven (A) and four (B) independent experiments. Statistical differences are indicated for groups with *p<0.05 by Kruskall-Wallis; *p<0.05 and **p<0.01 by Mann-Whitney test.
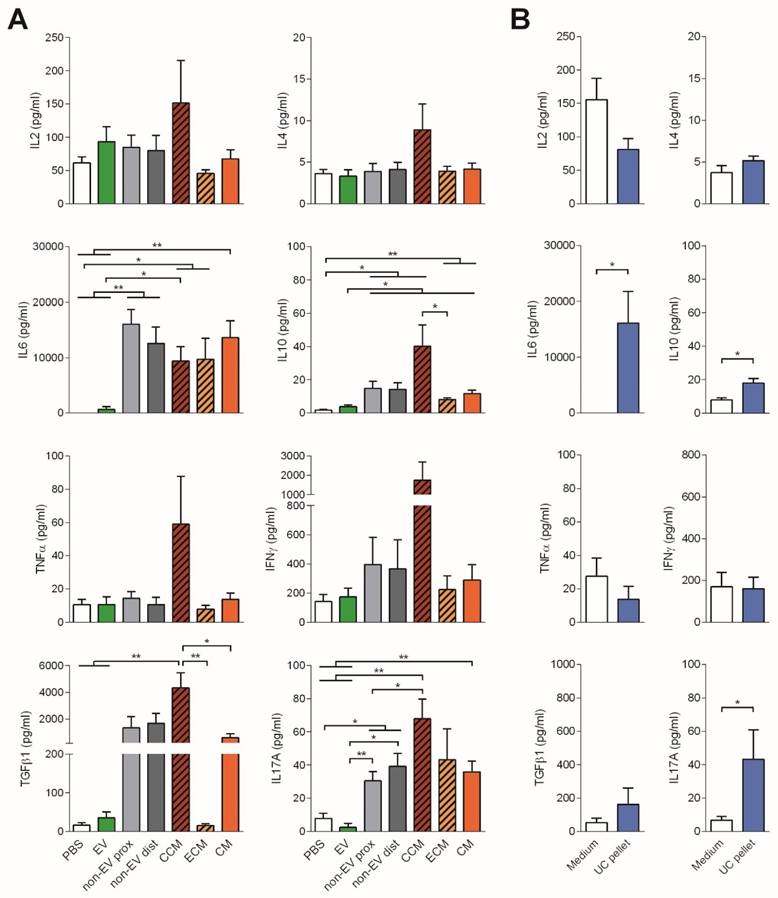
To our surprise, addition of full CCM to T cells promoted a cytokine rush, fostering the secretion of all the cytokines studied (IL-2, IL-4, IL-6, IL-10, TNF-α, IFNγ, TGF-β1 and IL-17A). On the other hand, addition of the UC pellet to T cells also fostered the production of IL-6 and IL-17A, resembling, rather than to SEC-EVs, the cytokine profile of T cells stimulated with CM (Figure 4B). These pro-inflammatory cytokines were also found in supernatants from MSC:T cell cultures at high cell ratios (Figure S4). Of note, all cytokines were also determined in CCM, ECM and CM, showing values always below the cytokines found in T cell proliferation supernatants (Figure S5), thus indicating that cytokine production in stimulated T cell cultures can be attributed genuinely to T cells.
Concentrated UCMSC-EVs further reduce T cell proliferation
In order to discard the possibility of a “dilution effect” of culture medium (volume added from SEC fractions) as an important factor influencing T cell proliferation, similar experiments were set up using vacuum concentration of the pooled fractions to minimize culture medium dilution (1.05-fold instead of 2-fold) (Figure 5A).
Concentrated EV and non-EV fractions but not CM inhibit T cell polyclonal proliferation. A: EV, proximal non-EV fractions and CM were vacuum-concentrated and checked for T cell proliferation suppression capacity. B: Proliferation of T cells stimulated with anti-CD2/CD3/CD28 beads (10:1 ratio) was analyzed by CFSE loss in the presence of vacuum-concentrated UCMSC-EVs, proximal non-EV fractions or CM. Quantities were adjusted to 2.5x105 initial UCMSC, and dosage dependency was studied diluting samples 1/2 and 1/10 in PBS. Bars represent means ± SD of proliferation relative to their PBS control, for three independent experiments. C: Viability of stimulated T cells assessed by FSC-A/SSC-A. Data represent means ± SD for five independent experiments. Statistical differences are indicated for groups with p<0.05 by Kruskall-Wallis; #p<0.01 by Wilcoxon Signed Rank test (to the 100%); *p<0.05 and **p<0.01 by Mann-Whitney test.
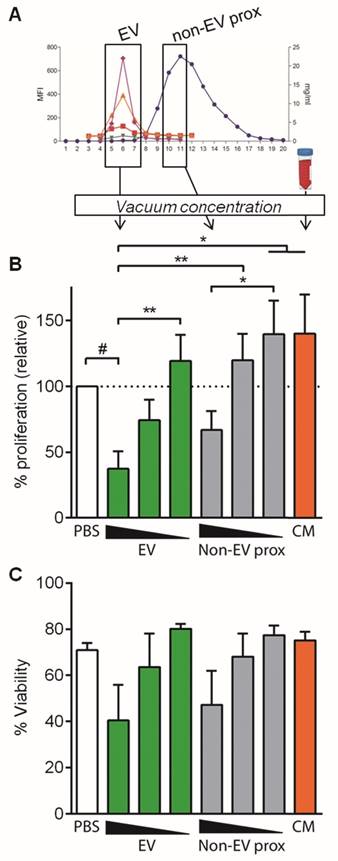
In these experimental conditions, concentrated EVs reduced polyclonal T cell proliferation more than a 60% (37.47% proliferation relative to control, Figure 5B), compared to the 34% reduction in non-concentrated conditions (Figure 3B). Intriguingly, in some experiments T cell viability was affected when concentrated EVs were added to the culture, although this effect was not statistically significant (Figure 5C). All these effects were lost upon EV dilution, confirming the EV-mediated T cell inhibition (Figure 5B and Figure S3C).
An unexpected result was to observe that the concentrated proximal non-EV fraction turned out to mildly reduce T cell proliferation in a dose-dependent manner (Figure 5B), a feature that was not seen before in non-concentrated samples. Nevertheless, when the cytokine profile of stimulated T cells was analyzed, an enormous production of IFNγ, IL-6 and TGF-β1 was uniquely detected in supernatants from T cells stimulated in the presence of the proximal non-EV fraction (Figure 6), alerting of a pro-inflammatory stimulation of T cells, a feature that was completely absent in T cells incubated with EVs. Moreover, in accordance to non-concentrated experiments, CM was unable to suppress T cell polyclonal activation (Figure 5B).
Non-EV fractions and CM induce an inflammatory response on stimulated T cells. Cytokines found in the supernatants of T cell proliferation assays corresponding to Figure 5 were analyzed by CBA (IL-2, IL-4, IL-6, IL-10, TNF-α and IFNγ) and TGF-β1 and IL-17 ELISA. Bars represent means ± SD of three independent experiments for IL-2, IL-4, IL-6, IL-10, TNF-α and IFNγ and five independent experiments for TGF-β1 and IL-17. Statistical differences are indicated for groups with *p<0.05, **p<0.01 and ***p<0.001 by One-way ANOVA with Tukey's post hoc analysis.
To summarize, these results indicate that only the isolated SEC-EV fraction managed to greatly immunosuppress polyclonal T cell activation, while non-EV fractions, CM and UC pellet surprisingly promoted an inflammatory milieu and Th17 polarization of T cells.
Effect of UCMSC-EVs on monocyte polarization
We also explored the effect of the pooled SEC fractions and full CM in the skewing of monocyte polarization. In this setting, we used three different well-determined monocyte polarizing stimuli as positive controls [29,30]: LPS plus IFNγ as an inflammatory “M1” phenotype inducer, and IL-4 or IL-10 to generate anti-inflammatory “M2a” or “M2c” phenotypes, respectively. As expected, M1 (LPS+IFNγ) macrophages highly increased CD80 mRNA, showed mild changes in CD163, and downregulated the expression of CD206 (Figure 7A). These cells also produced high levels of TNF-α and IL-10 (Figure 7B). In sharp contrast, M2a (IL-4) and M2c (IL-10) monocytes did not undergo changes in CD80 while upregulated CD206. Only M2c (IL-10) also incremented CD163 expression, and had IL-10 in supernatants (probably as carryover effect from the activation stimulus used). None of the M2-skewing stimuli induced the secretion of TNF-α (Figure 7B).
UCMSC-EVs do not affect CD80, CD163 and CD206 polarization marker expression in monocytes, while the non-EV fraction and CM induce increased expression of CD163 and CD206. Monocytes were cultured for 48h with polarizing cytokines to induce an inflammatory M1 (LPS+IFNγ) or anti-inflammatory M2a (IL-4) and M2c (IL-10) phenotypes, or with the EV, proximal non-EV fractions or full CM. A: mRNA fold change of the M1 marker CD80 and M2 markers CD163 and CD206, as analyzed by real time PCR. Values are relative to 18S and expressed as a fold change to the PBS-treated monocytes in a log2 scale. B: Levels of TNF-α and IL-10 in the supernatants of monocytes as measured by ELISA. Data are expressed as mean + SD and account for two different UCMSC and four monocyte donors. Statistical differences are indicated for groups with *p<0.05 by Kruskall-Wallis; *p<0.05 by Mann-Whitney test.
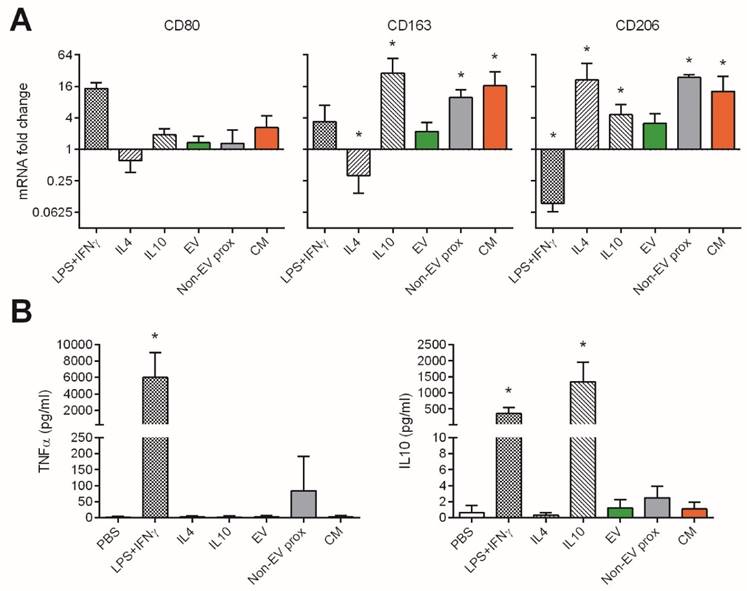
We then analysed the capacity of UCMSCs media to skew monocyte polarization. Culture with UCMSC-EVs maintained monocytes in a non-activated state, as cells did not substantially modify the expression of CD80, CD163, CD206 or IL-10 compared to the control (p>0.05 to non-activated monocytes). Moreover, no TNF-α was detected in supernatants, indicating that samples did not contain pro-inflammatory mediators.
Conversely, the proximal non-EV fraction and CM induced the expression of CD163 and CD206, while not altering that of CD80, thus resembling an M2c(IL-10) polarization (Figure 7A), although not relevant amounts of IL-10 were detected (Figure 7B). Of note, some TNF-α could be found when the proximal non-EV fraction was added.
Discussion
In this study, we demonstrate that EVs isolated from UCMSCs by SEC strongly immunomodulate activated T cells in vitro. In particular, our results indicate that the SEC-purified EV fraction greatly abrogates polyclonal T cell proliferation and cytokine production in comparison with non-EV fraction, CM and UC pellet, which conversely result in an inflammatory T cell response and foster the Th17 polarization of T cells. Additionally, EVs do not induce monocyte polarization or cytokine secretion, but the non-EV fraction induces the expression of CD163 and CD206 and some production of TNF-α by monocytes. These findings not only increase the growing evidence confirming that EVs are an active component of MSC's paracrine immunosuppressive function, but also highlight the importance of working with well purified EV preparations to specifically achieve this effect.
MSCs have been described to suppress the immune response affecting T cell proliferation and polarization, to induce regulatory T cells, and to module Antigen Presenting Cells (APCs) [11,31-34]. In our settings, UCMSCs were confirmed to have potent suppressive capabilities on T cell proliferation, but whereas IFNγ conditioning enhances immunosuppressive functions of both bone marrow- and adipose tissue-derived MSCs [7,9,35], no differences were found comparing IFNγ-primed to non-primed UCMSCs. This observation could be explained by the intrinsic capacity of UCMSCs to suppress T cell proliferation at low ratios compared to other studies (1:240 MSC to T cells), which may indicate that these cells own already a potent modulatory capacity of the T cell response. Another explanation might be the lack of APCs in our experimental setting, which have been described to partially mediate T suppression induced by MSCs [36]. In line, MSCs have been described to modulate the immune response by polarizing monocytes towards a M2 phenotype, which would in turn further modulate inflammation [37]. In any case, our results confirmed that non-primed UCMSCs showed a high immunomodulatory capacity at low MSC:T cell ratios.
Also in line with previous studies, IFNγ increased the expression of HLA-II molecules in UCMSCs [38-40], which may have detrimental effects in the allogeneic scenario. Expression of HLA-II molecules in MSCs would trigger the activation of the host's innate immune system which in turn would impede their potential immunomodulatory effect. In fact, it has been suggested that the increased HLA expression in EVs from umbilical cord blood-derived MSCs after treatment with IFNγ may be responsible of the loss of their protective effect against ischemic acute kidney injury [31]. Given that our results suggest that non-primed UCMSCs are as potent as primed MSCs in regulating T cell responses, we would consider using non-primed cells in therapeutic approaches.
In the context of cell therapy, transplantation of MSCs to induce immune suppression and tissue regeneration has still some limitations. Such drawbacks include the right homing and implantation -which is impaired by the entrapment of cells in the lung barrier upon systemic administration [41,42]-, the possible change in phenotype of the infused cells -influenced by the initial inflammatory phase occurring after in vivo infusion of MSCs [43]-, the requirement of cell viability after transplantation and ease of storage and availability. The majority of these caveats could be overcome by using EVs, resulting in a number of reports describing their benefits [22,44]. As MSCs exert their function through both cell contact and soluble mechanisms, it is expected that higher concentrations of EVs would be needed to reach the same level of inhibition obtained using MSCs, as we observed in our results. However, in the context of cell-free therapeutic approaches it is of relevance that one of the paracrine mechanisms of T cell inhibition by MSCs is based on well-defined and effectively enriched MSC-derived EVs.
Recent studies have shown that MSC-EV products can efficiently modulate inflammatory disorders in vivo, including rat and mouse models and also human clinical trials [17,21,45-47]. Yet, several controversial studies noticed reduced immunosuppressive functions of CM and EVs compared to their parental cells [26,27]. Nevertheless, most of the studies performed so far on MSC-EV effect in vitro and in vivo used ultracentrifugation or precipitation methods [47]. These methods lead to EV preparations containing high quantities of non-EV proteins [24,48,49], result in cytotoxicity [49] and might explain the incongruous results on EV effects. Importantly, our results highlight the need of using well characterized and efficiently purified EVs to obtain an optimal cell-free immunosuppressive product.
Isolation of EVs from MSC-CM by SEC yielded highly purified EVs that could be easily detected not only by tetraspanin markers, but also the MSC markers CD73 and CD90, confirming previous observations [28,50,51]. Importantly, SEC ensured the separation of EVs from the bulk of protein and impurities found in CM, as confirmed by cryo-EM, as it has been widely demonstrated before using other complex fluids such as plasma or urine [49,52-55].
Stressing the importance of using a well-defined cell-free product, it was extremely relevant to observe that SEC-purified EVs significantly differ from the non-EV fractions, full CM, and UC pellet in their functional capabilities. While isolated EVs successfully suppressed T cell proliferation and concomitantly inhibited the induction of cytokine production in a dose dependent manner, the non-EV fraction, full CM, and UC pellet displayed completely divergent properties. Further concentration of purified EVs using vacuum concentration resulted in an enhanced reduction of T cell proliferation compared to the control situation. This effect may be partially explained by a reduced T cell viability, which has been described before as a possible mechanism used by MSCs to constrain T cell activation [56,57]. Nevertheless, in our hands, reduced T cell viability was highly dependent on the EV batch, and was not observed when EVs were further diluted.
In sharp contrast, the non-EV fractions only reduced T cell proliferation when they were vacuum concentrated, but at the same time induced the production of high amounts of inflammatory IL-6 combined with TGFβ1. These two cytokines -along with IL-1β and IL-23-, are involved in the generation of Th17 responses [58,59], and in fact we could find the induction of Th17 cells in all conditions except SEC-EVs. Th17 polarization is known to be the cause of exacerbated inflammatory disorders and especially detrimental for autoimmune diseases such as multiple sclerosis, rheumatoid arthritis or to mediate GvHD and allergies [58,60]. In line with these results, recent studies delineate how EV products obtained by ultracentrifugation could lead to immune cell activation via NFκB [61]. Moreover, high levels of IL-6 are the cause of inflammatory diseases and also have been linked with the exacerbated activation of the immune response causing the cytokine-release syndrome, an unexpected dangerous side effect found in some cell therapy clinical trials [62-64].
Antigen Presenting Cells (APCs) may also be potential targets of the MSC regulatory effect. In this sense, monocytes have been claimed as necessary for Treg generation by MSCs and MSC-derived CM, through their skewing to an M2 phenotype [37,65] and secretion of IL-10 [66], a functional trait of M2 monocytes [29,67]. In line with these results, both CM and the non-EV fractions induced the expression of the M2 markers CD163 and CD206 on monocytes, with marginal amounts of IL-10 detected in supernatants. However, monocytes cultured with the non-EV fraction produced some levels of TNF-α, which may be indicative of unwanted cell activation. In sharp contrast, monocytes cultured in the presence of EVs did not show polarization or cytokine secretion, thus proving to be clean of polarizing stimuli or inflammatory mediators.
All these results strongly suggest the need to use well-defined, cell-free, highly purified EV products in therapeutic approaches, and put into further value the immunomodulatory role of SEC-derived EVs on T cells. Given the complexity of the immune response, in which many actors play fundamental roles, defining the specific effects of EVs and non-EV fractions on single cell populations is a fundamental step for the deciphering of the underlying mechanisms of EVs in the modulation of key players, such as T cells and monocytes.
Conclusion
In summary, the present study thoroughly characterizes the different fractions found in UCMSCs-CM in terms of immune modulation potential. We have proven the feasibility of a strategy based on SEC to effectively isolate nanosized EVs responsible at least in part of the genuine MSC immunomodulatory capacities. Most importantly, our results highlight the importance of purity and fine characterization of the EV product envisioned as a cell-free therapeutic approach to avoid unwanted inflammatory responses. In this sense, since EVs are apparently well-tolerated, their use paves the way for innovative and more efficient therapies based in nanomedicine avoiding the putative side effects associated to stem cell transplantation. Together with other potential uses such as targeting cell membranes, delivering bioactive molecules and being analyzed for biomarkers (i.e, theranostics), this natural source of nanoparticles may be crucial in future developments on nanomedicine.
Materials and Methods
UC collection, MSC isolation, culture and characterization
The study protocols were approved by the Clinical Research Ethics Committee of our institution (Comitè Ètic d'Investigació Clínica, HuGTiP, Refs. CEIC: EO-10-016 and EO-12-022) and conformed to the principles outlined in the Declaration of Helsinki. With the consent of the parents, fresh umbilical cords (n= 10) were obtained after birth and maintained in phosphate-buffered saline buffer (PBS; Gibco Life Technologies/Invitrogen, Carlsbad, CA) supplemented with 5,000 U heparin (Sigma Aldrich, St. Louis, MO) and 1% penicillin/streptomycin (Gibco) before tissue processing to isolate MSCs. UCs (10 g) were sectioned into 3-6 mm3 pieces and carefully washed in PBS to eliminate residual blood contained in arteries and vein. During mechanical disruption, elimination of UC vein and subendothelium was achieved. Further procedures include two enzymatic disaggregations at 37ºC with gentle agitation and a filtration step using Falcon Cell Strainers (BD Biosciences, San Diego, CA) in order to release uniform cell suspensions. First digestion was conducted using Collagenase type-I (880 U/mL; Gibco) plus Hyaluronidase II (3,960 U/mL; Sigma Aldrich) for 60 min, followed by a second digestion using Trypsin-EDTA (0,125%; Gibco) plus DNase I (0.2 mg/mL; Roche Diagnostics, Mannheim, Germany) for 30 min. The supernatants from both digestions were mixed together and centrifuged at 1,200 rpm for 10 min. Cell pellet was then resuspended in α-MEM (Sigma) supplemented with 10% heat-inactivated Foetal Bovine Serum (FBS; Life Technologies, Carlsbad, CA), 2mM L-glutamine and 1% penicillin/streptomycin (Gibco) and 5 µg/mL plasmocin (Invivogen, San Diego, CA). Adherent cells were maintained under standard culture conditions until third-passage cells, when cells were used to analyze their surface marker expression profile and multipotency, as previously described [68-70].
Cells were labelled with 7AAD for viability and the antibodies anti-CD73-PE, -CD90-PE-Cy7, -HLA-DR-APC-H7 or the corresponding IgG isotype control (all from BD) for immunophenotyping. Labelling was performed at room temperature for 15 min, washed with FACSFlow 2% FBS and centrifuged at 400g for 5 min. Data was acquired in a Canto II flow cytometer (BD) and analysed by FlowJo v.X software (TreeStar, Ashland, OR).
Generation of EV-depleted culture medium
Complete culture medium was composed of α-MEM (Sigma Aldrich) or TexMACS (Miltenyi Biotech, Bergisch Gladbach, Germany) medium supplemented with 2mM L-Glutamine (Sigma Aldrich), 100U/ml Penicillin (Cepa S.L., Madrid, Spain), 100µg/ml Streptomycin (Normon Laboratories S.A., Madrid, Spain) and 10% (v/v) Heat Inactivated-FBS or Human platelet lysate (Lonza, Basel, Switzerland) for MSC and T cell culture, respectively. Plasmocin (5 µg/mL; Invivogen) was added for MSC culture.
Culture medium was depleted of bovine/human EVs by ultracentrifugation of 2x complete medium in polypropylene ultracentrifugation tubes (Beckman coulter, Brea, CA) at 100,000g for 16 h (SW28 rotor, 28000 rpm, adjusted k-Factor= 253.96). The supernatant was collected and filtered through a 0.22 µm filter (Sarstedt, Germany) to sterilize the medium, which was finally diluted to 1x working concentration with αMEM/TexMACS medium alone for cell culture.
EV isolation
EVs were isolated from UCMSCs following the scheme in Figure 2A. For MSCs-CM generation, 5x106 UCMSCs were seeded in bovine EV-depleted culture medium with or without 200 ng/ml (120 IU/ml) IFNγ (Peprotech; cat#300-02, Rocky Hill, NJ) when indicated. Supernatant was collected after 48 h and sequentially centrifuged at 400g for 5 min and at 2,000g for 10 min to exclude cells and cell debris, respectively. This debris-cleared CM was then concentrated by 100 kDa ultrafiltration using Amicon Ultra (Millipore, Millerica MA) at 2,000g for 35 min, obtaining typically 250 µl concentrated CM (CCM). The eluted CM (ECM) was kept for additional experiments.
UCMSC-EVs were then isolated from the CCM by SEC using a modification of the previously published method [54]. Briefly, 1 ml of Sepharose CL-2B (Sigma Aldrich) was extensively washed with PBS (Oxoid, Hampshire, UK) and packed in a 1-ml syringe (BD). A 100 µl sample of CCM was loaded into the column and 100µl fractions (up to 20) were collected immediately after loading. Protein elution was checked by reading absorbance at 280 nm of each fraction using Nanodrop (Thermo Scientific, San Diego, CA).
In some experiments, CM was ultracentrifuged at 100,000g for 2 h (SW55Ti rotor, 32500 rpm, adjusted k-Factor= 138.67), and the UC pellet was used for comparative experiments. As a control, 15 ml of bovine EV-depleted culture medium alone was incubated and followed the same EV isolation procedure. All fractions were kept at 4ºC and used within 24h for in vitro experiments, or frozen (-1ºC/min) at -80ºC for NTA and cryo-EM analysis. In the indicated experiments, fractions were concentrated seven-fold by 90 min vacuum concentration at 30ºC using the miVac (GeneVac, Ipswich, UK).
EV characterization
Flow cytometry
The presence of EVs in the SEC fractions was determined according to their content in tetraspanins by bead-based flow cytometry. Briefly, EVs were coupled to 4 µm aldehyde/sulphate-latex microspheres (Invitrogen, Carlsbad, CA) for 15 minutes at RT and blocked in BCB buffer (PBS/0.1% BSA/0.01% NaN3; both from Sigma Aldrich) on overnight rotation. EV-coated beads were spun down at 2000g for 10 min, washed with BCB buffer and re-suspended in PBS.
EV-coated beads were then labelled with the fluorochrome-conjugated antibodies anti-CD73-PE, -CD90-PE-Cy7, -HLA-ABC-FITC or -HLA-DR-APC-H7 (all from BD) or indirectly labelled with the primary antibodies anti-CD9 (Clone VJ1/20) and -CD63 (Clone TEA3/18) or the IgG isotype control (Abcam, Cambridge, UK) and secondary antibody FITC-conjugated Goat F(ab')2 Anti-Mouse IgG (Bionova, Halifax, NS, Canada). Labelling was performed at room temperature for 30 min under mild shaking, and EV-coupled beads were washed after each step with BCB buffer and centrifuged at 2,000g for 10 min. Data was acquired in a FACSVerse flow cytometer (BD) and analysed by FlowJo v.X software (TreeStar).
Nanoparticle Tracking Analysis
Size distribution of particles on SEC fractions was determined by NTA in a NanoSight LM10-12 instrument (Malvern Instruments Ltd, Malvern, UK), equipped with a 638 nm laser and CCD camera (model F-033). Data was analyzed with the NTA software version 3.1. (build 3.1.46), with detection threshold set to 5, and blur, Min track Length and Max Jump Distance set to auto. Samples were diluted 10 or 20 times with PBS to reach optimal concentration for instrument linearity: 20-120 particles/frame as advised by the manufacturer. Readings were taken on triplicates of 60 s at 30 frames per second, at a camera level set to 16 and with manual monitoring of temperature.
Cryo-electron microscopy
SEC fractions were examined for EV size and morphology by cryo-electron microscopy (cryo-EM). Vitrified specimens were prepared by placing 3 μl of a sample on a Quantifoil® 1.2/1.3 TEM grid, blotted to a thin film and plunged into liquid ethane-N2(l) in the Leica EM CPC cryoworkstation (Leica, Wetzlar, Germany). The grids were transferred to a 626 Gatan cryoholder and maintained at -179ºC. Samples were analyzed with a Jeol JEM 2011 transmission electron microscope (Jeol, Tokyo, Japan) operating at an accelerating voltage of 200 kV. Images were recorded on a Gatan Ultrascan 2000 cooled charge-coupled device (CCD) camera with the Digital Micrograph software package (Gatan, Pleasanton, CA).
Proliferation assay
Whole blood was obtained from healthy donors after informed consent approved by the local Ethics Committee (Germans Trias i Pujol Universitary Hospital). PBMCs were obtained by Ficoll Hypaque PlusTM (GE Healthcare, Uppsala, Sweden) density centrifugation and T cells were then isolated using the negative selection EasySepTM Human T cell Enrichment Kit (StemCell Technologies, Grenoble, France) following manufacturer's instructions. Enriched T cells were then washed and stained with Carboxyfluorescein succinimidyl ester (CFSE, Molecular Probes, Leiden, The Netherlands) to assess cell proliferation. Succinctly, enriched T cells were resuspended in PBS for staining with an equal volume of 0.8µM CFSE for 10 minutes, after which unbound dye was quenched with RPMI + 10% FBS. Labeled cells were washed twice with RPMI + 10% FBS before resuspending in 1x human EV-depleted TexMACS complete medium. T cells were routinely >93% pure (CD3+) and >94% viable in all experiments performed.
CFSE-labeled T cells (3·105) were stimulated with anti-CD2/CD3/CD28 coated microbeads (Pan T Cell Activation Kit; Miltenyi Biotech) or uncoated microbeads as a negative control in a 1:10 bead:T cell ratio, in flat-bottomed well plates in which allogeneic UCMSCs had been previously seeded (20000, 5000, 1250, 625, 325 or 160 cells/well). In parallel experiments, CFSE-labeled T cells were plated at 5·104 cells/well in round-bottomed well plates and stimulated in the same way with anti-CD2/CD3/CD28 coated microbeads. T cells were co-cultured in the presence of 1:1 or 1:2 (v/v) EV, proximal or distal non-EV fractions, CCM, ECM or CM, corresponding to 2.5x105 or 1.25x105 initial UCMSCs, respectively. Alternatively, 1:20 (v/v) vacuum concentrated samples were added to stimulated T cells, corresponding to 2.5x105, 1.25x105 (1/2 dilution) or 2.5x104 (1/10 dilution) initial UCMSCs. T cell proliferation was measured after 3.5 days in a LSR Fortessa Analyzer (BD Biosciences) and expressed as the percentage of FSChighCFSElow cells out of the living cells gated by FSC/SSC using the proliferation module of the FlowJo V9.8.2.
Monocyte polarization
PBMCs were obtained from leukocyte residues from healthy donors from the Blood and Tissue Bank (Barcelona, Spain) by Ficoll Hypaque PlusTM density gradient centrifugation (GE Healthcare Biosciences), and CD3+ cells were depleted using the RosetteSepTM Human CD3 Depletion Cocktail (StemCell Technonogies). Monocytes were then isolated using the MagniSort Human CD14 Positive Selection kit (eBioscience) according the instructions supplied by manufacturer. Recovered cells were counted using PerfectCount Microspheres (Cytognos, Salamanca, Spain) and assessed for purity (>93% CD14+) and viability (≥97% by FSC/SSC and 7AAD- (BD) gating) in a Canto II flow cytometer (BD).
Monocytes were plated at 1·106cells/ml in RPMI medium containing 5% FBS and the polarizing stimuli for M1(LPS+IFNγ), 50ng/mL IFNγ (Preprotech) plus 100ng/mL LPS from E. coli O111:B4 (Sigma-Aldrich); M2a(IL-4), 40 ng/ml IL-4 (Preprotech); M2c(IL-10), 50 ng/ml IL-10 (Preprotech), or with the EV, proximal non-EV fractions or full CM, and PBS alone (non-activated control). After 48h, the supernatant was harvested for cytokine determination and whole RNA was extracted from cells using the RNeasy Mini Kit (Qiagen). cDNA was synthesized using random hexamers (Qiagen) and the iScriptTM One-Step RT-PCR Kit (BioRad Laboratories) according to supplier's protocol. Each cDNA was then amplified in a LightCycler® 480 PCR system (Roche Life Science) using the KAPA SYBR Fast Master Mix (KAPA Biosystems). Samples were incubated for an initial denaturation at 95°C for 5 min, and then 40 PCR cycles were performed at 95°C for 10 s, 60°C for 20 s and 72°C for 10 s. The values obtained by the “Fit point” method were correlated to a standard curve and normalized to the expression levels of the endogenous reference gene 18S. The gene expression levels of each stimulus were calculated as a fold change relative to non-activated monocytes.
Measurement of cytokine production
Cytokines present in supernatants from alloproliferation assays collected at day 3.5 were measured using the CBA human Th1/Th2 or the Th1/Th2/Th17 Cytokine kit (both from BD Biosciences), the TGF-β1 ELISA (eBioscience, San Diego, CA) and IL-17 ELISA (U-CyTech, Utrecht, The Netherlands) following manufacturer's instructions. Cytokines present in supernatants of 48h-cultured monocytes were measured using the human IL-10 and TNF-α ELISA (U-CyTech). Concentrations given by CBA were assessed in a LSR Fortessa Analyzer (BD) and concentrations of ELISA determinations in a Varioskan LUX multimode microplate reader (Thermo Scientific). The minimum detectable concentration (pg/ml) of each protein was 2.6 for IL-2 and IL-4, 3.0 for IL-6, 2.8 for IL-10 and TNF-α, 7.1 for IFNγ, 8 for TGF-β1, 2 for IL-17 and 1 for IL-10 and TNF-α.
Statistical Analysis
Values are expressed as mean ± standard deviation (SD). Kolmogorov-Smirnov analysis was used to check for normality of data. ANOVA one-way with Tukey's post-hoc analysis was applied to determine significance among more than two groups of parametric data. Paired T test and Wilcoxon matched-pairs signed rank test were used to analyze differences between two paired parametric and non-parametric data groups, respectively. Kruskall-Wallis analysis was used to determine significance among groups and Mann-Whitney test to find differences between two groups of non-parametric data. One sample T test and Wilcoxon Signed Rank test were used to determine differences of normalized parametric and non-parametric data, respectively. Analyses were performed using the GraphPad Prism software (6.01 version) and the SPSS statistic software (19.0.1 version, SPSS Inc., Chicago, IL), and differences were considered significant when p<0.05.
Supplementary Material
Figures S1, S2, S3, S4 and S5 and Table S1.
Abbreviations
MSC: mesenchymal stem cell; UCMSC: umbilical cord mesenchymal stem cell; EV: extracellular vesicle; SEC: size exclusion chromatography; CM: conditioned medium; CCM: concentrated conditioned medium; ECM: eluted conditioned medium; UC: ultracentrifuged; NTA: nanoparticle tracking analysis; cryo-EM: cryo-electron microscopy; MFI: median fluorescence intensity; APC: antigen-presenting cell.
Acknowledgements
Thanks to Marco A. Fernández (Flow Cytometry Unit, IGTP) and Pablo Castro Hartmann (Electron Microscopy Unit, UAB). Also to Hernando A. del Portillo (ICREA Research Professor at ISGLOBAL-IGTP) for access to NTA instrument. Antibodies anti-CD9 and anti-CD63 were both a gift from Dr. María Yáñez-Mó (Unidad de Investigación, Hospital Sta Cristina, IIS-IP; Departamento Biología Molecular/CBM-SO, UAM) and Dr. Francisco Sánchez-Madrid (Servicio de Inmunología, Hospital Universitario de la Princesa, IIS-IP, UAM; Cell-cell Communication Laboratory, CNIC). This work was supported in part by Grants from Instituto de Salud Carlos III (FIS PI13/00050) and (FIS PI14/01682), SGR programme of “Generalitat de Catalunya” (2014SGR804, Group REMAR) and (2014SGR699, Group ICREC), ISCIII-REDinREN (16/0009 Feder Funds), MINECO (SAF2014-59892-R), Fundació La Marató TV3 (201502 and 201516), Red de Terapia Celular-TerCel (RD12/0019/0029), and CIBER Cardiovascular (CB16/11/00403). This work has been developed in the context of AdvanceCat with the support of ACCIÓ (Catalonia Trade & Investment; Generalitat de Catalunya) under the Catalonian ERDF operational program (European Regional Development Fund) 2014-2020. MMT is sponsored by a Grant (2014FI B00649) and MF is sponsored by the “Beatriu de Pinós”-B contract (2014BP B00118), both from the “Agència de Gestió d'Ajuts Universitaris i de Recerca” (AGAUR) from the Catalan Government. FEB is sponsored by the “Researchers Stabilization Program” from the Spanish “Sistema Nacional de Salud” (SNS- ISCIII) and “Direcció d'Estratègia i Coordinació” Catalan Health Department (CES07/015).
Conflicts of Interest
None.
References
1. Roura S, Soler-Botija C, Bagó JR. et al. Postinfarction Functional Recovery Driven by a Three-Dimensional Engineered Fibrin Patch Composed of Human Umbilical Cord Blood-Derived Mesenchymal Stem Cells. Stem Cells Transl. Med. 2015;4:956-66
2. Le Blanc K, Rasmusson I, Sundberg B. et al. Treatment of severe acute graft-versus-host disease with third party haploidentical mesenchymal stem cells. Lancet. 2004;363:1439-1441
3. Watt SM, Gullo F, van der Garde M. et al. The angiogenic properties of mesenchymal stem/stromal cells and their therapeutic potential. Br. Med. Bull. 2013;108:25-53
4. Troyer DL, Weiss ML. Wharton's jelly-derived cells are a primitive stromal cell population. Stem Cells. 2008;26:591-9
5. Dominici M, Le Blanc K, Mueller I. et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006;8:315-7
6. Menard C, Pacelli L, Bassi G. et al. Clinical-Grade Mesenchymal Stromal Cells Produced Under Various Good Manufacturing Practice Processes Differ in Their Immunomodulatory Properties: Standardization of Immune Quality Controls. Stem Cells Dev. 2013;22:1789-1801
7. Krampera M, Cosmi L, Angeli R. et al. Role for interferon-gamma in the immunomodulatory activity of human bone marrow mesenchymal stem cells. Stem Cells. 2006;24:386-398
8. Polchert D, Sobinsky J, Douglas G. et al. IFN-gamma activation of mesenchymal stem cells for treatment and prevention of graft versus host disease. Eur. J. Immunol. 2008;38:1745-1755
9. Renner P, Eggenhofer E, Rosenauer A. et al. Mesenchymal stem cells require a sufficient, ongoing immune response to exert their immunosuppressive function. Transplant. Proc. 2009;41:2607-2611
10. Ryan JM, Barry F, Murphy JM, Mahon BP. Interferon-gamma does not break, but promotes the immunosuppressive capacity of adult human mesenchymal stem cells. Clin. Exp. Immunol. 2007;149:353-363
11. de Witte SFH, Franquesa M, Baan CC, Hoogduijn MJ. Toward Development of iMesenchymal Stem Cells for Immunomodulatory Therapy. Front. Immunol. 2015;6:648
12. Regateiro FS, Howie D, Nolan KF. et al. Generation of anti-inflammatory adenosine by leukocytes is regulated by TGF-β. Eur. J. Immunol. 2011;41:2955-2965
13. Madrigal M, Rao KS, Riordan NH. A review of therapeutic effects of mesenchymal stem cell secretions and induction of secretory modification by different culture methods. J. Transl. Med. 2014;12:260
14. Meisel R, Zibert A, Laryea M, Göbel U, Däubener W, Dilloo D. Human bone marrow stromal cells inhibit allogeneic T-cell responses by indoleamine 2,3-dioxygenase-mediated tryptophan degradation. Blood. 2004;103:4619-4621
15. Chen K, Wang D, Du WT. et al. Human umbilical cord mesenchymal stem cells hUC-MSCs exert immunosuppressive activities through a PGE2-dependent mechanism. Clin. Immunol. 2010;135:448-58
16. Li T, Yan Y, Wang B. et al. Exosomes derived from human umbilical cord mesenchymal stem cells alleviate liver fibrosis. Stem Cells Dev. 2013;22:845-854
17. Lener T, Gimona M, Aigner L. et al. Applying extracellular vesicles based therapeutics in clinical trials - an ISEV position paper. J. Extracell. vesicles. 2015;4:30087
18. Yáñez-Mó M, Siljander PR-M, Andreu Z. et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. vesicles. 2015;4:27066
19. Gatti S, Bruno S, Deregibus MC. et al. Microvesicles derived from human adult mesenchymal stem cells protect against ischaemia-reperfusion-induced acute and chronic kidney injury. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc. - Eur. Ren. Assoc. 2011;26:1474-1483
20. Lai RC, Arslan F, Lee MM. et al. Exosome secreted by MSC reduces myocardial ischemia/reperfusion injury. Stem Cell Res. 2010;4:214-222
21. Kordelas L, Rebmann V, Ludwig AK. et al. MSC-derived exosomes: a novel tool to treat therapy-refractory graft-versus-host disease. Leukemia. 2014;28:970-3
22. Katsuda T, Kosaka N, Takeshita F, Ochiya T. The therapeutic potential of mesenchymal stem cell-derived extracellular vesicles. Proteomics. 2013;13:1637-1653
23. Lötvall J, Hill AF, Hochberg F. et al. Minimal experimental requirements for definition of extracellular vesicles and their functions: a position statement from the International Society for Extracellular Vesicles. J. Extracell. Vesicles. 2014:3
24. Franquesa M, Hoogduijn MJ, Ripoll E. et al. Update on Controls for Isolation and Quantification Methodology of Extracellular Vesicles Derived from Adipose Tissue Mesenchymal Stem Cells. Front. Immunol. 2014:5
25. Fais S, O'Driscoll L, Borras FE. et al. Evidence-Based Clinical Use of Nanoscale Extracellular Vesicles in Nanomedicine. ACS Nano. 2016
26. Conforti A, Scarsella M, Starc N. et al. Microvescicles Derived from Mesenchymal Stromal Cells Are Not as Effective as Their Cellular Counterpart in the Ability to Modulate Immune Responses In Vitro. Stem Cells Dev. 2014
27. Gouveia de Andrade AV, Bertolino G, Riewaldt J. et al. Extracellular vesicles secreted by bone marrow- and adipose tissue-derived mesenchymal stromal cells fail to suppress lymphocyte proliferation. Stem Cells Dev. 2015;24:1374-6
28. Kim H-S, Choi D-Y, Yun SJ. et al. Proteomic analysis of microvesicles derived from human mesenchymal stem cells. J. Proteome Res. 2012;11:839-49
29. Murray PJJ, Allen JEE, Biswas SKK. et al. Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity. 2014;41:14-20
30. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 2014;6:13
31. Kilpinen L, Impola U, Sankkila L. et al. Extracellular membrane vesicles from umbilical cord blood-derived MSC protect against ischemic acute kidney injury, a feature that is lost after inflammatory conditioning. J. Extracell. Vesicles. 2013:2
32. Patel SA, Meyer JR, Greco SJ, Corcoran KE, Bryan M, Rameshwar P. Mesenchymal Stem Cells Protect Breast Cancer Cells through Regulatory T Cells: Role of Mesenchymal Stem Cell-Derived TGF-. J. Immunol. 2010;184:5885-5894
33. Le Blanc K, Mougiakakos D. Multipotent mesenchymal stromal cells and the innate immune system. Nat. Rev. Immunol. 2012;12:383-396
34. Regateiro FS, Cobbold SP, Waldmann H. CD73 and adenosine generation in the creation of regulatory microenvironments. Clin. Exp. Immunol. 2013;171:1-7
35. Mancheño-Corvo P, Menta R, del Río B. et al. T Lymphocyte Prestimulation Impairs in a Time-Dependent Manner the Capacity of Adipose Mesenchymal Stem Cells to Inhibit Proliferation: Role of Interferon γ, Poly I:C, and Tryptophan Metabolism in Restoring Adipose Mesenchymal Stem Cell Inhibitory Effect. Stem Cells Dev. 2015;24:2158-2170
36. Groh ME, Maitra B, Szekely E, Koç ON. Human mesenchymal stem cells require monocyte-mediated activation to suppress alloreactive T cells. Exp. Hematol. 2005;33:928-34
37. Melief SM, Schrama E, Brugman MH. et al. Multipotent stromal cells induce human regulatory T cells through a novel pathway involving skewing of monocytes toward anti-inflammatory macrophages. Stem Cells. 2013;31:1980-1991
38. Tipnis S, Viswanathan C, Majumdar AS. Immunosuppressive properties of human umbilical cord-derived mesenchymal stem cells: role of B7-H1 and IDO. Immunol. Cell Biol. 2010;88:795-806
39. Chan JL, Tang KC, Patel AP. et al. Antigen-presenting property of mesenchymal stem cells occurs during a narrow window at low levels of interferon-gamma. Blood. 2006;107:4817-24
40. Zhou C, Yang B, Tian Y. et al. Immunomodulatory effect of human umbilical cord Wharton's jelly-derived mesenchymal stem cells on lymphocytes. Cell. Immunol. 2011;272:33-8
41. Fischer UM, Harting MT, Jimenez F. et al. Pulmonary passage is a major obstacle for intravenous stem cell delivery: the pulmonary first-pass effect. Stem Cells Dev. 2009;18:683-92
42. Lee RH, Pulin AA, Seo MJ. et al. Intravenous hMSCs improve myocardial infarction in mice because cells embolized in lung are activated to secrete the anti-inflammatory protein TSG-6. Cell Stem Cell. 2009;5:54-63
43. Hoogduijn MJ, Roemeling-van Rhijn M, Engela AU. et al. Mesenchymal stem cells induce an inflammatory response after intravenous infusion. Stem Cells Dev. 2013;22:2825-2835
44. Lai RC, Chen TS, Lim SK. Mesenchymal stem cell exosome: a novel stem cell-based therapy for cardiovascular disease. Regen. Med. 2011;6:481-492
45. Zhao Y, Sun X, Cao W. et al. Exosomes Derived from Human Umbilical Cord Mesenchymal Stem Cells Relieve Acute Myocardial Ischemic Injury. Stem Cells Int. 2015;2015:761643
46. Yang J, Liu X-X, Fan H. et al. Extracellular Vesicles Derived from Bone Marrow Mesenchymal Stem Cells Protect against Experimental Colitis via Attenuating Colon Inflammation, Oxidative Stress and Apoptosis. PLoS One. 2015;10:e0140551
47. Yu B, Zhang X, Li X. Exosomes derived from mesenchymal stem cells. Int. J. Mol. Sci. 2014;15:4142-57
48. Baranyai T, Herczeg K, Onódi Z. et al. Isolation of Exosomes from Blood Plasma: Qualitative and Quantitative Comparison of Ultracentrifugation and Size Exclusion Chromatography Methods. PLoS One. 2015;10:e0145686
49. Gámez-Valero A, Monguió-Tortajada M, Carreras-Planella L, Franquesa M la, Beyer K, Borràs FE. Size-Exclusion Chromatography-based isolation minimally alters Extracellular Vesicles' characteristics compared to precipitating agents. Sci. Rep. 2016;6:33641
50. Yang Y, Bucan V, Baehre H, von der Ohe J, Otte A, Hass R. Acquisition of new tumor cell properties by MSC-derived exosomes. Int. J. Oncol. 2015;47:244-52
51. Amarnath S, Foley JE, Farthing DE. et al. Bone Marrow Derived Mesenchymal Stromal Cells Harness Purinergenic Signaling to Tolerize Human Th1 Cells In Vivo. Stem Cells. 2014;33:1-18
52. Böing AN, van der Pol E, Grootemaat AE, Coumans FAW, Sturk A, Nieuwland R. Single-step isolation of extracellular vesicles by size-exclusion chromatography. J. Extracell. vesicles. 2014:3
53. de Menezes-Neto A, Sáez MJF, Lozano-Ramos I. et al. Size-exclusion chromatography as a stand-alone methodology identifies novel markers in mass spectrometry analyses of plasma-derived vesicles from healthy individuals. J. Extracell. vesicles. 2015;4:27378
54. Lozano-Ramos I, Bancu I, Oliveira-Tercero A. et al. Size-exclusion chromatography-based enrichment of extracellular vesicles from urine samples. J. Extracell. vesicles. 2015;4:27369
55. Welton JL, Webber JP, Botos L-A, Jones M, Clayton A. Ready-made chromatography columns for extracellular vesicle isolation from plasma. J. Extracell. vesicles. 2015;4:27269
56. Plumas J, Chaperot L, Richard M-J, Molens J-P, Bensa J-C, Favrot M-C. Mesenchymal stem cells induce apoptosis of activated T cells. Leukemia. 2005;19:1597-604
57. Akiyama K, Chen C, Wang D. et al. Mesenchymal-stem-cell-induced immunoregulation involves FAS-ligand-/FAS-mediated T cell apoptosis. Cell Stem Cell. 2012;10:544-55
58. Volpe E, Servant N, Zollinger R. et al. A critical function for transforming growth factor-beta, interleukin 23 and proinflammatory cytokines in driving and modulating human T(H)-17 responses. Nat. Immunol. 2008;9:650-7
59. Benwell RK, Lee DR. Essential and synergistic roles of IL1 and IL6 in human Th17 differentiation directed by TLR ligand-activated dendritic cells. Clin. Immunol. 2010;134:178-87
60. Annunziato F, Cosmi L, Liotta F, Maggi E, Romagnani S. The phenotype of human Th17 cells and their precursors, the cytokines that mediate their differentiation and the role of Th17 cells in inflammation. Int. Immunol. 2008;20:1361-8
61. Anderson JD, Johansson HJ, Graham CS. et al. Comprehensive Proteomic Analysis of Mesenchymal Stem Cell Exosomes Reveals Modulation of Angiogenesis via NFkB Signaling. Stem Cells. 2016;34:601-13
62. Rossi J-F, Lu Z-Y, Jourdan M, Klein B. Interleukin-6 as a therapeutic target. Clin. Cancer Res. 2015;21:1248-57
63. Lee DW, Gardner R, Porter DL. et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014;124:188-95
64. Maude SL, Barrett D, Teachey DT, Grupp SA. Managing cytokine release syndrome associated with novel T cell-engaging therapies. Cancer J. 2014;20:119-22
65. Cutler AJ, Limbani V, Girdlestone J, Navarrete C V. Umbilical Cord-Derived Mesenchymal Stromal Cells Modulate Monocyte Function to Suppress T Cell Proliferation. J. Immunol. 2010;185:6617-6623
66. Melief SM, Geutskens SB, Fibbe WWE. et al. Multipotent stromal cells skew monocytes towards an anti-inflammatory interleukin-10-producing phenotype by production of interleukin-6. Haematologica. 2013;98:888-95
67. Martinez FO, Gordon S, Locati M, Mantovani A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J. Immunol. 2006;177:7303-11
68. Bayes-Genis A, Roura S, Soler-Botija C. et al. Identification of cardiomyogenic lineage markers in untreated human bone marrow-derived mesenchymal stem cells. Transplant. Proc. 2005;37:4077-9
69. Prat-Vidal C, Roura S, Farré J. et al. Umbilical cord blood-derived stem cells spontaneously express cardiomyogenic traits. Transplant. Proc. 2007;39:2434-7
70. Ullah M, Eucker J, Sittinger M, Ringe J. Mesenchymal stem cells and their chondrogenic differentiated and dedifferentiated progeny express chemokine receptor CCR9 and chemotactically migrate toward CCL25 or serum. Stem Cell Res. Ther. 2013;4:99
Author contact
![]() Corresponding author: Francesc E. Borràs, PhD. IVECAT Group Leader, Institut d'Investigació en Ciències de la Salut Germans Trias i Pujol (IGTP), Can Ruti campus Ctra. de Canyet s/n, Edifici “Escoles” 08916 Badalona (Barcelona) Spain. Phone: (+34) 93497 867 Fax: (+34) 934978668 E-mail: feborrascat.
Corresponding author: Francesc E. Borràs, PhD. IVECAT Group Leader, Institut d'Investigació en Ciències de la Salut Germans Trias i Pujol (IGTP), Can Ruti campus Ctra. de Canyet s/n, Edifici “Escoles” 08916 Badalona (Barcelona) Spain. Phone: (+34) 93497 867 Fax: (+34) 934978668 E-mail: feborrascat.