Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Materials and Methods
Supplementary Materials
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2016; 6(8):1176-1189. doi:10.7150/thno.12866 This issue Cite
Research Paper
Polymer-DNA Nanoparticle-Induced CXCR4 Overexpression Improves Stem Cell Engraftment and Tissue Regeneration in a Mouse Hindlimb Ischemia Model
Lorenzo Deveza1,2, Jeffrey Choi3, Jerry Lee4, Ngan Huang4, John Cooke4, Fan Yang1,5 ![]()
1. Department of Bioengineering, Stanford University, Stanford, CA 94305, USA.
2. Medical Scientist Training Program, Stanford University School of Medicine, Stanford, CA 94305, USA.
3. Department of Biology, Stanford University, Stanford, CA 94305, USA.
4. Division of Cardiovascular Medicine, Department of Medicine, Stanford University School of Medicine, Stanford, CA 94305, USA.
5. Department of Orthopaedic Surgery, Stanford University School of Medicine, Stanford, CA 94305, USA.
Received 2015-6-3; Accepted 2016-4-18; Published 2016-5-23
Abstract

Peripheral arterial disease affects nearly 202 million individuals worldwide, sometimes leading to non-healing ulcers or limb amputations in severe cases. Genetically modified stem cells offer potential advantages for therapeutically inducing angiogenesis via augmented paracrine release mechanisms and tuned dynamic responses to environmental stimuli at disease sites. Here, we report the application of nanoparticle-induced CXCR4-overexpressing stem cells in a mouse hindlimb ischemia model. We found that CXCR4 overexpression improved stem cell survival, modulated inflammation in situ, and accelerated blood reperfusion. These effects, unexpectedly, led to complete limb salvage and skeletal muscle repair, markedly outperforming the efficacy of the conventional angiogenic factor control, VEGF. Importantly, assessment of CXCR4-overexpressing stem cells in vitro revealed that CXCR4 overexpression induced changes in paracrine signaling of stem cells, promoting a therapeutically desirable pro-angiogenic and anti-inflammatory phenotype. These results suggest that nanoparticle-induced CXCR4 overexpression may promote favorable phenotypic changes and therapeutic efficacy of stem cells in response to the ischemic environment.
Keywords: CXCR4
Introduction
Peripheral arterial disease (PAD) affects nearly 202 million individuals worldwide [1]. It is characterized by reduced blood flow to the limbs, leading to pain, non-healing ulcers, or even limb amputation in severe cases [2, 3]. Treating severe PAD generally involves surgical revascularization [2, 3], but many patients suffering from PAD have comorbidities that prevent them from undergoing surgery. Therapeutic angiogenesis, an alternative treatment option for PAD, relies on the delivery of drugs or cells to promote blood vessel repair. Compared to conventional drug-based therapy, cell-based therapeutics may offer additional advantages given their ability to secrete paracrine signals and to respond dynamically to environmental stimuli at disease sites [4]. Stem cells are particularly attractive options for treating PAD, either by differentiating into cell types that make new blood vessels or indirectly by secreting paracrine signals to promote blood-vessel growth in situ [4]. However, transplanting stem cells alone has met with variable clinical success, and is often limited by poor cell engraftment and insufficient paracrine signaling.
Gene therapy has the potential to overcome these issues by overexpressing desirable therapeutic factors to promote angiogenesis. Upregulation of vascular endothelial growth factor (VEGF) has been shown to improve therapeutic angiogenesis achieved by endothelial progenitor cells [5] or adipose-derived stem cells (ADSCs) [6]. One limitation of previous strategies is the need for viral vectors to achieve efficient gene delivery; these vectors are associated with potential safety concerns for clinical translation [7]. Polymer-mediated gene delivery offers a potentially safer alternative, and has been shown to be effective in overexpressing VEGF in bone marrow-derived mesenchymal stem cells (BM-MSCs). Direct injection of VEGF-overexpressing MSC improved angiogenesis in a murine model of hindlimb ischemia, with up to 50% limb salvage [8]. However, VEGF exposure can yield leaky vasculature and promote exuberant inflammation [9], which may underlie incomplete tissue salvage. Further, patients with PAD often have comorbidities such as obesity and diabetes, and obtaining sufficient numbers of BM-MSCs is challenging [10]. Thus, there remains a strong need to identify alternative therapeutic gene targets and an alternative autologous cell population that is abundantly available from PAD patients, which promotes optimal therapeutic angiogenesis and tissue regeneration.
C-X-C chemokine receptor 4 (CXCR4) is a G protein-coupled receptor known to be involved in cell homing toward ischemia [11]. CXCR4 is expressed on progenitor and inflammatory cells, facilitating their migration toward ischemic tissues to participate in revascularization and tissue repair [12]. The ligand for CXCR4 is stromal cell-derived factor 1α, which is upregulated in hypoxic tissues and is secreted under the control of the transcription factor hypoxia-inducible factor 1α [11-13]. Due to its involvement in hypoxia signaling, the overexpression of CXCR4 by stem cells may enhance stem cell migration to tissues affected by ischemia in vivo. Notably, most prior approaches have focused on upregulating single growth factors, whereas overexpression of a cellular receptor such as CXCR4 may sensitize the stem cell to the environment and cause it to respond appropriately.
Compared to BM-MSCs, ADSCs are a much more abundant autologous cell source with similar phenotypes to BM-MSCs [14]. Clinically, ADSCs can be isolated in a minimally invasive manner using liposuction, with higher yields than can be achieved with BM-MSCs. ADSCs have been shown to promote therapeutic angiogenesis and tissue repair in animal models of hindlimb ischemia [15] and myocardial ischemia [16], possibly by secreting pro-angiogenic and anti-apoptotic cytokines [17]. Previously, we reported a polymer-mediated gene delivery method for overexpressing VEGF in ADSCs using poly(β-amino ester) (PBAE) nanoparticles, and demonstrated that paracrine release from these cells enhanced proliferation and tube formation by endothelial cells cultured in vitro [18].
In the present study, we sought to assess the efficacy of overexpressing CXCR4 in ADSCs for therapeutic angiogenesis and tissue regeneration using a murine model of PAD (Fig. 1A). We hypothesized that CXCR4 overexpression would promote ADSC survival and engraftment to the ischemic tissue in vivo, and that co-expression of CXCR4 and VEGF would further enhance angiogenesis. We first developed CXCR4- and/or VEGF-overexpressing ADSCs using PBAE nanoparticles, then assessed therapeutic efficacy in vivo by monitoring cell survival, blood reperfusion and limb salvage over time, tissue histology in the regenerated limb tissue, and the in vivo angiogenic and inflammatory response. Finally, the effects of CXCR4 overexpression on the paracrine signaling of ADSCs were characterized in vitro to help unravel the mechanisms underlying the observed tissue regeneration.
Development of CXCR4- and/or VEGF-overexpressing mouse ADSCs using PBAE nanoparticles. (A) Schematic of experimental design. Briefly, mouse ADSCs were transfected ex vivo with PBAE nanoparticles carrying plasmid DNA (pDNA) encoding human genes for hCXCR4 and/or hVEGF. Transfected ADSCs were transplanted into murine models of hindlimb ischemia to assess their ability to promote therapeutic angiogenesis. CXCR4 overexpression in mouse ADSCs was verified with qRT-PCR (B) and by western blot (C) at 48 h post-transfection. (D) qRT-PCR demonstrated that hCXCR4 mRNA overexpression was transient, peaking at 48 h and declining over 8 days. VEGF overexpression was also verified through qRT-PCR (E) and ELISA (F) at 48 h. (G) Similarly, increased VEGF release was transient as demonstrated by VEGF ELISA: peak release occurred over the first 48 h and declined to basal levels by 10 days. Abbreviations: C: CXCR4; CV: CXCR4/VEGF; V: VEGF; G: GFP; rel.: relative; conc.: concentration; WT: wild-type. All data are reported as mean ± standard error, n=3. *p<0.05 and **p<0.01 compared with GFP-ADSC controls. #p<0.05.
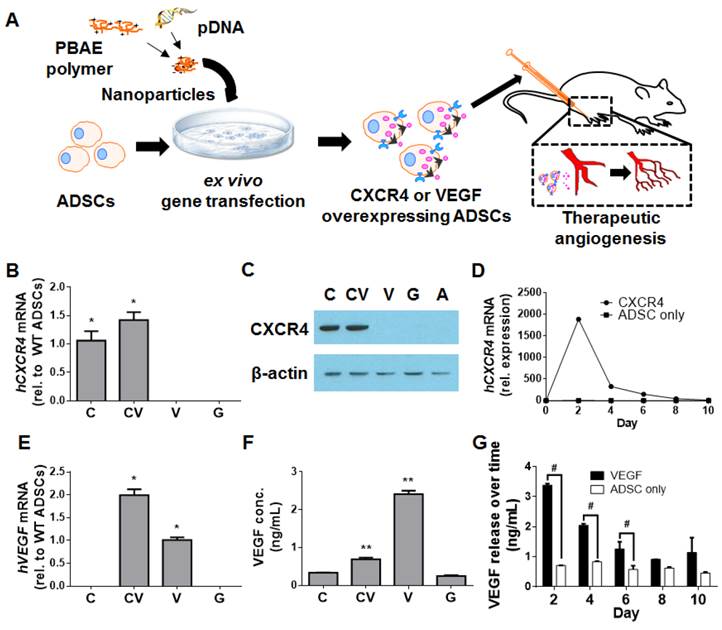
Results
Polymeric nanoparticles led to efficient CXCR4 and VEGF overexpression in ADSCs
To generate CXCR4- and/or VEGF-overexpressing ADSCs, poly(β-amino ester) (PBAE)-based nanoparticles containing plasmid DNA for these respective genes were applied to ADSCs ex vivo (Fig. 1A). Overexpression was verified at the mRNA level with the quantitative reverse transcription polymerase chain reaction (qRT-PCR) and at the protein level with the enzyme-linked immunosorbent assay (ELISA), western blotting, and fluorescence-activated cell sorting (FACS). The transfection efficiency achieved using PBAE nanoparticles in mouse ADSCs was assessed using wild-type mouse ADSCs with the reporter gene enhanced green fluorescent protein (GFP), which demonstrated that PBAE nanoparticles significantly improved transfection efficiency (19.9±2.0%) compared to controls (5.4±0.8%; p<0.05; Fig. S1A-S1B) with minimal effects on ADSC viability (Fig. S1C). For all remaining experiments, transfections were carried out with GFP-positive/luciferase-positive (GFP(+)/Luc(+)) mouse ADSCs; these cells enabled live cell tracking during animal experiments.
Successful CXCR4 overexpression in ADSCs transfected using PBAE nanoparticles was confirmed with qRT-PCR (Fig. 1B) and western blotting (Fig. 1C). Quantification of CXCR4 mRNA levels over 10 days revealed that hCXCR4 expression peaked at 48 h post-transfection and remained high over 8 days (Fig. 1D). To assess the level of cell-surface expression of the transmembrane receptor CXCR4, FACS was performed; surface expression was significantly greater in CXCR4-transfected cells than in GFP-transfected controls (1.41±0.02% vs. 0.9±0.11%, respectively; p<0.05; Fig. S2). This subtle but significant effect suggests that CXCR4 protein is located predominantly within the cell, as reported previously [19].
VEGF overexpression was similarly confirmed through qRT-PCR (Fig. 1E) and ELISA (Fig. 1F). As with CXCR4, VEGF release peaked at 48 h and was significantly higher in VEGF-transfected ADSCs (2.41±0.19 ng/mL) and in CXCR4/VEGF-cotransfected ADSCs (0.69±0.09 ng/mL) than in GFP-transfected controls (0.25±0.4 ng/mL; p<0.05, one-way analysis of variance; Fig. 1F). VEGF release from VEGF-overexpressing ADSCs remained significantly elevated over the first 6 days (p<0.05; Fig. 1G).
CXCR4 overexpression enhanced ADSC engraftment and prolonged cell survival in ischemic limb muscles
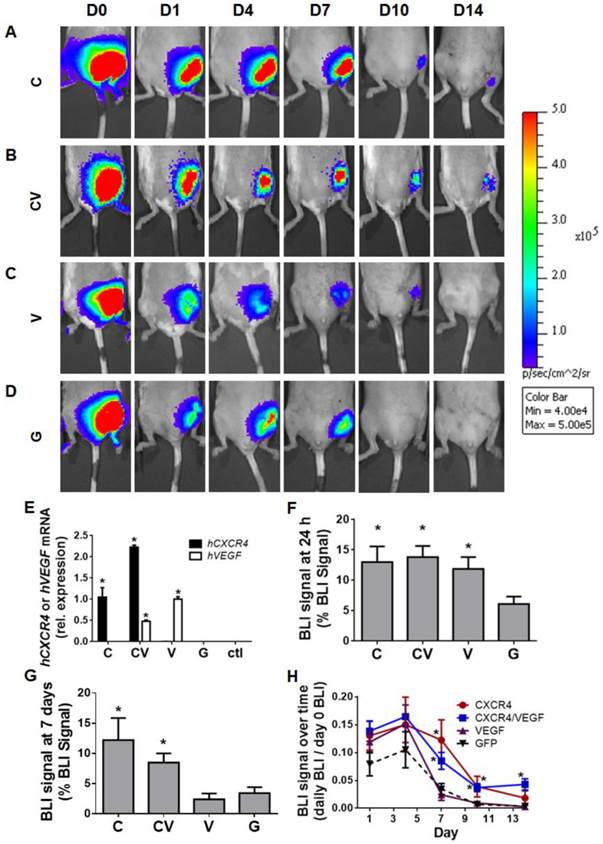
To assess the effects of CXCR4 overexpression on ADSC survival in vivo, GFP(+)/Luc(+) ADSCs were injected into the medial muscles of a murine model of hindlimb ischemia (Fig. 2A-2D). GFP-transfected ADSCs were used as a control for PBAE-mediated transfection. Genetic upregulation of the human genes encoding hCXCR4 and/or hVEGF in transplanted ADSCs was initially confirmed by harvesting ischemic tissues on day 4 and subjecting them to qRT-PCR (Fig. 2E). This verification further confirmed that transplanted ADSCs survived in vivo at least through day 4. Bioluminescence imaging (BLI) was performed to assess ADSC survival over time (Fig 2A-2D). The engraftment of CXCR4-, VEGF-, and CXCR4/VEGF-overexpressing ADSCs was significantly greater than that of GFP-transfected controls (13.00±6% combined for the CXCR4-, VEGF-, and CXCR4/VEGF-transfected groups vs. 6.11±3% for the GFP-transfected group at 24 h; p<0.05; Fig. 2F). For all groups, BLI signal peaked at day 4 (Fig. 2H), indicating that the transplanted cells recovered and proliferated. At day 7, BLI signal for all groups began to decline, but the signal in the CXCR4- and CXCR4/VEGF-transfected groups remained relatively high (11.20±8.8% combined for the CXCR4- and CXCR4/VEGF-overexpressing groups) compared to the GFP-transfected groups (2.96±2.6%; p<0.05; Fig. 2G). By day 10, BLI signal was only detected in the CXCR4- and CXCR4/VEGF-overexpressing groups (Fig. 2H), demonstrating improved cell survival due to CXCR4-overexpression.
CXCR4-overexpressing ADSCs displayed improved cell engraftment and survival in murine models of hindlimb ischemia. (A-D) Representative BLI of mouse ischemic hindlimbs transplanted with GFP(+)/Luc(+) mouse ADSCs transfected with CXCR4 (A), CXCR4/VEGF (B), VEGF (C), or GFP (D) over 14 days post-ischemia induction. (E) Confirmation of human CXCR4 and human VEGF overexpression in transplanted ADSCs by qRT-PCR performed on harvested tissue at 4 days post-procedure (data are expressed as mRNA expression relative to single transfection with hCXCR4 or hVEGF). Percentage of BLI signal remaining from transplanted cells measured 24 h (F) and 7 days (G) after cell transplantation. (H) Quantitative BLI signal of transplanted GFP(+)/Luc(+) ADSCs demonstrates prolonged cell survival by CXCR4-overexpressing groups over 14 days post-ischemia induction (expressed as percent of BLI signal relative to day 0 signal). Abbreviations: C: CXCR4; CV: CXCR4/VEGF; V: VEGF; G: GFP; rel.: relative; h: human. All data are reported as mean ± standard error, n=8. *p<0.05 compared with GFP-ADSC controls.
CXCR4-overexpressing ADSCs led to faster blood reperfusion and 100% limb salvage in vivo
Therapeutic efficacy was evaluated by monitoring functional blood reperfusion and limb outcome. To monitor functional blood reperfusion, laser Doppler perfusion imaging was performed weekly (Fig. 3A-3D, Fig. S3) beginning on the day of surgery to confirm the induction of hindlimb ischemia (Fig. 3A-3D). By day 7, CXCR4-overexpressing groups displayed faster blood reperfusion than VEGF- and GFP-transfected groups (p<0.05 compared to GFP; Fig. 3F). Relative reperfusion levels after 14 days did not significantly differ from each other (Fig. S3), indicating that the host regenerative response likely dominated the effects of the transplanted cells beyond this time point. Consequently, the faster blood reperfusion response exhibited by CXCR4 overexpression was associated with improved limb salvage.
Early reperfusion and enhanced therapeutic efficacy in ischemic hindlimbs. (A-D) Representative laser Doppler perfusion imaging of ischemic hindlimbs transplanted with mouse ADSCs transfected with CXCR4 (A), CXCR4/VEGF (B), VEGF (C), or GFP (D) over 14 days post-ischemia induction. (E) Gross observation of hindlimbs at day 14 post-procedure demonstrating enhanced limb salvage by CXCR4 overexpression (red circles indicate ischemic limbs). (F) Quantification of blood reperfusion at day 7 demonstrating significantly enhanced early perfusion by CXCR4-overexpressing ADSCs. (G) Quantification of limb outcome demonstrates 100% limb salvage by CXCR4 overexpression and improved limb salvage by VEGF and CXCR4/VEGF in comparison to GFP controls. Abbreviations: C: CXCR4; CV: CXCR4/VEGF; V: VEGF; G: GFP. All data are reported as mean ± standard error, n=8. *p<0.05 compared with GFP-ADSC controls.
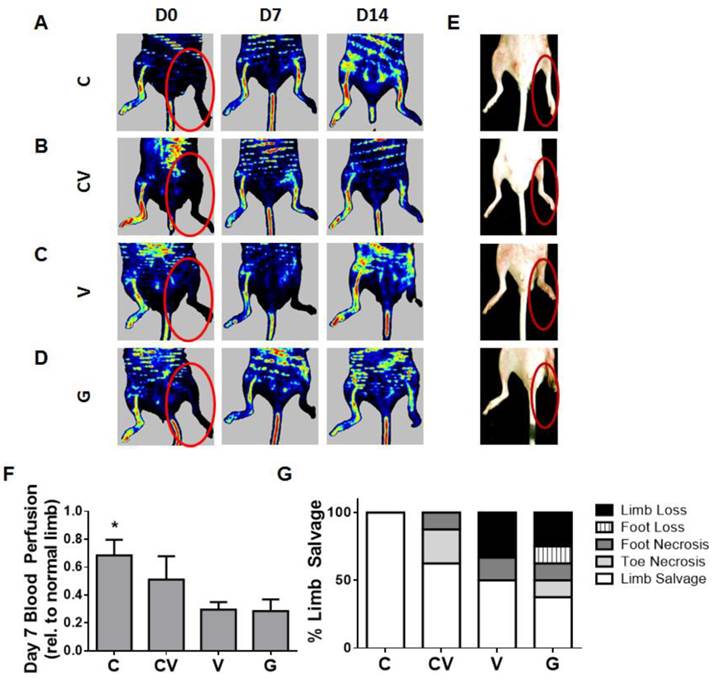
Importantly, CXCR4 overexpression led to 100% limb salvage in all tested animals (n=8), which was the best therapeutic response in all groups (Fig. 3E, 3G). Co-overexpression with CXCR4/VEGF also improved limb salvage compared to the VEGF- and GFP-overexpressing groups (Fig. 3G), although two mice suffered toe necrosis and one mouse suffered foot necrosis. VEGF overexpression led to 50% limb salvage (Fig. 3G), which was greater than the limb salvage exhibited by mice transfected with GFP-control constructs, but VEGF overexpression also led to the highest amount of limb loss (Fig. 3G).
Muscle regeneration improved in ischemic hindlimbs with CXCR4-overexpressing ADSCs
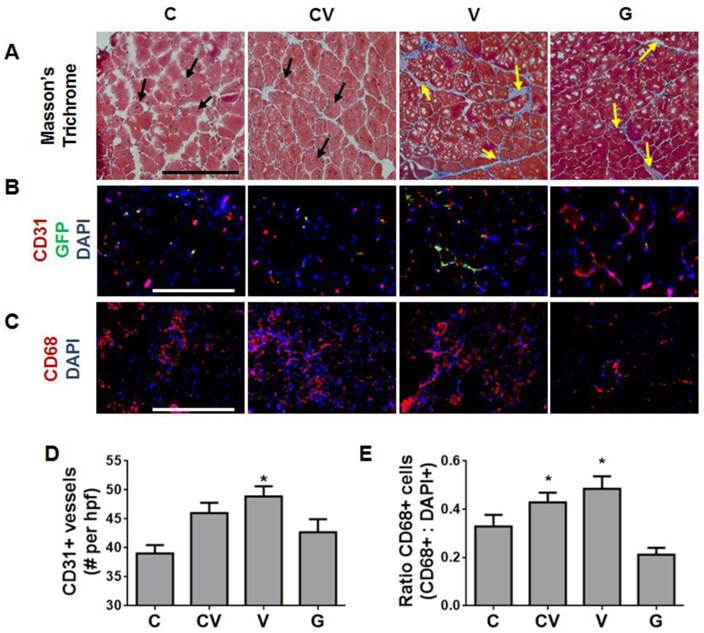
Muscle histology was performed to determine the health of ischemic limbs at 4 weeks after ischemia induction and cell transplantation (Fig. 4A). Sections were stained with Masson's trichrome and assessed for muscle regeneration or damage, which manifested as fibrosis. Regenerating muscle fibers were observed in the CXCR4- and CXCR4/VEGF-transfected groups, as indicated by the presence of centrally located nuclei (Fig. 4A). This observation suggested that these tissues had reached the late stage of muscle regeneration. In contrast, few regenerating muscle fibers were noted in the VEGF-transfected group; instead, classic fibrosis was observed throughout, as manifested by the increased collagen (stained blue) between muscle fibers (Fig. 4A). Similarly, much fibrosis was observed in the GFP-transfected group (Fig. 4A).
Enhanced muscle regeneration and modulated host angiogenic and inflammatory response by CXCR4-overexpressing ADSCs. (A) Representative tissue sections at 4 weeks after ischemia induction stained with Masson's trichrome demonstrating enhanced muscle fiber regeneration (black arrows) from CXCR4 overexpression and prolonged muscle fibrosis (yellow arrows) from VEGF and GFP overexpression (scale bar: 500 μm). (B) Immunostaining of tissue sections at 4 weeks after ischemia induction for capillaries (red, anti-CD31), transplanted cells (green, anti-GFP), and nuclei (blue, 4',6-diamidino-2-phenylindole (DAPI)) (scale bar: 250 μm). VEGF overexpression, not CXCR4 overexpression, was associated with enhanced capillary density. Transplanted stem cells that remained in tissues engrafted into capillaries, as demonstrated by overlapping GFP and CD31 staining. (C) Immunostaining of tissue sections at 4 days after transplantation for macrophages (red, anti-CD68) and nuclei (blue, DAPI) (scale bar: 500 μm). VEGF overexpression was associated with increased macrophage infiltrate. (D) Quantification of CD31-positive capillaries per high-powered field (hpf) showing increased capillary density in animals with ADSCs overexpressing VEGF. (E) Ratio of CD68-positive macrophages demonstrating increased inflammation due to VEGF overexpression. Abbreviations: C: CXCR4; CV: CXCR4/VEGF; V: VEGF; G: GFP. All data are reported as mean ± standard error, n=4 for week 4 histological analysis and quantification and n=3 for day 4 analysis. *p<0.05 compared with GFP-ADSC controls.
VEGF overexpression, but not CXCR4 overexpression, enhanced vessel density and inflammation in vivo
Given that angiogenesis and inflammation are important processes in muscle repair, we immunostained ischemic tissue sections for capillaries (anti-CD31) and macrophages (anti-CD68). Transplanted cells were immunostained with anti-GFP to determine their presence and distribution (Fig. 4B). By 4 weeks after transplantation, most regenerated nascent capillaries arose from endogeneous cells (Fig. 4B). Very few transplanted cells remained; those that did remain were located in close proximity to newly formed capillaries (Fig. 4B). These results suggest that transplanted cells likely contribute to new blood-vessel formation mostly via paracrine signaling rather than via direct differentiation and incorporation. To gauge the magnitude of the angiogenic response, capillaries were quantified at 4 weeks. VEGF-overexpressing ADSCs displayed the largest increase in vessel density (48.9±10 vessels per high-powered field) over GFP-transfected controls (42.7±2 vessels per high-powered field; p<0.05; Fig. 4D). Co-transfection with CXCR4 and VEGF also yielded increased vessel density, but this effect was not significant (Fig. 4D). Interestingly, vessel density in the CXCR4-transfected group did not significantly differ from density in the GFP-transfected controls (Fig. 4D).
Acute inflammation in response to ischemic injury has been reported to peak around days 3-7 [20]. As such, day-4 tissue sections were stained for CD68, a marker for macrophages, the major inflammatory cell type that responds to injuries. This analysis revealed that VEGF-overexpressing ADSCs substantially increased the acute host inflammatory response in ischemic muscles, as shown by the increased percentage of CD68-positive cells in mice transplanted with VEGF-overexpressing ADSCs (Fig. 4C). Both VEGF- and CXCR4/VEGF-overexpressing ADSCs prompted a significant increase in the number of CD68-positive macrophages compared to GFP-transfected controls (p<0.05; Fig. 4E). In contrast, CXCR4-overexpressing ADSCs did not significantly change the number of CD68-positive cells compared to GFP-transfected controls (Fig. 4E).
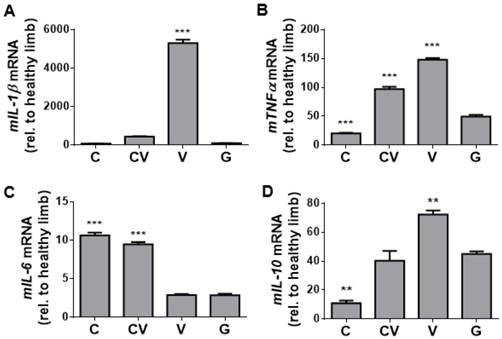
To complement our immunostaining-based assessment of inflammation, qRT-PCR for genes encoding the inflammatory cytokines mIL-1β (Fig. 5A), mTNF-α (Fig. 5B), mIL-6 (Fig. 5C), and mIL-10 (Fig. 5D) was performed on day-4 harvested muscle tissues. Consistent with the trend of staining for CD68 (Fig. 4E), the VEGF-transfected group displayed significant increases in the expression of genes encoding the pro-inflammatory cytokines mIL-1β (p<0.001; Fig. 5A) and mTNF-α (p<0.001; Fig. 5B) compared to GFP-transfected controls, while co-transfection with CXCR4/VEGF led to significant increases in the expression of mTNF-α only (p<0.001; Fig. 5B). Expression of mIL-6 was significantly upregulated in both CXCR4-transfected and CXCR4/VEGF-transfected groups compared to VEGF-transfected and GFP-transfected groups (p<0.001; Fig. 5C), indicating that CXCR4 plays a role in mIL-6 upregulation. Expression of the gene encoding the anti-inflammatory cytokine mIL-10 (Fig. 5D) followed trends that were similar to mTNF-α (Fig. 5B), suggesting that this upregulation may occur in response to increases in the expression of the genes encoding pro-inflammatory cytokines.
Inflammatory assessment of ischemic hindlimbs. (A-B) qRT-PCR of tissue harvested at day 4 for the genes encoding the inflammatory cytokines mIL-1β (A), mTNFα (B), mIL-6 (C), and mIL-10 (D). Significantly increased levels of mIL-1β and mTNF-α indicate an increased inflammatory response due to VEGF overexpression, but this increase was absent from CXCR4-overexpressing and GFP-control tissues. Increased levels of mIL-6 indicate that a more complex anti- and pro-inflammatory response may be occurring due to CXCR4 overexpression. The cytokine mIL-10 is an anti-inflammatory marker that may be elevated in response to the inflammatory response associated with VEGF overexpression. Abbreviations: C: CXCR4; CV: CXCR4/VEGF; V: VEGF; G: GFP; rel. relative. All data are reported as mean ± standard error, n=3. **p<0.01, ***p<0.001 compared with GFP-ADSC controls.
CXCR4 overexpression modulated ADSC paracrine signaling by promoting a pro-angiogenic and anti-inflammatory phenotype in hypoxic culture
To elucidate how CXCR4 overexpression may lead to the observed robust tissue regeneration under ischemia, we cultured transfected ADSCs under hypoxia in vitro and assessed changes in the gene expression of angiogenesis and inflammation markers. Interestingly, compared to GFP-transfected cells and untransfected cells, ADSCs overexpressing CXCR4 were associated with the upregulation of genes encoding the mouse angiogenic factors mVEGF (p<0.05), basic fibroblast growth factor (mFGF, p<0.05), and hepatocyte growth factor (mHGF, p<0.05; Fig. 6A). In terms of inflammatory cytokines, CXCR4 overexpression led to significant decreases in the expression of mIL-1β (p<0.05) and mIL-6 (p<0.05) compared to GFP-transfected cells and untransfected cells (Fig. 6B). These trends were not detected for the CXCR4/VEGF-transfected or VEGF-transfected groups (Fig. 6A, 6B).
CXCR4-based promotion of a pro-angiogenic and anti-inflammatory phenotype in ADSCs under hypoxia. Transfected mouse ADSCs were cultured under hypoxia in vitro and assessed after 48 h via qRT-PCR of genes encoding markers of angiogenesis (A) and inflammation (B) (measurements are relative to ADSCs cultured under normoxia). Hypoxia-cultured ADSCs overexpressing CXCR4 had increased expression of angiogenic genes (mVEGF, mFGF, mHGF) and decreased gene expression of inflammatory markers (mIL-1 and mIL-6). (C) Enhanced proliferation of human umbilical vein endothelial cells (HUVECs) occurred in vitro after exposure to paracrine release from ADSCs overexpressing CXCR4. (D) Paracrine release from VEGF-overexpressing, but not CXCR4-overexpressing ADSCs led to increased THP-1 macrophage migration in vitro. (E) Proposed model for a CXCR4-mediated therapeutic response to tissue ischemia. SDF-1α (red) is secreted from sites of ischemia, which activates CXCR4 (blue) on the surfaces of transfected ADSCs to promote ischemia-specific responses in angiogenesis and inflammation. Abbreviations: C: CXCR4; CV: CXCR4/VEGF; V: VEGF; G: GFP; rel.: relative; m, mouse. All data are reported as mean ± standard error, n=8. *p<0.05, **p<0.01, ***p<0.001 compared with GFP-ADSC controls.
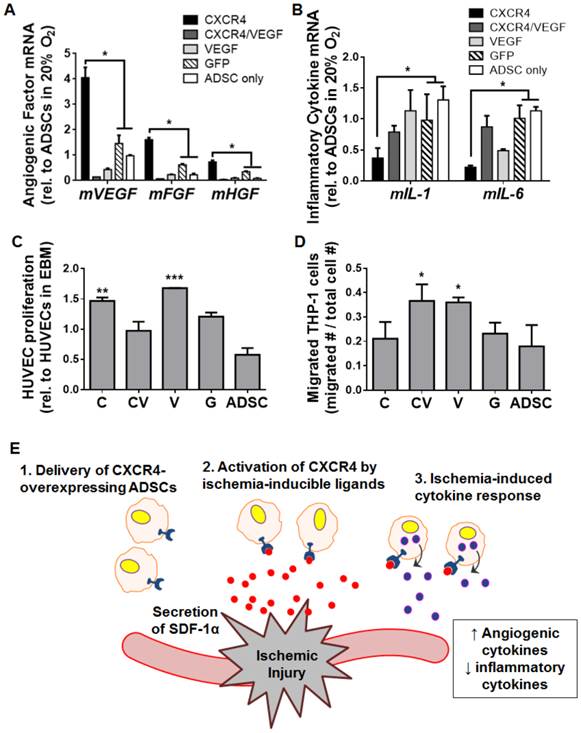
To further assess the changes in ADSC paracrine release due to CXCR4 overexpression, we applied paracrine molecules released from transfected ADSCs to human umbilical vein endothelial cells and a macrophage cell line (THP-1) in vitro. Both CXCR4-overexpressing ADSCs and VEGF-overexpressing ADSCs increased the proliferation of endothelial cells compared to controls (p<0.01 and p<0.001 for CXCR4- and VEGF-overexpressing ADSCs, respectively), which was not seen after co-transfection with CXCR4-VEGF (Fig. 6C). Macrophage migration was significantly enhanced by VEGF- and CXCR4/VEGF-transfected ADSCs (p<0.05) compared to controls (p<0.05). CXCR4-ADSCs did not lead to marked changes in macrophage migration (Fig. 6D). These results indicate that CXCR4 overexpression may promote a pro-angiogenic and anti-inflammatory phenotype of ADSCs in response to hypoxia (Fig. 6E), which may underlie a desirable therapeutic outcome.
Discussion
PAD is a significant problem worldwide that continues to increase in prevalence [1]. Effective treatment for PAD remains lacking due to co-morbidities in most PAD patients that prohibit them from undergoing surgical revascularization. Stem cells hold promise for treating PAD via stimulating therapeutic angiogenesis, but the use of stem cells alone remains inconsistently efficacious, likely due to poor cell engraftment and insufficient paracrine signaling [21]. Genetic engineering can improve stem cell therapy by enhancing cellular processes related to cell survival, migration, and paracrine signaling [4]. One interesting molecule known to mediate these processes is CXCR4, which contributes to stem cell homing in ischemia and other injuries [11, 12]. In this study, we found that CXCR4 overexpression enhanced the engraftment and survival of ADSCs (Fig. 2F, 2G), which subsequently led to more rapid restoration of blood flow (Fig. 3F) and 100% limb salvage in a murine model of PAD (Fig. 3G). Since our approach relied on a readily available stem cell population and a non-viral transfection agent, it represents a potentially clinically translatable method for treating PAD.
One of the main goals for treating PAD is the prevention of ischemic tissue damage; thus, the most interesting result of our study was that CXCR4-overexpressing ADSCs led to 100% limb salvage in all animals examined (Fig. 3G). In comparison, VEGF-overexpressing ADSCs resulted in 50% limb salvage (Fig. 3G), which is comparable to previously reported results using VEGF-transfected endothelial progenitor cells [5]. Histology (Fig. 4A) further demonstrated that CXCR4 overexpression accelerated the regeneration of new muscle fibers in affected ischemic limb tissues 28 days after cell transplantation, which was not observed in the VEGF- or GFP-overexpressing groups (Fig. 4A). These findings demonstrate that CXCR4-overexpressing ADSCs exert a beneficial effect in promoting tissue salvage in ischemic tissues in vivo.
In terms of the mechanism of repair of ischemic damage, CXCR4-overexpressing ADSCs likely promote an early angiogenic response and moderate inflammation. CXCR4-overexpressing ADSCs were associated with a robust reperfusion response by day 7 (Fig. 3F), which aligned with the host healing response by day 14 (Fig. S3). Histological sections at day 28 revealed fewer capillaries in the CXCR4-overexpression group than in the VEGF-overexpressing group (Fig. 4B), which may suggest that the tissue-repair process had returned to baseline in animals treated with CXCR4-overexpressing ADSCs. This phenomenon could be due to the host's natural response to ischemic injury: blood supply increases above normal levels during healing, and then gradually returns to pre-ischemic levels upon complete repair [22]. The early inflammatory phase of healing is a necessary process that can damage tissue if present at excessive levels. We found that CXCR4-transfected ADSCs tended to moderate inflammation, while VEGF-transfected ADSCs led to an exuberant and likely damaging inflammatory response, as indicated by increases in the expression of genes encoding the inflammatory cytokines mIL-1β (Fig. 5A) and mTNF-α (Fig. 5B) and in the number of CD68-positive macrophages (Fig. 4E). These findings were further supported by our in-vitro data, in which paracrine release from CXCR4-transfected ADSCs cultured under hypoxia promoted endothelial cell proliferation (Fig. 6C) and decreased THP-1 macrophage migration (Fig. 6D). Differences were evident in the inflammatory cytokine profile of IL-6 between mouse whole tissue (Fig. 5C) and transplanted cells (Fig. 6B), which likely reflects differences in the host response versus the phenotypic response of the cells.
Cell engraftment and survival after transplantation remain major challenges for stem cell-based therapeutics. Our study suggests that CXCR4 overexpression may improve both of these processes. Here, CXCR4-overexpressing ADSCs, including CXCR4- and CXCR4/VEGF-transfected ADSCs, exhibited high BLI signal compared to controls at 24 h, a trend that persisted over 14 days (Fig. 2B). CXCR4 is a G protein-coupled receptor known to activate multiple intracellular signaling cascades that lead to cell migration and cell survival [23]; thus, our findings suggest a strategy for utilizing CXCR4 to overcome issues related to cell engraftment and survival. The improvements in cell engraftment and survival observed here likely contributed to the early angiogenic response achieved by CXCR4-overexpressing ADSCs, which then promoted tissue salvage.
To examine transfection efficiency, we characterized the overexpression of CXCR4 and VEGF via mRNA expression and protein secretion (Fig. 1B-1H). Gene expression represents a single time point (48 h), while protein secretion represents accumulated protein production over 48 h. The total amount of released protein is the more relevant quantity, as it is directly related to the amount of available paracrine signals in vivo. For CXCR4 overexpression, FACS revealed that cell-surface expression remained comparable to controls (Fig. 1D). Prior studies of CXCR4 expression in MSCs also found low cell-surface expression, and instead discovered high intracellular levels of CXCR4 [24]. Further studies found that intracellular CXCR4 could be rapidly induced to the cell surface by a cytokine cocktail [25], suggesting that the timing of CXCR4 response could be improved by increasing intracellular levels of the protein. Similarly, CXCR4 overexpression in ADSCs likely remained intracellular in the present study, as suggested by qRT-PCR, western blotting, and FACS. Considering the results from prior studies, we propose that overexpression of the gene encoding CXCR4 in ADSCs may enable enhanced receptor turnover; the continued expression of CXCR4 may prolong the cell's response to high SDF-1α levels.
For successful clinical translation of stem cell- and gene delivery-based therapies, there are several important bottlenecks to overcome, including (1) the need for a readily available and abundant autologous cell source, (2) the need for an efficient non-viral method for gene delivery, and (3) identification of gene targets for therapeutic efficacy. This study has shown promising results for overcoming these bottlenecks by using ADSCs [15, 17, 26, 27], by employing polymeric nanoparticles to achieve efficient gene delivery, and by identifying CXCR4 as a novel gene target to enhance stem cell survival and to accelerate angiogenesis. First, we chose ADSCs given their ease of isolation, their abundance as an autologous cell source, and their demonstrated benefit for angiogenesis in vivo [15, 17, 26, 27]. Second, we selected PBAE-based gene delivery because it is potentially safer than viral vectors, circumventing risks such as immunogenicity and insertional mutagenesis [7, 28]. Using high-throughput screening of polymeric vectors, we previously identified optimal polymeric vectors with high transfection efficiency [29, 30] for various human stem cells that are known to be difficult to transfect [31]. Unlike viral vectors, polymeric vectors transiently upregulate gene expression; the DNA does not integrate into the host genome [32], decreasing potential risks in humans. Third, we identified CXCR4 as a promising therapeutic gene for enhancing stem cell survival, accelerating blood reperfusion, and maximizing limb salvage. Together, the findings of this study are of great significance given the large population of patients suffering from ischemic diseases and the well-documented difficulties in modifying stem cells to achieve satisfactory therapeutic outcomes.
In the present study, the transplanted cells were a heterogeneous population, with ~20% of ADSCs transfected and 80% of ADSCs remaining naive. Regardless of the gene delivery approach (viral or non-viral), transfected cells are inherently heterogeneous populations, and they are often used directly after transfection. While it is possible to use FACS to select transfected cells prior to transplantation, it is undesirable for clinical translation due to additional time and cost requirements, as well as due to the potential negative impacts on cell viability and phenotype during sorting. Further, the heterogeneity of transplanted cells may actually facilitate tissue regeneration, as it enables the combination of paracrine signals secreted by naive stem cells as well as the enhanced paracrine signals prompted by gene delivery. We showed that transplantation of a heterogeneous population of ADSCs after CXCR4 transfection led to 100% limb salvage in the mouse hindlimb ischemia model (Fig. 3G). Our results confirm that ADSCs transfected using PBAE/CXCR4 polymeric nanoparticles without further purification are sufficient to induce satisfactory healing, which is highly desirable for clinical translation.
In our study, we intentionally chose to use a half dose of VEGF and CXCR4 plasmids for the co-transfected group (Fig. 1) in order to determine the most therapeutically efficacious genes to deliver in a clinical setting, given a single total DNA dosage. To establish fair comparisons of target gene upregulation, we characterized CXCR4 (Fig. 1B, C, D) and VEGF (Fig 1E, F) expression at the level of the gene and at the level of the protein. While the DNA plasmid dosage was half in the CXCR4/VEGF co-transfected group compared to either CXCR4 or VEGF alone, it led to expression of the gene encoding CXCR4 at a level comparable to that prompted by transfection of CXCR4 alone (Fig. 1B-1C). The co-transfected group displayed higher expression of the gene encoding VEGF at 48 h (Fig. 1E), but less accumulated VEGF protein release (Fig. 1F). Since we have reported complete characterization data (Fig. 1), our functional conclusions are based upon changes illustrated in Figure 1. Further, the CXCR4 and VEGF signaling pathways can influence each other [33], as confirmed by our data (Fig. 1, Fig. 6).
We have demonstrated the promise of CXCR4-overexpressing ADSCs for treating PAD in a mouse model of hindlimb ischemia. We chose this model because it is a well-established small animal model of PAD and because it has been widely used for assessing the efficacy of novel therapies for therapeutic angiogenesis. The promising results from the present study support the feasibility of this strategy; future work should evaluate the efficacy of CXCR4-overexpressing ADSCs in a large animal model of PAD that better mimics the scale of the human condition. Further studies in large animals are warranted to uncover modifications to the present platform that may be more relevant for human therapy. One advantage of our platform is that it requires only one injection to achieve 100% limb salvage in a murine hindlimb ischemia model (Fig. 3G). If necessary, multiple local cell injections could be carried out, increasing the dosage of paracrine signaling required for healing larger ischemic tissues in larger animals. Further, the current platform is a multi-step process involving cell isolation, expansion, and transfection ex vivo prior to transplantation, which contributes to higher cost and is subject to more stringent regulatory standards (Fig. 1A). To further lower the barriers for clinical translation, future research may explore the possibility of a one-step approach that enables direct transfection of freshly isolated cells in the operating room followed by direct transplantation back to the patient. Alternatively, it would be interesting to explore the possibility of implanting a drug-delivery depot into ischemic tissues to directly recruit and modify endogenous progenitor cells in situ, which may eliminate the need for cell isolation and modification ex vivo.
In summary, this study demonstrates that CXCR4-overexpressing ADSCs engineered with biodegradable polymeric nanoparticles promote therapeutic angiogenesis for the treatment of PAD. CXCR4 overexpression improves cell engraftment, prolongs cell survival, and leads to complete limb salvage and tissue regeneration in a mouse model of hindlimb ischemia. The concept of modulating cell-membrane receptors to promote a therapeutically desirable phenotype of transplanted stem cells may be broadly applicable to repairing other ischemic diseases such as myocardial infarction and stroke.
Materials and Methods
Materials
Monomers for synthesizing PBAEs were purchased from Sigma-Aldrich, Scientific Polymer Products, and Molecular Biosciences. Anhydrous dimethyl sulfoxide was purchased from Sigma-Aldrich. A buffer solution (pH 5.2) of 25 mM NaOAc was prepared by diluting 3 M stock solution (Sigma-Aldrich). Plasmids pEGFP-N1 and pcDNA3.1(+)-CXCR4 were purchased from Elim Biopharmaceuticals, and pVEGF165 plasmid was obtained from Aldevron.
ADSC isolation and culture
ADSCs were collected by harvesting inguinal fat pads from 5-6-week-old, GFP-positive/luciferase-positive (GFP(+)/Luc(+)), transgenic male mice (kindly provided by Dr. Joseph Wu, Stanford University) and Friend Virus B wild-type male mice (Charles River). All animals were handled in accordance with the Stanford University Animal Care and Use Committee Guidelines; all protocols were approved by the Stanford University Institutional Animal Care and Use Committee. After serial betadine washes, the fat pads were finely minced in cold, sterile phosphate-buffered saline and digested with 0.075% collagenase II (Sigma-Aldrich) at 37 °C for 30 min in a shaking water bath. Digestion was neutralized with Dulbecco's Modified Eagle Medium (Invitrogen) supplemented with 10% fetal bovine serum (Invitrogen) and 1% penicillin-streptomycin (Sigma-Aldrich). Neutralized cells were centrifuged at 4 °C and 1,000 rpm for 5 min, and floating adipocytes and media were removed before pelleted ADSCs from the stromal-vascular portion were resuspended in culture medium, passed through a 100-μm Falcon nylon cell strainer (Becton Dickinson), and plated on a 10-cm dish. Cells were incubated at 37 °C and 5% atmospheric CO2. Twenty-four hours after isolation, the cells were washed in phosphate-buffered saline (Invitrogen) to remove debris and nonadherent cells, and fresh medium was added to the cells. Medium was changed every 2 days, and adherent spindle-shaped cells were grown to subconfluence, passaged using standard methods of trypsinization, and propagated through passage 2.
Synthesis of PBAEs
PBAE polymers were synthesized as previously described [31]. Briefly, acrylate-terminated C32 polymer was prepared by mixing butanediol (C) and aminopentanol (32) monomers using a diacrylate/amine monomer molar ratio of 1.2:1 at 90 °C for 24 h. Subsequently, amine-terminated C32 polymers were generated by reacting acrylate-terminated C32 polymer with diamine monomer (122) in dimethyl sulfoxide. End-capping reactions were performed overnight at room temperature with a 1.6-fold molar excess of amine over acrylate end groups.
Transfection
ADSCs were transfected using PBAE nanoparticles containing three plasmids including CXCR4, VEGF, or GFP (control) using our previously optimized conditions [18]. Briefly, PBAE nanoparticles were formed by mixing PBAE polymer C32-122 with plasmids at 30:1 polymer:DNA ratio with a DNA dose of 4.5 µg in 25 mM NaAc. For transfection with both CXCR4 and VEGF, the two plasmids were combined for a total dose of 4.5 µg (a half-dose of each plasmid). After 10 min of incubation, nanoparticles were added to ADSCs cultured in Dulbecco's Modified Eagle Medium containing 10% fetal bovine serum. After 4 h of incubation, the medium was replaced with fresh growth medium. For in vitro assays, ADSCs were transfected at 3.0 x 105 cells per well in 6-well plates (n=3/group). For animal studies, ADSCs were transfected in T150 tissue culture flasks at 3.5 x 106 cells per flask (1.0 x 106 cells were used per mouse). Cells were cultured for an additional 2 h in fresh growth medium prior to use for animal experiments. Cell viability/proliferation was evaluated at 48 h post-transfection using the Cell Titer 96 Aqueous One Solution Assay (Promega), and results were normalized by absorbance value from untransfected ADSCs (n=4 per sample).
FACS for ADSC transfection efficiency and CXCR4 cell-surface expression
To verify transfection efficiency with PBAE nanoparticles, GFP expression by transfected wild-type ADSCs was measured 48 h after transfection via FACS on a BD FACScalibur (BD Biosciences). Dead cells were excluded from the analysis by staining with propidium iodide (Life Technologies); 10,000 live cells per sample were included in the analysis. The percentage of GFP-positive cells was quantified using Flowjo 7.6 (Tree Star, Inc.).
qRT-PCR
To confirm the transfection of CXCR4 and VEGF, total RNA was harvested from ADSCs at 48 h post-transfection using the RNeasy Kit (Qiagen). cDNA was synthesized via reverse transcription with the Superscript First-Strand Synthesis System (Invitrogen) and 1 μg of total RNA. qRT-PCR was performed using Power SYBR Green PCR Master Mix (Life Technologies) following the manufacturer's protocol; all samples underwent 40 cycles on an Applied Biosystems 7900 Real-Time PCR System. The relative expression level of target genes was determined by the ΔΔCT method [34]. Target gene expression was first normalized to an endogenous housekeeping gene (the gene encoding mouse glyceraldehyde 3-phosphate dehydrogenase, mGAPDH), followed by a second normalization to the expression level measured in untransfected control cells. Upregulation of mouse angiogenic and inflammatory genes was confirmed by isolating total RNA from ADSCs cultured under 1% O2 for 48 h, then synthesizing cDNA and performing qRT-PCR. Measurement of mRNA extracted from ischemic muscle was performed by digesting muscle with TRIzol Reagent (Invitrogen) and proceeding through RNA extraction, cDNA synthesis, and qRT-PCR as described above. Four replicates were assayed per sample. All primers used in this study are listed in Table S1.
Measurement of VEGF production via ELISA
VEGF secretion by mouse ADSCs was measured in conditioned medium with the Human VEGF Standard ELISA Development Kit (Peprotech) according to the manufacturer's protocol. VEGF production was measured on days 2, 4, 6, and 8. Cumulative VEGF release was calculated by summing the amounts of VEGF released at all previous time points. Secreted VEGF (in vitro) was expressed as ng of VEGF per mL of supernatant. Three replicates were assayed per sample.
Determination of CXCR4 upregulation by western blotting
CXCR4 protein production was determined by western blotting. Total protein was isolated utilizing radioimmunoprecipitation assay buffer (Thermo Scientific). Proteins were separated via gel electrophoresis on NuPage Novex Bis-Tris Mini Gels (Invitrogen) at 200 V for 35 min. Separated proteins were transferred to a polyvinylidene difluoride membrane (Invitrogen) via the XCell II Blot Module (Invitrogen) at 30 V for 1 h. To assess CXCR4 expression, blotted membranes were stained for anti-human CXCR4 (product ab124824, 1:100 dilution, Abcam) utilizing a horseradish peroxidase-labeled secondary antibody (product ab6721, 1:10000 dilution, Abcam). Staining with anti-β actin (product ab8227, 1:2000 dilution, Abcam) served as a loading control. X-ray films were developed with the chemiluminescent reagent SuperSignal West Pico (Thermo Scientific). Three replicates were performed per assay.
Transplantation of stem cells into a mouse model of hindlimb ischemia
All animal experiments were performed in accordance with the Stanford University Animal Care and Use Committee Guidelines and approved Animal Panel on Laboratory Animal Care protocols. Hindlimb ischemia was induced in female Friend Virus B mice aged 10-12 weeks (Charles River), as previously described [35, 36]. Briefly, the femoral artery was ligated at two sites (distal to the external iliac artery and proximal to the popliteal artery) through a skin incision with 5-0 silk suture (Ethicon). The femoral artery was excised between the points of ligation. After arterial dissection, cells (1.0 x 106 cells per injection) were suspended in 100 µL phosphate-buffered saline and injected intramuscularly into two sites (the adductor muscle region in the medial thigh and the gastrocnemius) using 29-gauge tuberculin syringes. Four experimental groups (n=11/group) consisted of animals that received CXCR4-transfected ADSCs, CXCR4/VEGF-transfected ADSCs, VEGF-transfected ADSCs, or GFP-transfected ADSCs (control). All transfected genes were human species and all ADSCs were mouse species. Physiological status of ischemic limbs was followed up to 4 weeks after treatment. Animals were euthanized via cervical dislocation at two time points: after 4 days (n=3) and after 4 weeks (n=8). Limbs of the ischemic sides were retrieved for analysis.
BLI
ADSC survival was monitored after injection via BLI over 2 weeks (on the day of surgery and on days 1, 3, 4, 7, 10, and 14). Animals were anesthetized with 2-3% inhaled isoflurane and injected intraperitoneally with the reporter probe D-luciferin at 100 mg/kg body weight. The IVIS Spectrum system (Xenogen) was used to image the animals under 2% inhaled isoflurane anesthesia. Each animal was scanned until peak signal was reached. Radiance was quantified in photons/s/cm2/steradian.
In-vivo assessment of limb salvage and limb reperfusion
Ischemic limbs were evaluated via direct observation of limb status and via measurement of blood reperfusion. Limbs were assessed every week up to 4 weeks. Grading of limb ischemia was based on the severity of limb ischemia as: 1) limb salvage (no observable limb injury), 2) toe necrosis, 3) foot necrosis, 4) foot loss, and 5) limb loss. Blood reperfusion in ischemic limbs was measured using a PeriScan PIM3 laser Doppler system (Perimed AB) as previously described [35, 36]. Briefly, each animal was pre-warmed to a body temperature of 36 °C. Regions of interest were drawn around entire hindlimbs, and the levels of perfusion in ischemic and normal hindlimbs were quantified using the mean pixel value within each region of interest. Relative changes in blood flow were expressed as the ratio of the ischemic limb to the normal limbs.
Histology, immunofluorescence, capillary densitometry, and macrophage quantification
Mice were euthanized at 4 days and 4 weeks to retrieve muscle tissue from ischemic limbs for histology. Specimens were embedded in optimal cutting temperature (O.C.T; Tissue-Tek) compound for cryosectioning. Sections were cut at 8-µm thickness. General muscle health (regeneration and fibrosis) was assessed via staining with Masson's trichrome. Immunofluorescence was performed to assess features of angiogenesis and inflammation. Sections of day-28, transplanted, GFP(+)/Luc(+) cells were stained with anti-GFP (product #A-21312, 1:200 dilution, Invitrogen). Capillaries were detected with anti-CD31 (product #550274, 1:50 dilution, BD Pharmingen). Macrophages were detected with anti-CD68 (product #MCA1957, 1:100 dilution, AbD Serotec). Rhodamine anti-rat antibodies (product #31680, 1:200 dilution, Thermo Scientific) were used as secondary antibodies for both CD31 and CD68 staining. Sections were mounted with Vectashield Mounting Medium with 4',6-diamidino-2-phenylindole (DAPI; Vector Laboratories) to counterstain nuclei. Sections were imaged with a fluorescence microscope (Axio Observer, Zeiss). The number of CD31-positive cells was quantified to estimate capillary density per high-powered field using ImageJ v1.48 (NIH). Quantitative analysis of macrophages was performed by determining the number of DAPI-positive cells that overlapped with CD68-positive signals; analysis was performed using MATLAB R2010a (MathWorks).
Statistical analysis
GraphPad Prism 6 (Graphpad Software) was used for all statistical analyses. One-way analysis of variance with Dunnett's multiple comparison post-hoc test was performed to compare all experimental groups and to determine statistical significance. Comparisons between control groups and combined groups were carried out using Student's t-test. All quantitative data were expressed as mean ± standard error. A calculated p-value of less than 0.05 was used to reject the null hypothesis.
Supplementary Materials
Fig. S1. Improvement of transfection efficiency and low cytotoxicity with PBAE nanoparticles. Fig. S2 Cell-surface expression of human CXCR4 on transfected mouse ADSCs. Fig. S3 Blood reperfusion of ischemic hindlimbs. Table S1. List of primers used for quantitative real time-polymerase chain reaction.
Acknowledgements
The authors would like to thank the following funding sources for support including American Heart Association National Scientist Development Grant (10SDG2600001) (F.Y.), Stanford Chem-H Institute New Materials for Applications in Biology and Medicine Seed Grant (F.Y.), NIH R01DE024772-01 (F.Y.), NIH R01AR063717-01 (F.Y.), NIH R01AR055650-05A1 (F.Y.), National Science Foundation CAREER award program (CBET-1351289) (F.Y.), California Institute for Regenerative Medicine Tools and Technologies award ( RT3-07804) (F.Y.), Stanford Child Health Research Institute Faculty Scholar award (F.Y.), Stanford Bio-X Interdisciplinary program (F.Y.), and Alliance for Cancer Gene Therapy (F.Y.). L.D. would like to thank Stanford Medical Scientist Training Program for fellowship support. We thank Joseph Wu's lab at Stanford School of Medicine for kindly providing the GFP/luciferase transgenic mice for isolating cells. We thank Stanford Small Animal Imaging Facilities for support in bioluminescence imaging, and the Stanford Flow Cytometry Shared Resource for help with flow cytometry.
Author contributions
L.D., F.Y., N.H., and J.C. designed the research. L.D., J.C., and J.L. performed in vitro and in vivo experiments. L.D., J.C., J.L., N.H., J.C., and F.Y. analyzed the data. L.D. and F.Y. wrote the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Fowkes FG, Rudan D, Rudan I, Aboyans V, Denenberg JO, McDermott MM. et al. Comparison of global estimates of prevalence and risk factors for peripheral artery disease in 2000 and 2010: a systematic review and analysis. Lancet. 2013;382:1329-40
2. Criqui MH, Langer RD, Fronek A, Feigelson HS, Klauber MR, McCann TJ. et al. Mortality over a period of 10 years in patients with peripheral arterial disease. N Engl J Med. 1992;326:381-6
3. Selvin E, Erlinger TP. Prevalence of and risk factors for peripheral arterial disease in the United States: results from the National Health and Nutrition Examination Survey, 1999-2000. Circulation. 2004;110:738-43
4. Fischbach MA, Bluestone JA, Lim WA. Cell-based therapeutics: the next pillar of medicine. Sci Transl Med. 2013;5:179ps7
5. Iwaguro H, Yamaguchi J-i, Kalka C, Murasawa S, Masuda H, Hayashi S-i. et al. Endothelial Progenitor Cell Vascular Endothelial Growth Factor Gene Transfer for Vascular Regeneration. Circulation. 2002;105:732-8
6. Jabbarzadeh E, Starnes T, Khan YM, Jiang T, Wirtel AJ, Deng M. et al. Induction of angiogenesis in tissue-engineered scaffolds designed for bone repair: a combined gene therapy-cell transplantation approach. Proc Natl Acad Sci U S A. 2008;105:11099-104
7. Hacein-Bey-Abina S, Garrigue A, Wang GP, Soulier J, Lim A, Morillon E. et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. The Journal of Clinical Investigation. 2008;118:3132-42
8. Yang F, Cho SW, Son SM, Bogatyrev SR, Singh D, Green JJ. et al. Genetic engineering of human stem cells for enhanced angiogenesis using biodegradable polymeric nanoparticles. Proc Natl Acad Sci U S A. 2010;107:3317-22
9. Weis SM, Cheresh DA. Pathophysiological consequences of VEGF-induced vascular permeability. Nature. 2005;437:497-504
10. Dimmeler S. Regulation of bone marrow-derived vascular progenitor cell mobilization and maintenance. Arterioscler Thromb Vasc Biol. 2010;30:1088-93
11. Petit I, Jin D, Rafii S. The SDF-1-CXCR4 signaling pathway: a molecular hub modulating neo-angiogenesis. Trends Immunol. 2007;28:299-307
12. Ceradini DJ, Kulkarni AR, Callaghan MJ, Tepper OM, Bastidas N, Kleinman ME. et al. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med. 2004;10:858-64
13. Jin DK, Shido K, Kopp HG, Petit I, Shmelkov SV, Young LM. et al. Cytokine-mediated deployment of SDF-1 induces revascularization through recruitment of CXCR4+ hemangiocytes. Nat Med. 2006;12:557-67
14. Zuk PA, Zhu M, Mizuno H, Huang J, Futrell JW, Katz AJ. et al. Multilineage cells from human adipose tissue: implications for cell-based therapies. Tissue Eng. 2001;7:211-28
15. Miranville A, Heeschen C, Sengenes C, Curat CA, Busse R, Bouloumie A. Improvement of postnatal neovascularization by human adipose tissue-derived stem cells. Circulation. 2004;110:349-55
16. Cai L, Johnstone BH, Cook TG, Tan J, Fishbein MC, Chen PS. et al. IFATS collection: Human adipose tissue-derived stem cells induce angiogenesis and nerve sprouting following myocardial infarction, in conjunction with potent preservation of cardiac function. Stem Cells. 2009;27:230-7
17. Rehman J, Traktuev D, Li J, Merfeld-Clauss S, Temm-Grove CJ, Bovenkerk JE. et al. Secretion of angiogenic and antiapoptotic factors by human adipose stromal cells. Circulation. 2004;109:1292-8
18. Deveza L, Choi J, Imanbayev G, Yang F. Paracrine release from nonviral engineered adipose-derived stem cells promotes endothelial cell survival and migration in vitro. Stem Cells Dev. 2013;22:483-91
19. Kollet O, Petit I, Kahn J, Samira S, Dar A, Peled A. et al. Human CD34(+)CXCR4(-) sorted cells harbor intracellular CXCR4, which can be functionally expressed and provide NOD/SCID repopulation. Blood. 2002;100:2778-86
20. Paoni NF, Peale F, Wang F, Errett-Baroncini C, Steinmetz H, Toy K. et al. Time course of skeletal muscle repair and gene expression following acute hind limb ischemia in mice. Physiol Genomics. 2002Dec3;11(3):263-72
21. Ranganath Sudhir H, Levy O, Inamdar Maneesha S, Karp Jeffrey M. Harnessing the Mesenchymal Stem Cell Secretome for the Treatment of Cardiovascular Disease. Cell Stem Cell. 2012;10:244-58
22. Hong G, Lee JC, Jha A, Diao S, Nakayama KH, Hou L. et al. Near-infrared II fluorescence for imaging hindlimb vessel regeneration with dynamic tissue perfusion measurement. Circ Cardiovasc Imaging. 2014;7:517-25
23. Busillo JM, Benovic JL. Regulation of CXCR4 signaling. Biochim Biophys Acta. 2007;1768:952-63
24. Wynn RF, Hart CA, Corradi-Perini C, O'Neill L, Evans CA, Wraith JE. et al. A small proportion of mesenchymal stem cells strongly expresses functionally active CXCR4 receptor capable of promoting migration to bone marrow. Blood. 2004;104:2643-5
25. Shi M, Li J, Liao L, Chen B, Li B, Chen L. et al. Regulation of CXCR4 expression in human mesenchymal stem cells by cytokine treatment: role in homing efficiency in NOD/SCID mice. Haematologica. 2007;92:897-904
26. Kim Y, Kim H, Cho H, Bae Y, Suh K, Jung J. Direct comparison of human mesenchymal stem cells derived from adipose tissues and bone marrow in mediating neovascularization in response to vascular ischemia. Cell Physiol Biochem. 2007;20:867-76
27. Cao Y, Sun Z, Liao L, Meng Y, Han Q, Zhao RC. Human adipose tissue-derived stem cells differentiate into endothelial cells in vitro and improve postnatal neovascularization in vivo. Biochem Biophys Res Commun. 2005;332:370-9
28. Putnam D. Polymers for gene delivery across length scales. Nat Mater. 2006;5:439-51
29. Green JJ, Zugates GT, Tedford NC, Huang YH, Griffith LG, Lauffenburger DA. et al. Combinatorial Modification of Degradable Polymers Enables Transfection of Human Cells Comparable to Adenovirus. Advanced Materials. 2007;19:2836-42
30. Zugates GT, Peng W, Zumbuehl A, Jhunjhunwala S, Huang YH, Langer R. et al. Rapid optimization of gene delivery by parallel end-modification of poly(beta-amino ester)s. Mol Ther. 2007;15:1306-12
31. Yang F, Green JJ, Dinio T, Keung L, Cho SW, Park H. et al. Gene delivery to human adult and embryonic cell-derived stem cells using biodegradable nanoparticulate polymeric vectors. Gene Ther. 2009;16:533-46
32. Green JJ, Langer R, Anderson DG. A Combinatorial Polymer Library Approach Yields Insight into Nonviral Gene Delivery. Accounts of Chemical Research. 2008;41:749-59
33. Petit I, Jin D, Rafii S. The SDF-1-CXCR4 signaling pathway: a molecular hub modulating neo-angiogenesis. Trends in Immunology. 2007;28:299-307
34. Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative CT method. Nat Protocols. 2008;3:1101-8
35. Niiyama H, Huang NF, Rollins MD, Cooke JP. Murine model of hindlimb ischemia. J Vis Exp. 2009
36. Keeney M, Deveza L, Yang F. Programming stem cells for therapeutic angiogenesis using biodegradable polymeric nanoparticles. Journal of visualized experiments: JoVE. 2013 e50736
Author contact
![]() Corresponding author: Phone: 650-725-7128 Email: fanyangedu
Corresponding author: Phone: 650-725-7128 Email: fanyangedu