Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Experimental Section
Results
Discussion
Conclusion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2016; 6(11):2000-2014. doi:10.7150/thno.15102 This issue Cite
Research Paper
Magnetic Enrichment of Dendritic Cell Vaccine in Lymph Node with Fluorescent-Magnetic Nanoparticles Enhanced Cancer Immunotherapy
Honglin Jin1,2,3, Yuan Qian1,2, Yanfeng Dai1,2, Sha Qiao1,2, Chuan Huang1,2, Lisen Lu1,2, Qingming Luo1,2, Jing Chen3*, Zhihong Zhang1,2* ![]()
1. Britton Chance Center for Biomedical Photonics, Wuhan National Laboratory for Optoelectronics-Huazhong University of Science and Technology, Wuhan 430074, China;
2. MoE Key Laboratory for Biomedical Photonics, Department of Biomedical Engineering, Huazhong University of Science and Technology, Wuhan 430074, China;
3. Cancer Center, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, 1277 JieFang Avenue, Wuhan 430022, China.
*These authors are co-corresponding authors.
Received 2016-1-26; Accepted 2016-7-13; Published 2016-9-2
Abstract

Dendritic cell (DC) migration to the lymph node is a key component of DC-based immunotherapy. However, the DC homing rate to the lymphoid tissues is poor, thus hindering the DC-mediated activation of antigen-specific T cells. Here, we developed a system using fluorescent magnetic nanoparticles (α-AP-fmNPs; loaded with antigen peptide, iron oxide nanoparticles, and indocyanine green) in combination with magnetic pull force (MPF) to successfully manipulate DC migration in vitro and in vivo. α-AP-fmNPs endowed DCs with MPF-responsiveness, antigen presentation, and simultaneous optical and magnetic resonance imaging detectability. We showed for the first time that α-AP-fmNP-loaded DCs were sensitive to MPF, and their migration efficiency could be dramatically improved both in vitro and in vivo through MPF treatment. Due to the enhanced migration of DCs, MPF treatment significantly augmented antitumor efficacy of the nanoparticle-loaded DCs. Therefore, we have developed a biocompatible approach with which to improve the homing efficiency of DCs and subsequent anti-tumor efficacy, and track their migration by multi-modality imaging, with great potential applications for DC-based cancer immunotherapy.
Keywords: DC vaccine, immunotherapy, antigen delivery, DC migration tracking, magnetic targeting.
Introduction
Dendritic cell (DC)-based immunotherapy represents a promising approach to the treatment of human diseases, including cancer [1-4]. Progress has been achieved in the DC vaccination field by the use of ex vivo-generated, tumor antigen-pulsed DCs; however, clinical trials have demonstrated a poor efficacy [5]. One of the major factors restricting in vivo efficacy is the inefficient migration of DCs to the lymph node (LN), wherein DCs activate antigen-specific T cells [6]. To date, the most commonly used method to improve DC migration is the preinjection of pro-inflammatory cytokines, which helps to provide a more suitable inflammatory microenvironment for the migration of DCs through the lymphatic vessels [7]. The preinjection of mouse tumor necrosis factor (TNF) was reported to generate a 10-fold increase in the number of DCs that reached the LNs [8]. Alternatively, enhancing the expression levels of specific DC homing receptors can also facilitate migration [9]. For example, DCs that had been adenovirally transduced with the CC-chemokine receptor-7 (CCR-7) gene, which encodes a chemokine receptor responsible for DC migration toward the lymphatics, exhibited an approximately 6-fold increase in migration efficiency relative to control DCs [10]. Other factors, such as the route of administration, number of injected DCs, and maturation degree of the DCs, also affect the migration efficiencies of injected DCs [11]. Despite these efforts, the in vivo DC migration efficiency remains unsatisfactory with a typical rate of less than 4% when administered via intradermal injection, the most frequently employed method in DC-based immunotherapy [9]. In fact, the DC homing route comprises at least two rate-limiting steps, including the pathway from the injection site to the initial lymphatic vessels as well as the subsequent movement along the lymphatic vessels [12]. Current studies have generally emphasized improvements of the former; however, an ideal approach would consider both steps. As the first step has been addressed by previously described methods, such as TNF-α preinjection, the development of efficient approaches to accelerate the migration of injected DCs in the lymphatic vessels would be expected to dramatically augment DC migration and enhance subsequent therapeutic outcomes.
Recent progress in the development of non-invasive imaging technologies has made it possible for researchers to visually and even quantitatively study the migration and homing of injected DCs to LNs [13]. Bimodal imaging (e.g., the combination of magnetic resonance (MR) and near infrared (NIR) fluorescence imaging) is appealing and may be useful for in vivo DC tracking as this bimodal imaging approach can provide both anatomical background information and high-sensitivity detection [14-17]. Bimodal nanovehicles containing fluorescent agents as well as iron oxide particles have been designed to label DCs for both fluorescence and MR imaging [18-20]. However, clinical applications are often hampered by either the nanoparticle cytotoxicity or the absence of suitable fluorophores for in vivo NIR imaging [21]. Multifunctional indocyanine green (ICG) and iron oxide particle-loaded poly (lactic-co-glycolic acid) (PLGA) nanoparticles have demonstrated excellent biocompatibility, bimodal imaging ability, as well as strong cytotoxic T lymphocyte (CTL) responses. However, PLGA-loaded antigens might exhibit antigenicity and stability issues resulting from emulsification steps during preparation [16, 22]. More importantly, there is a lack of a second layer of driving force to facilitate DC migration toward the LNs. Accordingly, we expected that if optical probes, MRI contrast agents, and tumor antigens could be simultaneously and efficiently delivered into DCs to endow them with both antigen-presenting and magnetic pull force (MPF)-responsive capabilities, it might be possible to simultaneously promote their homing efficiency using MPF, track their migration using bimodal imaging, and ultimately exert potent antigen-specific immune responses. To reach this goal, the nanocarriers should be tailored to exhibit the following features: (1) an excellent biocompatibility; (2) an efficient uptake by DCs; (3) a sufficient capacity to simultaneously accommodate antigens, NIR probes, and magnetic substances when applied for tumor immunotherapy; and (4) a loaded antigen that will be delivered preferentially into the cytoplasm rather than the endo/lysosomal compartment as the MHC class I antigen-presenting pathway is required for the activation of antitumor CTL (CD8+) immune responses.
Here, we report a fluorescent magnetic nanoparticle (α-AP-fmNP) that was generated by coating iron oxide nanoparticles with phospholipids using a solvent exchange method followed by lipid layer occupation with ICG molecules and fusion peptides (α-AP), which contain α-helix peptide (α-peptide) and antigen peptide (AP) sequences. Using this nanoplatform, this study aimed to achieve the efficient delivery of ICG/iron oxide/AP to DCs to investigate the feasibility of applying MPF to enhance the migration efficiency of α-AP-fmNP-loaded DCs during an MR and NIR imaging evaluation and ultimately to enhance the subsequent antitumor efficacy. This study provides a valuable approach to improve the migration efficiency and antitumor efficacy of DCs and to monitor this migration process.
Experimental Section
Materials
1,2-Dimyristoyl-sn-Glycero-3-Phosphocholine (DMPC) and 1-myristoyl-2-hydroxy-sn-glycero-3-phosphocholine (MHPC) were purchased from Avanti Polar Lipids Inc. (Alabaster, AL, USA). Cholesteryl oleate (CO), Hoechst 33258, Iron Stain Kit, and ICG were obtained from Sigma-Aldrich Co. (St. Louis, MO, USA). The APgp100 peptide (gp10025-33, KVPRNQDWL), APOVA peptide (OVA257-264, SIINFEKL), α-APgp100 (Ac-FAEKFKEAVKDYFAKFWD-GSG-KVPRNQDWL), α-APOVA (Ac-FAEKFKEAVKDYFAKFWD-GSG-SIINFEKL), and FAM-α-APgp100 were synthesized by Bankpeptide Ltd. (Hefei, China). The oleic-acid-stabilized iron oxide nanocrystals, synthesized by a thermal decomposition method [23], were provided by Nanjing Nanoeast Biotech Co., LTD.
Synthesis of α-AP-fmNPs
To synthesize α-APgp100-fmNPs, oleic-acid-stabilized iron oxide nanocrystals were coated with DMPC and MHPC according to a dual solvent exchange method. Briefly, 5 mg each of DMPC and MHPC and 10 mg of oleic-acid-stabilized iron oxide nanocrystals were dissolved in 1 mL of a chloroform/methanol solution (20:1 (v: v)). Next, the solution was added dropwise over 30 min to 6 mL of distilled/deionized water at 80 ºC with magnetic stirring. After an extended period of heating at 80 ºC for 30 min, the solution was centrifuged three times at 3,000 rpm for 15 min to remove the undissolved iron oxide particles or other large particles. Subsequently, 8 mg of α-APgp100 in 2 mL of water and 5 mg of ICG in 1 mL of water were added. After overnight storage at 4 ºC, the solution was washed three times with Ultra-4 centrifugal filter devices (30,000 NMWL, Millipore Co., MA, USA) to remove unbound α-APgp100 and ICG molecules and then concentrated to the desired concentrations. FAM-α-APgp100-fmNP and α-APOVA-fmNP were prepared in an analogous manner by replacing α-APgp100 with FAM-α-APgp100 or α-APOVA, respectively.
Mice and Cells
6 weeks old female C57BL/6 mice were purchased from HFK BIOSCIENCE (Beijing, China). EGFP transgenic C57BL/6 mice were purchased from the second military medical university (Shanghai, China), and OT-I mice were purchased from the Jackson Lab (Bar Harbor, ME). All animal studies were performed in compliance with protocols that had been approved by the Hubei Provincial Animal Care and Use Committee, following the experimental guidelines of the Animal Experimentation Ethics Committee of the Huazhong University of Science and Technology (HUST). The DC2.4 cells were provided by the laboratory of Prof. Guanxin Shen (HUST). The cells were cultured in RPMI-1640 medium (HyClone, Beijing, China), containing 100 U/mL of penicillin-streptomycin (HyClone, Beijing, China) and 10% FBS, at 37 °C in an incubator with 5% CO2.
Measurement of α-AP-fmNPs Concentration
The α-AP-fmNPs concentration was represented by the iron content, which was determined with a ferrozine assay as previously reported [24]. The peptide concentration was measured with a CBQCA protein quantitation kit (Cat. # MP 0667, Invitrogen Corporation, CA, USA). To measure the ICG concentration, 3 mg/mL of ICG in water was serially diluted in methanol to produce standard samples. Using these standard samples, a standard curve correlating ICG concentration and UV absorbance was measured. α-AP-fmNPs were diluted in methanol, vortexed, and centrifuged to remove undissolved iron oxide particles. The supernatant was then collected to determine the ICG concentration by measuring its UV absorbance and comparing to the standard curve.
Prussian Blue Staining
Iron staining was performed using an iron stain kit obtained from Sigma-Aldrich Co. (St. Louis, MO, USA) according to the manufacturer's recommendations.
Nanoparticle Characterization
Transmission electron microscopy (TEM) was performed on a TECNAI G2 (FEI company, OR, USA) instrument to determine the α-APgp100-fmNPs morphology. Prior to imaging, α-AP-fmNPs were negatively stained with 1% phosphotungstic acid. The α-AP-fmNPs size distribution was measured via dynamic light-scattering photon correlation spectroscopy on a Zetasizer Nano-ZS90 (Malvern Instruments, Worcestershire, UK). The absorbance measurements were conducted on a Lambda 35 UV-Visible spectrophotometer (PerkinElmer, USA).
Generation of BMDCs
BMDCs were generated from the bone marrow of 7-10 week old C57BL/6 mice. The bone marrow cells were flushed out of the femurs and tibias with Hank's Balanced Salt Solution (HBSS; HyClone, Beijing, China). The cells were passed through a cell strainer to obtain single cell suspensions. Erythrocytes were lysed by treatment with ACK buffer. The remaining cells were washed twice with HBSS and cultured in RPMI-1640 medium (HyClone, Beijing, China) containing 100 U/mL of penicillin-streptomycin (HyClone, Beijing, China), 10% FBS, recombinant mouse GM-CSF (10 ng/mL; PeproTech, Rocky Hill, NJ), mouse IL-4 (10 ng/mL; PeproTech, Rocky Hill, NJ, USA), and β-mercaptoethanol (50 nM; Gibco Invitrogen, Carlsbad, CA, USA). Subsequently, these cells were seeded at a concentration of 1 × 106 cells per well in a 24-well plate (Corning, NY, USA) in a final volume of 1 mL, and half of the medium was replaced with an equal volume of culture medium at day 3 and 5, respectively. At day 7, the acquired DCs were induced to mature by treating with 1 μg/mL lipopolysaccharide (LPS) for 24 h, generating mature BMDCs. EGFP-BMDCs were isolated from the EGFP transgenic C57BL/6 mice and derived the same way as the BMDCs. In a very similar nanoparticle system, it is reported that LPS-stimulated mature BMDCs were superior to immature BMDCs in terms of activation of AP-specific CTL [25]. Thus, we selected mature BMDCs other than immature BMDCs as antigen recipient.
Confocal Imaging
DC2.4 cells (2 × 104 per well) were cultured in 8-well chambers with glass bottoms (Nunc Lab-Tek, Sigma-Aldrich, USA) for 24 h. The cells were then incubated with FAM-α-APgp100-fmNPs (16 μg/mL) for 6 h at 37 °C. Hoechst 33258 and LysoTracker (Beyotime, Jiangsu, China) were used for nuclear and lysosome organelle staining, respectively. Confocal imaging was performed with a Zeiss LSM 710 confocal microscope (Zeiss, Oberkochen, Germany) at excitation wavelengths of 488 nm for FAM-APgp100, 405 nm for Hoechst 33258, and 561 nm for LysoTracker, respectively. For LN imaging, the mice PLNs were dissected at 24 h post DC inoculation or after MPF treatment, then placed on a confocal dish and imaged with a Zeiss LSM 710 confocal microscopy (Zeiss, Oberkochen, Germany) at an excitation wavelength of 633 nm for ICG (Sigma-Aldrich, USA).
Cytotoxicity Measurement
The cell proliferation assays were performed with the Cell Counting Kit 8 (CCK-8, Dojindo Molecular Technologies, Inc., Kumamoto, Japan). Briefly, DC2.4 cells were cultured in 96-well plates for 24 h (8 × 103 cells/200 μL) in RPMI 1640 medium containing 10% FBS (complete medium). Next, the medium was replaced with 200 μL of fresh complete medium containing the designated concentrations of α-AP-fmNPs. After a 24-h incubation, the medium was removed and the cells were washed twice with PBS. Next, 200 μL of fresh complete medium was added to each well. After a 24-h incubation, the medium was replaced with 100 μL of fresh complete medium containing 10% CCK-8 assay solution. After a 1-h incubation at 37 °C, the absorbance in each well was measured with a 96-well plate reader (Model 525; TECAN Group Ltd., Mannedorf, Switzerland). The cytotoxicity to BMDCs was measured using the similar method with an initial cell number of 1.0 × 104 cells.
Flow Cytometry
To examine the cell labeling abilities of α-AP-fmNPs, DC2.4 cells were cultured in 24-well plates for 24 h (2 × 105 cells/500 μL) in complete culture medium. The medium was then replaced with 500 μL of fresh medium containing the designated concentrations of α-AP-fmNPs. After a 6-h incubation, the medium was removed and the cells were trypsin-digested, washed, and resuspended in PBS. Next, the cell fluorescent signals of ICG were detected by Guava easyCyteTM flow cytometry (Merck Millipore, Billerica, MA, USA), using an excitation wavelength of 640 nm and a filter of 785/70 nm. The uptake of FAM-α-AP-fmNPs by DC2.4 cells was determined using the same method with an excitation wavelength of 488 nm and a filter of 525/30 nm. Flow cytometry was utilized to investigate the effect of α-AP-fmNPs on the morphology changes of DC2.4 and BMDCs. DC2.4 cells and BMDCs (2 × 105 cells) were incubated with various concentration of α-AP-fmNPs for 24 h, and these cells were then subjected to digestion (for DC2.4 cell only) and washing to prepare α-AP-fmNP-loaded DC2.4 cells and α-AP-fmNP-loaded BMDCs. Subsequently, the forward-side distribution (FSC and SSC) of these cells was measured by flow cytometry. The percentage of morphologically normal cells was calculated as follows: (the number of α-AP-fmNP-loaded DCs in gated cells / the number of control DCs in gated area) × 100%.
In Vitro Migration Assay
DC2.4 cells were incubated with α-APgp100-fmNPs (16 μg/mL) in a culture dish for 6 h and subsequently digested, washed, and counted. Next, 1 × 106 cells were added to a 25-cm2 cell culture flask (Corning, NY, USA) with 10 mL of complete medium, and a magnet (N35, 49 mm × 9.3 mm × 4.7 mm) was attached to one side of the flask. After 24 h, the magnet was removed, and both sides of the flask were imaged with an upright confocal microscope (NIKON Ni-E, Nikon, Japan). Transwell migration studies were performed in 24-well transwell culture chambers (8 μm; Corning, NY, USA). Briefly, BMDCs were stimulated with 1 μg/mL LPS for 24 h to promote maturation before being incubated with α-APgp100-fmNPs for 6 h. Culture medium without FBS (600 μL) and with CCL19 (100 ng/mL; R&D Systems, Inc., MN, USA) was added to the lower chambers and the α-APgp100-fmNP-loaded BMDCs in serum-free culture medium (1 × 105 cells/100 μL) were placed into the upper chambers. As a positive control, we added 10% FBS to the bottom chamber, as this was reported to induce cell migration under starved conditions [26]. The MPF-treated group (without FBS) was further treated with a magnetic plate (130 × 85 × 11 mm; Gauss, 1.2 k; Nanoeast Biotech, Nanjing, China), which was placed under the 24-well culture chambers. After a 2-h incubation, the cells that had migrated to the bottom chambers were counted by flow cytometry.
In Vivo NIR and MR Imaging
C57BL/6 mice were preinjected with TNF-α in the hind-leg footpads (25 ng/leg). After 24 h, 1.2 × 106 cells loaded with or without α-AP-fmNPs (50 μL in PBS) were injected into the left and right mouse footpads, respectively, under isoflurane anesthetization. To provide continuous magnetic exposure around the right PLN, a special device containing a ring magnet (40 × 10 mm; Gauss, 11.7-12.1 k; Jingzheng, Shanghai, China), N35, and a wooden mat (57 × 42 × 18 mm) was designed. After a 24-h MPF treatment, the magnets were removed, and NIR and MR images were acquired. Fluorescence imaging was performed with a house-made whole-body imaging system equipped with an excitation filter (716/40 nm) and emission filter (800/40 nm). The MR images of the mice PLNs were acquired using the following sequence parameters: MSME 3500/12 ms (TR/TE), 20 slices with a thickness of 0.5 mm, a FOV of 3.5 × 3.5 cm, a matrix of 256 × 256, and NEX = 8. This study was performed for both DC2.4 cells and BMDCs.
T Cell Proliferation Assays
CD8+ T cells were isolated from the inguinal lymph nodes (ILNs) and spleens of the OT-1 mice with an EasySep Mouse CD8+ T cell isolation kit (Stemcell, Vancouver, BC, Canada). The collected T cells were then stained with 3 μM carboxyfluorescein succinimidyl ester (CFSE, Molecular Probes-Invitrogen, Carlsbad, CA, USA). The cells were subsequently co-cultured with DCs that had been loaded with PBS, APOVA, α-APOVA, or α-APOVA-fmNPs at a T cell: DC ratio of 50: 1 with a fixed T cell number of 1 × 105 cells. After a 3-day incubation, the culture supernatants were collected to measure the IFN-γ concentration via enzyme-linked immunosorbent assay (ELISA) with a Quick EIATM kit (Dakewe Biotech, Beijing, China) according to the manufacturer's instructions. T cell proliferation was evaluated by CFSE dilution using Guava easyCyteTM 8HT flow cytometry (Millipore Corporation, Billerica, MA, USA).
Measurement of CTL Activity
The CTL assay was evaluated by CFSE/PI (propidium iodide) double staining. Briefly, splenocytes collected from immunized mice (BMDCs alone or α-APOVA-fmNPs-loaded BMDCs) were cultured and re-stimulated in the presence of 10 μM APOVA for 5 days. Subsequently, these cells were washed and enriched by HISTOPAQUE 1083 (Sigma), following with co-culture of CFSE-stained E.G7-OVA target cells (5 × 104 cells/well) or non-targeted EL-4 cells (control cells) in 96-well cell culture plates at effector: target ratios of 10: 1, 50: 1, and 100: 1. After 4 h of incubation, the cells were stained with PI and then analyzed by Guava easyCyte 8HT using flow cytometry. The data were analyzed using FlowJo software. The percentage of specific lysis was calculated as follows: specific lysis = (experimental death% - spontaneous death%)/(1- spontaneous death%) × 100.
Calculation of DC Migration Efficiency in Vivo
EGFP-BMDCs were obtained from EGFP-transgenic mice. For calculating the EGFP-positive DCs in the LNs, α-AP-fmNP-loaded EGFP-BMDCs (1.2 × 106/50 μL in PBS) were injected into the right footpads of mice prestimulated with 25 ng TNF-α. These mice were divided into two groups - one group received MPF treatment (2-3 mice/group), whereas the other group was not treated with MPF (2-3 mice/group). After 24 h, the PLNs in each group were dissected, pooled, mechanically disrupted, and filtered to obtain a single cell suspension. The EGFP+ cells in these solutions were analyzed by flow cytometry using the formula: migration efficiency = EGFP+ cells/total injected cells × 100%. Migration efficiencies were calculated as the mean ± SEM of three independent experiments.
Immunofluorescence of LN
The collected fresh PLNs were dehydrated in 30% sucrose solution for 24 h and 10 µm-thick cryosections were prepared. The tissue slides were fixed by incubation with cold acetone for 10 min, air-dried, and subjected to three shaking-washing processes with PBS (each 10 min). After that, they were incubated with 1% bovine serum albumin (BSA), followed by incubation with Alexa Fluor@594-conjugated antibody against CD3 and Alexa Fluor@647-conjugated antibody CD45R/B220 (both BioLegend; San Diego, CA, USA) at 4 °C overnight for labeling T cells and B cells, respectively. After extensive washing with PBS, sections were immersed in a drop of 50% glycerol, covered with a cover slide, and directly observed using confocal imaging.
In Vivo Tumor Growth Inhibition
6 weeks old C57BL/6 mice were prestimulated with 25 ng TNF-α. After 24 h, these mice were immunized at the right hind-leg footpads with PBS control, BMDCs control, or α-APOVA-fmNP-loaded BMDCs (1.2 × 106 in 50 μL PBS). Half of the α-APOVA-fmNP-BMDCs-injected mice were treated with MPF and six mice were used for each group. At 7 days after DC injection, these mice were subjected to repeated immunization using the same method. At one week after the second treatment, these mice were injected subcutaneously with E.G7-OVA cells (5 × 106 in 100 μL PBS) at the right flank close to the PLN. Tumor size was measured by digital caliper and the tumor volume was calculated according to the formula
V = 0.5 × L (length) × W (width)2
Statistical Analysis
Student's two-tailed t-test was used for the in vitro and in vivo studies. Tumor growth data were analyzed applying the Kaplan-Meier method. Significant differences between the groups were indicated by * for P < 0.05, ** for P < 0.01, and *** for P < 0.001, respectively.
Results
Characterization of α-AP-fmNPs
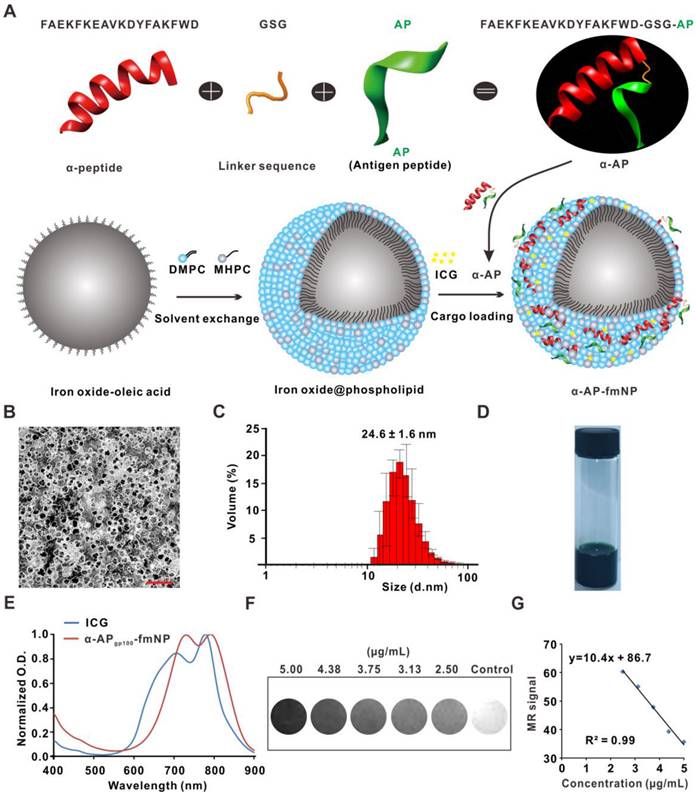
The synthesis of α-AP-fmNPs included coating oleic acid-stabilized iron oxide nanocrystals with the biocompatible assembled lipids 1-myristoyl-2-hydroxy-sn-glycero-3-phosphocholine (MHPC) and 1, 2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) according to a solvent exchange method (Figure 1a). This lipid-coating process imparted hydrophilic properties to the iron oxide nanocrystals and allowed the embedding of ICG molecules due to their high-affinity lipid-layer binding abilities. To load the AP, a fusion peptide comprising the melanoma AP gp10025-33 (APgp100) and an ApoA1 mimetic α-peptide was designed, which was denoted as α-APgp100; this fusion peptide facilitated strong attachments to lipid layers containing phosphatidylcholine (e.g., MHPC and DMPC). Thus, iron oxide nanocrystals, ICG, and α-APgp100 could be successfully loaded to form the α-APgp100-fmNP nanoparticles; the encapsulating efficiencies of these components were 18%, 78%, and 34% for iron oxide crystals, ICG, and α-APgp100, respectively. In Figure 1b, a transmission electron microscopy (TEM) image clearly shows the iron oxide core (dark appearance) and phospholipid layers (white appearance) with a negative stain. The resulting α-APgp100-fmNPs feature an average hydrodynamic diameter of approximately 25 nm as determined by dynamic light scattering (DLS; Figure 1c). The α-APgp100-fmNP solution had a dark-brown appearance due to the presence of ICG and iron oxide (Figure 1d) as well as absorption maxima in the NIR region at 731 and 792 nm; this indicates a slight red-shift when compared with ICG alone (707 nm and 778 nm) in phosphate buffered saline (PBS) solution (Figure 1e) and suggests the suitability of these particles for NIR imaging. Additionally, α-APgp100-fmNPs exhibited MR contrast signals (Figure 1f). The increase in the α-APgp100-fmNPs concentration dramatically decreased the signal intensity and relaxation time (Figure 1g) due to T2 shortening, demonstrating a good correlation between concentration and MR signal.
Synthesis and characterization of α-AP-fmNPs. a) Schematic representation of α-AP-fmNPs. The synthesis of α-AP-fmNPs involved coating oleic acid-stabilized iron oxide nanocrystals with phospholipids according to a solvent exchange method. To load AP, a fusion peptide comprising AP and α-peptide sequences was designed (α-AP). α-AP-fmNPs were formed by self-association between ICG, α-AP, and the iron oxide@phospholipid complexes. b) TEM image of α-APgp100-fmNPs. Scale bar represents 100 nm. c) Size distribution of α-APgp100-fmNPs determined with DLS. d) Photograph of the α-APgp100-fmNPs solution in PBS. e) Absorption measurement of α-APgp100-fmNPs and ICG in PBS. f) MR images of PBS solutions of α-APgp100-fmNPs with various concentrations. g) Analysis of MR signals versus α-APgp100-fmNPs concentration.
Dual-Labeling and MPF Treatment of DCs
We next used flow cytometry and Prussian blue staining to investigate the ability of α-AP-fmNPs to label the murine dendritic cell line DC2.4 and bone marrow-derived dendritic cells (BMDCs). To determine a safe concentration range, the impact of α-AP-fmNPs loading on the cell viability was assessed with the Cell Counting Kit-8 (CCK-8) assay. Incubation of DC2.4 cells over 6 h with α-AP-fmNPs of increasing concentrations (0-48 μg/mL) provoked a continuous increase in the mean fluorescence intensity (MFI; Figure 2a), suggesting an increasing cellular uptake of α-AP-fmNPs. In BMDCs, the MFI reached a maximum at a concentration of 16 μg/mL within 6 h (Figure 2a). Cell toxicity was not observed at any tested concentration (0-48 μg/mL; Figure 2b) for both BMDCs and DC2.4 cells. Flow cytometry analysis showed that more than 80% of BMDCs and DC2.4 cells maintained a normal side-scatter profile at α-AP-fmNP concentrations below 16 μg/mL (Figure S1), indicating that the uptake of α-AP-fmNPs had only a little effect on the DC morphology at these concentrations. Thus, we chose for the following studies to incubate the cells with α-AP-fmNPs in the moderate concentration of 16 μg/mL. The obtained data showed that nearly 100% of the BMDCs and DC2.4 cells were fluorescently labeled after incubation with α-AP-fmNPs in this concentration (Figure S2). In DC2.4 cells, as few as 1.0 × 103 cells (in 50 μL PBS) could be detected using NIR imaging (Figure S3) and the uptake of the α-AP-fmNPs-encapsulated iron oxide particles was confirmed by Prussian blue staining (Figure 2c). To compare the AP delivery efficiency between free AP and α-AP-fmNPs, the nanoparticles were labeled with the fluorescent dye FAM that was covalently conjugated with α-AP at its C-terminal (FAM-α-AP), denoted as FAM-α-AP-fmNPs. Compared with free FAM-AP, FAM-α-AP-fmNPs delivered AP more efficiently, generating a 2-4-fold enhanced uptake in both DC2.4 and BMDCs after incubation with different concentrations of α-AP-fmNPs (Figure 2d and 2e). These data demonstrate that the incorporation of AP into α-AP-fmNPs promoted the intracellular AP uptake by DCs. To further explore the subcellular localization of α-AP-fmNPs-carried α-AP, DC2.4 cells were incubated with FAM-α-AP-fmNPs, and confocal microscopy was used to determine whether the FAM signal was co-localized with LysoTracker. This revealed significant FAM-α-AP signals in the cell membrane and cytoplasm organelles after a 6-h incubation (Figure 2f), which were only partially co-localized with the LysoTracker signal. This finding suggests that AP delivered to DCs by α-AP-fmNPs may be processed and cross-presented through the MHC-I pathway.
Analysis of α-AP-fmNPs uptake by BMDCs and DC2.4 cells and in vitro evaluation of MPF-promoted DC migration. a) Flow cytometric assessment of cellular α-AP-fmNPs uptake. The mean fluorescence intensity (MFI) was recorded and presented as the mean ± SEM (n = 3). b) Effect of α-AP-fmNPs labeling on the viability of BMDCs and DC2.4 cells. c) Prussian blue staining of α-AP-fmNP-internalized DC2.4 cells. d-e) Comparison of intracellular uptake of FAM-α-AP-fmNPs and FAM-AP in DC2.4 cells (d) and BMDCs (e). All experiments were carried out in triplicate. f) Determination of the subcellular localization of α-AP-fmNPs. Confocal imaging of DC2.4 cells incubated with FAM-α-AP-fmNPs for 6 h and stained with LysoTracker and Hoechst 33258. g) Representative images of migrated cells in the absence (-MPF) or presence (+MPF) of MPF. α-AP-fmNP-loaded DC2.4 cells were resuspended in a cell culture flask with a magnet attached to one side of the flask for 24 h, and the photograph of the migrated cells was acquired. h) Calculations of the migrated cells from bright images. Data were acquired from nine fields of view of three independent images. i) Analysis of the migration ability of α-AP-fmNP-loaded BMDCs. The number of the migrated BMDCs in the lower chamber was counted via flow cytometry. All experiments were carried out in triplicate. Data are presented as mean ± SEM (n = 3).
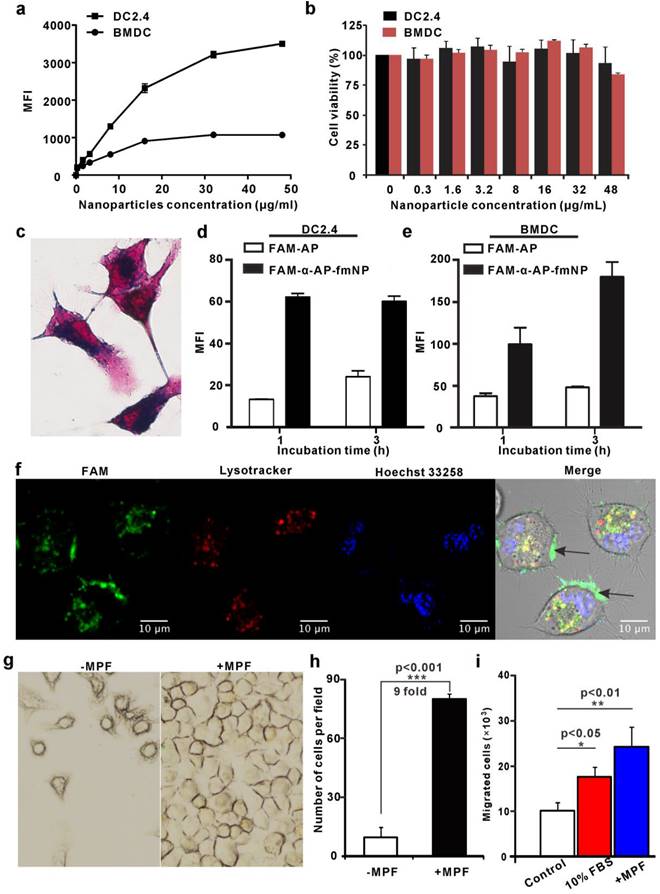
As the α-AP-fmNPs-loaded DCs contained large amounts of iron oxide particles, we tested whether the presence of MPF would guide and facilitate DCs migration. Prior to this experiment, we verified using a transwell migration assay that α-AP-fmNPs loading has no effect on the migration ability of DC2.4 cells (Figure S4). For evaluation of the MPF treatment, α-AP-fmNP-loaded DC2.4 cells were resuspended in a cell culture flask with complete medium, and a magnet was placed on one side of the flask. After 24 h, the cell numbers at each side of the flask were counted and compared to assess the effect of MPF on the DCs migration. This revealed that the number of cells on the side of the flask with the magnetic field was 9-fold higher than that on the non-MPF side (Figure 2g and 2h). Regarding the response of the BMDCs to MPF, a transwell migration assay revealed that the number of migrated cells in the bottom chambers of both the 10% FBS (fetal bovine serum, positive control) and MPF-treated groups (without FBS) were 1.8 and 2.5-fold, respectively, larger than that of the untreated groups after a 2-h incubation at 37 ºC (Figure 2i). These findings demonstrate that the application of MPF dramatically enhanced the migration of α-AP-fmNP-loaded DCs.
Bimodal Imaging of MPF-Improved DC Migration
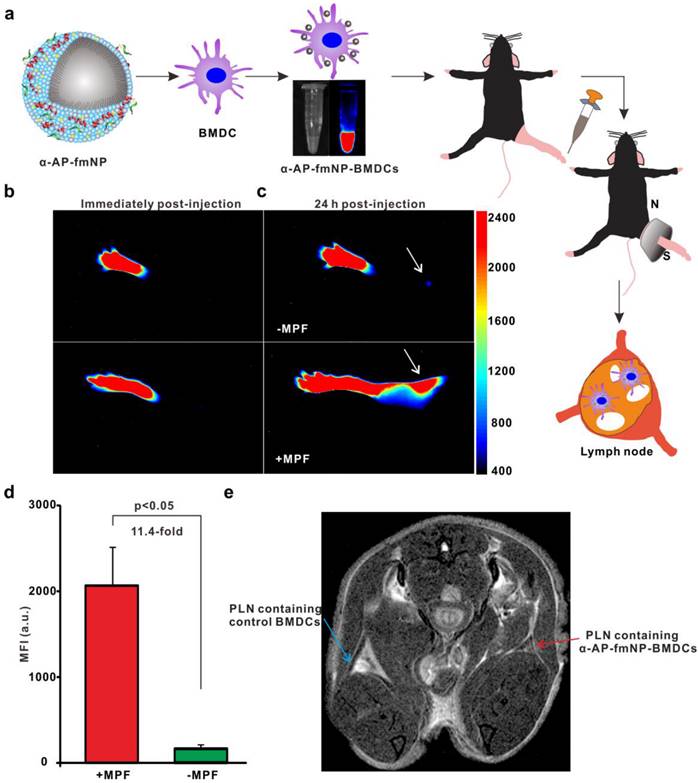
To ensure effective DC-based immunotherapy, sufficient amounts of DCs must migrate to LNs to activate T cells. For the evaluation of in vivo applications of MPF-promoted DCs migration, we chose the mouse footpad model - a well-characterized lymphatic drainage system. The mice were prestimulated at the injection site with TNF-α to generate an inflammatory microenvironment, followed by hind footpad inoculation of the α-AP-fmNP-loaded BMDCs (1.2 × 106 cells in 50 μL PBS) at 24 h after TNF-α prestimulation. In the MPF-treated group, a ring magnet was placed around the thigh of each mouse for 24 h (Figure 3a) while the control group was not treated with magnets. Whole-body fluorescence imaging and MRI were performed to track the migration of the injected BMDCs into the popliteal lymph nodes (PLNs) of the mice. This revealed that the α-AP-fmNP-loaded BMDCs displayed strong fluorescence signals in PBS solution (Figure 3a, middle panel), and the injection of these cells caused intense fluorescence signals in the footpads (Figure 3b). At 24 h post injection, the fluorescence signals detected in the PLN area were dramatically enhanced in the MPF-treated group compared with the control group (Figure 3c), leading to a 11.4-fold increase in the average fluorescence signal of the MPF-treated group (Figure 3d). Moreover, the MRI results clearly demonstrated a strong signal reduction in the PLN area (Figure 3e), thus indicating the accumulation of iron oxide particles.
Bimodal imaging of MPF-promoted DC migration to the LN. a) Schematic representation of the α-AP-fmNP/MPF-based strategy for improving BMDCs migration in mice. The α-AP-fmNP-loaded BMDCs were injected into the hind-leg footpad and subjected to MPF treatment for promotion of migration to PLN. b-c) Optical imaging of BMDCs migration at 0 h (b) and 24 h (c) post-injection in the presence (+MPF, bottom panel) or absence (-MPF, top panel) of magnetic field exposure. d) Average fluorescence measurements from c). Data are presented as the mean ± SEM (3-4 mice per group). The experiment was repeated at least three times with similar results. e) Representative MRI images of α-AP-fmNP-treated mice. MRI images reveal the accumulation of iron oxide particles in the PLN (blue arrow: BMDCs alone; red arrow: α-AP-fmNP-loaded BMDCs).
To ensure the suitability of this approach to dendritic cell lines, similar studies were also performed in DC2.4 cells. As revealed by in vivo NIR imaging, the fluorescence signal of MPF-treated group was enhanced by 16-fold as compared with the control group (Figure S5a and S5b). This difference was confirmed by fluorescence imaging of the dissected PLNs (Figure S5b, right panel) and further proved by in vivo MRI results (Figure S5c). To directly visualize the migrated DC2.4 cells in situ, the dissected LNs were observed via confocal imaging, and LN tissue sections were prepared and analyzed with Prussian blue staining. Confocal imaging revealed significant numbers of fluorescent cells in the PLNs of MPF-treated mice (Figure S6a). In contrast, only a limited number of fluorescent cells were observed in the control PLN (Figure S6b). Similarly, a histological analysis using Prussian blue staining suggested an increased presence of iron-loaded cells in the PLN of MPF-treated mice relative to the control mice (Figure S6c and S6d), which is consistent with the bimodal imaging results. Collectively, α-AP-fmNPs can deliver bimodal imaging agents for monitoring DCs trafficking toward the LNs and remarkably promote in vivo DCs migration through MPF.
Validation of MPF-Improved DC Migration
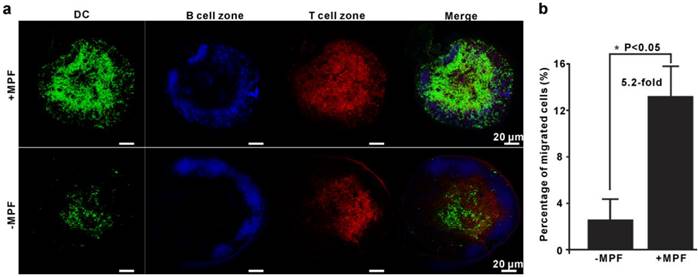
To further validate the above results and investigate the in situ biodistribution of α-AP-fmNP-loaded DCs, enhanced green fluorescent protein (EGFP)-expressing BMDCs (EGFP-BMDCs) generated from EGFP-transgenic mice were used. After loading with α-AP-fmNPs, EGFP-BMDCs were injected into the footpad of mice prestimulated with TNF-α for migration efficiency analysis. As shown in Figure 4a, the immunofluorescence of the dissected LN further confirmed the presence of a remarkably larger number of BMDCs in the MPF-treated group than in the control group (green signal). Flow cytometry analysis showed that the mean percentage of migrated BMDCs in the MPF-treated group accounted for 13.2% (+MPF) of the injected cells in contrast to 2.6% in control group (-MPF), corresponding to a more than 5-fold enhancement (Figure 4b). Moreover, these DCs were mostly found within the T-cell areas (red signal), but not in the B-cell follicles (blue signal; Figure 4a), which presents an essential step for DCs to initiate effective immune responses. Our findings demonstrate that the use of MPF can dramatically enhance the migration ability of DCs without altering their biodistribution pattern.
Validation of MPF-assisted in vivo DC migration. a) Representative immunofluorescence images of PLNs from either MPF-treated mice (+MPF, top panels) or control mice (-MPF, bottom panels). The green, blue, and red signals indicate EGFP-expressing BMDCs, B cell zone (Alexa fluor 647 anti-B220), and T cell zone (Alexa fluor 594 anti-CD3), respectively. b) Calculation of the average percentage of migrated cells to PLN by EGFP fluorescence. The EGFP-BMDCs were obtained from EGFP-transgenic mice. The PLNs were dissected and mechanically disrupted to obtain single cell suspensions, and the EGFP-positive cells in these solutions were analyzed by flow cytometry. Each group contained 2-3 mice and the indicated results represent the means ± SEM of three independent experiments.
Immune Function Assays
After confirming the important role of α-AP-fmNPs in DC labeling, AP delivery, and MPF-enhanced DC migration, we verified the antigen-presenting capacity of the α-AP-fmNP-loaded DCs by examining their ability to activate antigen-specific CD8+ T cells using nanoparticles carrying OVA257-264 peptide (α-APOVA-fmNPs). For this purpose, we carried out a T cell proliferation study and measured the killing activity of cytotoxic T lymphocytes (CTL). The CD8+ T cells isolated from the ILNs and spleens of the OT-I TCR transgenic mice (T cell receptors are specific for OVA257-264) were used for the proliferation test. Results show that α-APOVA-fmNP-loaded BMDCs, α-APOVA-loaded BMDCs and APOVA-loaded BMDCs induced a much higher CD8+ T cell proliferation rate than BMDCs alone (Figure 5a and 5b). Additionally, we measured the production of IFN-γ. Consistent with the T cell proliferation results, the α-APOVA-fmNP-loaded BMDCs group produced IFN-γ more effectively than the BMDCs groups, and this level was slightly higher than that of α-APOVA-loaded BMDCs and APOVA-loaded BMDCs (Figure 5c).
Immune function assessment. a-b) In vitro OVA-specific CD8+ T cell activation by α-AP-fmNP-loaded BMDCs. T cells collected from the OT-I mice were stained with CFSE and co-cultured with mature BMDCs loaded with PBS, APOVA, α-APOVA, or α-APOVA-fmNPs at a T cell: DC ratio of 50: 1. T cell proliferation was measured via CFSE fluorescence dilution using flow cytometry. c) Measurement of IFN-γ concentrations. d-e) Measurement of the killing activity of CTL. Data are presented as the mean ± SEM (n = 3).
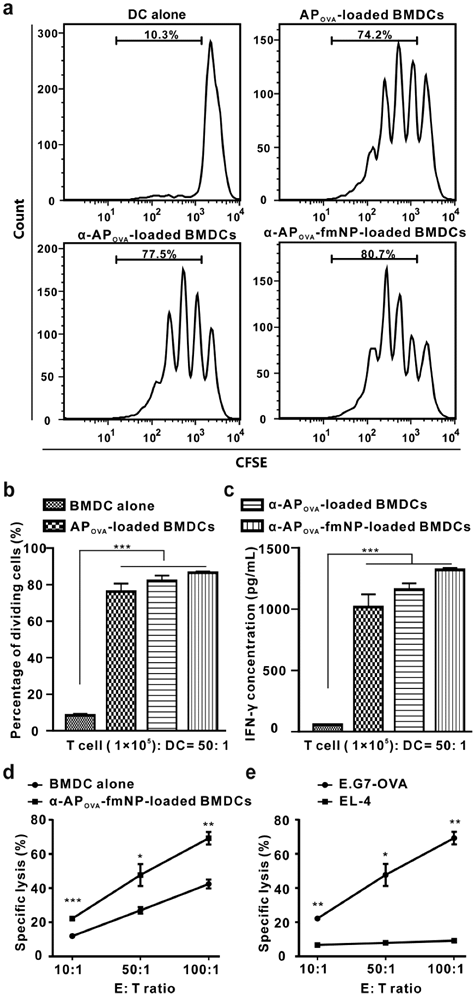
To examine the CTL activity elicited by α-APOVA-fmNP-loaded BMDCs, the mice were immunized with α-APOVA-fmNP-loaded BMDCs or BMDCs control twice at the tail base and then splenocytes were isolated. As depicted in Figure 5d, a CTL: E.G7-OVA (derivative OVA-expressing E.G7 lymphoma cell line) cells ratio-dependent response was clearly observed for α-APOVA-fmNP-loaded BMDCs. The killing activity of CTL against E.G7-OVA in α-APOVA-fmNP-BMDCs-immunized mice was significantly higher than that of BMDCs-immunized mice at all tested CTL: E.G7-OVA ratios. When this ratio increased to 50: 1 and 100: 1, the death rates of tumor cells reached 48% and 69%, respectively (Figure 5d). More importantly, CTL generated from α-APOVA-fmNP-BMDCs-immunized mice had only marginal lysis effect on EL-4 cells (a lymphoma cell line, OVA-; Figure 5e), suggesting the killing effect provided by α-APOVA-fmNP-loaded BMDCs was antigen-specific. Thus, α-AP-fmNPs endowed DCs with strong antigen-presenting capability.
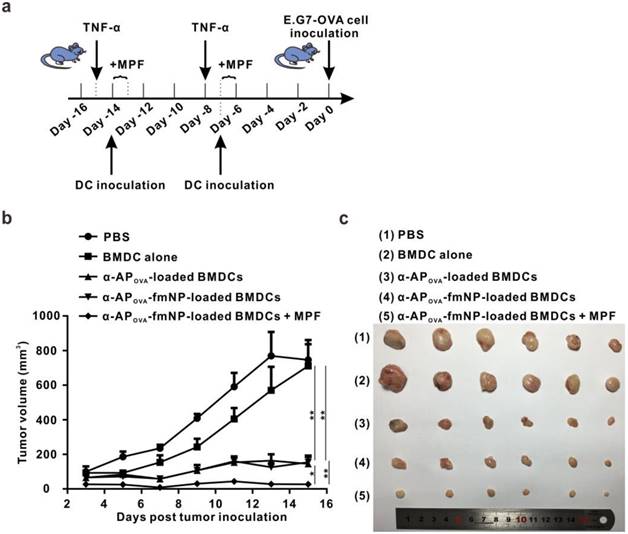
To investigate the effect of MPF treatment-enhanced DC migration on the outcomes of immunotherapy, we tested the antitumor ability of the established system for a possible use as prophylactic cancer vaccine in mice. For this purpose, α-APOVA-fmNPs and the tumor model using E.G7-OVA were adopted. The flow chart of the treatment schedule is depicted in Figure 6a. Compared with the control PBS and BMDCs group, the pretreatment with α-APOVA-fmNP-loaded BMDCs dramatically delayed the tumor growth within fifteen days (Figure 6b). At fifteen days post tumor inoculation, the tumor inhibitory efficiency for α-APOVA-fmNP-BMDC group, α-APOVA-BMDC group and α-APOVA-fmNP-BMDCs/MPF group were 79%, 80% and 96%, respectively (Figure 6c). The α-APOVA-fmNP-BMDCs/MPF-treated group showed a significantly lower average tumor size than the α-APOVA-fmNP-BMDCs group (without MPF treatment) (p = 0.0029; n = 6) and α-APOVA-BMDCs group (p = 0.0349; n = 6). Together, these results suggest that α-AP-fmNPs impart to DCs a strong ability to activate antigen-specific CD8+ T cells and permit the use of MPF as a powerful approach to improve the antitumor efficacy of DC-based immunotherapy stemming from the enhancement of the DC migration efficiency.
Evaluation of in vivo anti-tumor efficiency. a) Flow chart of the treatment schedule for α-AP-fmNPs/MPF-assisted, DC-based tumor immunotherapy. b) Tumor growth curve of immunized mice. c) Photograph of the dissected tumor tissues. Data are presented as the mean ± SEM (p1 = 0.0016; p2 = 0.0018; p3 = 0.0349; p4 = 0.0029; n = 6).
Discussion
For effective migration to LNs, the DCs must efficiently enter into the lymphatic vessels and successfully migrate along the lymphatic vessels [10]. Several approaches can effectively guide DC migration to the initial lymphatic vessels, such as inflammatory cytokine stimulation of the injection site and transfection of DCs with CCR7 [7, 10]. In this study, the mice were prestimulated with TNF-α to promote DC entry into the lymphatic vessels. Unlike previous studies that relied on chemokine gradients to guide DC migration, we showed that α-AP-fmNP-loaded DCs were sensitive to MPF, and their migration efficiency could be dramatically improved both in vitro and in vivo through MPF treatment. Bimodal imaging data revealed that the combination of MPF and inflammatory cytokine preinjection yielded a much more efficient migration of DCs to the PLN relative to that observed with inflammatory cytokine preinjection alone and suggested that MPF could drive DC migration along the lymphatic vessels (Figure S7). Flow cytometry analysis revealed DC migration efficiency up to 13.2% (calculated by the ratio of migrated cells to total injected cells), which corresponds to a more than 5-fold enhancement with respect to the inflammatory cytokine preinjection method. These findings indicate that the use of both chemokine and magnetic gradients could overcome two rate-limiting steps during DC migration in vivo. Moreover, these MPF-treated α-AP-fmNP-DCs were mainly distributed in the T cell zone of LNs, which is an essential prerequisite for DCs to exert their immune functions. Additionally, to ensure effective MPF treatment, sufficient exposure time, which often vary from several hours to days depending on the types of drugs (nanoparticles or cells) injected, must be guaranteed [27-29]. In our case, a short time exposure (such as 2 h) did not result in significant differences when compared with the control group and a relatively longer time (at least 12 h) of continuous external MPF exposure was found to be effective (data not shown), thus demonstrating that MPF-directed DC migration is not a rapid process.
Enhancement of therapeutic index is the ultimate goal that the magnetic targeting strategy should be achieved. A previous study by Schreibera et al demonstrated that magnetic targeting could be possibly utilized for enrichment of tail-base injected DCs in mouse ILN [30]. However, they have not found a consistent method to direct DCs migration in vivo. In our study, we documented the success of magnetic targeting to enrich α-AP-fmNP-loaded DCs in PLNs from several aspects including bimodal imaging, confocal imaging and flow cytometry analysis. More importantly, we reported for the first time that MPF treatment significantly augmented antitumor efficacy of the nanoparticle-loaded DCs.
Nanoparticles offer great potential for cancer immunotherapy because of their large surface areas, which allow the incorporation of both antigens and imaging agents, and their abilities to efficiently deliver these bioactive molecules to immune cells [31, 32]. A diverse of nanoparticles and bioconjugated molecules have been applied for either DC-based immunotherapy through manipulation of ex vivo-generated DCs or nanovaccine-based immunotherapy by direct targeting of immune cells in LN [25, 33-35]. However, the design of biocompatible nanoparticles is indispensable for such applications. The components of α-AP-fmNPs comprise DMPC, MHPC, iron oxide particles, ICG and peptide. Of these materials, similar iron oxide nanoparticle formulations have already been used in patients [36], ICG is the only FDA-approved fluorescence dye used in the clinic [37], DMPC is a primary component of several FDA-approved liposomal formulations [38], MHPC was proven to be highly biocompatible in a few mouse studies [39], and several type of AP are currently under clinical investigation [40]. Additionally, magnetic resonance targeting technical has been developed to promote the infiltration of iron oxide nanoparticle loaded-macrophages to tumor cells, providing a more clinically relevant source of magnetic gradient [41]. Regarding in vivo DC migration tracking, the bimodal imaging capability provided by α-AP-fmNPs allows both high sensitivity and high spatial resolution detection. Therefore, these features render α-AP-fmNPs great potential for clinical translation.
To elucidate the mechanisms mediating the uptake of α-AP-fmNP-carried FAM-α-AP by BMDCs, an inhibition study was performed with high concentrations of a previously developed HDL-mimicking peptide-phospholipid scaffold (HPPS) nanocarrier [42, 43]. In the presence of high concentration of HPPS, the uptake of α-AP-fmNP-delivered ICG and FAM-α-AP molecules was remarkably decreased by approximately 70% within a 1-h incubation period (Figure S8), suggesting that the scavenger receptor class B type I (SR-B1) might play a mediatory role in the uptake of α-AP-fmNPs.
As a proof-of-concept, gp100 and OVA peptide were incorporated into these fluorescent magnetic nanoparticles but could be easily replaced with other selected peptide antigens. In a model antigen of OVA, α-APOVA-fmNP-loaded DCs displayed strong and specific antigen-presenting capability but it induced only a slightly higher CD8+ T cell proliferation rate than that induced by OVA peptide-loaded DCs. Interesting, in a clinically relevant model of melanoma AP (gp10025-33), α-APgp100-fmNP-loaded DCs induced a much higher CD8+ T cell proliferation rate than APgp100-loaded DCs at various CD8+ T cell: DC ratios (Figure S9). These might be attributed to the following two reasons: the role of the α-peptide (advantageous factor), which not only promotes the uptake of fusion peptide (α-AP) but also endows the AP with an intracellular cytoplasm distribution pattern that is an essential prerequisite for efficient MHC class I presentation; the AP described here was short-sequence peptide antigen, which may have the potential to directly bind to MHC-I molecules on the cell surface to be more efficiently presented without undergoing intracellular enzymatic degradation process required for antigen presentation. Finally, the majority of functional studies reported here are limited in an ideal model antigen of OVA and further studies would take advantage of this MPF treatment-enhanced DC migration to investigate the possibilities of applying this system for other clinically relevant tumor models with varying administration routes (such as intravenous and intradermal injection) in DC-based immunotherapy.
Conclusion
As DC-based immunotherapy requires the efficient migration and precise tracking of DCs to the LNs, we developed a fluorescent magnetic nanoparticle that was loaded with antigen peptide, iron oxide nanoparticles and ICG. The resulting nanoparticle endowed DCs with the capabilities of optical and magnetic resonance tracking. More importantly, the deposition of iron oxide particles within the DCs provided them with MPF-responsive properties. In the context of DC-based immunotherapy, α-AP-fmNP-loaded DCs revealed to possess antigen presentation capability and their in vivo migration efficiency was dramatically enhanced under MPF, resulting in a significantly improved antitumor efficacy, thus possibly bypassing the inefficient, distribution-mediated antitumor activity of DCs in vivo.
Supplementary Material
Supplementary figures.
Acknowledgements
We thank Dr. Gang Zheng (University of Toronto, Toronto, ON, Canada) for paper discussion and Dr. Yu Zhang lab (Southeast University) for providing oleic-acid stabilized iron oxide nanocrystals. We thank the Optical Bioimaging Core Facility of WNLO-HUST and the Center for Nanoscale Characterization & Devices (CNCD, Tecnai G20 U-Twin) of WNLO-HUST for the support in data acquisition, and the Analytical and Testing Center of HUST for spectral measurements. We also thank the Wuhan Center for Magnetic Resonance, State Key Laboratory of Magnetic Resonance and Atomic and Molecular Physics, Wuhan Institute of Physics and Mathematics, Chinese Academy of Sciences for the assistance of MRI studies. This study was supported by the Major Research plan of the National Natural Science Foundation of China (Grant No. 91442201), the China Postdoctoral Science Foundation funded project (Grant No. 2013T60721), the Open Research Fund of State Key Laboratory of Bioelectronics of Southeast University, the Fundamental Research Funds for the Central Universities (HUST: 2015ZDTD014), and the Director Fund of WNLO.
Conflict of Interest
The authors declare no competing financial interests.
References
1. Sabado RL, Bhardwaj N. Cancer immunotherapy: dendritic-cell vaccines on the move. Nature. 2015;519:300-1
2. Tacken PJ, de Vries IJ, Torensma R, Figdor CG. Dendritic-cell immunotherapy: from ex vivo loading to in vivo targeting. Nat Rev Immunol. 2007;7:790-802
3. van den Ancker W, van Luijn MM, Westers TM, Bontkes HJ, Ruben JM, de Gruijl TD. et al. Recent advances in antigen-loaded dendritic cell-based strategies for treatment of minimal residual disease in acute myeloid leukemia. Immunotherapy. 2010;2:69-83
4. Zhou FF, Nordquist RE, Chen WR. Photonics immunotherapy — A novel strategy for cancer treatment. J Innov Opt Health Sci. 2016;9:1630001
5. Kastenmuller W, Kastenmuller K, Kurts C, Seder RA. Dendritic cell-targeted vaccines-hope or hype? Nat Rev Immunol. 2014;14:705-11
6. Ridolfi R, Riccobon A, Galassi R, Giorgetti G, Petrini M, Fiammenghi L. et al. Evaluation of in vivo labelled dendritic cell migration in cancer patients. J Transl Med. 2004;2:27
7. Aarntzen EH, Srinivas M, Schreibelt G, Heerschap A, Punt CJ, Figdor CG. et al. Reducing cell number improves the homing of dendritic cells to lymph nodes upon intradermal vaccination. Oncoimmunology. 2013;2:e24661
8. MartIn-Fontecha A, Sebastiani S, Hopken UE, Uguccioni M, Lipp M, Lanzavecchia A. et al. Regulation of dendritic cell migration to the draining lymph node: impact on T lymphocyte traffic and priming. J Exp Med. 2003;198:615-21
9. Verdijk P, Aarntzen EH, Punt CJ, de Vries IJ, Figdor CG. Maximizing dendritic cell migration in cancer immunotherapy. Expert Opin Biol Ther. 2008;8:865-74
10. Okada N, Mori N, Koretomo R, Okada Y, Nakayama T, Yoshie O. et al. Augmentation of the migratory ability of DC-based vaccine into regional lymph nodes by efficient CCR7 gene transduction. Gene Ther. 2005;12:129-39
11. Xu H, Cao X. Dendritic cell vaccines in cancer immunotherapy: from biology to translational medicine. Front Med. 2011;5:323-32
12. Randolph GJ, Angeli V, Swartz MA. Dendritic-cell trafficking to lymph nodes through lymphatic vessels. Nat Rev Immunol. 2005;5:617-28
13. Baumjohann D, Lutz MB. Non-invasive imaging of dendritic cell migration in vivo. Immunobiology. 2006;211:587-97
14. Chen YC, Wen S, Shang SA, Cui Y, Luo B, Teng GJ. Magnetic resonance and near-infrared imaging using a novel dual-modality nano-probe for dendritic cell tracking in vivo. Cytotherapy. 2014;16:699-710
15. Cruz LJ, Tacken PJ, Bonetto F, Buschow SI, Croes HJ, Wijers M. et al. Multimodal imaging of nanovaccine carriers targeted to human dendritic cells. Mol Pharm. 2011;8:520-31
16. Lim YT, Noh YW, Han JH, Cai QY, Yoon KH, Chung BH. Biocompatible polymer-nanoparticle-based bimodal imaging contrast agents for the labeling and tracking of dendritic cells. Small. 2008;4:1640-5
17. Noh YW, Jang YS, Ahn KJ, Lim YT, Chung BH. Simultaneous in vivo tracking of dendritic cells and priming of an antigen-specific immune response. Biomaterials. 2011;32:6254-63
18. Chou LY, Ming K, Chan WC. Strategies for the intracellular delivery of nanoparticles. Chem Soc Rev. 2011;40:233-45
19. Cormode DP, Skajaa T, Fayad ZA, Mulder WJ. Nanotechnology in medical imaging: probe design and applications. Arterioscler Thromb Vasc Biol. 2009;29:992-1000
20. Janib SM, Moses AS, MacKay JA. Imaging and drug delivery using theranostic nanoparticles. Adv Drug Deliv Rev. 2010;62:1052-63
21. Mackay PS, Kremers GJ, Kobukai S, Cobb JG, Kuley A, Rosenthal SJ. et al. Multimodal imaging of dendritic cells using a novel hybrid magneto-optical nanoprobe. Nanomedicine. 2011;7:489-96
22. Cho NH, Cheong TC, Min JH, Wu JH, Lee SJ, Kim D. et al. A multifunctional core-shell nanoparticle for dendritic cell-based cancer immunotherapy. Nat Nanotechnol. 2011;6:675-82
23. Xie J, Zhang Y, Yan C, Song L, Wen S, Zang F. et al. High-performance PEGylated Mn-Zn ferrite nanocrystals as a passive-targeted agent for magnetically induced cancer theranostics. Biomaterials. 2014;35:9126-36
24. Sarradin PM, Le Bris N, Le Gall C, Rodier P. Fe analysis by the ferrozine method: Adaptation to FIA towards in situ analysis in hydrothermal environment. Talanta. 2005;66:1131-8
25. Qian Y, Jin H, Qiao S, Dai Y, Huang C, Lu L. et al. Targeting dendritic cells in lymph node with an antigen peptide-based nanovaccine for cancer immunotherapy. Biomaterials. 2016;98:171-83
26. Akasaka K, Akasaka N, Di Luozzo G, Sasajima T, Sumpio BE. Homocysteine promotes p38-dependent chemotaxis in bovine aortic smooth muscle cells. J Vasc Surg. 2005;41:517-22
27. Bai J, Wang JT, Rubio N, Protti A, Heidari H, Elgogary R. et al. Triple-Modal Imaging of Magnetically-Targeted Nanocapsules in Solid Tumours In Vivo. Theranostics. 2016;6:342-56
28. Foy SP, Manthe RL, Foy ST, Dimitrijevic S, Krishnamurthy N, Labhasetwar V. Optical imaging and magnetic field targeting of magnetic nanoparticles in tumors. ACS Nano. 2010;4:5217-24
29. Luciani A, Wilhelm C, Bruneval P, Cunin P, Autret G, Rahmouni A. et al. Magnetic targeting of iron-oxide-labeled fluorescent hepatoma cells to the liver. Eur Radiol. 2009;19:1087-96
30. Schreiber HA, Prechl J, Jiang H, Zozulya A, Fabry Z, Denes F. et al. Using carbon magnetic nanoparticles to target, track, and manipulate dendritic cells. J Immunol Methods. 2010;356:47-59
31. Shao S, Geng J, Ah Yi H, Gogia S, Neelamegham S, Jacobs A. et al. Functionalization of cobalt porphyrin-phospholipid bilayers with his-tagged ligands and antigens. Nat Chem. 2015;7:438-46
32. Zhang P, Chen Y, Zeng Y, Shen C, Li R, Guo Z. et al. Virus-mimetic nanovesicles as a versatile antigen-delivery system. Proc Natl Acad Sci U S A. 2015;112:E6129-38
33. Jewell CM, Lopez SC, Irvine DJ. In situ engineering of the lymph node microenvironment via intranodal injection of adjuvant-releasing polymer particles. Proc Natl Acad Sci U S A. 2011;108:15745-50
34. Liu H, Moynihan KD, Zheng Y, Szeto GL, Li AV, Huang B. et al. Structure-based programming of lymph-node targeting in molecular vaccines. Nature. 2014;507:519-22
35. Reddy ST, van der Vlies AJ, Simeoni E, Angeli V, Randolph GJ, O'Neil CP. et al. Exploiting lymphatic transport and complement activation in nanoparticle vaccines. Nat Biotechnol. 2007;25:1159-64
36. Xie J, Jon S. Magnetic nanoparticle-based theranostics. Theranostics. 2012;2:122-4
37. Desmettre T, Devoisselle JM, Mordon S. Fluorescence properties and metabolic features of indocyanine green (ICG) as related to angiography. Surv Ophthalmol. 2000;45:15-27
38. Pinto AC, Moreira JN, Simoes S. Liposomal imatinib-mitoxantrone combination: formulation development and therapeutic evaluation in an animal model of prostate cancer. Prostate. 2011;71:81-90
39. Skajaa T, Cormode DP, Jarzyna PA, Delshad A, Blachford C, Barazza A. et al. The biological properties of iron oxide core high-density lipoprotein in experimental atherosclerosis. Biomaterials. 2011;32:206-13
40. Suzuki H, Fukuhara M, Yamaura T, Mutoh S, Okabe N, Yaginuma H. et al. Multiple therapeutic peptide vaccines consisting of combined novel cancer testis antigens and anti-angiogenic peptides for patients with non-small cell lung cancer. J Transl Med. 2013;11:97
41. Muthana M, Kennerley AJ, Hughes R, Fagnano E, Richardson J, Paul M. et al. Directing cell therapy to anatomic target sites in vivo with magnetic resonance targeting. Nat Commun. 2015;6:8009
42. Zhang Z, Chen J, Ding L, Jin H, Lovell JF, Corbin IR. et al. HDL-mimicking peptide-lipid nanoparticles with improved tumor targeting. Small. 2010;6:430-7
43. Zheng Y, Liu Y, Jin H, Pan S, Qian Y, Huang C. et al. Scavenger receptor B1 is a potential biomarker of human nasopharyngeal carcinoma and its growth is inhibited by HDL-mimetic nanoparticles. Theranostics. 2013;3:477-86
Author contact
![]() Corresponding author: Zhihong Zhang (czyzzhhust.edu.cn).
Corresponding author: Zhihong Zhang (czyzzhhust.edu.cn).