Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2016; 6(11):1792-1809. doi:10.7150/thno.14584 This issue Cite
Research Paper
Peripheral Blood Cell Gene Expression Diagnostic for Identifying Symptomatic Transthyretin Amyloidosis Patients: Male and Female Specific Signatures
Sunil M. Kurian1, Marta Novais2, Thomas Whisenant1, Terri Gelbart1, Joel N. Buxbaum1, Jeffery W. Kelly1,3 ![]() , Teresa Coelho2, Daniel R. Salomon1
, Teresa Coelho2, Daniel R. Salomon1 ![]()
1. Department of Molecular and Experimental Medicine, The Scripps Research Institute, 10550 N. Torrey Pines Road, La Jolla, CA 92037.
2. Hospital de Santo António, Porto, Portugal.
3. Department of Chemistry and The Skaggs Institute for Chemical Biology, The Scripps Research Institute, La Jolla, CA 92037.
Received 2015-12-2; Accepted 2016-6-7; Published 2016-7-18
Abstract

Background: Early diagnosis of familial transthyretin (TTR) amyloid diseases remains challenging because of variable disease penetrance. Currently, patients must have an amyloid positive tissue biopsy to be eligible for disease-modifying therapies. Endomyocardial biopsies are typically amyloid positive when cardiomyopathy is suspected, but this disease manifestation is generally diagnosed late. Early diagnosis is often difficult because patients exhibit apparent symptoms of polyneuropathy, but have a negative amyloid biopsy. Thus, there is a pressing need for an additional early diagnostic strategy for TTR-aggregation-associated polyneuropathy and cardiomyopathy.
Methods and Findings: Global peripheral blood cell mRNA expression profiles from 263 tafamidis-treated and untreated V30M Familiar Amyloid Neuropathy patients, asymptomatic V30M carriers, and healthy, age- and sex-matched controls without TTR mutations were used to differentiate symptomatic from asymptomatic patients. We demonstrate that blood cell gene expression patterns reveal sex-independent, as well as male- and female-specific inflammatory signatures in symptomatic FAP patients, but not in asymptomatic carriers. These signatures differentiated symptomatic patients from asymptomatic V30M carriers with >80% accuracy. There was a global downregulation of the eIF2 pathway and its associated genes in all symptomatic FAP patients. We also demonstrated that the molecular scores based on these signatures significantly trended toward normalized values in an independent cohort of 46 FAP patients after only 3 months of tafamidis treatment.
Conclusions: This study identifies novel molecular signatures that differentiate symptomatic FAP patients from asymptomatic V30M carriers as well as affected males and females. We envision using this approach, initially in parallel with amyloid biopsies, to identify individuals who are asymptomatic gene carriers that may convert to FAP patients. Upon further validation, peripheral blood cell mRNA expression profiling could become an independent early diagnostic. This quantitative gene expression signature for symptomatic FAP could also become a biomarker to demonstrate significant disease-modifying effects of drugs and drug candidates. For example, when new disease modifiers are being evaluated in a FAP clinical trial, such surrogate biomarkers have the potential to provide an objective, quantitative and mechanistic molecular diagnostic of disease response to therapy.
Keywords: familial transthyretin
Introduction
One of more than 100 mutations in the plasma protein transthyretin (TTR) destabilizes it, enabling aggregation, which appears to cause the amyloid disease familial amyloid polyneuropathy (FAP) [1, 2]. FAP is an autosomal dominant long fiber sensory-motor and autonomic neuropathy, often accompanied by cardiomyopathy [3]. The most common FAP-associated mutation is V30M. FAP is usually fatal 10 to 15 years after onset of symptoms if untreated [4-7].
The tetrameric TTR protein is secreted largely by the liver [8]. In heterozygotic FAP patients, the tetramers are composed of mutant and/or wild-type (WT) TTR subunits in the statistically predicted stoichiometries. Rate-limiting tetramer dissociation, followed by partial TTR monomer denaturation, facilitates concentration-dependent aggregation of predominantly mutant TTR [9, 10]. There is strong genetic and pharmacologic evidence that extracellular TTR aggregation causes degeneration in tissues not synthesizing TTR, including the peripheral and autonomic nervous systems, through cell non-autonomous mechanisms that are still not well understood [9, 11-15].
Liver transplantation from a donor with two normal TTR genes replaces the mutant allele in FAP heterozygotes [8, 14, 16, 17]. This surgical form of gene therapy has been performed on > 2000 patients, slowing FAP progression and extending lifespan [16, 18, 19]. Clinical trial results demonstrate that tafamidis and diflunisal are also effective for the amelioration of FAP [20-22]. These TTR kinetic stabilizers bind to the unoccupied thyroxine binding sites within TTR tetramers, dramatically slowing tetramer dissociation and inhibiting TTR aggregation [23, 24].
The amyloid hypothesis posits that TTR aggregation causes amyloidosis. Though the genetic and pharmacologic evidence is convincing [20-22, 25, 26], we still do not understand the variable clinical penetrance of these diseases. We envision that there are additional triggers or modifiers of the onset and rate of progression of FAP. For clinicians, it remains difficult to identify those TTR mutation carriers who are at highest risk for developing symptomatic FAP. A facile method for monitoring TTR mutation carriers who convert from being asymptomatic to symptomatic is important because there is evidence that earlier treatment results in better outcomes [8, 17-22].
A definitive diagnosis of symptomatic FAP requires the presence of TTR amyloid via biopsy, which can be challenging to detect as amyloid deposition is not uniform [27] [29]. There are currently no accepted early diagnostic approaches that can be used in parallel with amyloid biopsies to identify carriers who have transitioned to FAP patients. A minimally invasive (i.e., blood) molecular diagnostic for the sensitive and early detection of symptomatic FAP in individuals at risk is the first envisioned application of our peripheral blood cell transcriptional signature approach. A potential second use would be in symptomatic patients, wherein a quantitative molecular signature could reflect both the burden of disease (upon correlation with clinical symptoms) and the response of each FAP patient to therapy through normalization of the signature. The latter could be especially useful for determining the minimal effective dosage of currently available drugs, and for use as a surrogate biomarker in clinical trials evaluating novel agents [20-22].
Herein, we tested three hypotheses: 1) that peripheral blood cell gene expression profiling of symptomatic FAP patients vs. asymptomatic carriers would reveal genomic signatures reflective of clinical status, 2) that tafamidis treatment would normalize these signatures, and 3) that genes exhibiting transcriptional changes in blood can be mapped to functional pathways that may provide further mechanistic insights into FAP etiology. This study was made possible by the availability of blood samples from clinically well characterized FAP patients being cared for by experienced clinicians at the Hospital de Santo António, Porto, Portugal. The FAP patients all carry the same autosomal dominant V30M mutation, whose prevalence in that population is estimated to be 1/1000 [30]. The response to therapy component of this study was enabled because tafamidis was approved for use by the European Medicines Agency in November 2011 and in Portugal shortly thereafter [21].
Given that male V30M FAP patients have earlier onset and more aggressive disease progression than female subjects with the same mutation, we also asked if there were any gender-specific features of the disease that might be revealed by these transcriptional signatures. It could be that males would benefit from a higher dose of tafamidis relative to females, and individuals with highly destabilizing mutations would also benefit from a higher dose of tafamidis.
Materials and Methods
Study Subjects: Tafamidis-treated and untreated V30M FAP patients, asymptomatic V30M carriers, and healthy, age- and sex-matched controls without TTR mutations provided informed consent at the Hospital de Santo António, Porto, Portugal. Whole blood (5 ml) was collected in PAXgene tubes (Qiagen) from a total of 309 human subjects. The study comprised 7 subject groups: Normal Male Controls (n=37); Normal Female Controls (n=43); Symptomatic Males with V30M FAP (n=46); Symptomatic Females with V30M FAP (n=50); Asymptomatic Males carrying the V30M mutation linked to FAP (n=41); and Asymptomatic Females carrying the V30M mutation linked to FAP (n=46). These 6 subject groups, representing 263 total expression arrays, were randomized into two independent cohorts with roughly similar proportions of each subject group: Discovery (n=150) and Validation (n=113). In addition, tafamadis-treated patients (n=46; males = 23, females = 23) were analyzed as a single validation cohort for studying response to therapy.
RNA Extraction and Microarray Processing: RNA was extracted from whole blood with the PAXgene RNA Whole Blood Kit. Each RNA sample (100 ng) was amplified and labeled with the Ambion WT Express Kit and hybridized to Affymetrix HuGene 1.1 ST Arrays using the Affymetrix GeneTitan Multi-Channel (MC) platform. Normalized signals were generated using Robust Multichip Average (RMA) in Partek Genomics Suite software version 6.6. Probesets on the DNA microarrays with low expressed signals were determined using a kernel density plot and signals <Log2 of 6 were excluded from analysis.
Microarray Analysis: We performed statistical power and sample size calculations for the microarray profiling of whole blood using the sample size estimation tool implemented in Partek genomics Suite [31, 32]. To detect a low fold-change of 1.3-fold at a p-value <0.005 that we would accept for a biological signal or biomarker gene at a power of 85%, a sample size of 45 samples per group for whole blood is necessary. Therefore, we were more than adequately powered even for the Discovery cohort used in this study. None of the samples were excluded based on the standard Affymetrix Quality Control metrics and by Principal Components Analysis plots derived in Partek Genomics Suite. Class comparisons were performed using a 1-way ANOVA model using the Method of Moments [33]. The one-way ANOVA that we employed to detect differentially expressed genes is also robust and tolerates reasonable levels of experimental data variance. Moreover, the danger of violating the normal distributions corresponds with the possibility to detect false positives, which we have controlled for with a stringent False Discovery Rate (FDR) of <5%. Class predictions were performed with the Support Vector Machines (SVM) algorithm. To correct for the possibility of over-fitting of the predictors, we used the bootstrapping method of Harrell et al. where the original data set is sampled 500 times with replacement and the Area Under the Curves (AUCs) calculated for each resampling or bootstrap iteration [34, 35]. Receiver Operating Characteristic (ROC) curves were generated using pROC in R [36]. ROC curves were plotted using the posterior probabilities of a sample being called symptomatic or not based on SVM or Logistic Regression models. Diagnostic metrics were calculated based on the number of true and false positives using the formulae given below: Sensitivity = (TP/(TP+FN)), Specificity = (TN/(FP+TN)), Positive Predictive Value = (TP/(TP+FP)) and Negative Predictive Value = (TN/(FN+TN)). The STARD flowchart describing study design and diagnostic marker pipeline is shown in Additional file 1: Supplemental Methods. We developed molecular scores based on gene expression signals for the highest value predictive signatures. Details about the development of the molecular scores are given in Additional file 1: Supplemental Methods.
Scores were tested for differences using the two-sample t-test for different subjects in each group. The two groups are assumed to be normally distributed and have equal variance (for the equal variance t-test). Significant differential gene expression for each of the different group comparisons were mapped to functionally significant biological pathways using Ingenuity Pathways Analysis (Qiagen). Clinical study parameters were tested using paired t-test for continuous variables and McNemar's chi-square test for categorical variables, including prevalence. CEL files and normalized signal intensities are posted at the NIH Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo; accession number GSE 67784).
Plasma Proteomic Analysis: A multi-analyte panel of 45 Luminex bead assays (Human InflammationMAP v1.0; Myriad/Rules Based Medicine, Austin, TX) was used to profile a selected subset of 40 plasma samples to validate the pro-inflammatory state indicated by analysis of the microarray data. These comprised 10 samples each from the four studied groups of patients with FAP mutations: symptomatic vs. asymptomatic, males vs. females.
Results
Patient Characteristics
All of our plasma samples, including those from genetically similar and locally-collected healthy controls, asymptomatic V30M carriers, symptomatic V30M FAP patients, and tafamidis-treated patients were collected from one location, Hospital de Santo António in Porto, Portugal. Patients were classified as symptomatic if they had symptoms and an irregular physical exam as well as abnormal laboratory exams that confirmed cardiac and/or neuropathic involvement. The severity of peripheral nerve and cardiac involvement is defined in the legend to Table 1. The demographics for the discovery and validation cohorts are provided (Table 1, controls and patients; Table 2, asymptomatic vs. symptomatic patients). Controls for the Discovery and Validation sets (n=46 and 34, respectively) were local Portuguese, healthy, age- and sex-matched subjects. Symptomatic patients were significantly older than asymptomatic patients, had higher white blood cell counts and had significantly increased levels of proteinuria despite normal creatinine levels. Symptomatic alkaline phosphatase levels were higher but not those of aspartate aminotransferase (AST) or alanine aminotransferase (ALT). Although we acknowledge that measures of high-sensitivity C-reactive protein (hsCRP) are often employed in inflammation studies, we did not assess hsCRP levels since our inflammation findings emerged during analysis and long after the clinical samples had been collected in Portugal. Symptomatic patients had significant clinical peripheral nerve and cardiac involvement. The degree of neuropathic involvement was assessed using the Neuropathy Impairment Score-Lower Limbs (NIS-LL) and the Norfolk Quality of Life Diabetic Neuropathy Total Score (TQOL). The presence of cardiomyopathy was assessed by echocardiography, electrocardiography and measurement of B-type Natriuretic Peptide (BNP) to gauge congestive heart failure. The treated patients profiled in this study had received 3 months of commercially available and self-administered tafamidis therapy (20 mg once daily) at the time of blood sampling [22]. Among the symptomatic FAP patients 29/96 (30%) were on neuropathy medication (predominantly pregabalin or gabapentin) and none in the asymptomatic group were on these medications. However, there was no significant differential expression of genes among our classifier molecular signatures based on the medications as determined by ANOVA and supervised hierarchical clustering (Supplemental Methods).
Patient and control Discovery and Validation set Demographics*
| Characteristic | Discovery Set (n = 150) | Validation Set (n = 113) | ||
|---|---|---|---|---|
| Control (n=46) | Patient (n=104) | Control (n=34) | Patient (n=79) | |
| Sex—Female (n (%)) | 25 (54.4) | 55 (52.9) | 18 (52.9) | 41 (51.9) |
| Age (years) (mean(SD)) | 39.4 (11.4) | 40.4 (14.3) | 39.1 (15.0) | 38.3 (12.6) |
| BMI (kg/m2) (mean(SD)) | 24.5 (5.0) | 24.4 (4.8) | ||
| (n=95) | (n=75) | |||
| Parental Inheritance (n (%)) | ||||
| Mother | 47 (45.2) | 41 (51.9) | ||
| Father | 48 (46.2) | 33 (41.8) | ||
| Both | 1 (1.0) | 5 (6.3) | ||
| Unknown | 8 (7.7) | |||
| WBC (mean (SD)) | 7.1 (1.9) | 7.2 (1.9) | ||
| (n=97) | (n=76) | |||
| Platelets (mean (SD)) | 236.7 (55.5) | 248.5 (54.5) | ||
| (n=97) | (n=76) | |||
| Alkaline phosphatase (mean (SD)) | 60.0 (16.1) | 64.7 (25.6) | ||
| (n=88) | (n=66) | |||
| AST (mean (SD)) | 24.5 (12.9) | 21.8 (6.8) | ||
| (n=89) | (n=66) | |||
| ALT (mean (SD)) | 25.9 (19.8) | 22.6 (10.9) | ||
| (n=89) | (n=66) | |||
| Creatinine (mean (SD)) | 0.78 (0.25) | 0.77 (0.33) | ||
| (n=102) | (n=79) | |||
| Significant Proteinuria (n (%)) | 15/103† (14.6) | 7/72† (8.9) | ||
| Peripheral Nerve Involvement (n (%)) | 51/51† (100) | 41/42† (97.6) | ||
| Characteristic | Discovery Set | Validation Set | ||
| Control (n=46) | Patient (n=104) | Control (n=34) | Patient (n=79) | |
| Peripheral Nerve Severity (n (%)) | n=51 | n=41 | ||
| 1 | 8 (15.7) | 5 (12.2) | ||
| 2 | 20 (39.2) | 20 (48.8) | ||
| 3 | 10 (19.6) | 7 (17.1) | ||
| 4 | 7 (13.7) | 3 (7.3) | ||
| 5 | 6 (11.8) | 6 (14.6) | ||
| Cardiac Involvement (n (%)) | 28/51† (54.9) | 31/43† (72.1) | ||
| Cardiac Severity (n (%)) | n=28 | n=31 | ||
| Mild | 10 (35.7) | 13 (41.9) | ||
| Moderate | 9 (32.1) | 9 (29.0) | ||
| Severe | 9 (32.1) | 9 (29.0) | ||
| Diabetes (n (%)) | 4/53† (7.6) | |||
†denominator is number with a yes or no answer to the question
*t-test for continuous variables, chi-square for categorical variables
Patients are classified as symptomatic if they have constant and progressive symptoms as well as abnormal signs in the physical exam, particularly in the neurologic exam and abnormal laboratory exams that confirm cardiac and/or neuropathic involvement. The sensitivy of the biopsy is ≈ 80-90% A small subset of patients with negative amyloid biopsies were classified as asymptomatic with negative amyloid biopsies.
We classified the severity of neuropathic involvement according to the commonly used 5-stage polyneuropathy disability score: stage 1 = sensory neuropathy without motor dysfunction; stage 2 = sensory and motor neuropathy, walking without assistance; 3 = sensory and motor neuropathy, patient needs unilateral support to walk; stage 4 = sensory and motor neuropathy, patient needs bilateral support to walk; stage 5 = patient is wheelchair bound or bedridden.
There are two different types of cardiac involvement: patients may have myocardial infiltration with cardiomyopathy, alternatively they may present subendocardial infiltration with conduction disturbances, invariably leading to the utilization of a pacemaker. The first presentation is not common among Portuguese patients, the last is invariable present after a disease course of several years. We classified both types of cardiac involvement together and divided patients into three categories:
Mild patients exhibit objective signs of cardiac involvement with abnormal EKG and/or abnormal proBNP and /or abnormal echocardiogram, but no symptoms.
Moderate patients exhibit symptoms that interfere with physical exertion.
Severe patients exhibit life-threatening conditions, such as conduction disturbances requiring a pacemaker or cardiomyopathy with symptoms of cardiac failure impairing regular daily activities.
Demographic characteristics for all study patients and carriers.
| Discovery Set of FAP Patients and asymptomatic carriers (n=104) | Validation Set of FAP Patients and asymptomatic carriers (n=79) | |||||
|---|---|---|---|---|---|---|
| Characteristic | Asymptomatic (n=51) | Symptomatic (n=53) | p | Asymptomatic (n=35) | Symptomatic (n=44) | p |
| Sex—Female (n (%)) | 27 (52.9) | 28 (52.8) | 18 (51.4) | 23 (52.3) | ||
| Age (years) (mean (SD)) | 35.4 (11.9) | 45.2 (14.8) | .0003 | 31.1 (9.4) | 44.0 (12.0) | <.0001 |
| BMI (kg/m2) (mean (SD)) | 25.5 (4.9) | 23.5 (5.0) | .05 | 24.4 (3.4) | 24.3 (5.8) | .90 |
| (n=48) | (n=49) | (n=35) | (n=40) | |||
| Parental Inheritance (n (%)) | .27 | .70 | ||||
| Mother | 26 (51.0) | 21 (39.6) | 19 (45.7) | 22 (50.0) | ||
| Father | 24 (47.1) | 24 (45.3) | 16 (54.3) | 17 (38.6) | ||
| Both | 1 (2.0) | 0 | 5 (11.4) | |||
| Unknown | 8 (15.1) | |||||
| WBC (mean (SD)) | 6.6 (1.7) | 7.6 (2.0) | .01 | 6.8 (2.0) | 7.6 (2.4) | .06 |
| (n=48) | (n=49) | (n=33) | (n=44) | |||
| Platelets (mean (SD)) | 236.7 (49.7) | 236.8 (61.2) | .99 | 233.1 (48.9) | 260.2 (56.2) | .03 |
| (n=48) | (n=49) | (n=33) | (n=43) | |||
| Alkaline phosphatase (mean (SD)) | 55.7 (16.5) | 63.7 (14.9) | .02 | 53.3 (13.0) | 70.9 (28.5) | .007 |
| (n=41) | (n=47) | (n=23) | (n=43) | |||
| AST (mean (SD)) | 22.3 (8.7) | 26.3 (15.6) | .15 | 20.9 (6.5) | 22.3 (6.9) | .42 |
| (n=41) | (n=48) | |||||
| ALT (mean (SD)) | 24.2 (14.8) | 27.3 (23.3) | .46 | 20.5 (9.3) | 23.7 (11.6) | .26 |
| (n=41) | (n=48) | (n=23) | (n=43) | |||
| Note: one outlier with ALT = 159 | ||||||
| Creatinine (mean (SD)) | 0.73 (0.16) | 0.83 (0.32) | .04 | 0.75 (0.13) | 0.78 (0.42) | .67 |
| (n=51) | (n=51) | (n=35) | (n=44) | |||
| Significant Proteinuria (n (%)) | 1/51† (2.0) | 14/52† (26.9) | <.0001 | 0/35 (0) | 7/44 (15.9) | .01 |
| Peripheral Nerve Involvement (n (%)) | 1/1† (100) | 50/50† (100) | 0/1† (0) | 41/41† (100) | ||
| Peripheral Nerve Severity (n (%)) | ||||||
| 1 | 1 (100) | 7 (14) | 5 (12.2) | |||
| 2 | 20 (40) | 20 (48.8) | ||||
| 3 | 10 (20) | 7 (17.1) | ||||
| 4 | 7 (14) | 3 (7.3) | ||||
| 5 | 6 (12) | 6 (14.6) | ||||
| Cardiac Involvement (n (%)) | 0/1† (0) | 28/51† (56.0) | 1/2† (50.0) | 30/41† (73.2) | ||
| Cardiac Severity (n (%)) | n=28 | n=1 | n=30 | |||
| Mild | 10 (35.7) | 13 (43.3) | ||||
| Moderate | 9 (32.1) | 1 (100) | 8 (26.7) | |||
| Severe | 9 (32.1) | 9 (30.0) | ||||
| Diabetes (n (%)) | 1/1† (100) | 3/52† (5.8) | 0/0† | 0/40† (0) | ||
†denominator is number with a yes or no answer to the question.
*t-test for continuous variables, chi-square for categorical variables.
Discovery and validation of a molecular diagnostic in peripheral blood cells
We first queried whether there is a gene expression signature that could distinguish asymptomatic V30M TTR carriers from symptomatic V30M FAP patients. To identify a robust and unbiased candidate diagnostic signature for all study subjects (both male and female) we split the 183 symptomatic V30M TTR FAP patients and asymptomatic V30M carriers into a Training and Test cohort (~85% of samples) and an external Validation cohort of samples (~15%). Both cohorts had equal representation of asymptomatic V30M TTR carriers and symptomatic V30M FAP subjects. It is important to note that the external cohort of samples was “blinded” to the training and validation of the signature, which was done exclusively in the first cohort. In this way a true and unbiased estimate of the accuracy and the Area Under the Curve (AUC) of a “locked” classifier derived from the Training and Test cohort was possible. To further remove bias from a single randomization, we randomized the 183-sample core dataset into 10 total randomizations and report the mean AUCs from all randomizations.
For each randomization, a class comparison of symptomatic V30M TTR FAP patients and asymptomatic V30M carriers was performed in the Training and Test cohort using a False Discovery rate (FDR) of 5%. Signatures with feature (probeset) sizes of 30-200 were tested using the Support Vector Machines (SVM) algorithm for all randomizations with a 70 Training/30 Test split of the samples using bootstrapping with 100 iterations of the randomization experiments. The best performing models from the Training and Test for 10 Randomizations were each “locked” and performance was tested on the External cohort of “blinded” samples. The Receiver Operating Characteristic (ROC) curves plotted on the 10 randomly selected external validation sets shown in Figure 1a had a mean AUC of 0.81 ± 0.05. The diagnostic metrics for all 10 randomizations are shown in Table 3.
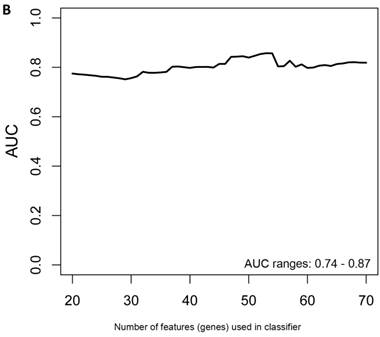
Class comparison of symptomatic V30M TTR FAP patients and asymptomatic V30M carriers. (a) ROC curves for 10 randomly selected external validation data sets derived from signatures with feature (probeset) sizes of 30-200 using the Support Vector Machines (SVM) algorithm with a 70 Training/30 Test split of the samples and bootstrapping with 100 iterations. The best performing models from the Training and Test cohorts for 10 Randomizations were each “locked” and performance was tested on the External cohort of “blinded” samples. (b) Bias-corrected plot of the AUCs for sex-independent classifiers of different feature sizes (20-70) using a logistic regression model. The classifier models tested had a range of AUCs from 0.74 to 0.87 using 20-70 genes. The optimal results (AUCs >0.80) were obtained with gene sets greater than 50.
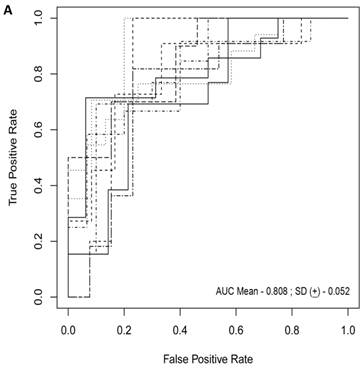
The diagnostic metrics of the SVM classifiers for all 10 randomizations of the symptomatic vs. asymptomatic subjects in the study.
| Diagnostic Metric | Mean | Range | SD | Lower CI | Upper CI |
|---|---|---|---|---|---|
| Sensitivity (TP/(TP+FN)) | 0.844 | 0.169 | 0.065 | 0.798 | 0.891 |
| Specificity (TN/(FP+TN)) | 0.805 | 0.308 | 0.139 | 0.731 | 0.933 |
| Positive Predictive Value (TP/(TP+FP)) | 0.829 | 0.312 | 0.126 | 0.739 | 0.919 |
| Negative Predictive Value (TN/(FN+TN)) | 0.825 | 0.173 | 0.069 | 0.782 | 0.886 |
| Matthews Correlation Coefficient ((TP*TN-FP*FN)sqrt(P*N*P'*N')) | 0.652 | 0.406 | 0.127 | 0.561 | 0.743 |
| Area Under Curve (((TP/(TP+FN))+ (TN/(FP+TN))*0.5) | 0.819 | 0.192 | 0.057 | 0.778 | 0.859 |
| Likelihood Ratio | 8.920 | 12.340 | 3.550 | 6.381 | 11.468 |
CI=Confidence Interval.
Another rigorous method to test whether the obtained classifier is subject to statistical over-fitting (this would inflate the claimed predictive results) can be done with the method of Harrell et al. [37]. This method uses bootstrapping to get bias-corrected (overfitting corrected) estimates of predicted vs. observed values of diagnostic accuracy using a logistic regression model. This is accomplished by sampling with replacement, where the sample labels are randomly reassigned for 500 bootstrap iterations. Using this method we ran classifier models with features (genes) ranging from 20-70 genes, chosen from the differential expression analysis of all asymptomatic vs. symptomatic FAP subjects ranked by p-values. All models were tested for 500 bootstrap iterations in the regression modeling strategies (rms) R package [34]. We then created a plot of the “bias corrected” AUCs for models using the different feature sizes (20-70) as shown in Figure 1b. The results show that the classifier models tested had a range of AUCs from 0.74 to 0.87 using 20-70 genes. The optimal results (AUCs >0.80) were obtained with gene sets greater than 50, consistent with the importance of using a larger number of genes for a robust classifier to accurately distinguish symptomatic and asymptomatic FAP subjects.
Evidence for sex-specific signatures in symptomatic vs. asymptomatic carriers
The second question addressed was whether there are gender-specific genomic signatures that can differentiate symptomatic male and female V30M FAP patients from asymptomatic V30M TTR carriers. This inquiry was motivated by the fact that male V30M FAP patients have earlier onset and more aggressive disease progression than female V30M FAP patients. This is particularly true for males that inherit the mutant gene from their mothers. The same tools and comparisons as described above for the sex independent signature were applied to the males and females as separate cohorts The only exception was that due to the smaller sample sizes (females; n = 96 and males; n = 87) only one random internal 70% Training and 30% Test data split was performed with 500 iterations by bootstrapping. The purpose of this work was to establish a proof-of-concept for sex-specific signatures. Independent data set validations of these signatures will be required before any claims of a diagnostic are considered.
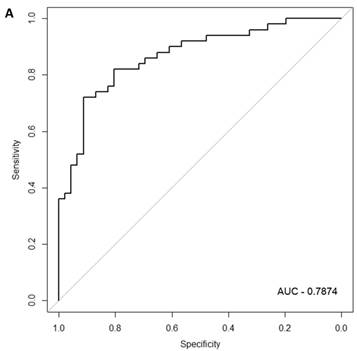
In the females, there were 125 genes that were differentially expressed between the symptomatic and asymptomatic subjects. A SVM “locked” best model comprising 80 genes and 500 bootstrap iterations gave an AUC of 0.7874 (Figure 2a) with a sensitivity, specificity, Positive Predictive Value (PPV) and Negative Predictive Value (NPV) of 76%, 80%, 81% and 75%, respectively. The Harrell bootstrapping plot of the “bias corrected” AUCs for models using different feature sizes (20-70) using a logistic regression model in the females is shown in Figure 2b. The results again show that models with >50 genes/features give AUCs >80%.
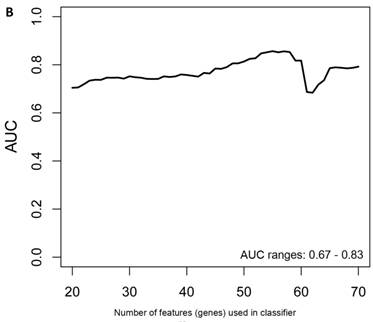
Class comparison of female symptomatic V30M TTR FAP patients and asymptomatic V30M carriers. (a) ROC curve for symptomatic vs. asymptomatic FAP female subjects using an SVM “locked” best model comprised of 80 genes and 500 bootstrap iterations with sensitivity, specificity, PPV and NPV of 76%, 80%, 81% and 75%, respectively. (b) The Harrell bootstrapping plot of the “bias-corrected” AUCs for female specific classifiers of different feature sizes (20-70) using a logistic regression model. The results again show that models with >50 genes/features give AUCs >80%.
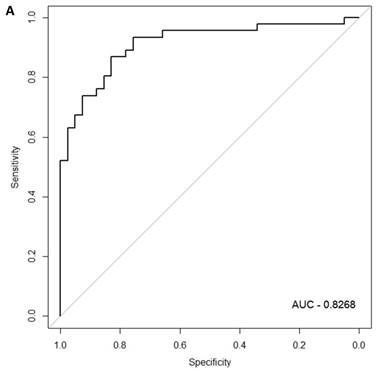
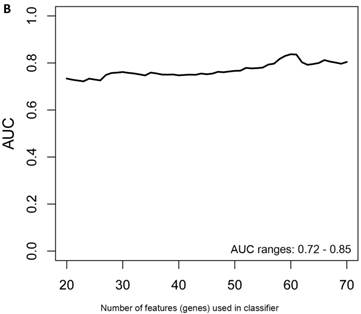
When the same comparisons were made with the male symptomatic V30M FAP and asymptomatic V30M TTR carriers, there were 353 differentially expressed genes between symptomatic and asymptomatic males. We tested classifier models using 20-200 of the differentially expressed genes ranked by p-value. The SVM best model best-fit classifier comprised all 200 genes. This classifier had an AUC of 0.8268 and sensitivity, specificity, PPV and NPV of 80%, 82%, 84% and 79%, respectively (Figure 3a). The Harrell bootstrapping plot of the “bias corrected” AUCs for models using different feature sizes (20-70) is shown in Figure 3b. The male signatures achieved AUCs >80% when more than 50 genes were used.
Class comparison of male symptomatic V30M TTR FAP patients and asymptomatic V30M carriers. (a) ROC curve for the SVM best model best-fit classifier for symptomatic vs. asymptomatic FAP male patients. This classifier comprised 200 genes had an AUC of 0.8268 and sensitivity, specificity, PPV and NPV of 80%, 82%, 84% and 79%, respectively. (b) The Harrell bootstrapping plot of the “bias-corrected” AUCs for male specific classifiers of different feature sizes (20-70) using a logistic regression model. The male signatures achieved AUCs >80% when more than 50 genes were used.
Developing molecular scores and evaluating tafamidis-treated samples
A simple classifier validated for differentiating between symptomatic V30M FAP patients vs. asymptomatic V30M carriers does not reflect disease severity or change in response to therapy. Serial monitoring requires translation of the classifier's output to a continuous variable or molecular score based on the underlying gene expression derived from the classifier. Therefore, we developed a method for calculating molecular scores (see Supplemental Methods). A desirable future study would be to allow clinicians to serially monitor disease progression, and response to therapy.
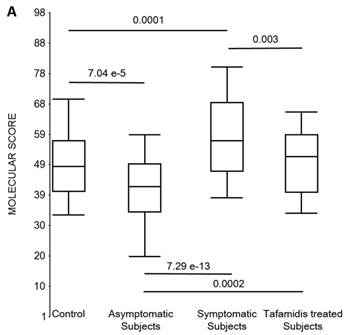
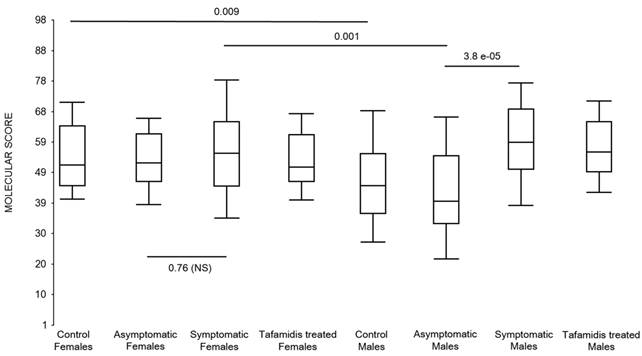
First, the molecular scores were calculated for symptomatic FAP vs. asymptomatic V30M carriers and analyzed as a single cohort, independent of sex. Since we had tested signatures using 10 different randomizations for the sex-independent molecular diagnostic, we chose a core set of 96 genes that were common to all 10 randomizations to calculate the molecular scores. As shown in Figure 4a, it is clear from the discovery set that the symptomatic patients had significantly higher molecular scores than both the asymptomatic V30M carriers (p=7.2E-13) and healthy controls (0.0001). Interestingly, the molecular scores of the asymptomatic FAP carriers are significantly lower than the healthy controls (7.1E-05).
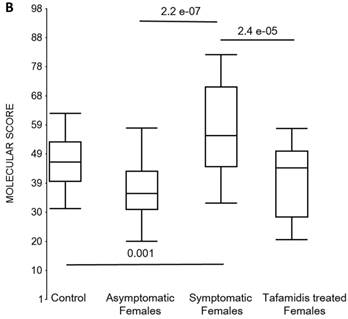
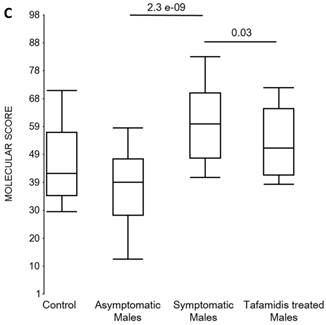
Box plots of the molecular scores. (a) Molecular scores calculated for symptomatic FAP vs. asymptomatic V30M carriers with a core set of 96 genes common to all 10 randomizations, used to generate the sex-independent molecular diagnostic. The symptomatic patients had significantly higher molecular scores than both the asymptomatic V30M carriers (p=7.2E-13) and healthy controls (p=0.0001). The molecular scores of the asymptomatic FAP carriers are significantly lower than the healthy controls (p=7.1E-05). Tafamidis treatment of the symptomatic patients significantly (p=0.003) lowered the molecular scores. (b) Molecular scores for female-specific signatures which clearly separated the female symptomatic and asymptomatic V30M carriers (p=2.2E-07) and the healthy controls (p=0.001). The scores also significantly separated the female symptomatic and tafamidis-treated female patients (p=2.4E-05). (c) Molecular scores for male-specific signatures which clearly separated the male symptomatic and asymptomatic V30M carriers (p=2.3E-09). The scores also separated the male symptomatic and tafamidis-treated male patients (p=0.03).
Scores were then generated for the 46 tafamidis-treated FAP patients as another kind of validation. The scores for symptomatic FAP patients after 3 months of tafamidis therapy showed significant improvement (p=0.003), dropping to levels roughly comparable to the healthy controls (p=0.29; NS) but still significantly higher than asymptomatic FAP subjects (p=0.0002). These results were obtained after only 3 months of tafamidis treatment (20 mg once daily) of symptomatic FAP patients.
We also calculated the molecular scores for female and male-specific signatures. The molecular scores clearly separated the female symptomatic and asymptomatic V30M carriers (p=2.2E-07) and the healthy controls (p=0.001) (Figure 4b). The scores also effectively separated the symptomatic and tafamidis-treated female patients (p=2.4E-05). We confirmed the score's female specificity by calculating the scores for these female-specific genes in the males and the result was not significant (p=0.16). Similarly, the male-specific molecular scores demonstrated clear separations between male symptomatic patients and asymptomatic V30M carriers (p=2.3E-09; Figure 4c) as well as healthy male controls (p=0.001). There were also significant differences in the scores between the tafamidis-treated males and the symptomatic males (p=0.03), consistent with a response to therapy, although the scores did not normalize to the level of asymptomatic V30M male carriers.
Mapping the biology of gender-independent and gender-specific genes
One value of using global gene expression profiling is the ability to use bioinformatic tools to map the expression of differentially expressed genes to known biological functions. The key is to correlate this mapping with the clinical phenotypes. First, a p-value cut-off of <0.005 and an FDR of <3% was set for comparing all symptomatic FAP to asymptomatic V30M carriers. There were 1534 significantly differentially expressed probesets representing 1426 annotated genes. These genes were associated with 36 significant pathways (Benjamini-Hochberg corrected p-value <0.05) (Table S1). 285 genes (18%) mapped to immune/inflammatory processes. There was a marked down-regulation of pathways such as eIF2, primary immunodeficiency signaling, and purine nucleotide biosynthesis in symptomatic patients, whereas signaling networks for FCγ, TREM1, NK cells, and cytokines IL3, IL15, and IL22 were upregulated.
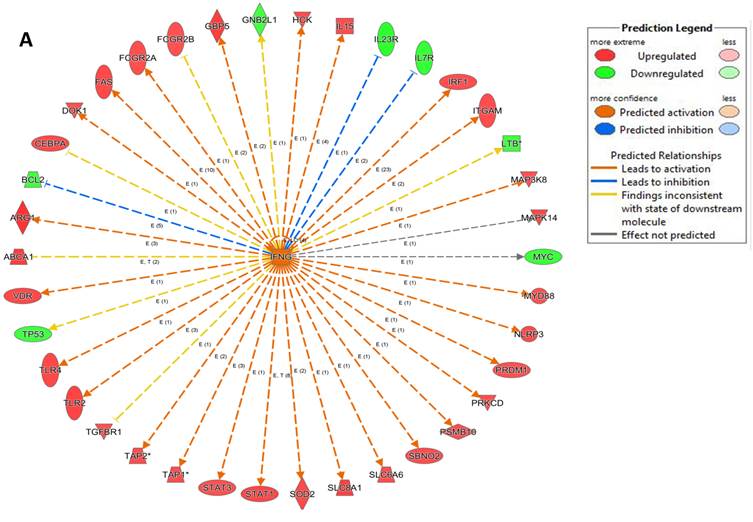
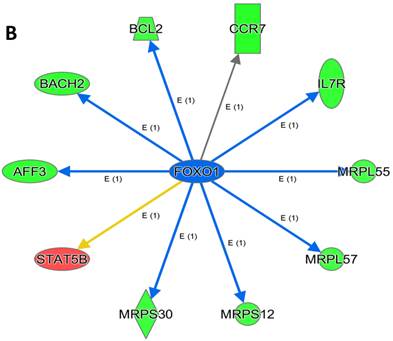
An analysis of upstream regulators was done to identify genes that are putative drivers of the downstream expression changes. This analysis identified a set of 23 candidate driver genes, with 11 of the 23 (48%) being known transcriptional regulators (Table S2). Two powerful candidate drivers based on upstream activation scores were IFNG and FOXO1 that mapped to the expression of 38 target molecules known in the literature to be upregulated by IFNG and 10 known to be downregulated by FOXO1 (Figures 5a & b and Table S2).
Expression of two candidate driver genes and their target genes. IFNG and FOXO1 are the two most powerful genes (by activation scores) that were identified by an analysis of upstream regulators known to be putative drivers of downstream expression changes. (a) 38 target molecules known in the literature to be upregulated by IFNG, and (b) 10 molecules known to be downregulated by FOXO1.
To map the biology of the male- and female-specific genes, class comparisons were made between the asymptomatic and symptomatic males and females separately. At p<0.005 there were 588 differentially expressed genes in the females (FDR<10%; Table S3) and 894 differently expressed genes in the males (FDR<6%; Table S4). Only 49 genes (3%) were shared between the male and female signatures, confirming that male and female FAP patients have very specific and different patterns of gene expression. Of the 588 differentially expressed genes in the females, there were only 66 molecules associated with immune/inflammation processes, and of these, only 11 (17%) were upregulated in the symptomatic females as compared to the asymptomatic females. The majority of all genes (438 genes; 75%) were downregulated in symptomatic females. In contrast to the females, 691 (78%) of the 894 differentially expressed genes in the symptomatic males were upregulated and 179 were associated with immune/inflammatory processes. Of the latter, 146 (82%) were upregulated.
In the females, there were only 4 significant canonical signaling pathways: eIF2, primary immunodeficiency, T-helper cell differentiation, and iCOS signaling (Table S5). Strikingly, almost all the genes that were associated with these pathways were downregulated in symptomatic females. In the males, there were 29 significant canonical pathways linked to immunity such as Fcγ receptor, natural killer cell, Toll-like receptor, B Cell receptor, leukocyte extravasation, and IL-12 Signaling (Table S5). As noted previously, the majority of genes associated with these pathways were upregulated in symptomatic males.
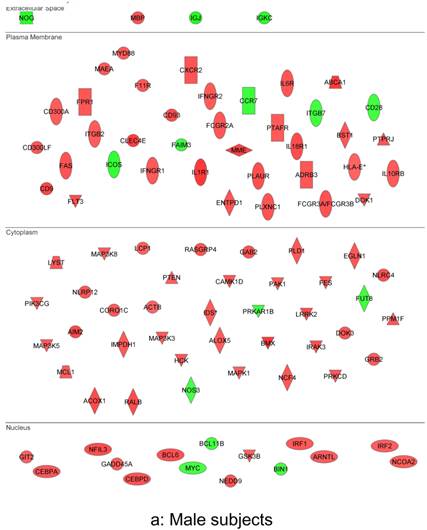
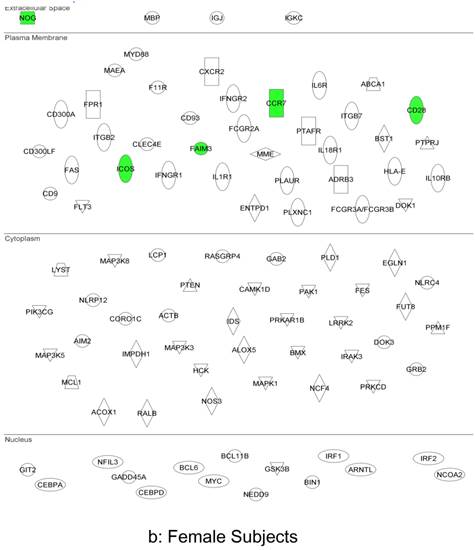
In summary, the symptomatic males reveal upregulated inflammation-associated genes and pathways (Figure 6a). The male-specific inflammatory genes are not differentially expressed in symptomatic females (Figure 6b).
Expression of the male-specific inflammatory genes in FAP subjects. (a) Symptomatic males reveal upregulated inflammation-associated genes and pathways. (b) Male-specific inflammatory genes are not differentially expressed in symptomatic females.
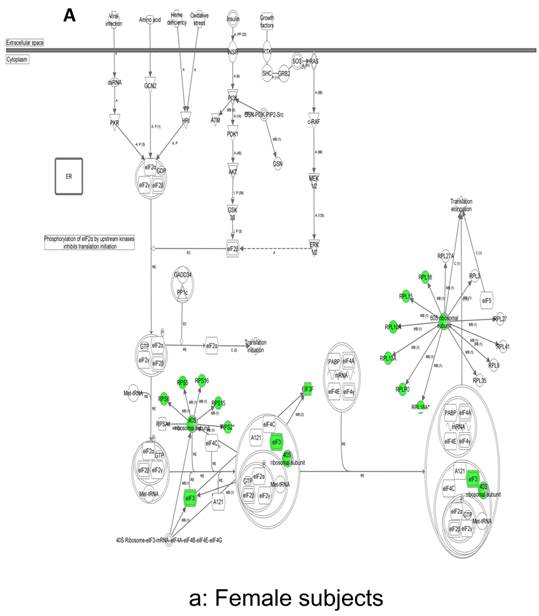
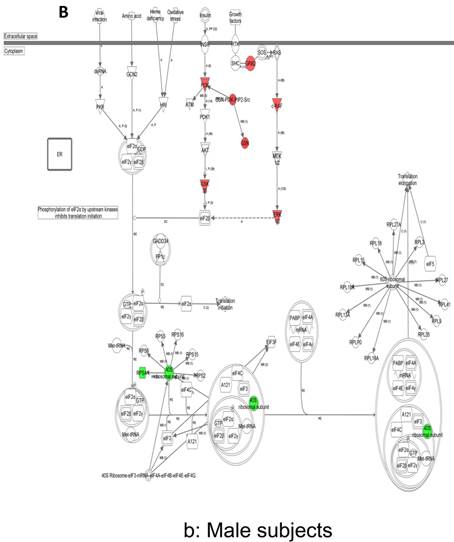
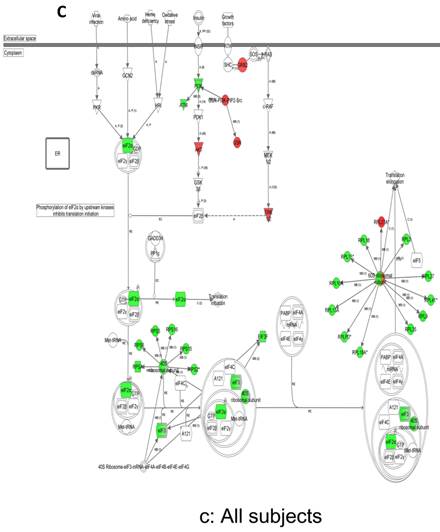
In contrast, there is a marked downregulation of the eIF2α signaling pathway in symptomatic females (Figure 7a), and that difference is not seen in males, who actually show upregulated expression of some proximal signaling molecules (Figure 7b). The eIF2 pathway is also globally downregulated in all symptomatic patients compared to asymptomatic V30M carriers (Figure 7c). Finally, it is important to emphasize that the genes identified by analysis of all the patients as differentially expressed and independent of sex reveals another set of potential mechanistic pathways for FAP disease that should be simply added to the candidates revealed by sex-specific analysis (Tables S1 and S2; Figure 5a and 5b).
Expression of eIF2α signaling pathway genes. (a) There is a marked downregulation of the eIF2α signaling pathway in symptomatic females. (b) Similar differences are not seen in males, who actually show upregulated expression of some proximal signaling molecules. (c) The eIF2 pathway is also globally downregulated in all symptomatic patients compared to asymptomatic patients.
Because there was a correlation of upregulated immune/inflammatory genes in symptomatic FAP males but not symptomatic females, we analyzed the 146 male-specific immune/inflammatory genes and determined their expression in the females by molecular scores (Figure 8). Indeed, the basal level of gene expression for these male-specific inflammatory genes was relatively high in all women including the healthy female controls.
Box-plots of the molecular scores of the 146 male-specific immune/inflammatory genes. Expression of the immune/inflammatory genes differentially upregulated in symptomatic males was scored in female FAP subjects and healthy controls and in male FAP subjects and healthy controls. The basal level of gene expression for these male-specific inflammatory genes was relatively high in all women including the healthy female controls.
Among the 894 differentially expressed genes in the males, 47 proteins are targets of 95 known drugs (Table S5). These included tocilizumab, a humanized monoclonal antibody against the interleukin-6 receptor (IL-6R); anakinra, an interleukin-1 (IL-1) receptor antagonist that blocks the biologic activity of naturally occurring IL-1; talmapimod, a selective inhibitor of p38 mitogen-activated protein kinase (MAPK), a potential agent with immunomodulating, anti-inflammatory and antineoplastic activities. Another interesting drug was lexipafant (BB-882), a platelet activating factor receptor antagonist, which has been identified as an agent that may reduce the severity of the inflammatory responses to liver injury in animal models.
Multi-analyte plasma protein panel for inflammatory genes
A commercial plasma proteomic panel for inflammatory molecules was used to profile 44 different analytes in plasma samples from 40 selected V30M carriers, comprised of equal numbers of male and female subjects, and equal numbers of symptomatic FAP and asymptomatic subjects (Table S6). 31 analytes were consistently detected in the plasma above threshold. There were 5 inflammation-linked plasma proteins that were significantly differentially upregulated among the symptomatic males (p<0.05): Alpha-2-Macroglobulin, Ferritin, Tissue Inhibitor of Metalloproteinases-1 (TIMP1) and Vascular Cell Adhesion Molecule-1. Interleukin-7 (IL7) was significantly downregulated in symptomatic males. IL7 is an important growth factor for T and B cells produced through TLR4 signaling by the liver, dendritic, stromal and intestinal epithelial cells and low expression has been linked in mouse models to pathogenesis of Experimental Autoimmune Encephalitis [38]. Among the symptomatic females, 4 proteins were significantly upregulated (p<0.05): Macrophage Inflammatory Protein-1 beta (MIP1b), Monocyte Chemotactic Protein-1 (MCP1), Complement C3 and Beta 2 Microglobulin. Analysis of the males and females together found 6 significantly upregulated inflammatory markers in the plasma: eotaxin 1, ferritin, TIMP1, MIP1b, and MCP1.
Discussion
Using global gene expression profiling of peripheral blood cells from two independent cohorts of asymptomatic V30M carriers, symptomatic V30M FAP patients and age- and sex-matched local healthy controls, we identified three different signatures: a signature for differentiating symptomatic vs. asymptomatic FAP male subjects, a different signature for distinguishing symptomatic vs. asymptomatic FAP female subjects, and a third sex-independent signature for differentiating symptomatic vs. asymptomatic FAP. The latter signature as a continuous variable or molecular score showed a reduction in response to tafamidis treatment in a third completely independent cohort of symptomatic FAP patients, demonstrating the robustness of our classifier and scoring methodology.
There have been very few studies that look at the global expression of genes that might drive post-mitotic tissue loss in FAP patients. One microarray study in clinical FAP looked at salivary glands of 4 symptomatic patients [39]. Only two genes were identified as differentially expressed and validated using qPCR; biglycan and neutrophil gelatinase-associated lipocalin (NGAL). In our gene expression profiling, NGAL (LCN2) mRNA levels did not differ between symptomatic and asymptomatic patients, despite being moderately expressed (raw signals ~200 across all samples studied). We did not measure NGAL protein levels in this study. Another gene profiling study in symptomatic V30M Swedish TTR amyloidosis patients was done with biopsies of adipose tissue (n=12) and liver (n=9) vs. normal healthy controls (n=16) [40]. While there were no significant differences found in adipose tissue biopsies (TTR target tissue), evidence for an impaired ER/protein folding pathway in the livers was revealed.
A recent study investigated the biological effects of TTR proteins on angiogenesis using primary human umbilical vein endothelial cells (HUVECs; n=6) exposed to the WT or V30M TTR tetramer [41]. Exposure to the V30M tetramer down-regulated many pro-angiogenic genes such as VEGFR1, VEGFR2, FGF2, TGFB2, and ANGPT2 and also inhibited migration and decreased HUVEC survival by inducing apoptosis. Consistent with these results, we demonstrated differential expression of the VEGF and FGF pathways in the symptomatic patients (p=0.025 and 0.03, respectively).
The results presented above corroborate our recent study of a murine model of wild type human TTR cardiac amyloidosis. Microarray analysis of the TTR synthesizing tissue (liver) and a target of TTR deposition (heart) revealed substantial transcriptional evidence of inflammation in the target tissue prior to the time of deposition, which diminished (relative to mice without deposits) after deposition. There was also transcriptional evidence in the hearts consistent with hypoxia and possible heart failure, including expression of VEGF and genes downstream in the VEGF pathway and evidence of B cell and immune activation via T cell receptor pathways [42].
There is limited evidence demonstrating a link between FAP and inflammation. Chronic administration of an IL-1 antagonist (Anakinra) that alleviates inflammation in mice that deposit TTR, but do not exhibit FAP phenotypes, reveal that Anakinra prevents extracellular deposition of TTR in the sciatic nerve and inhibits apoptosis [43]. In a separate study, altering the threshold of the inflammatory response in injured mice reveal that Anakinra differentially regulates TTR expression and deposition [44]. Amyloid fibrils have been shown to bind and signal through the cell surface Receptor for Advance Glycation End-products (RAGE), to upregulate NFkB that is linked to a number of cell stress and inflammatory pathways including IL6 and TNF [45]. Immunohistochemical analysis of nerve biopsies from 16 symptomatic FAP patients showed upregulated expression of RAGE, IL1b, TNF, and iNOS [46]. Interestingly, diflunisal, a generic nonsteroidal anti-inflammatory drug has been successfully repurposed for the treatment of FAP. In a global, multicenter clinical trial of 130 patients with FAP, diflunisal treatment reduced the rate of progression of neurological impairment [20]. Diflunisal was assessed in the clinical trial not because of its potential anti-inflammatory properties, but rather because diflunisal, like tafamidis, binds to TTR, kinetically stabilizing the circulating TTR tetramers and thereby inhibiting the release of the TTR monomer required for amyloidogenesis [47, 48]. It is tempting to hypothesize that some of the alleviation of FAP symptoms might be due to the anti-inflammatory effect of diflunisal, though metrics for inflammation were not measured. It would be interesting to compare the transcriptional profiles of patients treated with diflunisal to those of patients treated with tafamidis, which has no known anti-inflammatory properties, to look for similar (or potentially different) mechanisms. In such an envisioned clinical trial comparison, it is imperative that the doses of tafamidis and diflunisal be adjusted so that they kinetically stabilize transthyretin equally. This has not been done in the trials carried out to date.
Sex-specific differences in immune and inflammatory responses are believed to be responsible for differences in the risks for various disease states, such as increased cardiovascular disease in males [49, 50] and increased risk for autoimmune disease in females [51, 52]. This is also true in the response to bacterial infections, which is impaired in males relative to females [53, 54]. Basal cell surface expression levels of the LPS-binding Toll-like receptor-4 (TLR4) and its co-receptor CD14 are higher in macrophages of males relative to females [55]. In our data both TLR4 and CD14 were differentially upregulated in symptomatic males compared to asymptomatic males, and neither was differentially expressed in females. Other TLRs such as TLR2, TLR6 and TLR8 also showed a male-specific upregulation in our study.
The correlation of upregulated immune/inflammatory genes in symptomatic FAP males, but not in symptomatic females, suggests different roles for immune/inflammatory pathways in men vs. women. Notably, the absence of upregulation in females should not be interpreted as inflammation being absent in females. Indeed, the basal level of gene expression for the male-specific inflammatory genes was relatively high in all women regardless whether they were symptomatic, asymptomatic or healthy controls. Thus, it is possible that certain pathways of inflammation may be a factor in symptomatic FAP in both sexes but since females have higher baseline levels of expression of these inflammatory genes, there is no significant differential change when symptomatic vs. asymptomatic subjects are compared. It is also possible that inflammation is better tolerated by females. Thus, inflammation may be triggered in both symptomatic and asymptomatic females and not be the only mechanism driving the conversion to a symptomatic state in females.
The most differentially expressed pathway, the eIF2 pathway, was downregulated in all V30M FAP patients relative to asymptomatic carriers. Multiple signaling pathways drive the integrated stress response pathways that phosphorylate eIF2, slowing the rate of translational initiation. The eIF2 kinase, PERK, senses accumulation of misfolded or aggregated proteins in the lumen of the endoplasmic reticulum where TTR is biosynthesized [56-58]. Activation of the PERK arm of the unfolded protein response reduces global protein synthesis and remediates the overloaded endoplasmic reticulum secretory pathway via eIF2 phosphorylation. Downregulation of the eIF2 family of transcripts is seen irrespective of gender and could be the major mechanism involved in the females in the lack of a differential expression of inflammatory signals. In the symptomatic subjects, the eIF2 pathway showed 33/38 differentially expressed molecules as being downregulated, including a host of ribosomal proteins.
Surprisingly, we show that the molecular scores for asymptomatic FAP carriers are lower than healthy controls for all groups evaluated, but this is not unprecedented. Down-regulation of host defense response genes and selective up-regulation of genes associated with protection in the peripheral blood of asymptomatic dengue patients was shown using microarrays [59]. Another study showed that high levels of migration inhibitory factor (MIF), a cytokine implicated in the pathogenesis of a number of human inflammatory diseases, were found in the peripheral blood of cerebral malaria patients and may be associated with fatal outcomes. MIF protein levels, as measured by ELISA, were lower in patients with a mild form of malaria than even healthy controls [60]. This inverse correlation between protection and MIF levels was observed previously, where significantly reduced circulating MIF levels were seen in children with acute malaria infection compared to matched healthy controls [61].
A common question for any molecular diagnostic is how specifically it can diagnose the disease of interest relative to maladies with similar clinical phenotypes. In the case of FAP, patients harbor a genetic mutation, which along with an amyloid positive biopsy and abnormal physical and laboratory exams constitutes a definitive FAP diagnosis (see Table 1 legend). We envision that the peripheral blood cell FAP signature could become a complementary diagnostic strategy, however we recognize that the prevalence of V30M FAP in the Portuguese population is high (0.001% in the endemic areas). The clinical presentation and the age of onset are more variable in the Swedish and Japanese populations [30, 62, 63]. We acknowledge that the prevalence of FAP in the Portuguese population studied could influence the PPV and NPV of the test. However, we envision that our diagnostic signature would initially be validated in other FAP populations where the prevalence is also high, such as the Japanese and Swedish populations of allele carriers. Whether the signatures identified will be specific for FAP in populations with lower disease prevalence and higher genetic diversity like the US population remains to be established.
We also acknowledge that we need to study other related peripheral neuropathies to test the specificity of expression profile as an early diagnostic strategy. The fact that some of the major pathways identified by our signatures are related to immune/inflammatory activation gives us confidence that our signatures are probably not similar to idiopathic polyneuropathy, but that will need to be tested.
Another question is how such a potential molecular diagnostic would be used in clinical practice. We envision that once V30M carriers are identified, serial monitoring of the molecular scores could give clinicians early warning of increasing risk for developing symptomatic FAP, at which point these patients could be brought back to the clinic for amyloid biopsy. There is compelling data already available that the earlier treatment is started, the better the outcome [17-22]. Whether the molecular scores will correlate well enough with response to therapy to guide individualized dosing decisions will require a prospective, properly powered clinical study that includes serial observations of patients over time. It is also possible that these molecular scores will be considered in parallel with Technetium Pyrophosphate Scanning [64-66] and magnetic resonance imaging [67] for diagnosing symptomatic transthyretin aggregation-associated cardiomyopathy and polyneuropathy, respectively.
In conclusion, our data from the peripheral blood cells of FAP patients reveal a first generation sex-independent diagnostic molecular signature for symptomatic FAP that correlates with response to tafamidis therapy. Equally importantly, this data generates several testable hypotheses about putative driver and compensatory mechanisms in FAP that will require further study. We also demonstrate that there are sex-specific FAP signatures that are different than the sex-independent profiles. While these sex-dependent differences will need further validation in independent cohorts in the next stage of work, they offer the potential to explain why male patients generally progress faster in FAP. The trend toward normalization of the scores of all groups of FAP patients treated with tafamidis motivate longer prospective studies to determine whether this strategy of blood gene expression profiling could become a useful biomarker for response to treatment.
Supplementary Material
Additional File 1Supplemental Methods.
Table S1.
Table S2.
Table S3.
Table S4.
Table S5.
Table S6.
Acknowledgements
The authors would like to thank Dr. Jill Waalen for multivariable statistical analyses of the patients' clinical characteristics.
Funding
We acknowledge the following sources of research funding: NIH U19 A1063603 (DRS, SMK), NIH DK46335 (JWK) and NIH R01AG19259 (JNB).
Competing Interests
JWK receives royalties from tafamidis..
References
1. Rowczenio DM, Noor I, Gillmore JD, Lachmann HJ, Whelan C, Hawkins PN. et al. Online registry for mutations in hereditary amyloidosis including nomenclature recommendations. Human mutation. 2014;35:E2403-12
2. Saraiva MJM. Transthyretin Mutations in Health and Disease. Human mutation. 1995;5:191-6
3. Benson MD. Familial Amyloidotic polyneuropathy. TIBS. 1989;12:88-92
4. Ando Y, Coelho T, Berk JL, Cruz MW, Ericzon BG, Ikeda S. et al. Guideline of transthyretin-related hereditary amyloidosis for clinicians. Orphanet journal of rare diseases. 2013;8:31
5. Coelho T. Familial amyloid polyneuropathy: new developments in genetics and treatment. Current opinion in neurology. 1996;9:355-9
6. Sekijima Y, Kelly JW, Ikeda S. Pathogenesis of and therapeutic strategies to ameliorate the transthyretin amyloidoses. Current pharmaceutical design. 2008;14:3219-30
7. Suhr OB, Ericzon BG. Selection of hereditary transthyretin amyloid patients for liver transplantation: the Swedish experience. Amyloid: the international journal of experimental and clinical investigation: the official journal of the International Society of Amyloidosis. 2012;19(Suppl 1):78-80
8. Holmgren G, Steen L, Ekstedt J, Groth CG, Ericzon BG, Eriksson S. et al. Biochemical effect of liver transplantation in two Swedish patients with familial amyloidotic polyneuropathy (FAP-met30). Clinical genetics. 1991;40:242-6
9. Hammarstrom P, Jiang X, Hurshman AR, Powers ET, Kelly JW. Sequence-dependent denaturation energetics: A major determinant in amyloid disease diversity. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(Suppl 4):16427-32
10. Hurshman Babbes AR, Powers ET, Kelly JW. Quantification of the thermodynamically linked quaternary and tertiary structural stabilities of transthyretin and its disease-associated variants: the relationship between stability and amyloidosis. Biochemistry. 2008;47:6969-84
11. Eisele YS, Monteiro C, Fearns C, Encalada SE, Wiseman RL, Powers ET. et al. Targeting protein aggregation for the treatment of degenerative diseases. Nature reviews Drug discovery. 2015;14:759-80
12. Hammarstrom P, Wiseman RL, Powers ET, Kelly JW. Prevention of transthyretin amyloid disease by changing protein misfolding energetics. Science. 2003;299:713-6
13. Herlenius G, Wilczek HE, Larsson M, Ericzon BG. Ten years of international experience with liver transplantation for familial amyloidotic polyneuropathy: results from the Familial Amyloidotic Polyneuropathy World Transplant Registry. Transplantation. 2004;77:64-71
14. Jiang X, Smith CS, Petrassi HM, Hammarstrom P, White JT, Sacchettini JC. et al. An engineered transthyretin monomer that is nonamyloidogenic, unless it is partially denatured. Biochemistry. 2001;40:11442-52
15. Schneider F, Hammarstrom P, Kelly JW. Transthyretin slowly exchanges subunits under physiological conditions: A convenient chromatographic method to study subunit exchange in oligomeric proteins. Protein science. 2001;10:1606-13
16. Holmgren G, Ericzon BG, Groth CG, Steen L, Suhr O, Andersen O. et al. Clinical improvement and amyloid regression after liver transplantation in hereditary transthyretin amyloidosis. Lancet. 1993;341:1113-6
17. Suhr OB, Ericzon BG, Friman S. Long-term follow-up of survival of liver transplant recipients with familial amyloid polyneuropathy (Portuguese type). Liver transplantation: official publication of the American Association for the Study of Liver Diseases and the International Liver Transplantation Society. 2002;8:787-94
18. Wilczek HE, Larsson M, Ericzon BG, Fapwtr. Long-term data from the Familial Amyloidotic Polyneuropathy World Transplant Registry (FAPWTR). Amyloid. 2011;18:193-5
19. Yamashita T, Ando Y, Okamoto S, Misumi Y, Hirahara T, Ueda M. et al. Long-term survival after liver transplantation in patients with familial amyloid polyneuropathy. Neurology. 2012;78:637-43
20. Berk JL, Suhr OB, Obici L, Sekijima Y, Zeldenrust SR, Yamashita T. et al. Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. Jama. 2013;310:2658-67
21. Coelho T, Maia LF, da Silva AM, Cruz MW, Plante-Bordeneuve V, Suhr OB. et al. Long-term effects of tafamidis for the treatment of transthyretin familial amyloid polyneuropathy. Journal of neurology. 2013;260:2802-14
22. Coelho T, Maia LF, Martins dSA, Waddington CM, Plante-Bordeneuve V, Lozeron P. et al. Tafamidis for transthyretin familial amyloid polyneuropathy: A randomized, controlled trial. Neurology. 2012;79:785-92
23. Bulawa CE, Connelly S, DeVit M, Wang L, Weigel C, Fleming JA. et al. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc Natl Acad Sci U S A. 2012;109:9629-34 S/1-S/9
24. Razavi H, Palaninathan SK, Powers ET, Wiseman RL, Purkey HE, Mohamedmohaideen NN. et al. Benzoxazoles as transthyretin amyloid fibril inhibitors: synthesis, evaluation, and mechanism of action. Angewandte Chemie. 2003;42:2758-61
25. Coelho T, Carvalho M, Saraiva MJ, Alves I, Almeida MR, Costa PP. A strikingly benign evolution of FAP in an individual found to be a compound heterozygote for two TTR mutations: TTR MET 30 and TTR MET 119. J Rheumatol. 1993;20:179
26. Hammarstrom P, Schneider F, Kelly JW. Trans-suppression of misfolding in an amyloid disease. Science. 2001;293:2459-62
27. Olsen KE, Sletten K, Westermark P. The use of subcutaneous fat tissue for amyloid typing by enzyme-linked immunosorbent assay. American Journal of Clinical Pathology. 1999;111:355-62
28. Fernandez-Escamilla A-M, Rousseau F, Schymkowitz J, Serrano L. Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nat Biotechnol. 2004;22:1302-6
29. Westermark P, Bergstrom J, Solomon A, Murphy C, Sletten K. Transthyretin-derived senile systemic amyloidosis: clinicopathologic and structural considerations. Amyloid: the international journal of experimental and clinical investigation: the official journal of the International Society of Amyloidosis. 2003;10(Suppl 1):48-54
30. Sousa A, Coelho T, Barros J, Sequeiros J. Genetic epidemiology of familial amyloidotic polyneuropathy (FAP)-type I in Povoa do Varzim and Vila do Conde (north of Portugal). American journal of medical genetics. 1995;60:512-21
31. Muller KE, Benignus VA. Increasing scientific power with statistical power. Neurotoxicology and teratology. 1992;14:211-9
32. Muller KE, Lavange LM, Ramey SL, Ramey CT. Power Calculations for General Linear Multivariate Models Including Repeated Measures Applications. Journal of the American Statistical Association. 1992;87:1209-26
33. Eisenhart C. The assumptions underlying the analysis of variance. Biometrics. 1947;3:1-21
34. Harrell FEJr. rms: Regression Modeling Strategies. R package version 4.3-0. http://CRAN.R-project.org/package=rms. 2015
35. Harrell FE Jr, Lee KL, Mark DB. Multivariable prognostic models: issues in developing models, evaluating assumptions and adequacy, and measuring and reducing errors. Statistics in medicine. 1996;15:361-87
36. Robin X, Turck N, Hainard A, Tiberti N, Lisacek F, Sanchez JC. et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC bioinformatics. 2011;12:77
37. Steyerberg EW, Harrell FE Jr, Borsboom GJ, Eijkemans MJ, Vergouwe Y, Habbema JD. Internal validation of predictive models: efficiency of some procedures for logistic regression analysis. Journal of clinical epidemiology. 2001;54:774-81
38. Sawa Y, Arima Y, Ogura H, Kitabayashi C, Jiang JJ, Fukushima T. et al. Hepatic interleukin-7 expression regulates T cell responses. Immunity. 2009;30:447-57
39. Sousa MM, do Amaral JB, Guimaraes A, Saraiva MJ. Up-regulation of the extracellular matrix remodeling genes, biglycan, neutrophil gelatinase-associated lipocalin, and matrix metalloproteinase-9 in familial amyloid polyneuropathy. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2005;19:124-6
40. Norgren N, Olsson M, Nystrom H, Ericzon BG, de Tayrac M, Genin E. et al. Gene expression profile in hereditary transthyretin amyloidosis: differences in targeted and source organs. Amyloid: the international journal of experimental and clinical investigation: the official journal of the International Society of Amyloidosis. 2014;21:113-9
41. Nunes RJ, de Oliveira P, Lages A, Becker JD, Marcelino P, Barroso E. et al. Transthyretin proteins regulate angiogenesis by conferring different molecular identities to endothelial cells. The Journal of biological chemistry. 2013;288:31752-60
42. Buxbaum JN, Tagoe C, Gallo G, Walker JR, Kurian S, Salomon DR. Why are some amyloidoses systemic? Does hepatic "chaperoning at a distance" prevent cardiac deposition in a transgenic model of human senile systemic (transthyretin) amyloidosis? FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2012;26:2283-93
43. Goncalves NP, Vieira P, Saraiva MJ. Interleukin-1 signaling pathway as a therapeutic target in transthyretin amyloidosis. Amyloid: the international journal of experimental and clinical investigation: the official journal of the International Society of Amyloidosis. 2014;21:175-84
44. Goncalves NP, Teixeira-Coelho M, Saraiva MJ. Protective role of anakinra against transthyretin-mediated axonal loss and cell death in a mouse model of familial amyloidotic polyneuropathy. Journal of neuropathology and experimental neurology. 2015;74:203-17
45. Sousa MM, Yan SD, Stern D, Saraiva MJ. Interaction of the receptor for advanced glycation end products (RAGE) with transthyretin triggers nuclear transcription factor kB (NF-kB) activation. Laboratory investigation; a journal of technical methods and pathology. 2000;80:1101-10
46. Sousa MM, Du Yan S, Fernandes R, Guimaraes A, Stern D, Saraiva MJ. Familial amyloid polyneuropathy: receptor for advanced glycation end products-dependent triggering of neuronal inflammatory and apoptotic pathways. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2001;21:7576-86
47. Miller SR, Sekijima Y, Kelly JW. Native state stabilization by NSAIDs inhibits transthyretin amyloidogenesis from the most common familial disease variants. Laboratory investigation; a journal of technical methods and pathology. 2004;84:545-52
48. Sekijima Y, Dendle MA, Kelly JW. Orally administered diflunisal stabilizes transthyretin against dissociation required for amyloidogenesis. Amyloid: the international journal of experimental and clinical investigation: the official journal of the International Society of Amyloidosis. 2006;13:236-49
49. Njolstad I, Arnesen E, Lund-Larsen PG. Smoking, serum lipids, blood pressure, and sex differences in myocardial infarction. A 12-year follow-up of the Finnmark Study. Circulation. 1996;93:450-6
50. Tunstall-Pedoe H, Kuulasmaa K, Amouyel P, Arveiler D, Rajakangas AM, Pajak A. Myocardial infarction and coronary deaths in the World Health Organization MONICA Project. Registration procedures, event rates, and case-fatality rates in 38 populations from 21 countries in four continents. Circulation. 1994;90:583-612
51. Dooley MA, Hogan SL. Environmental epidemiology and risk factors for autoimmune disease. Current opinion in rheumatology. 2003;15:99-103
52. Gleicher N, Barad DH. Gender as risk factor for autoimmune diseases. Journal of autoimmunity. 2007;28:1-6
53. Brabin L, Brabin BJ. Parasitic infections in women and their consequences. Advances in parasitology. 1992;31:1-81
54. Grossman CJ. Interactions between the gonadal steroids and the immune system. Science. 1985;227:257-61
55. Marriott I, Bost KL, Huet-Hudson YM. Sexual dimorphism in expression of receptors for bacterial lipopolysaccharides in murine macrophages: a possible mechanism for gender-based differences in endotoxic shock susceptibility. Journal of reproductive immunology. 2006;71:12-27
56. Hershey JW. Protein phosphorylation controls translation rates. The Journal of biological chemistry. 1989;264:20823-6
57. Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nature cell biology. 2000;2:326-32
58. Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annual review of biochemistry. 2005;74:739-89
59. Yeo AS, Azhar NA, Yeow W, Talbot CC Jr, Khan MA, Shankar EM. et al. Lack of clinical manifestations in asymptomatic dengue infection is attributed to broad down-regulation and selective up-regulation of host defence response genes. PLoS One. 2014;9:e92240
60. Jain V, McClintock S, Nagpal AC, Dash AP, Stiles JK, Udhayakumar V. et al. Macrophage migration inhibitory factor is associated with mortality in cerebral malaria patients in India. BMC research notes. 2009;2:36
61. Awandare GA, Hittner JB, Kremsner PG, Ochiel DO, Keller CC, Weinberg JB. et al. Decreased circulating macrophage migration inhibitory factor (MIF) protein and blood mononuclear cell MIF transcripts in children with Plasmodium falciparum malaria. Clin Immunol. 2006;119:219-25
62. Holmgren G, Costa PM, Andersson C, Asplund K, Steen L, Beckman L. et al. Geographical distribution of TTR met30 carriers in northern Sweden: discrepancy between carrier frequency and prevalence rate. Journal of medical genetics. 1994;31:351-4
63. Ikeda S, Nakazato M, Ando Y, Sobue G. Familial transthyretin-type amyloid polyneuropathy in Japan: clinical and genetic heterogeneity. Neurology. 2002;58:1001-7
64. Castano A, Grogan M, Johnson GB, Dispenzieri A, Ruberg FL, Berk JL. et al. Multicenter Experience of Technetium Pyrophosphate Scanning for Diagnosing TTR Cardiac Amyloid: A Revival in Nuclear Cardiology. Journal of Cardiac Failure. 2015;21:S85-S
65. Rapezzi C, Quarta CC, Guidalotti PL, Pettinato C, Fanti S, Leone O. et al. Role of Tc-99m-DPD Scintigraphy in Diagnosis and Prognosis of Hereditary Transthyretin-Related Cardiac Amyloidosis. Jacc-Cardiovascular Imaging. 2011;4:659-70
66. Pilebro B, Suhr OB, Naslund U, Westermark P, Lindqvist P, Sundstrom T. Tc-99m-DPD uptake reflects amyloid fibril composition in hereditary transthyretin amyloidosis. Upsala Journal of Medical Sciences. 2016;121:17-24
67. Kollmer J, Hund E, Hornung B, Hegenbart U, Schonland SO, Kimmich C. et al. In vivo detection of nerve injury in familial amyloid polyneuropathy by magnetic resonance neurography. Brain. 2015;138:549-62
Author contact
![]() Corresponding authors: Daniel R. Salomon, M.D.: Telephone: 858-784-9381, email: dsalomonedu. Or Jeffery W. Kelly: Telephone: 858-784-9880, email: jkellyedu.
Corresponding authors: Daniel R. Salomon, M.D.: Telephone: 858-784-9381, email: dsalomonedu. Or Jeffery W. Kelly: Telephone: 858-784-9880, email: jkellyedu.