Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Experimental Sections
Results and Discussion
Conclusions
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2016; 6(7):1065-1074. doi:10.7150/thno.15420 This issue Cite
Research Paper
Supramolecular Crafting of Self-Assembling Camptothecin Prodrugs with Enhanced Efficacy against Primary Cancer Cells
Hao Su1*, Pengcheng Zhang1*, Andrew G Cheetham1, Jin Mo Koo1, Ran Lin1, Asad Masood1, Paula Schiapparelli2, Alfredo Quiñones-Hinojosa2, 4, Honggang Cui1, 3, 4 ![]()
1. Department of Chemical and Biomolecular Engineering, and Institute for NanoBioTechnology, The Johns Hopkins University, 3400 North Charles Street, Baltimore, Maryland 21218 United States.
2. Department of Neurosurgery, Johns Hopkins University School of Medicine, Baltimore, Maryland 21224, United States.
3. Center for Nanomedicine, The Wilmer Eye Institute, Johns Hopkins University School of Medicine, 400 North Broadway, Baltimore, Maryland 21231, United States.
4. Department of Oncology and Sidney Kimmel Comprehensive Cancer Center, Johns Hopkins University School of Medicine, Baltimore, Maryland 21205, United States.
* These authors contribute equally to this work.
Received 2016-3-1; Accepted 2016-3-21; Published 2016-4-28
Abstract

Chemical modification of small molecule hydrophobic drugs is a clinically proven strategy to devise prodrugs with enhanced treatment efficacy. While this prodrug strategy improves the parent drug's water solubility and pharmacokinetic profile, it typically compromises the drug's potency against cancer cells due to the retarded drug release rate and reduced cellular uptake efficiency. Here we report on the supramolecular design of self-assembling prodrugs (SAPD) with much improved water solubility while maintaining high potency against cancer cells. We found that camptothecin (CPT) prodrugs created by conjugating two CPT molecules onto a hydrophilic segment can associate into filamentous nanostructures in water. Our results suggest that these SAPD exhibit much greater efficacy against primary brain cancer cells relative to that of irinotecan, a clinically used CPT prodrug. We believe these findings open a new avenue for rational design of supramolecular prodrugs for cancer treatment.
Keywords: CPT Prodrug, high potency, self-assembly, nanomedicine, brain cancer, peptides.
Introduction
Many small molecule drugs are not directly suitable for clinical use due to their poor water solubility, premature degradation, and/or rapid renal clearance [1, 2]. One effective strategy is to chemically modify the drug to create a prodrug with much improved bioavailability, pharmacokinetic profile, and thus enhanced treatment efficacy. This prodrug approach has led to successful translations of many drugs into clinic [1]. In one example, camptothecin (CPT), a pharmaceutically active compound isolated from the Chinese tree Camptotheca acuminate, exhibits extraordinary anticancer activity by interfering DNA replication through topoisomerase I inhibition [3]. The clinical development of CPT was not successful due to its poor aqueous solubility (2-3 μg/mL), premature hydrolysis of the E-ring lactone, and strong binding affinity to plasma proteins [4-8]. After chemical modification [9, 10], the prodrug irinotecan (CPT-11) can be formulated to have a pharmacokinetic profile suitable for treating colon cancer and other cancer types. However, similar to many other water-soluble prodrug designs, irinotecan is still limited by its unpredictable, patient-dependent conversion to the parent drug SN-38, unwanted degradation in the physiological environment, and variable dose-related toxicities [4, 11, 12]. Furthermore, the small molecule and lipophobicity nature contributes to rapid rental clearance and poor intracellular uptake that further reduces irinotecan's therapeutic efficacy [13].
It has now been recognized that conjugation of an auxiliary segment onto a drug of interest not only improves its water solubility and bioavailability, but could also imbue the resulting prodrug with amphiphilic character so as to be able to assemble into a variety of discrete nanostructures in aqueous environment [14-16]. This creation of well-defined nanostructures using amphiphilic prodrugs offers a means to further manipulate the drug's pharmacokinetic profiles and cellular uptake efficiency by tuning the physicochemical properties of their assemblies, and also provides an innovative strategy to develop carrier-free one-component nanomedicines with a high and quantitative drug loading [14-30]. Therefore, from the perspectives of both fundamental prodrug design and clinical translation, these self-assembling prodrugs (SAPDs) could present a new platform to address the inherent drawbacks of water soluble small molecule prodrugs and those of multicomponent nanomedicine [31-34].
In this study, we report three self-assembling CPT prodrugs with enhanced efficacy against primary human brain cancer cells, one of the most aggressive cancer types to affect humans [35-38]. The principal concept demonstrated herein is to construct prodrug-like therapeutic agents with self-assembling feature to avoid premature degradation and rapid clearance, while not compromising the therapeutic efficacy of parent drugs (Figure 1a). Towards this end, we conjugated two CPT molecules to a short hydrophilic segment of three different types to give three model drugs including dCPT-K2, dCPT-OEG5-K2 and dCPT-Sup35-K2 (Figure 1b). The chosen hydrophilic segments are dipeptide (-Lys-Lys-Am) solely, or with water soluble oligo ethylene glycol (OEG) or a β-sheet-forming peptide sequences (Sup35) [39, 40] that are placed in the middle, respectively. The two C-terminal lysines served as the hydrophilic block facilitating the self-assembly of the CPT prodrugs into water soluble nanostructures. The β-sheet-forming peptide sequence was employed to enhance the intermolecular association by providing intermolecular hydrogen bonding while the OEG segment was used to weaken their intermolecular interactions through water solvation. For diCPT conjugates, the drug loading content is precisely controlled and independently tuned by altering the auxiliary segment, giving rise to the respective drug loadings of 43%, 36% and 28%. Disulfanyl-ethyl carbonate linker (etcSS) [41-43] was used as the biodegradable linker to bridge the peptide and the drug, which is expected to break down in the presence of a cancer-relevant intracellular reducing agent glutathione (GSH) [44]. This etcSS linker has been shown to be able to negate the effect of nanostructure-promoted disulphide formation to more effectively release bioactive CPT [45-50].
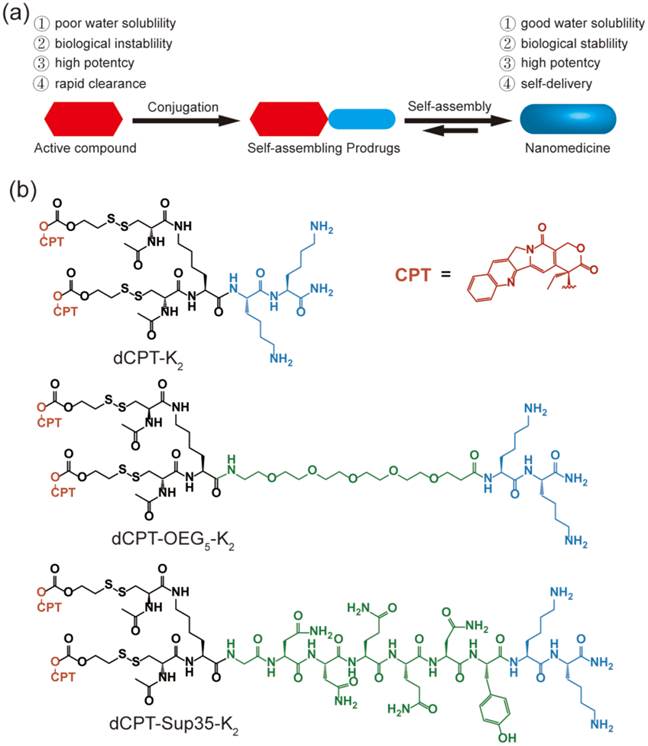
(a) Schematic illustration of the concept for rationally designed self-assembling prodrugs (SAPD) with high potency. (b) Chemical structures of the studied three model drugs: dCPT-K2, dCPT-OEG5-K2 and dCPT-Sup35-K2. Two CPTs were conjugated to three different hydrophilic auxiliary segments through disulfanyl-ethyl carbonate linker (etcSS).
Experimental Sections
Materials and instruments
Fmoc amino acids (except Fmoc-Lys(Fmoc)-OH), Fmoc-NH-OEG5-Propionic Acid and coupling reagents (HBTU or HATU) were purchased from Advanced Automated Peptide Protein Technologies (AAPPTEC, Louisville, KY, USA). Rink aminde MBHA resin and Fmoc-Lys(Fmoc)-OH which was obtained from Novabiochem (San Diego, CA, USA). Camptothecin were purchased from AvaChem Scientific (San Antonio, TX, USA) and all other reagents were sourced from Sigma-Aldrich (St. Louis, MO) or VWR (Radnor, PA, USA), unless otherwise stated.
RP-HPLC was performed on a Varian ProStar Model 325 HPLC (Agilent Technologies, Santa Clara, CA) equipped with a fraction collector. Preparative separations utilized a Varian PLRP-S column (100 Å, 10 µm, 150 × 25 mm), whilst analytical HPLC used a Varian Pursuit XRs C18 column (5 µm, 150 × 4.6 mm). Water and acetonitrile containing 0.1% v/v TFA were used as the mobile phase. Purified molecules were lyophilized using a FreeZone -105 °C 4.5 L freeze dryer (Labconco, Kansas City, MO). ESI-MS mass spectrometric data for characterization was acquired on a Finnigan LDQ Deca ion-trap mass spectrometer (Thermo-Finnigan, Waltham, MA).
Peptide synthesis
All peptide conjugates here were synthesized on the Rink Amide MBHA resins using standard 9-fluorenylmethoxycarbonyl (Fmoc) solid phase synthesis techniques at a 0.25 mmol synthesis scale (see detailed synthetic route in Scheme S1(a)). An automated peptide synthesizer (Focus XC, AAPPTec, Louisville, KY) was employed to build the (Ac-Cys)2KGN2Q2NYK2-NH2 (dCys-Sup35-K2) sequence before manual protocols were used to furnish the branching motif. The other two peptides (Ac-Cys)2KK2 (dCys-K2) and (Ac-Cys)2KOEG5 K2 (dCys- OEG5-K2) were both synthesized manually using the standard Fmoc chemistry protocols before the coupling of the camptothecin. Fmoc deprotections were performed using a 20% 4-methylpiperidine in DMF solution for 15 min and repeated once. The amino acid coupling cycle was performed after Fmoc deprotection, by adding a mixture of Fmoc-amino acids, HBTU and DIEA (4:4:6 molar equiv to resin) in DMF for 2 h. The addition of Fmoc-NH-OEG5-Propionic Acid was realized by the same protocols as amino acids coupling. The branching point was achieved by coupling Fmoc-Lys(Fmoc)-OH to yield two primary amine groups for the further coupling of Fmoc-Cys(Trt)-OH. Acetylation was performed on α-amino groups of N-terminus amino acids using a 20% acetic anhydride in DMF solution with 100 μL DIEA, shaken for 15 min, and the coupling was repeated twice. In all cases, reactions were monitored by the ninhydrin test (Anaspec Inc., Fremont, CA) for free amines. Completed peptides were cleaved from the solid support using a mixture of TFA/TIS/H2O at a ratio of 95:2.5:2.5 for 3 h. Excess TFA was removed by rotary evaporation and the concentrated solution was precipitated in cold diethyl ether to get the crude product, which were collected and dried under vacuum overnight.
Self-assembling prodrug (SAPD) synthesis and characterization
The synthesis of SAPDs was carried out by mixing CPT-etcSS-Pyr and the corresponding crude peptides in N2-purged DMSO (2 mL) with a molar ratio of 2:1 (see detailed synthetic route in Scheme S1(b)). After reacting for 5 days, the mixture was diluted to 10 mL with 0.1% TFA in acetonitrile/water and purified by preparative RP-HPLC using a Varian ProStar model 325 HPLC (Agilent Technologies, Santa Clara, CA, USA) equipped with a fraction collector. Separations were performed using a Varian PLRP-S column (100 Å, 10 μm, 150 × 25 mm) monitoring at 370 nm. Collected fractions were analyzed by ESI-MS (LDQ Deca ion-trap mass spectrometer, Thermo Finnigan, USA), and the appropriate fractions were collected, concentrated and lyophilized on a FreeZone -105 °C 4.5 L freeze dryer (Labconco, Kansas City, MO, USA). The powders obtained were then re-dissolved, calibrated and aliquotted into cryo-vials before re-lyophilization.
The purity of the conjugates was analyzed by HPLC using the following conditions: Agilent Zorbax-C18 column (5 μm, 4.6 × 150 mm); the flow rate was 1 mL/min, with the mobile phase starting from 15% MeCN (with 0.1% TFA) to 80% MeCN (with 0.1% TFA) at 25 min, hold for 5 min, gradient back to the initial condition in 3 min; the monitored wavelength was 370 nm. Molecular masses were determined by ESI-MS.
Calibration of the Concentration
The concentrations of purified self-assembling prodrugs (SAPDs) were calibrated by analyzing the reduced product free CPT from the SAPDs. Briefly, a stock solution of the SAPD was prepared by dissolving in MeCN/H2O (1:1). 5 μL of the stock solution was then diluted to 20 μL by addition of 15 μL MeCN/H2O (1:1) and mixed with 20 μL of fresh prepared 1 M aqueous TCEP for 1 h with periodic vortexing. 25 μL solution was then injected into the HPLC (so as to completely fill the 20 μL loop) to measure the area of the peak due to free CPT at 370nm. The CPT concentration was obtained by comparing the area under peak from treated solution with the standard calibration of CPT. The SAPD concentration was calculated based on the applied dilutions and number of CPT molecules. Finally, the stock solution was diluted to 100 μM according to the calibrated concentration and aliquotted into cryo-vials before re-lyophilization.
Transmission electron microscopy
100 μM stock solutions of corresponding samples in water were prepared by direct dissolution of the respective lyophilized powders, and allowed to age overnight. TEM Samples were prepared by depositing 7 μL of the appropriate solution onto a carbon-coated copper grid (Electron Microscopy Services, Hatfield, PA, USA), wicking away the excess solution with a small piece of filter paper. Then, 7 μL of a 2 wt% uranyl acetate aqueous solution was deposited on the surface for 30 seconds, wicking away the excess solution with filter paper. The specimens were air-dried at room temperature for at least 3 h prior to imaging. Bright-field TEM imaging was performed on a FEI Tecnai 12 TWIN Transmission Electron Microscope operated at an acceleration voltage of 100 kV. All TEM images were required by a SIS Megaview III wide-angle CCD camera.
Circular dichroism
All the CD spectra were recorded on a Jasco J-710 spectropolarimeter (JASCO, Easton, MD, USA) from 190 to 480 nm using a 1 mm (for 100 μM and 50 μM) or 10mm (for 12.5 μM and 3.125 μM) path length quartz UV-Vis absorption cell (Thermo Fisher Scientific, Pittsburgh, PA, USA). Background spectra of the solvents were acquired and subtracted from the sample spectra. Collected data was normalized from ellipticity (mdeg) to molar ellipticity (deg· cm2 ·dmol-1).
Drug release protocol
The release of dCPT-K2, dCPT-OEG5-K2 and dCPT-Sup35-K2 with or without GSH was evaluated using RP-HPLC. 50 μM stock solutions in deionized water were prepared and diluted to 25 μM with 20 mM PBS buffer with or without GSH (20 mM) (each has three parallel experiments). The solutions were incubated at 37℃ and samples were collected at 0 min, 5 min, 10 min, 15 min, 30 min, 1 h for those treated with GSH, and 0 h, 1 h, 6 h, 12 h, 24 h and 72 h for those without GSH. The samples were acidified with addition of 0.2 μL of 2M HCl, frozen with liquid nitrogen and stored at -30℃ until analysis. The amounts of released CPT were monitored by RP-HPLC using the following conditions: Varian Pursuit XRs C18 (5 μm, 150 × 4.6 mm); 362 nm detection wavelength; 1 ml/min flow rate; the gradient began at 85% of mobile phase A (0.1% aqueous TFA) and 15% of mobile phase B (acetonitrile containing 0.1% TFA) to 45% mobile phase A and 55% mobile phase B at 10 minutes, then to 15% mobile phase A and 85% mobile phase B at 13 minutes and held for another 2 minutes. Selected time points were characterized and data were plotted as a percentage of the total CPT concentration.
Cell culture
Human brain cell line U87 MG was a gift from Dr. Wirtz (ChemBE, JHU). Primary intraopreative human brain cancer cells (Primary 612 and 965) from primary patient tumors were provided by Dr. Quiñones-Hinojosa (Department of Neurosurgery, School of Medicine, JHU). DMEM (Invitrogen) containing 10% fetal bovine serum (FBS, Invitrogen) and 1% antibiotics (Invitrogen) was used for the culture of the U87 MG cells. Two primary cells were cultured in dishes coated with Laminin 1 μg/cm2 with following medium composition: DMEM/F12 (Invitrogen) with 1% Antimycotics (Invitrogen), 2% Gem 12 Neuroplex (Gemini), 20 ng/mL human fibroblast growth factor-basic (PeproTech) and 20 ng/mL human epidermal growth factor (PeproTech). All cell types were incubated at 37 ℃ in a humidified incubator (Oasis, Caron, Marietta, OH, USA) with a atmosphere of 5% CO2.
Cytotoxicity
The cytotoxicity of dCPT-K2, dCPT-OEG5-K2 and dCPT-Sup35-K2 was evaluated using the SRB method. U87 MG was seeded onto 96-well plates (5000 cells/well) and allowed to attach overnight. Primary 612 and primary 965 cells were seeded onto laminin-coated (1 μg/cm2) 96-well plates (5000 cells/well) and allowed to attach overnight. 100 μM stock solutions of three different SAPDs were prepared at the same time and aged overnight. The stock solutions were then diluted with fresh medium to achieve final CPT concentration of 0.1, 1, 10, 100, 500, 1000, 5000 and 10000 nM. After dilution, the SAPD-containing mediums were incubated with cells immediately. Medium containing the same concentration of free CPT ranging from 0.1 to 10000 nM was also used to incubate the cells, with non-treated cells (solvent only) as the control group. After 72 h incubation, the cell viability was evaluated using the SRB method according to the manufacturer's protocols (TOX-6, Sigma, St. Louis, MO).
Results and Discussion
Synthesis of self-assembling camptothecin prodrugs
The synthesis was achieved in a modular and efficient fashion using established synthetic strategies. Briefly, the peptide segments with two N-terminal cysteine residues (dCys-K2, dCys-OEG5-K2 and dCys-Sup35-K2) were first synthesized using standard Fmoc solid-phase synthesis protocols. The disulfide formation reaction to incorporate CPTs onto the peptide was carried out in nitrogen-purged DMSO as reported previously [45]. The final products were purified using preparative HPLC, and characterized using ESI-MS and analytical HPLC, respectively (ESI, Figure S1-S3).
Self-assembly of camptothecin prodrugs
The self-assembly of the three CPT conjugates was promoted by directly dissolving the corresponding molecule into deionized water to reach a final concentration of 100 μM. After aging for 24 h, transmission electron microscopy (TEM) images revealed that both dCPT-K2 and dCPT-OEG5-K2 assembled into nanotubes (> 1μm) of similar widths: 8.2 ± 0.8 nm for the former (Figure 2a and Figure S4 in SI) and 7.5 ± 0.7 nm for the latter (Figure 2b and Figure S5 in SI), respectively. In contrast, dCPT-Sup35-K2 formed nanofibers of a diameter of 6.9 ± 1.4 nm and lengths up to a few micrometers (Figure 2c and Figure S6 in SI). The formation of nanotubes versus nanofibers is likely due to the incorporation of the β-sheet-forming Sup35 sequence that facilitates the arrangement of intermolecular hydrogen bonding [51-53]. Circular dichroism (CD) spectrometry (Figure 2d) was then used to investigate the molecular packing within the assemblies of three CPT prodrugs. The observed signals at 250 nm (n-π*) and between 320 and 400 nm (π-π*) are all attributed to CPT absorptions, supporting a high degree of chiral packing of the CPT moieties in the assembled form [54-58]. In addition, a strong negative signal at 212 nm of dCPT-Sup35-K2 (Figure 2d, green curve) suggested the β-sheet conformation of the assembled Sup35 segments [59, 60]. But for both dCPT-K2 and dCPT-OEG5-K2 (Figure 2d, blue curve and red curve), the negative peaks slightly below 200 nm cannot be assigned to molecular adsorptions associated with any particular peptide secondary structures. This observation also highlights the role of the π-π interactions between CPT units in determining the formation of nanotubes [58, 61, 62]. It is likely that lack of the strong, ordered intermolecular hydrogen bonding among Sup35 segment allows for higher degree of packing between the CPT units into the tubular morphology. In the presence of the β-sheet-forming Sup35, the assembly of dCPT-Sup35-K2 into nanofibers is reminiscent of that of peptide amphiphiles reported by the Stupp laboratories [59, 63, 64]. Clearly, these results demonstrated that our self-assembling CPT prodrugs can indeed aggregate into well-defined nanostructures, with morphology tunable by the rational design of the auxiliary segment.
Molecular assembly and characterization of designed SAPDs. Representative TEM images of nanotubes formed by dCPT-K2 with diameter of 8.2 ± 0.8 nm (a) and dCPT-OEG5-K2 with diameter of 7.5 ± 0.7 nm (b). (Insert) High resolution TEM images display the tubular morphology. (c) TEM image of nanofibers formed by dCPT-Sup35-K2 with diameter of 6.9 ± 1.4 nm. (d) Normalized CD spectra of the three studied supramolecular prodrugs in water. All the samples were prepared at concentration of 100 μM in water and aged over three days before measurements. TEM samples were negatively stained with 2 wt% uranyl acetate and all scale bars = 100 nm. More TEM images can be found in the SI (Figure S4-S6).
Physical stability of self-assembling nanostructures
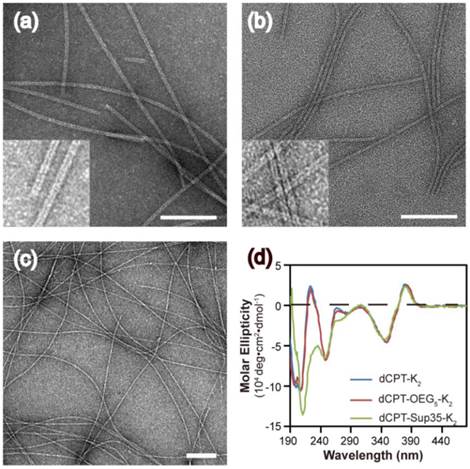
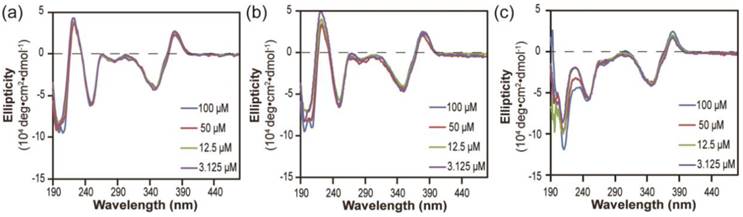
Next, we investigated the stabilities of these nanostructures upon dilution to determine whether the lacking of hydrogen bonding segment in dCPT-K2 and dCPT-OEG5-K2 would result in reduced stability [65, 66]. The three aged solutions containing 100 μM each conjugate were diluted to 50 μM, 12.5 μM and 3.125 μM, respectively, and then incubated for another three days before CD measurements. To assess the stabilities of these nanoassemblies, CD spectra were recorded to determine if there exists any change of molecular packing in response to dilution. Figure 3a and 3b show that no significant changes on CD results were observed for both dCPT-K2 and dCPT-OEG5-K2, indicating the stability of the self-assembled nanostructures at the concentrations as low as ~3 µM. Surprisingly, For dCPT-Sup35-K2 nanofibers (Figure 3c), a gradual decrease in β-sheet signal around 212 nm can be clearly seen and is accompanied with a slight loss of signal at 380 nm, suggesting dissociation of these self-assembled nanofibers into smaller aggregates and/or prodrug monomers. The reduced stability of naonfibers relative to nanotubes leads us to draw the conclusion that CPT stacking is the primary contributor to the formation of the observed one dimensional nanostructures and their stability, and introducing a segment capable of forming a different type of strong associative interactions may disrupt the CPT-CPT stacking thus reducing the overall stability of the nanostructures.
Stability study of nanostructures formed by dCPT-K2 (a), dCPT-OEG5-K2 (b) and dCPT-Sup35-K2 (c) after dilution to 50 μM, 12.5 μM and 3.125 μM in water, as monitored by CD. All the diluted solutions were aged over three days before CD measurements. Corresponding CD results showed no significant changes for both dCPT-K2 and dCPT-OEG5-K2. However, a slight decrease in signal of nanofibers formed by dCPT-Sup35-K2 was observed, indicating partial dissociation of the supramolecular nanostructures into smaller aggregates and/or prodrug monomers.
Drug release
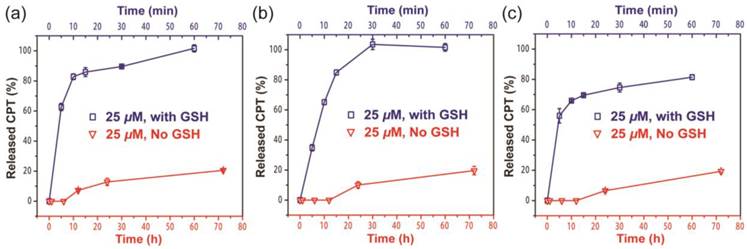
Since the ectSS linker used in our design is expected to be responsive to glutathione (GSH) to effectively release the free CPT (Figure S7a), we investigated how it could impact the release behavior of SAPDs. The release of active compound from three prodrugs were evaluated in PBS at 37℃ at the concentration of 25 μM with or without 10 mM GSH (Figure 4). We found that all three prodrugs demonstrated a much faster release profile in the presence of GSH and a remarkable amount of CPT can be released within 10 minutes for all three studied conjugates (Figure S7b, c and d). As shown in Figure 4a and 4b, dCPT-K2 and dCPT-OEG5-K2 released all the available CPT molecules within 1 h of incubation. Similarly, dCPT-Sup35-K2 (Figure 4c) released around 80% of the CPTs within 1 h. Under the GSH-free condition, only around 20% of bioactive CPT released out for all the three prodrugs at 72 h, since hydrolysis of ester bond is solely responsible for the release. These results indicated that the designed CPT prodrugs can maintain in the inactive form for a long period of time under the GSH-free physiological condition but is capable of releasing the bioactive CPT in the biologically relevant condition.
Cumulative drug release from dCPT-K2 (a), dCPT-OEG5-K2 (b) and dCPT-Sup35-K2 (c) in PBS at 37℃ at 25 μM with (blue curve) or without (red curve) 10 mM GSH. Data were given as mean ± s.d. (n = 3). For samples with 10 mM GSH, they presented ultra-fast release profiles (100% CPT released out for dCPT-K2 (a), dCPT-OEG5-K2 (b) and 81.4% CPT released out for dCPT-Sup35-K2 (c) within 1h). For samples without GSH, around 20% CPT released out at 72 h.
Cytotoxicity against human brain cancer cell lines
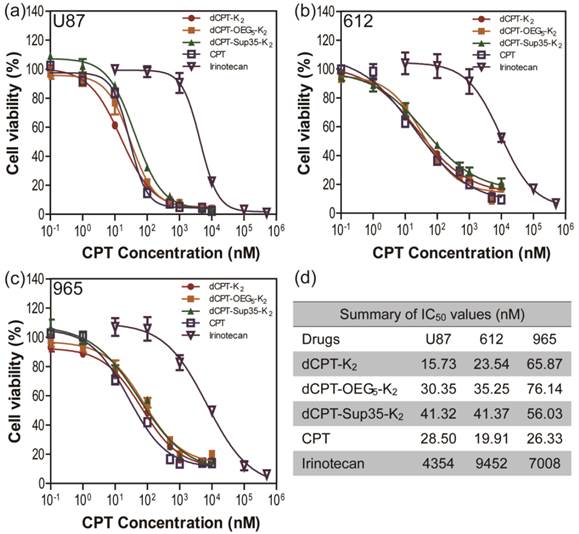
To further explore whether the fast release would affect their anticancer efficacy, the in vitro toxicities of three model prodrugs were assessed against three human brain cancer cell lines (glioblastoma) through a dose-response relationship based on CPT concentration using SRB method. We used one commercial cell line (U87 MG), and two primary cancer cells (612 and 965) derived from primary patient tumors, which are more clinically relevant, also known to be more resistant to chemical treatments [67]. All the cancer cells were incubated with dCPT-K2, dCPT-OEG5-K2 or dCPT-Sup35-K2 for 72 h, separately, with CPT and clinically used irinotecan as control drugs [8]. Very promisingly, we found that all three CPT prodrugs reveal much higher potency than irinotecan in the U87 brain cancer cell line and the two primary cells from two different patients, with their IC50 values two orders of magnitude lower than that of irinotecan. This is possibly due to the fact that irinotecan must be converted into its active form to exert its anticancer activity [4, 68, 69], and the slow hydrolysis of the irinotecan's ester bond, an essential step to release the bioactive form of CPT, could largely compromise its efficacy. The pharmacologically potent drugs, such as CPT and SN-38, however, are unable to be directly used in clinic due to their low aqueous solubility, instability at physiological pH (pH 7.4) and strong binding with plasma proteins.[4-8] In the case reported here, all the three CPT prodrugs showed even comparable efficacies to the parental CPT. The observed remarkable cytotoxicity could arise in part from the rapid reductive degradation of etcSS linker by intracellular GSH and in part from the self-assembling potential of the designed CPT drug amphiphiles that promote effective cellular internalization. In addition, no significant differences in toxicity between three CPT prodrugs were observed, suggesting that the surface chemistry and prompt release of drugs are more important in determining the efficacy rather than their detailed molecular packing and structural integrity, which could be important factors for in vivo applications.
In vitro cytotoxicity study of the SAPDs against human brain cancer cell lines U87 MG (a) and intraoperative derived human cancer cell lines (612 (b) and 965 (c)). All cancer cells were incubated with the appropriate SAPDs for 72 h and cell viability was determined by SRB assay. Data were given as mean ± s.d. (n = 3) and IC50 values were calculated according to CPT concentration. All the SAPDs exhibited greater efficacy against three human brain cancer cell lines than irinotecan.
Conclusions
In summary, we developed a generic and versatile strategy to construct self-assembling CPT prodrugs with improved solubility, enhanced stability, and a high and fixed drug loading content. The payload and nanostructure morphology are tunable by the conjugated auxiliary segment. Most importantly, by incorporating the biodegradable disulfanyl-ethyl carbonate linker, the bioactive CPT can be effectively released from the conjugates without compromising the high toxicity of the parental drug. We believe that this typical design of self-assembling drug amphiphiles provide a new platform for the development of chemotherapeutic agents with high potency and tunable physicochemical properties. This platform would also allow for the incorporation of other hydrophilic functional auxiliaries (e.g. tumor targeting peptides, cell penetrating peptides, enzyme cleavable peptides, zwitterionic segments, imaging agents) in the molecular design [70, 71], or even to create SAPD hydrogels, for use in both systemic and local chemotherapy.
Supplementary Material
Supplementary figures.
Acknowledgements
This work was supported by the National Science Foundation (DMR 1255281) and the National Institutes of Health (NIH/R21CA191740, and NIH/R01NS070024). We acknowledge JHU Integrated Imaging Center (IIC) for the use of the TEM facility, and Prof. Kalina Hristova at JHU Department of Material Science and Engineering for the use of the CD spectropolarimeter. We also acknowledge the JHU Department of Chemistry for the use of mass spectrometer (NSF CHE-0840463).
Conflict of Interest
The authors declare no competing financial interest.
References
1. Rautio J, Kumpulainen H, Heimbach T, Oliyai R, Oh D, Jarvinen T. et al. Prodrugs: design and clinical applications. Nat Rev Drug Discov. 2008;7:255-70
2. Venkatesh S, Lipper RA. Role of the development scientist in compound lead selection and optimization. J Pharm Sci. 2000;89:145-54
3. Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer. 2006;6:789-802
4. Bala V, Rao SS, Boyd BJ, Prestidge CA. Prodrug and nanomedicine approaches for the delivery of the camptothecin analogue SN38. J Control Release. 2013;172:48-61
5. Koizumi F, Kitagawa M, Negishi T, Onda T, Matsumoto S, Hamaguchi T. et al. Novel SN-38-incorporating polymeric micelles, NK012, eradicate vascular endothelial growth factor-secreting bulky tumors. Cancer Res. 2006;66:10048-56
6. Roger E, Lagarce F, Benoit JP. Development and characterization of a novel lipid nanocapsule formulation of Sn38 for oral administration. Eur J Pharm Biopharm. 2011;79:181-8
7. Thakur R, Sivakumar B, Savva M. Thermodynamic Studies and Loading of 7-Ethyl-10-hydroxycamptothecin into Mesoporous Silica Particles MCM-41 in Strongly Acidic Solutions. J Phys Chem B. 2010;114:5903-11
8. Kim SH, Kaplan JA, Sun Y, Shieh A, Sun HL, Croce CM. et al. The Self-Assembly of Anticancer Camptothecin-Dipeptide Nanotubes: A Minimalistic and High Drug Loading Approach to Increased Efficacy. Chem-Eur J. 2015;21:101-5
9. Lu JQ, Liu C, Wang PC, Ghazwani M, Xu JN, Huang YX. et al. The self-assembling camptothecin-tocopherol prodrug: An effective approach for formulating camptothecin. Biomaterials. 2015;62:176-87
10. Li XQ, Wen HY, Dong HQ, Xue WM, Pauletti GM, Cai XJ. et al. Self-assembling nanomicelles of a novel camptothecin prodrug engineered with a redox-responsive release mechanism. Chem Commun. 2011;47:8647-9
11. Newton M, Wetzstein G, Sullivan D. Topoisomerase I Inhibitors - The Camptothecins. Cancer Grow Prog-Dor. 2011;13:103-23
12. Slichenmyer WJ, Rowinsky EK, Donehower RC, Kaufmann SH. The Current Status of Camptothecin Analogs as Antitumor Agents. J Natl Cancer I. 1993;85:271-91
13. Eloy JO, de Souza MC, Petrilli R, Barcellos JPA, Lee RJ, Marchetti JM. Liposomes as carriers of hydrophilic small molecule drugs: Strategies to enhance encapsulation and delivery. Colloid Surface B. 2014;123:345-63
14. Cheetham AG, Zhang PC, Lin YA, Lock LL, Cui HG. Supramolecular Nanostructures Formed by Anticancer Drug Assembly. J Am Chem Soc. 2013;135:2907-10
15. Lin R, Cheetham AG, Zhang PC, Lin YA, Cui HG. Supramolecular filaments containing a fixed 41% paclitaxel loading. Chem Commun. 2013;49:4968-70
16. Su H, Koo JM, Cui HG. One-component nanomedicine. J Control Release. 2015;219:383-95
17. Zhao F, Ma ML, Xu B. Molecular hydrogels of therapeutic agents. Chem Soc Rev. 2009;38:883-91
18. MacKay JA, Chen MN, McDaniel JR, Liu WG, Simnick AJ, Chilkoti A. Self-assembling chimeric polypeptide-doxorubicin conjugate nanoparticles that abolish tumours after a single injection. Nat Mater. 2009;8:993-9
19. Desmaele D, Gref R, Couvreur P. Squalenoylation: A generic platform for nanoparticular drug delivery. J Control Release. 2012;161:609-18
20. Kataoka K, Harada A, Nagasaki Y. Block copolymer micelles for drug delivery: Design, characterization and biological significance. Adv Drug Deliver Rev. 2012;64:37-48
21. Lin R, Cui H. Supramolecular nanostructures as drug carriers. Curr Opin Chem Eng. 2015;7:75-83
22. Jeannine MC, Kaplan DL. Engineering Biomaterial-Drug Conjugates for Local and Sustained Chemotherapeutic Delivery. Bioconjug Chem. 2015
23. Bhattacharyya J, Bellucci JJ, Weitzhandler I, McDaniel JR, Spasojevic I, Li XH. et al. A paclitaxel-loaded recombinant polypeptide nanoparticle outperforms Abraxane in multiple murine cancer models. Nat Commun. 2015:6
24. Zhang SY, Zou J, Elsabahy M, Karwa A, Li A, Moore DA. et al. Poly(ethylene oxide)-block-polyphosphester-based paclitaxel conjugates as a platform for ultra-high paclitaxel-loaded multifunctional nanoparticles. Chem Sci. 2013;4:2122-6
25. Zou J, Zhang FW, Zhang SY, Pollack SF, Elsabahy M, Fan JW. et al. Poly(ethylene oxide)-block-Polyphosphoester-graft-Paclitaxel Conjugates with Acid-Labile Linkages as a pH-Sensitive and Functional Nanoscopic Platform for Paclitaxel Delivery. Adv Healthc Mater. 2014;3:441-8
26. Tong R, Cheng JJ. Paclitaxel-initiated, controlled polymerization of lactide for the formulation of polymeric nanoparticulate delivery vehicles. Angew Chem Int Edit. 2008;47:4830-4
27. Tong R, Cheng JJ. Ring-Opening Polymerization-Mediated Controlled Formulation of Polylactide-Drug Nanoparticles. J Am Chem Soc. 2009;131:4744-54
28. Gao Y, Kuang Y, Guo ZF, Guo ZH, Krauss IJ, Xu B. Enzyme-Instructed Molecular Self-assembly Confers Nanofibers and a Supramolecular Hydrogel of Taxol Derivative. J Am Chem Soc. 2009;131:13576 -+
29. Li XM, Li JY, Gao YA, Kuang Y, Shi JF, Xu B. Molecular Nanofibers of Olsalazine Form Supramolecular Hydrogels for Reductive Release of an Anti-inflammatory Agent. J Am Chem Soc. 2010;132:17707-9
30. Tan XY, Li BB, Lu XG, Jia F, Santori C, Menon P. et al. Light-Triggered, Self-Immolative Nucleic Acid-Drug Nanostructures. J Am Chem Soc. 2015;137:6112-5
31. Wang M, Alberti K, Sun S, Arellano CL, Xu QB. Combinatorially Designed Lipid-like Nanoparticles for Intracellular Delivery of Cytotoxic Protein for Cancer Therapy. Angew Chem Int Edit. 2014;53:2893-8
32. Bastings MMC, Koudstaal S, Kieltyka RE, Nakano Y, Pape ACH, Feyen DAM. et al. A Fast pH-Switchable and Self-Healing Supramolecular Hydrogel Carrier for Guided, Local Catheter Injection in the Infarcted Myocardium. Adv Healthc Mater. 2014;3:70-8
33. Khlebtsov N, Bogatyrev V, Dykman L, Khlebtsov B, Staroverov S, Shirokov A. et al. Analytical and Theranostic Applications of Gold Nanoparticles and Multifunctional Nanocomposites. Theranostics. 2013;3:167-80
34. Kumar R, Kulkarni A, Nagesha DK, Sridhar S. In Vitro Evaluation of Theranostic Polymeric Micelles for Imaging and Drug Delivery in Cancer. Theranostics. 2012;2:714-22
35. Abbadi S, Rodarte JJ, Abutaleb A, Lavell E, Smith CL, Ruff W. et al. Glucose-6-phosphatase Is a Key Metabolic Regulator of Glioblastoma Invasion. Mol Cancer Res. 2014;12:1547-59
36. Chaichana KL, Jusue-Torres I, Lemos AM, Gokaslan A, Cabrera-Aldana EE, Ashary A. et al. The butterfly effect on glioblastoma: is volumetric extent of resection more effective than biopsy for these tumors? J Neuro-Oncol. 2014;120:625-34
37. Chaichana KL, Jusue-Torres I, Navarro-Ramirez R, Raza SM, Pascual-Gallego M, Ibrahim A. et al. Establishing percent resection and residual volume thresholds affecting survival and recurrence for patients with newly diagnosed intracranial glioblastoma. Neuro-Oncology. 2014;16:113-22
38. McGirt MJ, Chaichana KL, Gathinji M, Attenello FJ, Than K, Olivi A. et al. Independent association of extent of resection with survival in patients with malignant brain astrocytoma. J Neurosurg. 2009;110:156-62
39. Nelson R, Sawaya MR, Balbirnie M, Madsen AO, Riekel C, Grothe R. et al. Structure of the cross-beta spine of amyloid-like fibrils. Nature. 2005;435:773-8
40. Hamley IW. Peptide fibrillization. Angew Chem Int Edit. 2007;46:8128-47
41. Dubikovskaya EA, Thorne SH, Pillow TH, Contag CH, Wender PA. Overcoming multidrug resistance of small-molecule therapeutics through conjugation with releasable octaarginine transporters. P Natl Acad Sci USA. 2008;105:12128-33
42. Henne WA, Doorneweerd DD, Hilgenbrink AR, Kularatne SA, Low PS. Synthesis and activity of a folate peptide camptothecin prodrug. Bioorg Med Chem Lett. 2006;16:5350-5
43. Kularatne SA, Venkatesh C, Santhapuram HKR, Wang K, Vaitilingam B, Henne WA. et al. Synthesis and Biological Analysis of Prostate-Specific Membrane Antigen-Targeted Anticancer Prodrugs. J Med Chem. 2010;53:7767-77
44. Estrela JM, Ortega A, Obrador E. Glutathione in cancer biology and therapy. Crit Rev Cl Lab Sci. 2006;43:143-81
45. Cheetham AG, Ou YC, Zhang PC, Cui HG. Linker-determined drug release mechanism of free camptothecin from self-assembling drug amphiphiles. Chem Commun. 2014;50:6039-42
46. Zhang QQ, He JL, Zhang MZ, Ni PH. A polyphosphoester-conjugated camptothecin prodrug with disulfide linkage for potent reduction-triggered drug delivery. J Mater Chem B. 2015;3:4922-32
47. Hu XL, Hu JM, Tian J, Ge ZS, Zhang GY, Luo KF. et al. Polyprodrug Amphiphiles: Hierarchical Assemblies for Shape-Regulated Cellular Internalization, Trafficking, and Drug Delivery. J Am Chem Soc. 2013;135:17617-29
48. Wang H, Tang L, Tu CL, Song ZY, Yin Q, Yin LC. et al. Redox-Responsive, Core-Cross-Linked Micelles Capable of On-Demand, Concurrent Drug Release and Structure Disassembly. Biomacromolecules. 2013;14:3706-12
49. Liu JY, Liu WE, Weitzhandler I, Bhattacharyya J, Li XH, Wang J. et al. Ring-Opening Polymerization of Prodrugs: A Versatile Approach to Prepare Well-Defined Drug-Loaded Nanoparticles. Angew Chem Int Edit. 2015;54:1002-6
50. Lee MH, Yang Z, Lim CW, Lee YH, Dongbang S, Kang C. et al. Disulfide-Cleavage-Triggered Chemosensors and Their Biological Applications. Chem Rev. 2013;113:5071-109
51. Versluis F, Marsden HR, Kros A. Power struggles in peptide-amphiphile nanostructures. Chem Soc Rev. 2010;39:3434-44
52. Cavalli S, Albericio F, Kros A. Amphiphilic peptides and their cross-disciplinary role as building blocks for nanoscience. Chem Soc Rev. 2010;39:241-63
53. Hamley IW. Self-assembly of amphiphilic peptides. Soft Matter. 2011;7:4122-38
54. Fleury F, Ianoul A, Berjot M, Feofanov A, Alix AJP, Nabiev I. Camptothecin-binding site in human serum albumin and protein transformations induced by drug binding. Febs Lett. 1997;411:215-20
55. Fleury F, Kudelina I, Nabiev I. Interactions of lactone, carboxylate and self-aggregated forms of camptothecin with human and bovine serum albumins. Febs Lett. 1997;406:151-6
56. Person RV, Monde K, Humpf HU, Berova N, Nakanishi K. A New Approach in Exciton-Coupled Circular-Dichroism (Eccd) - Insertion of an Auxiliary Stereogenic Center. Chirality. 1995;7:128-35
57. Watanabe K, Akagi K. Helically assembled pi-conjugated polymers with circularly polarized luminescence. Sci Technol Adv Mat. 2014:15
58. Aida T, Meijer EW, Stupp SI. Functional Supramolecular Polymers. Science. 2012;335:813-7
59. Cui HG, Webber MJ, Stupp SI. Self-Assembly of Peptide Amphiphiles: From Molecules to Nanostructures to Biomaterials. Biopolymers. 2010;94:1-18
60. Haines-Butterick L, Rajagopal K, Branco M, Salick D, Rughani R, Pilarz M. et al. Controlling hydrogelation kinetics by peptide design for three-dimensional encapsulation and injectable delivery of cells. P Natl Acad Sci USA. 2007;104:7791-6
61. Gopal R, Park JS, Seo CH, Park Y. Applications of Circular Dichroism for Structural Analysis of Gelatin and Antimicrobial Peptides. Int J Mol Sci. 2012;13:3229-44
62. Korevaar PA, Newcomb CJ, Meijer EW, Stupp SI. Pathway Selection in Peptide Amphiphile Assembly. J Am Chem Soc. 2014;136:8540-3
63. Ortony JH, Newcomb CJ, Matson JB, Palmer LC, Doan PE, Hoffman BM. et al. Internal dynamics of a supramolecular nanofibre. Nat Mater. 2014;13:812-6
64. Tantakitti F, Boekhoven J, Wang X, Kazantsev RV, Yu T, Li J. et al. Energy landscapes and functions of supramolecular systems. Nat Mater. 2016
65. Meijer JT, Roeters M, Viola V, Lowik DWPM, Vriend G, van Hest JCM. Stabilization of peptide fibrils by hydrophobic interaction. Langmuir. 2007;23:2058-63
66. Meijer JT, Henckens MJAG, Minten IJ, Lowik DWPM, van Hest JCM. Disassembling peptide-based fibres by switching the hydrophobic-hydrophilic balance. Soft Matter. 2007;3:1135-7
67. Smith CL, Chaichana KL, Lee YM, Lin BJ, Stanko KM, O'Donnell T. et al. Pre-Exposure of Human Adipose Mesenchymal Stem Cells to Soluble Factors Enhances Their Homing to Brain Cancer. Stem Cell Transl Med. 2015;4:239-51
68. Pitot HC, Adjei AA, Reid JM, Sloan JA, Atherton PJ, Rubin J. et al. A phase I and pharmacokinetic study of a powder-filled capsule formulation of oral irinotecan (CPT-11) given daily for 5 days every 3 weeks in patients with advanced solid tumors. Cancer Chemoth Pharm. 2006;58:165-72
69. Schoemaker NE, Kuppens IELM, Huinink WWT, Lefebvre P, Beijnen JH, Assadourian S. et al. Phase I study of an oral formulation of irinotecan administered daily for 14 days every 3 weeks in patients with advanced solid tumours. Cancer Chemoth Pharm. 2005;55:263-70
70. Choi KY, Swierczewska M, Lee S, Chen XY. Protease-Activated Drug Development. Theranostics. 2012;2:156-78
71. Chen K, Chen XY. Integrin Targeted Delivery of Chemotherapeutics. Theranostics. 2011;1:189-200
Author contact
![]() Corresponding author: hcui6edu.
Corresponding author: hcui6edu.