Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Supplementary Material
Acknowledgements
References
Author biography
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(1):106-116. doi:10.7150/thno.16911 This issue Cite
Research Paper
Heparin improves BMSC cell therapy: Anticoagulant treatment by heparin improves the safety and therapeutic effect of bone marrow-derived mesenchymal stem cell cytotherapy
Li Liao1,2, Bingzheng Shi1,3, Heran Chang1,3, Xiaoxia Su4, Lichao Zhang1,3, Chunsheng Bi1, Yi Shuai 1,2, Xiaoyan Du5, Zhihong Deng 6 ![]() , Yan Jin1,2
, Yan Jin1,2 ![]()
1. State Key Laboratory of Military Stomatology & National Clinical Research Center for Oral Diseases & Shaanxi International Joint Research Center for Oral Diseases, Center for Tissue Engineering, School of Stomatology, The Fourth Military Medical University, Xi'an, Shaanxi 710032, China;
2. Research and Development Center for Tissue Engineering, Fourth Military Medical University, Xi'an, Xi'an, Shaanxi 710032, China;
3. Stomatological Hospital Affiliated to Jia Mu Si University, JiaMuSi, Heilongjiang 154007, China;
4. Department of Orthodontics, Stomatology Hospital of Xi'an Jiaotong University College of Medicine, Xi'an, Shaanxi 710032, China;
5. Chinese Association of Plastics and Aesthetics, Beijing 100039, China;
6. Department of Otolaryngology, Xijing Hospital, Fourth Military Medical University, Xi'an, Shaanxi 710032, China.
Received 2016-7-20; Accepted 2016-9-14; Published 2017-1-1
Abstract

Systemic infusion of bone marrow-derived mesenchymal stem cells (BMSCs) has become a promising strategy for disease treatment and tissue regeneration. Strategies to enhance the efficiency of BMSC cell therapy are crucial to promote its clinical application. Here, we aimed to improve BMSC cell therapy by inhibiting the BMSC-induced coagulation reaction. Intravenous injection of gradient BMSCs into mice showed that BMSCs were not fully compatible with blood. Large doses of BMSCs induced a series of symptoms of respiratory failure and heart failure. Histological and homeostasis analysis confirmed that large doses of BMSCs induced disseminated intravascular thrombosis, exhaustion of platelets and coagulation factors, and prolonged prothrombin time (PT) and activated partial thromboplastin time (APTT). Similar to mouse BMSCs, goat and human BMSCs also induced coagulation reactions in vitro and in vivo. The coagulation was induced mostly by tissue factor, the overexpression of which enhanced the procoagulant activity of BMSCs during in vitro culture. Notably, clinical doses of BMSCs in cell therapy also induced mild and reversible coagulation, which increased BMSC lung embolism and clearance. Anticoagulation treatment by heparin (400 U/kg) prevented BMSC-induced coagulation and the acute adverse effects of large-dose BMSCs infusion efficiently. Importantly, heparin treatment led to decreased BMSC lung embolism and enhanced migration and maintenance of BMSCs to target organs in cell therapy. Based on an experimental colitis model, we confirmed that heparin treatment enhanced the effect of BMSC therapy efficiently to reduce mortality, prevent weight loss, suppress inflammation reaction and alleviate tissue injury. In conclusion, BMSCs possess procoagulant activity that could induce disseminated coagulation and thrombosis in recipients. Anticoagulation treatment by heparin is a practical strategy to improve both the safety and therapeutic effect of BMSC therapy.
Keywords: Anticoagulant, Cell therapy, Coagulation, Heparin, Mesenchymal stem cell.
Introduction
Systemic administration of BMSCs to regulate the immune response and enhance endogenous regeneration is becoming a new paradigm of BMSC cell therapy. Intravascular injection of BMSCs has proved to be a promising treatment for autoimmune diseases [1, 2], vascular diseases [3], graft vs. host disease (GVHD) [4] and diabetes [5]. Hundreds of clinical trials of BMSC cell therapy are now being carried out world-wide (www.clinicaltrials.gov).
A major challenge for BMSC therapy is the low-efficiency in clinical treatment. In most cases, recipients are administered intravascularly with 1 × 106/kg BMSCs. The large quantity of BMSCs leads to a number of issues, such as in vitro cell expansion and adverse effects induced by the infused BMSCs. Moreover, some phase Ⅲ clinic trials failed because of the low efficiency of BMSCs [6]. Therefore, identifying strategies to improve the therapeutic effect is becoming a crucial issue for BMSC cell therapy.
The majority of previous studies attempted to improve MSC therapy by regulating the biological characteristics of MSCs directly. For example, MSCs were transfected with Akt to promote their survival after infusion, therefore enhancing the therapeutic effect on infarcted heart [7]. MSCs were transfected with vasculoprotective gene angiopoietin 1 (ANGPT1) to enhance their effect on preventing LPS-induced acute lung injury [8]. MSCs were stimulated with TNF-α to enhance its therapeutic effect on tumors [9]. However, because of the limitations of these methods, no practical strategy is available to improve MSC cell therapy in clinic until now.
Notably, recent studies have found incompatible reactions between MSCs and the blood of recipients. MSCs cultured in vitro express procoagulant factors such as tissue factor (TF), collagen1A and fibronectin1, which could initiate coagulation cascades when MSCs are infused into blood [10, 11]. The instant blood-mediated inflammatory reaction induced by systemically infused islet cells and hepatocytes have been reported to compromise transplanted cell survival and function [10], suggesting that the incompatible reactions initiated by BMSCs after intravenous infusion have negative effects on MSC cell therapy. Therefore, targeting the factors affecting BMSC cell therapy might be an alternative approach to improve BMSC therapy.
In this study, we aimed to confirm the factors leading to incompatibility between infused BMSCs and the recipient, and explore a strategy to improve MSC cell therapy by targeting the adverse reactions.
Methods
Animal experiments
All animal experiments were approved by the Animal Use and Care Committee of the Fourth Military Medical University (Xi'an, China) (License Number: 2012 KQ-031). All animal procedures were performed according to the guidelines of the Animal Care Committee of the Fourth Military Medical University, which meet the NIH guidelines for the care and use of laboratory animals. Female C57BL/6J mice (eight weeks old, 24.8 ± 3.5 g) and female mountain goats (3.2 ± 0.6 years old, 59.3±7.3 kg) were purchased from the Animal Center of the Fourth Military Medical University. All mice were housed under specific pathogen-free conditions (22°C, 12-hour light/12-hour dark cycles, and 50%-55% humidity) with free access to food pellets and tap water. The goats were housed in pens and had free access to food and water.
Cell culture
Mouse BMSCs were isolated from hind limbs and cultured as previously described [12]. Briefly, after euthanasia by intraperitoneal injection of 300mg/kg pentobarbital sodium, the mice were sacrificed by cervical dislocation. The hind limbs were removed aseptically and bones were dissected free of soft tissues. Marrow cavities of both the femur and tibia were flushed with α-MEM using a 1 ml injector with (Gibco, Gaithersburg, MD, USA) supplemented with 20% fetal bovine serum (FBS) (Sijiqing, Hangzhou, China), 1% penicillin and streptomycin (Invitrogen, Carlsbad, CA, USA). The cell suspension was seeded in 10-cm tissue culture dishes and incubated in a humidified atmosphere of 5% CO2 at 37°C. The medium was changed every 2-3 days to remove non-adherent cells, and the adherent cells were cultured until confluent.
Human bone marrow samples were collected from five healthy female volunteers aged 42.4 ± 2.4 years with informed consent of the donors following the guidelines of the Ethics Committee of the University of the Fourth Military Medical University. Human BMSCs (hBMSCs) were isolated using monocyte separation medium (Sigma). Isolated monocytes were cultured in 90-mm dishes containing α-MEM (Gibco) supplemented with 10% FBS (Sijiqing) until confluent.
Goat BMSCs (gBMSCs) were isolated from the bone marrow of goat ilium using monocyte separation medium (Sigma), and cultured following the protocols of hBMSC culture.
All cultured BMSCs underwent routine characterization before use in subsequent experiments. BMSCs used in our study were positive for mesenchymal stem cells markers and negative for hematopoietic cell markers, and showed potent of self-renewal and multipotent differentiation. BMSCs at 4-6 passages were used in most of the experiments.
Intravenous injection of BMSCs
For BMSC injection, BMSCs digested with 0.25% trypsin were washed with PBS twice and resuspended in saline. The cell suspension was filtered through a 150μm mesh strainer and a 75μm mesh strainer, counted using a cell counting plate, and diluted to the designated concentration. For BMSC infusion in mice, the designated number of cells suspended in 1ml saline was slowly injected via the tail vein at 1 ml/min. For BMSC infusion in goats, a gBMSCs suspension (1×106/ml) was injected via the jugular vein at a final concentration of 1×106/kg. The infusion velocity was 5 ml/min in the goat experiment.
Systemic acute toxicity test of BMSCs
To determine the acute toxicity of BMSCs, 54 C57BL/6J mice (24.83 ± 3.51 g) were equally and randomly divided into nine groups. According to the clinical dosage (1×106/kg), 2.5×104 mBMSCs were used as the basal dosage. After 2.5×104 mBMSCs injection, the mice were observed for 30 min to evaluate the acute toxicity. If no severe adverse effect was observed, the dose of BMSCs was doubled for the next group. According to this protocol, the dosage of BMSCs was gradually increased until the absolute lethal dose (LD100) was reached.
Histological analysis
Mice were sacrificed 10 min after injection of 4×106 BMSCs or an equal volume of PBS. After opening the chest, the aorta was perfused with saline to wash out the blood, and then the mouse was instilled with 10% formalin. The lungs, hearts, livers, kidneys and spleens were removed and immersed in 10% formalin for 24 h. All organs were embedded in paraffin and cut into 5 mm sections. The sections were then deparaffinized and stained with hematoxylin and eosin (H&E) following standard protocols. Thrombosis formation in the vasculature was observed under a microscope. Photographs were taken under 40× magnification of five consecutive microscopic fields, and the thrombosis number per visual field was measured.
BMSC migration assay
To detect the migration of injected BMSCs in vivo, BMSCs were labeled with 5 μg/ml Hoechst 33342 (Sigma) for 2 h before injection. After BMSC administration, mice were sacrificed at specific times. To detect BMSC in circulation, mouse blood was collected through the angular vein and the red blood cells were eliminated using red blood cell lysis buffer (Sigma, USA). Hoechst+ cells in blood were detected by flow cytometry (FCM). To detect BMSC migration into internal organs, the organs were isolated and fixed immediately with 4% paraformaldehyde for 24 h and dehydrated with 10% sucrose solution for 24 h. The organs were then embedded in optimum cutting temperature compound (Sakura, USA) and sectioned at 10 μm with a freezing microtome (Leica, Wetzlar, Germany). Sections were examined under a fluorescence microscope (Olympus Optical, Tokyo, Japan) to detect Hoechst-labeled BMSCs. Photos were taken under 20× magnification on five consecutive microscopic fields. The number of labeled cells in photos was measured using Image Pro Plus 6.0 Software (Media Cybernetics, Rockville, MD, USA).
Homeostasis analysis
Mouse blood was isolated via the angular vein under anesthesia, while goat blood was isolated via the jugular vein after BMSC injection. For coagulation analysis, 1-2 ml of whole blood was anticoagulated with 0.1 volume of 3.8% sodium citrate (Sigma). Plasma was isolated by centrifugation at 1500 rpm for 5 min. Prothrombin time (PT), activated partial thromboplastin time (APTT), Factor VIII (FVIII) and fibrinogen (Fbg) were detected automatically using an automated blood coagulation analysis system (Siemens, Munich, Germany) according to the manufacturer's protocols. Notably, values of PT or APTT larger than the detection range of the coagulation analyzer were recorded as “120 sec” or “180 sec”, respectively.
Hematological analysis
For hematological analysis, citrated whole blood was analyzed using an automatic hematology analyzer (Sysmex, Kobe, Japan), following standard protocols. Red blood cells (RBCs), white blood cells (WBCs), platelets and hemoglobin were quantified automatically.
In vitro analysis of procoagulant activity of BMSCs
To determine the procoagulant activity of BMSCs in vitro, whole blood collected from mice, goats or healthy human volunteers were anticoagulated with 3.8% sodium citrate. mBMSCs, gBMSCs and hBMSCs were digested with trypsin, washed twice with PBS and resuspended at a final concentration of 5×105 cells/ml in PBS. BMSC's procoagulant activity was determined by mixing 10 μl of cell suspension, 90 μl of allogenic blood, and 7 μl of CaCl2 (0.2 M), and measuring the time of clot formation. An equal volume of PBS mixed with blood and CaCl2 was used as the vehicle control.
Flow cytometry analysis of TF
For FCM analysis, P2 and P12 mBMSCs were digested with trypsin and washed with PBS twice. 1×106 BMSCs were stained with a primary antibody against TF (USCN, Houston, TX, USA, PA524Mu01, 1:1000) for 2 h and with FITC-conjugated secondary antibody (CWbiotech, CW0114, 1:1000) for 1 h. Expression of TF in BMSCs was analyzed using an Elite ESP flow cytometry (Beckman Coulter, Fullerton, CA, USA), according to the manufacturer's instructions.
Real-time RT-PCR
Real-time reverse transcription polymerase chain reaction (RT-PCR) of mRNA was performed as previously reported [13]. In brief, total RNA of P2 and P12 mBMSCs was isolated using the Trizol reagent (Invitrogen). Random-primed cDNA was synthesized from 1 mg of RNA using an RT reagent kit (Takara, Dalian, China). Real-time PCR analysis was performed using a SYBR Premix Ex Taq II kit (Takara) and detected on the Bio-rad sequence detection system (Bio-rad, Hercules, CA, USA).
The primers for TF were:
Forward: 5′-AACGTGGTTGTAAAAGACTCACT-3′;
Reverse: 5′-GCTTGAGCCTTTCCGATAAGTAA-3′;
Primers for β-actin:
Forward: 5′-CTGGCACCACACCTTCTACA-3′; Reverse: 5′- GGTACGACCAGAGGCATACA-3′.
Western blotting
Western blotting was performed as described previously [13]. Primary antibodies for mouse TF (USCN, PA524Mu01, 1:1000) and β-actin (CWbiotech, CW0096A, 1:4000) were used in this study. β-actin was used as the loading control.
BMSC therapy for experimental colitis
Inflammatory colitis was induced in C57BL/6 mice by feeding them with 3% dextran sulfate sodium salt (DSS) (MP Biomedicals, Irvine, CA, USA) in water for 13 days [14]. At day 3, 1×106 BMSCs suspended in 1 ml PBS were injected into colitis mice via the tail vein. Mice administered with 1 ml PBS were used as the positive control for colitis. All mice were monitored daily for the occurrence of body weight change, diarrhea, fecal bleeding and death. Ten days later, the colons of the mice were harvested and their lengths recorded. For histopathological analysis, colon segments were embedded, sectioned and stained with H & E. The disease index and histological score were assessed according to previously published criteria [15].
Heparin pretreatment of BMSCs for cytotherapy
For heparin pretreatment, BMSCs were cultured in culture medium containing 4U/ml heparin sodium (Tianjin Biochem Pharmaceutical Co, Tianjin, China) for 12 h. BMSCs were then washed with PBS twice to remove residual heparin and digested with trypsin for cell therapy.
Statistical analysis
Data were analyzed using SPSS 19.0 (www.ibm.com/software/analytics/spss) or GraphPad 6 (http://www.graphpad.com). Continuous data (mean ± SD) were compared using Student's t test where two groups were compared, and parametric or non-parametric analysis of variance (ANOVA) where more than two groups were compared. Survival was analyzed by the Log-rank (Mantel-Cox) test. Significance was confirmed at P<0.05. No statistical method was used to predetermine the sample size. The experiments were randomized. The investigators were not blinded to the allocation during experiments and outcome assessment.
Results
Intravenous infused mBMSCs induce coagulation reaction in blood
To explore whether infused BMSCs are compatible with the recipient, we injected 2.5×104 cells/mouse (1×106 cells/kg) as basal dosage via the tail vein into mice and gradually increased the dose. Consistent with most animal tests [16], less than 1×106 mBMSCs caused no obvious acute symptom in mice (Table 1). However, 3×106 mBMSCs induced a number of severe symptoms, including dyspnea, cyanosis, tetraplegia, coolness of the extremities and exophthalmos, indicating respiratory and circulatory failure. Half of mice died within 3-50 min. The incidence of severe adverse effects was increased to 100% after infusion of 4×106 mBMSCs (Table 1). The different numbers of mBMSCs were suspended in a final volume of 1 ml, we explored whether the adverse effect is related to the high concentration of the cell suspension. By injecting different concentration of mBMSCs suspensions, we confirmed that it was the total cell number, not the concentration, which determined the acute adverse effects (Supplementary Table S1).
Acute adverse symptoms of mice injected with different dosages of mBMSCs.
| PBS | mBMSCs | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 2.5×104 | 5×104 | 1×105 | 5×105 | 1×106 | 2×106 | 3×106 | 4×106 | ||
| (n=6) | (n=6) | (n=6) | (n=6) | (n=6) | (n=6) | (n=6) | (n=6) | (n=6) | |
| Dyspnea | 0 | 0 | 0 | 0 | 0 | 0 | 2(33.3) | 6(100) | 6(100) |
| Cyanosis | 0 | 0 | 0 | 0 | 0 | 0 | 1(16.7) | 6(100) | 6(100) |
| Slow response | 0 | 0 | 0 | 0 | 0 | 0 | 1(16.7) | 5(83.3) | 6(100) |
| Exophthalmus | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3(50) | 6(100) |
| Tetraplegia | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 4(66.7) | 6(100) |
| Coolness of extremities | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 5(83.3) | 6(100) |
| Mortality | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3(50) | 6(100) |
Data are shown as n (%).
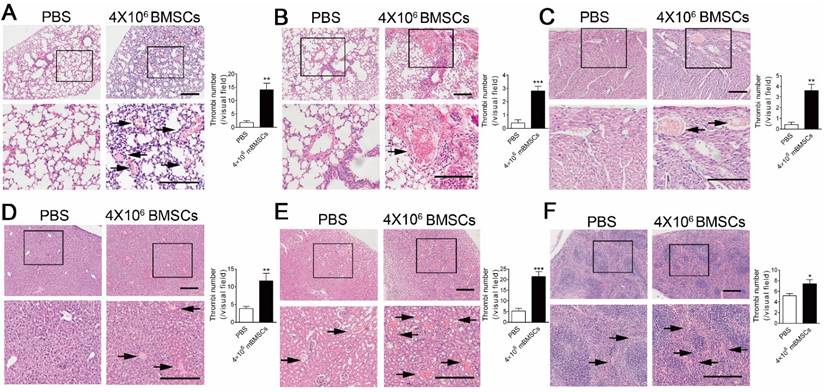
We performed histological examinations to explore the cause of BMSC-induced respiratory and circulatory failure. H&E staining showed marked micro-thrombus formation in microvessels and arterioles of the lung 10 min after 4×106 mBMSCs infusion (Figure 1A and B). Moreover, micro-thrombi were also formed in the heart (Figure 1C), liver (Figure 1D), kidney (Figure 1C) and spleen (Figure 1F). To further confirm that BMSCs are involved in thrombosis, we marked mBMSCs by Hoechst staining before injection. Fluorescence analysis showed that Hoechst+ Cells were found in the micro-thrombus in the microvessels (Supplementary Figure S1A) and arterioles (Supplementary Figure S1B) of the lung.
mBMSCs infusion induces disseminated intravascular coagulation in vivo. H&E staining of lung (A and B), heart (C), liver (D), kidney (E) and spleen (F) of mice 10 min after PBS or 4×106 mBMSCs infusion. Representative images are shown. Note that the images at the bottom are higher magnifications of the black-boxed regions at the top. Scale bar, 200 μm. The number of thrombi (indicated with black arrows) per visual field in each organ was calculated under a microscope. n=5. *P<0.05, **P<0.01, ***P<0.001 versus PBS group (t-test).
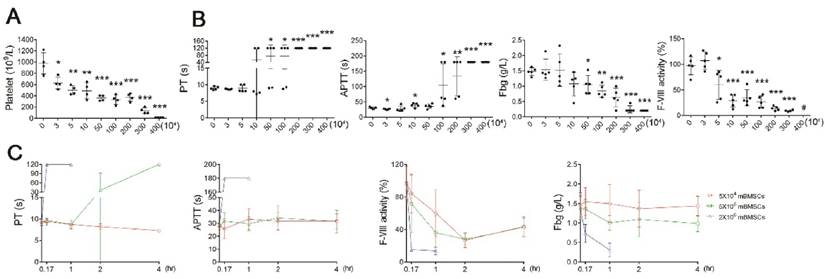
To confirm that BMSCs induce coagulation, we performed hemostasis analysis after BMSC infusion. Platelet numbers were significantly decreased after mBMSCs infusion, in a concentration-dependent manner (Figure 2A), whereas RBCs, WBCs and hemoglobin (HGB) levels were not affected by mBMSCs (Supplementary Figure S2). Moreover, mBMSCs (≥1×106) infusion resulted in a significant elongation of PT and APTT, and a decrease in FVIII and Fbg within 10 min (Figure 2B). Notably, infusion of a lethal dose of mBMSCs decreased platelet levels by over 80% (Figure 2A), prolonged the PT and APTT beyond the measurement range of the coagulation analyzer, and decreased the Fbg and FVIII levels by 90% (Figure 2B), indicating extreme exhaustion of coagulation factors. Continued examination confirmed that the rate of coagulation was positively associated with cell dosage (Figure 2C). 1×105 mBMSCs were sufficient to induce a mild coagulation reaction (Figure 2A-C).
mBMSCs infusion induces acute coagulation in vivo. A, Platelet number in blood isolated from mice 10 min after different number of mBMSCs injection. n=5. B, Hemostasis analysis of blood plasma of mice injected with different number of mBMSCs for 10 min. n=5. *P<0.05, **P<0.01, ***P<0.001 versus 0 mBMSC group (PBS injection as vehicle control) (ANOVA). #, indicates that the value was lower than the measurement range of the analyzer. C, Hemostasis analysis of blood plasma isolated at different time points from mice injected with mBMSCs. n=5. Data shown as mean ± SD.
Procoagulant activity of cultured BMSCs is conserved among species
To further confirm that BMSCs of other species possess procoagulant activity, we examined the procoagulant activity of hBMSCs and gBMSCs. Similar to mBMSCs, large doses of hBMSCs and gBMSCs induced symptoms of respiratory and circulatory failure (Table 2) and systemic coagulation in mice (Figure 3A). Both hBMSCs and gBMSCs significantly shortened the clotting time of homogenous plasma, ruling out a heterogeneous coagulation reaction (Figure 3B).
Mortality of mice injected with different generations of mBMSCs, gBMSCs and hBMSCs.
| P2 BMSCs | P6 BMSCs | P12 BMSCs | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 2×106 (n=4) | 3×106 (n=4) | 4×106 (n=4) | 2×106 (n=4) | 3×106 (n=4) | 4×106 (n=4) | 2×106 (n=4) | 3×106 (n=4) | 4×106 (n=4) | |
| mBMSCs | 0 | 0 | 1(25) | 0 | 2(50) | 4(100) | 4(100) | 4(100) | 4(100) |
| gBMSCs | 0 | 0 | 0(0) | 0 | 1(25) | 3(75) | 4(100) | 4(100) | 4(100) |
| hBMSCs | 0 | 0 | 2(50) | 2(50) | 4(100) | 4(100) | 4(100) | 4(100) | 4(100) |
Data are shown as n (%).
Long term in vitro expansion enhances procoagulant activity of BMSCs of different species. A, Hemostasis analysis of mice 10 min after PBS, 2×106 hBMSCs or 2×106 gBMSCs infusion. n=4. **P<0.01, ***P<0.001 versus PBS group (ANOVA). B, Clotting time of human serum and goat serum mixed with PBS or 1×105 homogenous BMSCs in vitro. n=4. ***P<0.001 (t-test). C, Hemostasis analysis of goats at different time points after 1×106/kg gBMSCs infusion. The value of each index was normalized to the values before gBMSCs infusion (0 h). n=5. **P<0.01, ***P<0.001 versus 0 h group (ANOVA). D, Clotting time of mouse serum mixed with different doses of mBMSCs in vitro. n=3. *P<0.05, ***P<0.001 versus 0 mBMSC group (ANOVA). E, Clotting time of mouse serum mixed with PBS, 1×105 P2 or P12 mBMSCs in vitro. n=5. ***P<0.001 versus PBS group (ANOVA). F, Hemostasis analysis of blood isolated from mice 10 min after 2×106 P6/P12 gBMSCs or hBMSCs injection. n=4. *P<0.05, **P<0.01, ***P<0.001 (t-test). G, Realtime RT-PCR analysis of TF mRNA levels in P2 or P12 mBMSCs. n=3. ***P<0.001 (t-test). H, Western blotting of TF protein levels in P2 or P12 mBMSCs. β-actin was used as the loading control. The gray levels of blots were quantified using Image J software. n=3. **P<0.01 (t-test). I, FCM analysis of TF expression in P2 or P12 mBMSCs. n=3. ***P<0.001 (t-test). J, Clotting time of mouse serum mixed with 1×105 mBMSCs (P2 or P12) alone or mBMSCs combined with 400ng TFPI. n=4. *P<0.05, **P<0.01 (ANOVA). Data shown as mean ± SD.
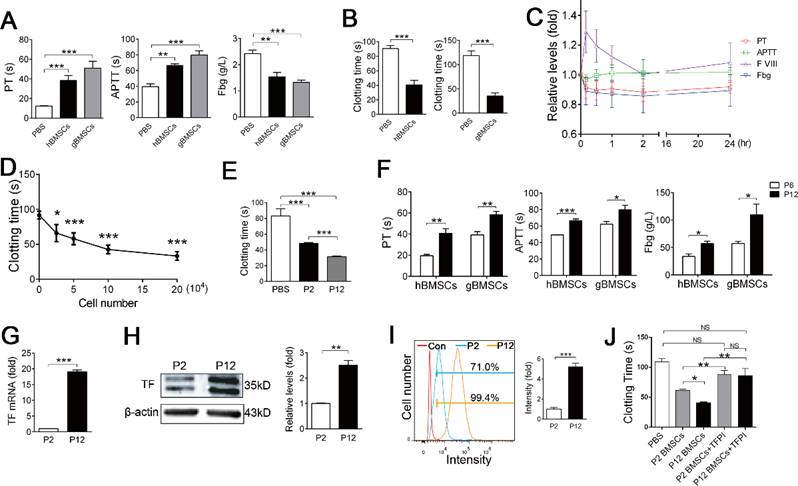
To confirm whether a clinical dose of BMSCs induces coagulation in large animals, we intravenously injected 1×106/kg gBMSCs, a dosage usually used in clinical therapy, into goats. Hemostasis analysis confirmed that 1×106/kg gBMSCs induced hypercoagulable states within 2 h, as shown by increased PT and decreased Fbg and FVIII (Figure 3C). No severe symptoms were observed in goats receiving gBMSCs injection, and the hypercoagulability gradually recovered with 24 h (Figure 3C).
We also compared the procoagulant activity of BMSCs cultured for different passages. The coagulation assay showed that the procoagulant activity increased during the expansion of mBMSCs, gBMSCs and hBMSCs in vitro (Figure 3E and F, Table 2), suggesting that in vitro culture triggers the procoagulant activity of BMSCs.
mBMSCs induce coagulation directly through Tissue Factor (TF)
We then investigated the mechanism by which BMSCs activate coagulation. The above coagulation assay showed that infusion of a small dose of mBMSCs affected the PT but not the APTT (Figure 2B and C), suggesting BMSCs activated mainly the extrinsic coagulation pathway. Tissue Factor (TF, also known as coagulation factor III) is the primary initiator of the extrinsic coagulation cascade [17], and has been reported to be expressed in human ADSCs and BMSCs [10, 11, 18]. FCM, real-time RT-PCR and western blotting confirmed that mBMSCs expressed TF (Figure 3G-I). Inhibition of TF function using a tissue factor pathway inhibitor (TFPI) significantly prevented mBMSC-induced coagulation (Figure 3J). Moreover, the levels of TF significantly increased during mBMSC passaging, which was in accordance with the enhanced procoagulant activity of BMSCs during in vitro culture. (Figure 3G-I). The procoagulant activity of long-term cultured mBMSCs was largely inhibited by TFPI (Figure 3J), suggesting that in vitro cultures increase TF expression to enhance the procoagulant activity of BMSCs.
Heparin prevents efficiently the coagulation reaction of BMSCs after intravenous infusion
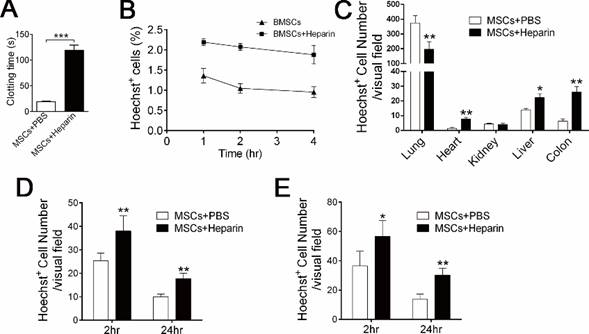
We investigated whether anticoagulant treatment could prevent the adverse effect of mBMSC infusion. Heparin, a strong anticoagulant, is the first-choice drug for disseminated intravascular coagulation (DIC) in the clinic. Therefore, we chose heparin to prevent the coagulation reaction after BMSCs infusion. Hemostasis analysis confirmed that heparin (4 U/ml) strongly inhibited the BMSC-induced coagulation reaction in vitro (Figure 4A). As expected, either pre-injection of heparin (400 U/kg) or injection of heparin (400 U/kg) with mBMSCs completely prevented all symptoms of respiratory and circulatory failure in mice that received 4×106 or 8×106 mBMSCs injection (Table 3). 1.6×107 BMMSCs were needed to induce 100% mortality after heparin treatment (Table 3), indicating that heparin treatment strongly inhibits coagulation and the thrombosis induced by high doses of BMSC administered in vivo. Furthermore, heparin treatment also largely prevented the adverse effect of long-term cultured BMSCs (Supplementary Table S2). Real-time RT-PCR analysis confirmed that heparin did not directly suppress the expression of TF in BMSCs (Supplementary Figure S3), suggesting that heparin functioned through blocking TF-induced coagulation.
Heparin treatment improves mBMSCs maintenance and migration. A, Clotting time of mouse serum mixed with mBMSCs and PBS or heparin (4 U/ml) in vitro. n=3. ***P<0.001 (t-test). B and C, Hoechst-labeled mBMSCs (1×106) alone or combined with 400 U/kg Heparin were injected into mice via tail vein. The mice were sacrificed after 24 h. Hoechst+ cells in blood were detected by FCM (B), and Hoechst+ cells in several organs were detected by fluorescence microscopy (C). n=5. *P<0.05, **P<0.01 (t-test). D and E, Mice were fed with drinking water containing 3% DSS for 14 days. At the third day, Hoechst-labeled 1×106 mBMSCs alone or combined with 400 U/kg Heparin were injected into the mice via tail vein. The migration of mBMSCs to the colon (D) and mesenteric lymph node (E), at 2 or 24 h after BMSC injection, was analyzed using fluorescence microscopy. n=4 *P<0.05, **P<0.01. (t-test).
Acute symptoms of mice injected with different dosages of mBMSCs and heparin.
| PBS | Heparin pre-injection+mBMSCs | Heparin combined with mBMSCs | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 4×106 | 8×106 | 12×106 | 16×106 | 4×106 | 8×106 | 12×106 | 16×106 | ||
| (n=6) | (n=6) | (n=6) | (n=6) | (n=6) | (n=4) | (n=4) | (n=4) | (n=4) | |
| Dyspnea | 0 | 0 | 2(33.3) | 5(83.3) | 6(100) | 0 | 2(50) | 4(100) | 4(100) |
| Cyanosis | 0 | 0 | 0 | 4(66.7) | 6(100) | 0 | 0 | 3(75) | 4(100) |
| Slow response | 0 | 0 | 0 | 4(66.7) | 6(100) | 0 | 0 | 4(100) | 4(100) |
| Exophthalmus | 0 | 0 | 0 | 3(50) | 6(100) | 0 | 0 | 2(50) | 4(100) |
| Tetraplegia | 0 | 0 | 0 | 3(50) | 6(100) | 0 | 0 | 3(75) | 4(100) |
| Coolness of extremities | 0 | 0 | 0 | 3(50) | 6(100) | 0 | 0 | 3(75) | 4(100) |
| Mortality | 0 | 0 | 0 | 3(50) | 6(100) | 0 | 0 | 2(50) | 4(100) |
Data are shown as n (%).
Anticoagulant treatment enhances the survival and distribution of BMSCs to target organs
Infusion with a clinical dose of BMSCs induced mild and reversible coagulation (Figure 2A-C, 3C); therefore, we questioned whether this reaction affects BMSC cell therapy. Coagulation might hinder the distribution and accelerate the clearance of exogenous BMSCs [19, 20]. Accordingly, 400 U/kg heparin treatment significantly increased the level of exogenous mBMSCs in the blood (Figure 4B), liver, colon and spleen (Figure 4C), while it decreased the number of mBMSCs in the lung (Figure 4C). To examine whether anticoagulant treatment enhances BMSCs maintenance in pathological conditions, we used an experimental colitis mouse model, a widely used inflammatory disease model. Hoechst-labeled BMSCs were infused with or without heparin into colitis mice. Histological analysis confirmed that heparin treatment significantly promoted mBMSC migration and maintenance in the colons and mesenteric lymph nodes (MLN) of the colitis mice (Figure 4D and E).
Heparin treatment promotes the therapeutic effect of BMSC immune therapy
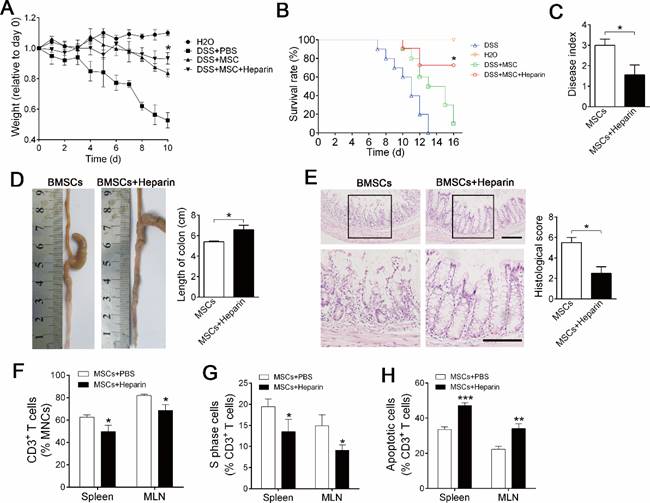
Sufficient migration and maintenance are crucial for the success of BMSC cell therapy [21, 22]; therefore, we explored whether heparin treatment improves the therapeutic effect of BMSC cell therapy on inflammatory colitis. mBMSCs infusion alleviated the mortality and body weight loss of experimental colitis mice compared with PBS injection group (Figure 5A, B). Ten days after treatment, analysis of symptoms confirmed that heparin treatment significantly improved the therapeutic effect of mBMSCs, as shown by reduced mortality, body weight loss and disease index compared with the PBS+BMSCs group (Figure 5A-C). Histological analysis revealed that heparin treatment improved the effect of mBMSCs in preventing colon inflammation and tissue injury (Figure 5D, E). Accordingly, FCM showed that heparin treatment promoted BMSCs' property of suppressing T cells in the blood and mesenteric lymph nodes (Figure 5F-H).
Heparin treatment improves BMSC cell therapy of experimental colitis. Mice were injected with 1×106 mBMSCs, alone or combined with Heparin (400 u/kg) into the mice via tail vein at day 3 of DSS feeding. A, Body weight was recorded daily for 10 days. The body weight of each mouse was normalized by its weight at day 0. n=7. *P<0.05 versus DSS+MSC group at critical day (t-test). B, Mortality of mice was recorded for 14 days. n=10. *P<0.05 versus DSS+MSC group (Log-rank (Mantel-Cox) test). C, Disease index was measured at day 10 of DSS feeding. n=5. *P<0.05 (t-test). D, Colons of each group were collected after 10 days and their lengths were measured. n=5. *P<0.05. (t-test). E, Histological structure of the colon was detected by H&E staining, and the histological score was measured. Representative images are shown. The images at the bottom are higher magnifications of the images at the top. Scale bar, 200 μm. n=5. *P<0.05 (t-test). F, CD3+ T cells percentage in the spleen and MLN were analyzed by flow cytometry. n=4. *P<0.05 (t-test). G, Cell cycle of CD3+ T cells was analyzed by flow cytometry. n=5. *P<0.0 (t-test). H, Apoptosis of CD3+ T cells was analyzed by flow cytometry. n=5. *P<0.05 (t-test). Data shown as mean ± SD.
To rule out the possibility that heparin directly suppresses the immune reaction, we injected heparin alone into colitis mice and found no improvement compared with the PBS injection group (Supplementary Figure S2A-F). To rule out the possibility that heparin directly improves immunoregulatory properties of BMSCs, we also precultured mBMSCs with heparin before cell therapy. As expected, mBMSCs cultured with heparin showed similar effects on colitis treatment as untreated mBMSCs (Supplementary Figure S4A-F).
In conclusion, our results revealed that in vitro cultured BMSCs possess procoagulant activity. Injection of a clinical dose of BMSCs induced a mild coagulant reaction, which reduced the migration and maintenance of BMSCs during cell therapy. Importantly, anticoagulation treatment is an effective strategy to prevent the BMSC-induced coagulation reaction and improve the therapeutic effect of BMSC cell therapy, indicating the necessity and importance of anticoagulation treatment in BMSC cell therapy.
Discussion
A number of strategies have been applied to improve BMSC cell therapy. Most of these strategies have focused on promoting the cellular properties of BMSCs. For example, certain genes were transfected into BMSCs to overexpress key regulators of cell therapy. In this study, we proposed a novel strategy to improve cell therapy by preventing the incompatible reaction between BMSCs and recipients. We found that heparin treatment could promote BMSCs migration and survival to the target organ efficiently, resulting in an improved therapeutic effect. Compared with previous strategies, our method has a number of advantages. First, our method does not change the biological characteristics of the BMSCs, which avoids the potential side effect of changes to BMSCs. Second, heparin is used widely and is much safer than molecular regulation methods. Third, heparin treatment improved not only the therapeutic effect, but also the safety of BMSC cell therapy. Fourth, our method could be combined with other strategies targeting BMSCs to promote MSC cell therapy.
Researchers hold high promise for BMSC cell therapy partly because BMSCs are considered as “safe cells” [16]. However, in most clinical cases, 1-2×106/kg BMSCs were administered intravenously into patients within a short time [23, 24]. Such a large amount of exogenous cells would be expected to induce wide reactions beyond the therapeutic effect in the recipients [16]. Increasing numbers of studies have disclosed that BMSC injection induces a number of acute adverse events, including fever [16], instant blood-mediated inflammatory reaction [10], microvascular embolism [19, 20, 25] and impaired heart function [26-28]. Notably, two recent cases of sudden death caused by lung embolism after MSC infusion focused worldwide attention on the acute toxicity of MSCs [29, 30]. In this study, we observed that injection of a clinical dose of BMSCs also induced mild and reversible coagulation, which would usually be ignored in clinic trials. Notably, infusion of a large dose of BMSCs induced severe symptoms of respiratory and circulatory failure. Although more convincing clinical data are necessary to confirm that BMSCs induce coagulation, our findings suggested that preventative measures are necessary to ensure the safety of BMSC cell therapy.
It was presumed that the coagulation reaction would increase BMSC obstruction in the lung and accelerate BMSC elimination, which prompted us to investigate the effect of anti-coagulant treatment on BMSC therapy. We found that anticoagulant treatment increased the number of BMSCs in the blood, colon, mesenteric lymph node, liver and heart, while it decreased BMSCs in the lung. Moreover, heparin treatment improved the distribution of BMSCs to the target organ in an experimental colitis mouse model. These findings indicated that more BMSCs could penetrate through lung capillary network and distribute to other organs after the coagulation reaction was prevented. BMSC migration and maintenance are the preconditions for successful cell therapy; therefore, our strategy might be a useful method to improve BMSC cell therapy for various diseases.
Heparin is the top choice for the clinical treatment of disseminated intravascular coagulation. Stephenne and colleagues found that anticoagulants such as heparin and bivalirudin inhibit potently the procoagulant activity of human adult liver-derived mesenchymal progenitor cells in vitro [31]. Gleeson and colleagues reported that heparin improved the safety of BMSCs infusion via the coronary artery for heart infarction [18]. In this study, we also found that heparin was inhibited BMSC-induced coagulation potently. Either pretreatment or co-administration with heparin prevented efficiently most of the acute adverse effect of infusion with a large dose of BMSCs. Notably, the absolute lethal dose was increased by four times after heparin treatment, indicating that heparin is an ideal drug to prevent the adverse effect induced by BMSCs.
Safety and efficacy are two essential factors to be considered for clinical treatment. Generally, a larger dose could achieve higher efficacy but increase the risk of side effects. Our data showed that infusion with a large dose of BMSCs infusion induced severe side effects, suggesting it is not feasible to enhance the therapeutic effect by increasing the dose. In our study, we found that heparin treatment improved both safety and therapeutic effect of BMSC therapy. Accordingly, a recent study showed that heparin reduced thrombosis and improved heart infarct after acute intracoronary MSC delivery in acute myocardial infarction [18]. These results suggested that the effect of anticoagulation treatment is universal for both local and systemic infusion of MSCs. Heparin is a widely used, safe drug; therefore, our strategy is practical to improve BMSC cell therapy in the clinic.
Supplementary Material
Supplementary tables and figures.
Acknowledgements
This work was supported by grants from the Nature Science Foundation of China 81470710 (to Y.J.), 31030033 (to Y.J.), 81470679 (to Z.D.), and the National Major Scientific Research Program of China 2011CB964700 (to Y.J.).
Authorship Contributions
Y.J., Z.D., and L.L. designed the study. L.L., B.S., H.C., X.S., L.Z. and C.B. performed experiments and collected the data. Y.J., Z.D., L.L., B.S., H.C., X.S., Y.S. and X.D. analyzed the data. Y.J., Z.D., and L.L. wrote the manuscript.
Competing Interests
The authors declare no competing financial interests.
References
1. Singer NG, Caplan AI. Mesenchymal stem cells: mechanisms of inflammation. Annual review of pathology. 2011;6:457-78
2. Shi Y, Hu G, Su J, Li W, Chen Q, Shou P. et al. Mesenchymal stem cells: a new strategy for immunosuppression and tissue repair. Cell research. 2010;20:510-8
3. Hare JM, Fishman JE, Gerstenblith G, DiFede Velazquez DL, Zambrano JP, Suncion VY. et al. Comparison of allogeneic vs autologous bone marrow-derived mesenchymal stem cells delivered by transendocardial injection in patients with ischemic cardiomyopathy: the POSEIDON randomized trial. JAMA. 2012;308:2369-79
4. Le Blanc K, Rasmusson I, Sundberg B, Gotherstrom C, Hassan M, Uzunel M. et al. Treatment of severe acute graft-versus-host disease with third party haploidentical mesenchymal stem cells. Lancet. 2004;363:1439-41
5. Lee RH, Seo MJ, Reger RL, Spees JL, Pulin AA, Olson SD. et al. Multipotent stromal cells from human marrow home to and promote repair of pancreatic islets and renal glomeruli in diabetic NOD/scid mice. Proc Natl Acad Sci U S A. 2006;103:17438-43
6. Ankrum J, Karp JM. Mesenchymal stem cell therapy: Two steps forward, one step back. Trends Mol Med. 2010;16:203-9
7. Mangi AA, Noiseux N, Kong D, He H, Rezvani M, Ingwall JS. et al. Mesenchymal stem cells modified with Akt prevent remodeling and restore performance of infarcted hearts. Nat Med. 2003;9:1195-201
8. Mei SH, McCarter SD, Deng Y, Parker CH, Liles WC, Stewart DJ. Prevention of LPS-induced acute lung injury in mice by mesenchymal stem cells overexpressing angiopoietin 1. PLoS medicine. 2007;4:e269
9. Lee RH, Yoon N, Reneau JC, Prockop DJ. Preactivation of human MSCs with TNF-alpha enhances tumor-suppressive activity. Cell Stem Cell. 2012;11:825-35
10. Moll G, Rasmusson-Duprez I, von Bahr L, Connolly-Andersen AM, Elgue G, Funke L. et al. Are therapeutic human mesenchymal stromal cells compatible with human blood? Stem Cells. 2012;30:1565-74
11. Tatsumi K, Ohashi K, Matsubara Y, Kohori A, Ohno T, Kakidachi H. et al. Tissue factor triggers procoagulation in transplanted mesenchymal stem cells leading to thromboembolism. Biochem Biophys Res Commun. 2013;431:203-9
12. Liao L, Su X, Yang X, Hu C, Li B, Lv Y. et al. TNF-alpha Inhibits FoxO1 by Upregulating miR-705 to Aggravate Oxidative Damage in Bone Marrow-Derived Mesenchymal Stem Cells during Osteoporosis. Stem Cells. 2016;34:1054-67
13. Liao L, Yang X, Su X, Hu C, Zhu X, Yang N. et al. Redundant miR-3077-5p and miR-705 mediate the shift of mesenchymal stem cell lineage commitment to adipocyte in osteoporosis bone marrow. Cell death & disease. 2013;4:e600
14. Okayasu I, Hatakeyama S, Yamada M, Ohkusa T, Inagaki Y, Nakaya R. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology. 1990;98:694-702
15. Naito Y, Takagi T, Kuroda M, Katada K, Ichikawa H, Kokura S. et al. An orally active matrix metalloproteinase inhibitor, ONO-4817, reduces dextran sulfate sodium-induced colitis in mice. Inflammation research: official journal of the European Histamine Research Society [et al]. 2004;53:462-8
16. Lalu MM, McIntyre L, Pugliese C, Fergusson D, Winston BW, Marshall JC. et al. Safety of cell therapy with mesenchymal stromal cells (SafeCell): a systematic review and meta-analysis of clinical trials. PloS one. 2012;7:e47559
17. Owens AP 3rd, Mackman N. Tissue factor and thrombosis: The clot starts here. Thromb Haemost. 2010;104:432-9
18. Gleeson BM, Martin K, Ali MT, Kumar AH, Pillai MG, Kumar SP. et al. Bone Marrow-Derived Mesenchymal Stem Cells Have Innate Procoagulant Activity and Cause Microvascular Obstruction Following Intracoronary Delivery: Amelioration by Antithrombin Therapy. Stem Cells. 2015;33:2726-37
19. Toma C, Wagner WR, Bowry S, Schwartz A, Villanueva F. Fate of culture-expanded mesenchymal stem cells in the microvasculature: in vivo observations of cell kinetics. Circ Res. 2009;104:398-402
20. Fischer UM, Harting MT, Jimenez F, Monzon-Posadas WO, Xue H, Savitz SI. et al. Pulmonary passage is a major obstacle for intravenous stem cell delivery: the pulmonary first-pass effect. Stem cells and development. 2009;18:683-92
21. Parekkadan B, Milwid JM. Mesenchymal stem cells as therapeutics. Annual review of biomedical engineering. 2010;12:87-117
22. Caplan AI, Correa D. The MSC: an injury drugstore. Cell Stem Cell. 2011;9:11-5
23. Le Blanc K, Frassoni F, Ball L, Locatelli F, Roelofs H, Lewis I. et al. Mesenchymal stem cells for treatment of steroid-resistant, severe, acute graft-versus-host disease: a phase II study. Lancet. 2008;371:1579-86
24. Burt RK, Loh Y, Pearce W, Beohar N, Barr WG, Craig R. et al. Clinical applications of blood-derived and marrow-derived stem cells for nonmalignant diseases. Jama. 2008;299:925-36
25. Janowski M, Lyczek A, Engels C, Xu J, Lukomska B, Bulte JW. et al. Cell size and velocity of injection are major determinants of the safety of intracarotid stem cell transplantation. J Cereb Blood Flow Metab. 2013;33:921-7
26. Hare JM, Traverse JH, Henry TD, Dib N, Strumpf RK, Schulman SP. et al. A randomized, double-blind, placebo-controlled, dose-escalation study of intravenous adult human mesenchymal stem cells (prochymal) after acute myocardial infarction. J Am Coll Cardiol. 2009;54:2277-86
27. Vulliet PR, Greeley M, Halloran SM, MacDonald KA, Kittleson MD. Intra-coronary arterial injection of mesenchymal stromal cells and microinfarction in dogs. Lancet. 2004;363:783-4
28. Breitbach M, Bostani T, Roell W, Xia Y, Dewald O, Nygren JM. et al. Potential risks of bone marrow cell transplantation into infarcted hearts. Blood. 2007;110:1362-9
29. Cyranoski D. Korean deaths spark inquiry. Nature. 2010;468:485
30. State of Florida Board of Medicine. Florida Department of Health v. Zannos G. Grekos. DOH-13-0914-FOF-MQA. 2013
31. Stephenne X, Nicastro E, Eeckhoudt S, Hermans C, Nyabi O, Lombard C. et al. Bivalirudin in combination with heparin to control mesenchymal cell procoagulant activity. PloS one. 2012;7:e42819
Author biography
Dr. Yan Jin is the professor and director of the research and development center of tissue engineering of Fourth Military Medical University, deputy direct of State Key Laboratory of Military Stomatology, Director-general of Collaborative innovation center of tissue engineering and regenerative medicine, leader of Innovative Research Team in University of Ministry of Education of China, director of maxillofacial repair and regeneration engineering center of Ministry of Education of China. His research focuses on the role of stem cells in degenerative diseases, application of stem cells in regenerative medicine, and tissue engineering of skin, cornea and bone. He has published more than 130 articles in SCI journals and rewarded the first prize of national scientific and technological progress award of China.
Dr. Zhihong Deng is the professor of the Department of Otolaryngology, Xijing Hospital, Fourth Military Medical University, Xi'an, Shaanxi, China. Her research focuses on the role of mesenchymal stem cells in pharyngolaryngeal diseases and tissue regeneration. She proved the existence of mesenchymal stem cells in laryngeal mucosa and confirmed their role in vocal fold regeneration. She also found several strategies of tissue engineering for mucosa and skin repair.
Dr. Li Liao received his doctor degree in the Fourth Military Medical University. His research focuses on the role of mesenchymal stem cell in age-related bone diseases and application of stem cells in disease treatment. He found that abnormal expression of microRNA lead to dysfunction of mesenchymal stem cells during osteoporosis and proposed a number of strategies to improve stem cell-based cytotherapy. He has coauthored over 15 publications including Theranostics, Stem cells, Molecular Therapy, and FASEB J.
![]() Corresponding authors: Prof. Yan Jin, M.D., Ph.D., MS-State Key Laboratory, Fourth Military Medical University, 145 West Changle Road, Xi'an, Shaanxi 710032, People's Republic of China. Telephone: +86-29-84776147; Fax: +86-29-83218039; E-mail: yanjinedu.cn. Or Prof. Zhihong Deng, M.D., Ph.D., Xijing Hospital, Fourth Military Medical University, 145 West Changle Road, Xi'an, Shaanxi 710032, People's Republic of China. Telephone: +86-29-84776472; Fax: +86-29-83218039; E-mail: dengzhedu.cn.
Corresponding authors: Prof. Yan Jin, M.D., Ph.D., MS-State Key Laboratory, Fourth Military Medical University, 145 West Changle Road, Xi'an, Shaanxi 710032, People's Republic of China. Telephone: +86-29-84776147; Fax: +86-29-83218039; E-mail: yanjinedu.cn. Or Prof. Zhihong Deng, M.D., Ph.D., Xijing Hospital, Fourth Military Medical University, 145 West Changle Road, Xi'an, Shaanxi 710032, People's Republic of China. Telephone: +86-29-84776472; Fax: +86-29-83218039; E-mail: dengzhedu.cn.