Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Intracellular ATP-dependent DOX...
Results and Discussion
Conclusions
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2016; 6(7):1053-1064. doi:10.7150/thno.14843 This issue Cite
Research Paper
ATP-Responsive and Near-Infrared-Emissive Nanocarriers for Anticancer Drug Delivery and Real-Time Imaging
Chenggen Qian1,2,3*, Yulei Chen1*, Sha Zhu1, Jicheng Yu2,3, Lei Zhang1, Peijian Feng1, Xin Tang1, Quanyin Hu2,3, Wujin Sun2,3, Yue Lu2,3, Xuanzhong Xiao1, Qun-Dong Shen1 ![]() , Zhen Gu2,3,4
, Zhen Gu2,3,4 ![]()
1. Department of Polymer Science & Engineering and Key Laboratory of High Performance Polymer Materials & Technology of MOE, Collaborative Innovation Center of Chemistry for Life Sciences, School of Chemistry & Chemical Engineering, Nanjing University, Nanjing 210023, China;
2. Joint Department of Biomedical Engineering, University of North Carolina at Chapel Hill and North Carolina State University, Raleigh, NC 27695, USA;
3. Division of Molecular Pharmaceutics and Center for Nanotechnology in Drug Delivery, Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA;
4. Department of Medicine, University of North Carolina School of Medicine, Chapel Hill, NC 27599, USA.
*These authors contributed equally.
Received 2015-12-29; Accepted 2016-3-13; Published 2016-4-28
Abstract

Stimuli-responsive and imaging-guided drug delivery systems hold vast promise for enhancement of therapeutic efficacy. Here we report an adenosine-5'-triphosphate (ATP)-responsive and near-infrared (NIR)-emissive conjugated polymer-based nanocarrier for the controlled release of anticancer drugs and real-time imaging. We demonstrate that the conjugated polymeric nanocarriers functionalized with phenylboronic acid tags on surface as binding sites for ATP could be converted to the water-soluble conjugated polyelectrolytes in an ATP-rich environment, which promotes the disassembly of the drug carrier and subsequent release of the cargo. In vivo studies validate that this formulation exhibits promising capability for inhibition of tumor growth. We also evaluate the metabolism process by monitoring the fluorescence signal of the conjugated polymer through the in vivo NIR imaging.
Keywords: drug delivery, nanomedicine, stimuli-responsive, ATP-responsive, theranostics, conjugated polymers.
Introduction
Nanocarrier-based drug delivery systems (DDSs) offer new opportunities to enhance therapeutic efficacy due to their improved bioactivity and enhanced bioavailability of drugs.[1-5] To further improve pinpoint therapeutic efficacy, various efforts have been devoted to explore stimuli-responsive[6, 7] and imaging-guided DDSs[8-11], which are specifically stimulated to release drugs within target sites and achieve real-time imaging-guided therapeutics. A broad range of stimuli have been explored to design smart DDSs, including the external triggers, such as light[12, 13], ultrasound[14], magnetism[15, 16], temperature[17], and electric field[18], as well as internal stimuli, such as pH[19, 20], redox potential[21], oxidative stress[22, 23], enzyme[24, 25], and glucose levels[26, 27]. Imaging-guided DDSs have been rapidly developed by inorganic nanosystems with contrast properties made of carbon dots[28], quantum dots[29], metals[30], and metal oxide frameworks[31]. In addition, conjugated polymer nanoparticles, as special organic nanosytems, have remarkable photophysical properties which are also suitable for biological fluorescence imaging.[32-37]
Herein, we report an innovative conjugated polymer-based nanocarrier capable of achieving near infrared (NIR) imaging and adenosine-5'-triphosphate (ATP)-responsive anticancer drug release (Figure 1). ATP, as the essential biogenic biomolecule for cellular energy metabolism and signaling[38-41], is recently attracting considerable interest as an effective stimulus due to the distinct difference in the ATP levels between extracellular and intracellular milieu.[42-45] This doxorubicin (DOX) encapsulated nanocarrier (designated DOX/PFFP NP) comprises of ATP-responsive and NIR-emissive FPBA- and PEG-grafted conjugated polymer (designated PFFP) (Figure 1). The NIR fluorescent core of the NP is hydrophobic conjugated polymer, which contains dithiophene-thienopyrazine[46] moiety as an NIR imaging agent. The 3-fluoro-4-carboxyphenylboronic acid (FPBA) tags on the nanocarrier surface are designed as binding sites to ATP through a specific recognition[47, 48]. At the intracellular ATP concentration, the FPBA groups interact with ATP to yield water-soluble conjugated polyelectrolytes[49], which promotes the disassembly of the drug nanocarrier.
Schematic representation of the ATP-triggered DOX release system. (A) Chemical structure of the conjugated polymer (PFFP) described in the text. (B) The main components of DOX/PFFP nanoparticles (NPs): near-infrared (NIR) fluorescent core and flexible hydrophilic PEG chains as shell by self-assembly of amphiphilic conjugated polymer (PFFP), and the 3-fluoro-4-carboxyphenylboronic acid (FPBA) tags on the surface designed as binding sites for ATP. (C) ATP-responsive delivery of DOX by DOX/PFFP NPs to nuclei for the targeted cancer therapy.
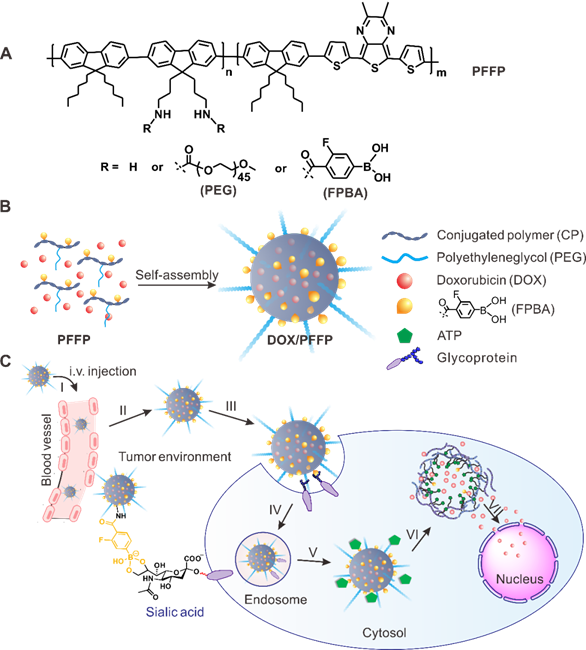
After intravenous (i.v.) injection, DOX/PFFP NPs preferentially accumulate at the tumor sites by the enhanced permeability and retention (EPR) effect[1], and the binding affinity toward the overexpressed glycans or sialic acids on the surface of cancer cells[50-53], followed by cellular internalization through endocytosis (Figure 1C). The hydrolysis of borate ester component promotes endosomal escape of the NPs, allowing the efficient transport of DOX/PFFP into the cytosol. At a significantly higher ATP level in the cytosol compared with that in the extracellular fluid[42, 45], DOX/PFFP NPs dissociate with the subsequent release of the cargo. The released DOX could accumulate into the nuclei to induce DNA damage-mediated cytotoxicity and apoptosis[54].
Methods
Materials
All chemicals were purchased from Sigma-Aldrich unless otherwise specified and were used as received. Monomer 2 and 3 were purchased from Beijing Allmers Chemical S&T Co., Ltd. (Beijing, China).
Synthesis of CP-NH2
The synthesis route to the conjugated polymers containing amine groups (CP-NH2) was shown in Figure S1. Briefly, 2,7-dibromo-9,9-bis(3-aminopropyl) fluorene (1) was prepared according to the reported reference [55]. Monomer 1 (0.22 g, 0.5 mmol), monomer 2 (0.62 g, 1 mmol), monomer 3 (0.24 g, 0.5 mmol), and Pd(PPh3)4 (50 mg) were dissolved in a degassed mixture of toluene (25 mL) and K2CO3 aqueous solution (2 mol/L, 12.5 mL). The mixture was stirred at 85-90 °C for 2 days under an argon atmosphere. After cooling to room temperature, the mixture was extracted with chloroform, washed with brine and distilled water, and then precipitated from acetone to afford a brown solid with 57% yield. 1H NMR (300 MHz, CDCl3, δ): 7.36-7.78 (m, Ar-H), 2.75 (s, pyrazine-CH3), 1.90-2.20 (m, Fluorene-CH2, N-CH2), 0.72-0.90 (m, -CH2CH2CH3). 13C NMR (75 MHz, CDCl3, δ): 152.12, 149.38, 143.39, 141.05, 139.91, 137.72, 133.9, 130.5, 129.70, 128.74, 127.63, 123.78, 120.57, 53.14, 50.19, 48.55, 46.31, 37.10, 32.17, 30.06, 29.43, 27.33, 26.42, 23.22, 19.44, 15.72. The molecular weight of CP-NH2 was determined by GPC (calibrated by polystyrene standard). The weight-average molecular weight was 11 000 g/mol, and the polydispersity was 1.2. MS (MALDI-TOF, m/z): 12995 (Figure S2).
Synthesis of PFFP and PFP
CP-NH2 was allowed to react with 3-fluoro-4-carboxyphenylboronic acid and methoxypoly(ethylene glycol) succinimidyl ester (PEG-SC), coupling their carboxyl groups with the amino groups of the conjugated polymer to yield conjugated polymer-g-FPBA/PEG (PFFP) (Figure S1). Briefly, CP-NH2 (0.12 mmol) and 4-dimethylaminopyridie (DMAP) (0.04 mmol) were added in anhydrous DMSO (25 mL) solution containing FPBA (0.15 mmol) and N, N'-dicyclohexylcarbodiimide (DCC) (0.15 mmol) in an ice bath with argon. The mixture was firstly stirred for 1 h at 0 °C, and further 24 h at room temperature. Then, PEG-SC (0.09 mmol) was added and further stirred at room temperature for 3 days. After being placed in a fridge, 1,3-dicyclohexylurea (DCU) was filtered off, and the filtrate was further dialyzed against distilled water using a dialyser with a cutoff molecular weight of 8000-10 000 g/mol for 2 days. The resulting aqueous solution was then freeze-dried and the product was vacuum-dried overnight to yield PFFP. 1H NMR (300 MHz, CDCl3, δ): 7.42-8.00 (m, Ar-H), 6.83 (s, -NH-CO), 3.56-3.64 (m, CH3-O-CH2-CH2-O-), 2.75 (s, pyrazine-CH3), 2.0-2.20 (m, Fluorene-CH2, N-CH2), 0.74-0.88 (m, -CH2CH2CH3). 13C NMR (75 MHz, CDCl3, δ): 161.55, 157.64, 152.25, 146.88, 141.39, 139.54, 138.25, 136.26, 132.38, 130.22, 129.11, 128.96, 127.84, 125.79, 123.50, 113.8, 66.63, 65.87, 55.06, 53.28, 46.95, 45.76, 36.13, 31.52, 30.44, 29.43, 28.20, 25.89, 23.08, 19.93, 14.28. 11B NMR (160.4 MHz, DMSO-d6, δ): 26.9. In addition, conjugated polymer-g-PEG (PFP) (without FPBA) was prepared by the same methods.
Preparation and characterization of DOX-loaded nanoparticles DOX/PFFP NPs
DOX/PFFP nanoparticles were prepared by a solvent-exchange method[56]. Briefly, PFFP (1.5 mg) was dissolved in THF (2 mL) and stirred for 3 h at room temperature for further use. A mixture solution of dimethyl sulfoxide (DMSO) (0.5 mL) and THF (1 mL) containing triethanolamine-treated doxorubicin hydrochloride (4 mg/mL) was stirred for 30 min at room temperature for further use. Then 0.1 mL of DOX was added into 2 mL of PFFP solution for stirring 10 min. 4 mL of deionized water was slowly added into the PFFP and DOX solution by syringe with stirring (850 rpm) at speed of 2 mL/h. The mixture solution was dialyzed against distilled water for 24 h, using a membrane with a cutoff molecular weight of 8000-10000 g/mol. The resulting nanoparticles (DOX/PFFP NPs) were cleaned and collected by repeating a procedure of centrifugation at 10 000 rpm and re-suspension in distilled water three times. The product was stored at 4 °C. The loading amount of DOX was determined by measuring the fluorescence intensity of DOX via microplate reader (Infinite M200 PRO, Tecan). DOX/PFFP NPs was characterized by the Zetasizer (Nano ZS, Malvern), TEM (JEM-2000FX, Hitachi), and SEM (Hitachi s-4800 high resolution SEM). Ultraviolet-visible (UV-vis) absorption spectra were collected on a Mapada Model UV-1800 spectrophotometer. Fluorescence spectra were measured on Horiba Jobin Yvon Model FM-4NIR spectrophotometer. In addition, DOX/PFP NPs were prepared by the same methods.
In vitro ATP-triggered DOX release
DOX/PFFP NPs (0.6 mL) with 4.8 μg of DOX were added into a dialysis tube (3.5K MWCO) (Slide-A-Lyzer, Thermo Scientific) embedded into 1 mL of PBS buffer solution (pH=7.4) containing different concentrations of ATP, and gently shaken at 37 °C in a shaker (New Brunswick Scientific). At predetermined time intervals, the total buffer solution was withdrawn, followed by replacing with 1mL of fresh buffer solution with the same ATP concentration. The fluorescence intensity of DOX released was measured at 595 nm with an excitation wavelength of 480 nm by a microplate reader (Infinite M200 PRO, Tecan).
Cell culture
HepG2 cells were obtained from Tissue Culture Facility of UNC Lineberger Comprehensive Cancer Center and cultured in Dulbecco's Modified Eagle Medium (DMEM) with 10% (v:v) fetal bovine serum (FBS), 100 U/mL penicillin, and 100 μg/mL streptomycin.
Intracellular ATP-dependent DOX release
HepG2 cells (1×105 cells per well) were seeded in a confocal microscopy dish. After cultured for 24 h, the cells were incubated with DOX/PFFP NPs (2 μg/mL DOX concentration) at 37 °C for 2 h, and then washed by PBS twice, following by incubation with fresh FBS-free culture medium for additional 0 h or 4 h. Subsequently, the cells were stained by Ly-so-Tracker Green (50 nM) (Life Technologies) at 37 °C for 30 min and Hoechst 33342 (1 μg/mL) (Life Technologies) at 37 °C for 10 min. Finally, the HepG2 cells were washed by PBS twice and observed via confocal laser scanning microscope (LSM710, Zeiss). Additionally, low temperature (4 °C) and iodoacetic acid (IAA) at 37 °C were used to inhibit ATP production in the cells. After incubated with DOX/PFFP NPs (2 μg/ml DOX concentration) at 37 °C for 2 h and then washed by PBS twice, the cells were incubated at 4 °C or with IAA (100 mM) at 37 °C for additional 4 h. For Hoechst 33342 detection, the excitation wavelength was 405 nm, and the emission filter was 455±20nm; For LysoTracker Green, DOX, PFFP detection, the excitation wavelength was 488 nm, and the emission filter band was 510±10nm, 605±20nm, and 710±30nm, respectively.
Flow cytometry quantification of DOX in HepG2 cell nuclei
HepG2 cells (1×105 cells per well) were seeded in six-well plates. After cultured for 48 h, the cells were incubated with DOX/PFFP NPs (2 μg/ml DOX concentration) at 37 °C for 2 h, and then washed by PBS twice, following by incubation with fresh FBS-free culture medium at different conditions for additional 4 h to ensure the released DOX to migrate into the cell nuclei, including normal temperature (37 °C), low temperature (4 °C), and with IAA (100 mM) at 37 °C. Finally, after trypsinization, HepG2 cells were suspended at a concentration of 5 ×106 cells/mL for 10 min at 4 °C in a 100 mM NaCl solution with 1 mM EDTA, 1% Triton X-100 and 10 mM Tris buffer (pH 7.4). The suspension was then centrifuged (15 min, 800 g), and the resulting precipitate of cell nuclei was separated from the supernatant cell cytosol, finally re-suspended in 0.2 mL of 1× PBS. The fluorescence intensity of DOX in the cell nuclei was measured using flow cytometry (CytoFLEX Flow Cytometry, Beckman Coulter Ltd.). In addition, untreated group, PFFP NPs, DOX/PFP NPs, and free DOX as control were analyzed by the same methods.
In vitro cytotoxicity[57]
HepG2 cells (5 × 103 cells/well) were seeded in 96-well plates. After incubation for 24 h, the cells were then treated with different samples at different DOX concentrations and further incubated for 24 h at 37 °C, including free DOX solution, DOX/PFP NPs, and DOX/PFFP NPs. Followed 20 μL of the MTT (5 mg/mL) was added into the HepG2 cells. After incubation for 4 h, the medium was replaced with DMSO (150 μL). The absorbance was measured at wavelength of 570 nm by the microplate reader (Infinite M200 PRO, Tecan).
Animals and tumor model
The animals were treated in accordance with the Guide for Care and Use of Laboratory Animals, approved by the Model Animal Research Center (MARC) of Nanjing University. HepG2 tumor model was established by subcutaneous injection of HepG2 cells (1 × 106) into the selected positions of the nude mice (6 weeks of age, purchased from MARC of Nanjing University). The tumor size was monitored by a vernier caliper, and the tumor volume (V) was calculated as V=L*W2/2, where L and W were the length and width of the tumor, respectively.
In vivo imaging study
When the HepG2 tumors reached 200-400 mm3, the mice were intravenously injected by DOX/PFFP NPs at PFFP dose of 3.6 mg/kg. The real-time NIR fluorescence imaging was performed using a MaestroTM EX fluorescence imaging system (Cambridge Research & Instrumentation, USA) after 4, 12, and 24 h post-injection. After the 24 h scanning, the mice were euthanized. The tumors and major organs were harvested, and subjected for ex vivo imaging. Region-of-interests were circled around the organs, and the fluorescence intensities were analyzed by Living Image Software. The excitation wavelength is 595 nm, and in vivo fluorescence imaging was collected from 680 to 780 nm (with 10 nm step). The autofluorescence of mice was removed by spectral unmixing software. Besides, DOX/PFP NPs were analyzed by the same methods.
To evaluate the metabolism process of DOX/PFFP NPs in mice by CLSM, the mice were intravenously injected by DOX/PFFP NPs at PFFP dose of 3.6 mg/kg, and then real-time NIR fluorescence imaging was performed within 5 days.
In vivo antitumor efficacy
The tumor-bearing mice were weighed and randomly divided into different groups when the tumor volume reached 50 mm3, and subjected to the following treatments: 1. Saline; 2. DOX solution; 3. DOX/PFP NPs; 4. DOX/PFFP NPs. The mice were intravenously injected with same DOX concentration (DOX of 2 mg kg-1) in all DOX formulations every other day for 12 days, meanwhile the tumor sizes were measured. At day 14, the mice were euthanized, and the tumors were collected, weighed, washed with saline thrice and fixed in the 10% neutral-buffered formalin. For the hematoxylin and eosin (HE) and the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) apoptosis staining, the formalin-fixed tumors were prepared by Nanjing Drum Tower Hospital, and observed by the fluorescence microscope (IX71, Olympus).
Statistical analysis
Data are given as Mean ± SD. Statistical significance was performed using two-tailed Student's t-test. Statistical significance was set at *P < 0.05, and extreme significance was set at **P < 0.01.
Results and Discussion
Synthesis and characterization of DOX/PFFP NPs
To substantiate our design, we firstly synthesized an ATP-responsive and NIR-emissive conjugated polymer PFFP (Figure 1A) by alternating copolymerization of fluorene- (a high-efficient blue emitting unit) and dithiophene-thienopyrazine- (a unit to effectively shift emission maximum to NIR wavelength[46]) containing monomers via the Suzuki cross-coupling reaction. Then the branched chains were further modified with FPBA and PEG by amidation reactions[55] (Figure S1). The obtained PFFP was an amphiphilic polymer, which was utilized to encapsulate DOX by self-assembly (Figure 1B). The fluorescent core of the nanoparticles was hydrophobic π-conjugated backbone, and the flexible hydrophilic PEG chains were selected due to its biocompatibility. The electron withdrawing amide carbonyl and fluorine on the FPBA moiety reduce the pKa of boronic acid monomer, which allows its response to ATP at a physiological pH. At the intracellular ATP concentration, the FPBA groups on the branched chains were expected to interact with ATP, resulting in water-solubility of the conjugated polymer, thus promoting disassembly of DOX/PFFP NPs. The signal at 26.9 ppm in 11B NMR spectrum of PFFP is characteristic of trigonal boronic acid, indicated the successful conjugation. The ratio of FPBA group and the PEG group was determined as 2/1 via 1H-NMR.
The rigid π-conjugated backbone is hydrophobic, while the grafted PEG chains easily dissolve in water. Thus this amphiphilic copolymer tends to form robust and spherical nanoparticles (NPs) in the aqueous solution. The NPs in aqueous suspension shown an average hydrodynamic diameter of 116 nm with a narrow size distribution measured by dynamic light scattering (DLS) (Figure 2A). The morphology of the conjugated polymer nanoparticles was observed using TEM and SEM (inset of Figure 2A and Figure 2C). The spherical shape of the nanoparticles was clearly distinguished with a diameter of slightly smaller than that in aqueous suspension due to the collapse of outmost PEG layers during the drying process. The DOX loading capacity was measured to be 20 % (wt/wt). The UV/Vis absorption spectrum of PFFP NPs in aqueous suspension displayed two absorption bands at 392 and 583 nm, respectively (Figure 2B), which arise from the light absorption by the π-electron system of the conjugated backbone. The maximum emission wavelength of PFFP was 720 nm, indicating that the NPs have NIR emission suitable for in vivo imaging, with a Stokes shift of approximately 140 nm (excited at 480 nm) (Figure 2B and Figure S3).
Characterization and in vitro ATP-responsive DOX release from DOX/PFFP. (A) Hydrodynamic size distribution and TEM image (inset) of DOX/PFFP NPs. Scale bar is 200 nm. (B) Normalized UV/Vis absorption and emission spectra of PFFP NPs and DOX/PFFP NPs (excitation at 480 nm). (C) SEM images and (D) size distribution of DOX/PFFP NPs at different ATP concentration. Scale bars are 200 nm. (E) Average size changes of DOX/PFFP NPs when incubated in DI water, PBS buffer, DMEM, and glucose for 7 days. (F) In vitro release of DOX from DOX/PFFP NPs in 0.4 mM, 4 mM ATP, and DOX/PFP NPs (without ATP sensitivity) in 4 mM ATP. Error bars indicate SD (n = 3).
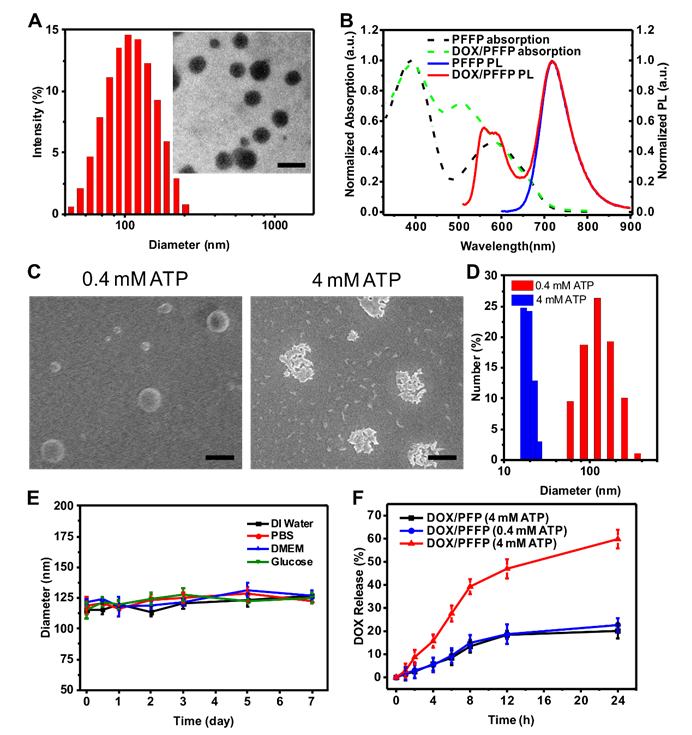
In vitro ATP-triggered DOX release from DOX/PFFP
To investigate the ATP-responsive capability, PFFP and DOX/PFFP NPs were incubated at 0.4 mM and 4 mM ATP concentrations, which represent the typical ATP level in the extracellular fluid and intracellular cytosol, respectively[42-45]. As shown in Figure 2C and Figure S4, a remarkable disassembly of the NPs was observed in the presence of 4 mM ATP concentration. In contrast, the NPs were nearly intact when treated with 0.4 mM ATP. DLS measurement also confirmed that the NPs were responsive to the high concentrations of ATP (Figure 2D and Figure S4). To evaluate their colloidal stability, DOX/PFFP NPs were incubated in the DI water, phosphate-buffered saline (PBS, pH 7.4), and Dulbecco's Modified Eagle Medium (DMEM). The average sizes of the nanoparticles had no significant change after incubation for 7 days (Figure 2E). Of note, the nanoparticles can maintain high stability in the glycemic concentration (5 mM). Moreover, the average sizes and morphology of the nanocarriers have no significant change after incubation with 1 mM H2O2 for 24 h at 37 °C (Figure S5).
The ATP-triggered DOX release behaviors of DOX/PFFP NPs were further tested (Figure 2F). In the presence of 0.4 mM ATP solution (pH=7.4), only 5% of DOX was released from DOX/PFFP NPs in the first 4 h and about 20% was released within 24 h. In the same buffer solution with 4 mM ATP, comparable to intracellular ATP levels, the release of DOX was dramatically accelerated. About 18% of DOX was released from DOX/PFFP NPs in the first 4 h and more than 55% was released within 24 h. It is also noteworthy that the release of DOX from DOX/PFP NPs, where the lack of FPBA groups in PFP led to the nanoparticles without ATP sensitivity, is slow even in the presence of 4 mM ATP. It indicated that FPBA groups were essential for the fast release of the intercalated DOX from the nanoparticles in an ATP-rich environment.
Intracellular ATP-triggered DOX release from DOX/PFFP
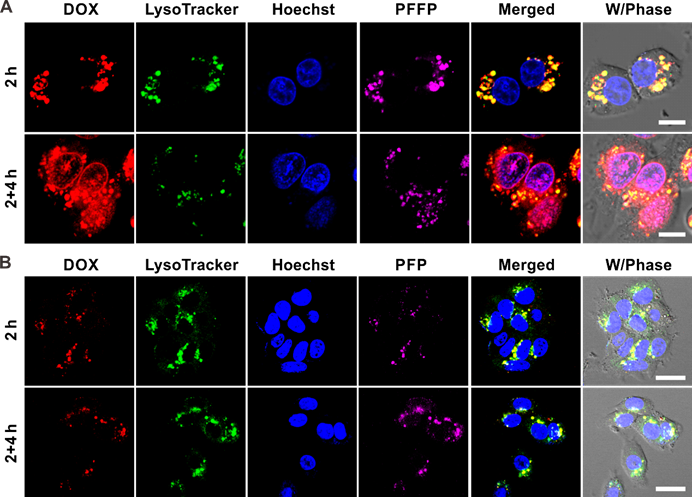
The intracellular delivery of DOX/PFFP NPs was explored in the human liver carcinoma (HepG2) cells using confocal laser scanning microscopy (CLSM). The HepG2 cells were incubated with DOX/PFFP NPs for 2 h, and then incubated with fresh culture medium for an additional 4 h at 37 °C. As shown in Figure 3A, most of the endocytic DOX/PFFP NPs were located in the endolysosomes judged by the yellow fluorescence during the first 2h of the cellular uptake of DOX/PFFP NPs. Notably, as evidenced by the magenta fluorescence, a significant increase of DOX fluorescence intensity in the nuclei was observed after an additional incubation for 4h. Moreover, we studied the microscopy images of DOX/PFP NPs as control, as shown in Figure 3B. Most of the endocytic DOX/PFP NPs were located in the endolysosomes, and no significant DOX fluorescent signal in the nuclei was observed after an additional incubation for 4h.
Intracellular delivery of (A) DOX/PFFP NPs and (B) DOX/PFP NPs into HepG2 cells at different time observed by CLSM. The endosomes and lysosomes were stained by LysoTracker Green, and the nuclei were stained by Hoechst 33342. Merged (DOX/ LysoTracker/ Hoechst). Scale bars are 20 μm.
To further demonstrate DOX/PFFP NPs-mediated intracellular ATP-triggered DOX release, HepG2 cells were incubated with DOX/PFFP NPs at a low temperature (4 °C) or in the presence of iodoacetic acid (IAA)[42, 58], which can inhibit the ATP generation. Cell incubation at 4 °C or with IAA at 37 °C both led to a significant decrease in ATP generation within the cells. The cells incubated both at 4 °C and with IAA at 37 °C showed insignificant DOX fluorescence signals in the nuclei (Figure 4A). We also tracked the intracellular ATP-triggered hydrolysis of PFFP NPs. As shown in Figure S6, PFFP were mainly distributed in the cytoplasm, and as contrast, the cells incubated both at 4 °C and with IAA at 37 °C showed that PFFP was mainly located in the endolysosomes indicated by the white fluorescence. In addition, the quantitative analysis of the cellular uptake of DOX by HepG2 cell nuclei was carried out using the flow cytometry. As shown in Figure 4B, more DOX molecules were found within DOX/PFFP NPs-treated cells incubated at 37 °C. However, at 4 °C and with IAA at 37 °C, cellular uptake of DOX was inhibited. Of note, for DOX/PFP NPs without ATP sensitivity, no remarkable DOX fluorescent signal was observed. Collectively, these results substantiate that the DOX release from DOX/PFFP NPs is an ATP level-dependent process.
Intracellular DOX/PFFP NPs-mediated ATP-triggered DOX release. (A) Intracellular delivery of DOX/PFFP NPs in HepG2 cells treated with different formulations observed by CLSM, including at 37 °C, 4 °C, and with the ATP inhibitor iodoacetic acid (IAA) at 37 °C. The endosomes and lysosomes were stained by LysoTracker Green, and the nuclei were stained by Hoechst 33342. Merged (DOX/ LysoTracker/Hoechst). Scale bar is 20 μm. (B) Flow cytometry analysis of the DOX fluorescence intensity in HepG2 cell nuclei. (C) In vitro cytotoxicity of HepG2 cells incubated for 24 h with free DOX, DOX/PFP NPs (without ATP sensitivity), and DOX/PFFP NPs. Error bars indicate SD (n = 3).
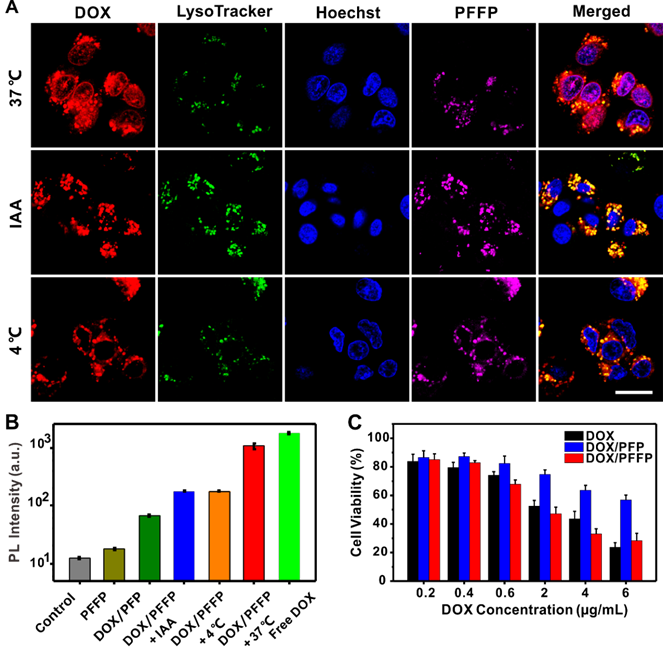
The in vitro cytotoxicity of DOX/PFFP NPs against HepG2 cells was evaluated by using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay[57]. Compared with ATP-insensitive DOX/PFP NPs, DOX/PFFP NPs exhibited obviously enhanced cytotoxicity towards HepG2 cells at whole studied DOX concentrations (Figure 4C). Notably, the cytotoxicity of DOX/PFFP NPs increased along with the concentration of DOX with a half lethal dose (IC50) of 2.1 μg mL-1 at incubation time of 24 h. Moreover, efficient intracellular release of DOX from DOX/PFFP NPs provided higher cytotoxic activity than that of the DOX solution (IC50 = 3.8 μg mL-1), indicating that an enhanced potency could be achieved using ATP-responsive chemotherapy.
In vivo NIR imaging and antitumor efficacy of DOX/PFFP
The therapeutic efficacy of DOX/PFFP was investigated in the mice bearing human liver carcinoma tumors. To explore the in vivo NIR imaging capability of PFFP, PFFP and DOX/PFFP NPs were administrated intravenously into the HepG2 tumor-bearing mice via tail vein. As shown in Figure S7 and 5A, a strong fluorescence signal of PFFP was detected at the tumor site after administration for 4 h. As time extended, an enhanced PFFP signal was found in the tumor than that in the normal tissues, suggesting a notable tumor targeting ability of DOX/PFFP NPs (Figure 5A). After 24 h post injection, the tumor and normal tissues were harvested for ex vivo imaging. The strongest fluorescence signal of PFFP was observed in the tumor and liver compared with other organs (Figure 5B). The quantitative region-of-interest (ROI) analysis (Figure 5C) indicated that DOX/PFFP NPs allowed high accumulation of the drugs at the tumor site via a combination of passive and active targeting effects, since phenylboronic acid tags on DOX/PFFP NPs can selectively bind to the over-expressed glycans or sialic acids on the surface of HepG2 cells[50-53]. Additionally, the enhanced accumulation of DOX/PFFP NPs at tumor site was further validated as compared with DOX/PFP NPs (without phenylboronic acid) by ex vivo imaging and ROI analysis, and the fluorescence signal at the tumor site of DOX/PFFP-treated mice was approximately 2.5- times as much as that of DOX/PFP-treated mice 24 h after injection (Figure 5D and 5E).
In vivo NIR imaging of DOX/PFFP NPs. (A) In vivo fluorescence images of the HepG2 tumor-bearing mice at 4, 12, and 24 h after intravenous injection of DOX/PFFP NPs. Arrow indicates the sites of tumor. (B) Ex vivo fluorescence images of the tumor and normal organs collected from the mice 24 h after administration. 1: heart, 2: liver, 3: spleen, 4: lung, 5: kidney, 6: tumor. (C) The quantitative region-of-interest (ROI) analysis of fluorescent signals of the tumor and normal organs. The relative fluorescence signal biodistribution of DOX/PFFP and DOX/PFP NPs in the tumor and liver: (D) Ex vivo fluorescence images of the tumor and liver collected from the mice 24 h after administration. (E) The quantitative region-of-interest (ROI) analysis of fluorescent signals of the tumor and liver. Error bars indicate SD (n = 3).
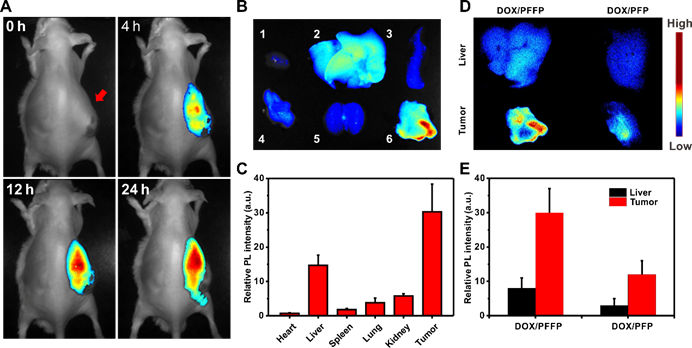
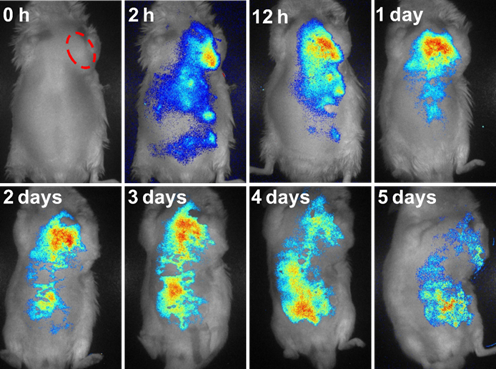
We also evaluated the metabolism process of DOX/PFFP NPs administered intravenously by monitoring the PFFP fluorescence signals (Figure 6). The NPs were accumulated in the tumor site within 1 day, and reached the maximum around 1-3 days' post-injection, then started to be discharged from the tumor at day 4, which suggested that the successive administration every other day was able to maintain a high drug concentration in the systemic circulation within a prolonged period. These results indicate that PFFP has potential to be exploited as an efficient drug-delivery system for imaging-guided therapeutics[59-61]. Moreover, we further studied the biodistribution of DOX and PFFP in various organs by quantitative analysis. The mice bearing the tumors were intravenously treated with DOX/PFFP NPs at a DOX-equivalent dose of 2 mg kg-1. After 24 h post injection, the tumor and normal tissues were harvested, following by tissue homogenization, acidic isopropanol extraction, and fluorometric analysis. As shown in Figure S8, DOX/PFFP NPs allowed high accumulation of the drugs at the tumor site after 24 h of intravenous administration.
In vivo fluorescence images of the HepG2 tumor-bearing mice in 5 days after intravenous injection of DOX/PFFP NPs. The red ellipse indicates the site of the tumor.
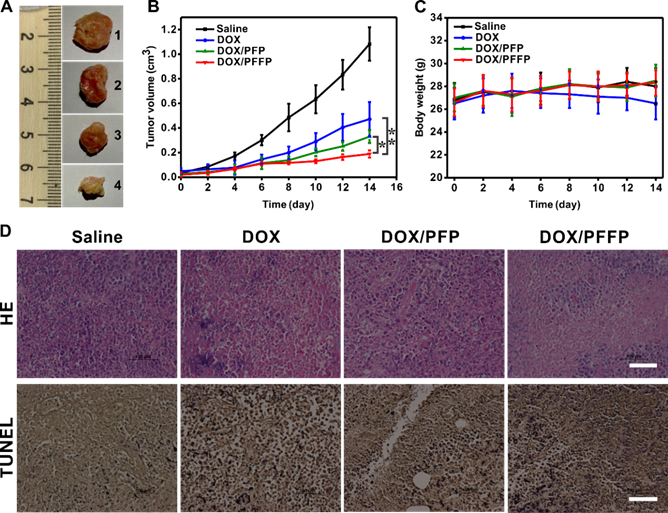
Next, to assess the in vivo efficacy of DOX/PFFP NPs for antitumor treatment, the HepG2 tumor-bearing mice were exposed to treatment with different samples, including the saline as a negative control, DOX, DOX/PFP NPs and DOX/PFFP NPs. The growth of the tumor was significantly inhibited after the treatment with different DOX formulations (Figure 7A and 7B). DOX/PFFP NPs and DOX/PFP NPs showed the remarkably higher inhibition efficacy towards tumor growth than free DOX, which could be attributed to the EPR effect of the NPs. Of note, DOX/PFFP NPs showed the smaller tumor volume compared with that of DOX/PFP NPs, which indicated that the efficient intracellular delivery and ATP-responsive DOX release of DOX/PFFP NPs enabled the reinforcement on the anticancer efficacy. Meanwhile, the body weight of the mice treated with different samples remained stable except free DOX, because of the strong side-effect of DOX to the normal organs[54] (Figure 7C). We further applied the hematoxylin and eosin (HE) and the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining assay to evaluate the treatment efficacy (Figure 7D). The images of HE-stained tumor tissues treated by DOX/PFFP NPs presented prominent necrosis of the tumor cells. The images obtained from the TUNEL staining showed the highest level of cell apoptosis in the tumor tissue of the mice receiving DOX/PFFP NPs. Furthermore, no obvious pathological abnormalities were observed in normal organs (Figure S9). Taken together, these results validated that ATP-responsive DOX/PFFP NPs efficiently accumulated at the tumor site, and thereby achieved optimal therapeutic efficacy in vivo through effective intracellular delivery and stimuli-triggered drug release.
Antitumor efficacy of DOX/PFFP NPs. (A) Representative images of the HepG2 tumors after treatment with different samples at day 14 (from top to bottom, 1: saline, 2: DOX, 3: DOX/PFP NPs, 4: DOX/PFFP NPs. (B) The HepG2 tumor growth curves after treatment with different samples. (C) The body weight variation of HepG2 tumor-bearing mice during treatment. (D) Histological observation of the tumor tissues stained with HE after treatment with different samples, and detection of apoptosis in the tumor tissues after treatment with the TUNEL staining assay. Scale bars are 100 μm. Error bars indicate SD (n = 5); *P < 0.05, **P < 0.01.
Conclusions
In summary, we have developed an ATP-responsive and NIR-imaging drug delivery system for imaging-guided chemotherapy. This conjugated polymer-based delivery system presents both targeting and controlled release behaviors associated with the surrounding ATP levels. This system provides guideline to design conjugated polymer-based theranostic formulation for enhancing efficacy of pinpoint cancer therapy. Further investigation includes detailed assessment of systemic and long-term toxicity as well as the bio-environment-associated photoluminescence property of the nanocarriers.
Supplementary Material
Figures S1-9.
Acknowledgements
This work was supported by the grants from National Natural Science Foundation of China (No. 21174060), NC TraCS, NIH's Clinical and Translational Science Awards (CTSA, NIH Grant No. 1UL1TR001111) at UNC-CH, and Program for Changjiang Scholars and Innovative Research Team in University (No. IRT1252). The authors acknowledge the use of the Analytical Instrumentation Facility (AIF) at NC State, which is supported by the State of North Carolina and the National Science Foundation (NSF). C. Qian greatly thanks the China Scholarship Council for financially supporting.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol. 2007;2:751-60
2. Chow EK, Ho D. Cancer nanomedicine: from drug delivery to imaging. Sci Transl Med. 2013;5:216rv4
3. Mitragotri S, Burke PA, Langer R. Overcoming the challenges in administering biopharmaceuticals: formulation and delivery strategies. Nat Rev Drug Discov. 2014;13:655-72
4. Mitragotri S, Anderson DG, Chen X, Chow EK, Ho D, Kabanov AV. et al. Accelerating the Translation of Nanomaterials in Biomedicine. ACS nano. 2015;9:6644-54
5. Zhu G, Zheng J, Song E, Donovan M, Zhang K, Liu C. et al. Self-assembled, aptamer-tethered DNA nanotrains for targeted transport of molecular drugs in cancer theranostics. Proc Natl Acad Sci U S A. 2013;110:7998-8003
6. Lu Y, Sun W, Gu Z. Stimuli-responsive nanomaterials for therapeutic protein delivery. J Control Release. 2014;194:1-19
7. Li S, Gaddes ER, Chen N, Wang Y. Molecular encryption and reconfiguration for remodeling of dynamic hydrogels. Angew Chem Int Ed Engl. 2015;54:5957-61
8. Xie J, Lee S, Chen X. Nanoparticle-based theranostic agents. Adv Drug Deliv Rev. 2010;62:1064-79
9. Zheng J, Zhang C, Dickson RM. Highly fluorescent, water-soluble, size-tunable gold quantum dots. Phys Rev Lett. 2004;93:077402
10. Xie J, Liu G, Eden HS, Ai H, Chen X. Surface-engineered magnetic nanoparticle platforms for cancer imaging and therapy. Acc Chem Res. 2011;44:883-92
11. Niu G, Chen X. Lymphatic imaging: focus on imaging probes. Theranostics. 2015;5:686-97
12. Nomoto T, Fukushima S, Kumagai M, Machitani K, Arnida, Matsumoto Y. et al. Three-layered polyplex micelle as a multifunctional nanocarrier platform for light-induced systemic gene transfer. Nat Commun. 2014;5:3545
13. Carter KA, Shao S, Hoopes MI, Luo D, Ahsan B, Grigoryants VM. et al. Porphyrin-phospholipid liposomes permeabilized by near-infrared light. Nat Commun. 2014;5:3546
14. Di J, Kim J, Hu Q, Jiang X, Gu Z. Spatiotemporal drug delivery using laser-generated-focused ultrasound system. J Control Release. 2015;220:592-9
15. Oliveira H, Perez-Andres E, Thevenot J, Sandre O, Berra E, Lecommandoux S. Magnetic field triggered drug release from polymersomes for cancer therapeutics. J Control Release. 2013;169:165-70
16. Chakravarty R, Goel S, Cai W. Nanobody: the “magic bullet” for molecular imaging. Theranostics. 2014;4:386
17. Choi SW, Zhang Y, Xia Y. A temperature-sensitive drug release system based on phase-change materials. Angew Chem Int Ed Engl. 2010;49:7904-8
18. Sirivisoot S, Pareta R, Webster TJ. Electrically controlled drug release from nanostructured polypyrrole coated on titanium. Nanotechnology. 2011;22:085101
19. Wang Y, Zhou K, Huang G, Hensley C, Huang X, Ma X. et al. A nanoparticle-based strategy for the imaging of a broad range of tumours by nonlinear amplification of microenvironment signals. Nat Mater. 2014;13:204-12
20. Sun CY, Shen S, Xu CF, Li HJ, Liu Y, Cao ZT. et al. Tumor Acidity-Sensitive Polymeric Vector for Active Targeted siRNA Delivery. J Am Chem Soc. 2015;137:15217-24
21. Cheng R, Meng F, Deng C, Zhong Z. Bioresponsive polymeric nanotherapeutics for targeted cancer chemotherapy. Nano Today. 2015;10:656-70
22. Broaders KE, Grandhe S, Frechet JM. A biocompatible oxidation-triggered carrier polymer with potential in therapeutics. J Am Chem Soc. 2011;133:756-8
23. Wang M, Sun S, Neufeld CI, Perez-Ramirez B, Xu Q. Reactive oxygen species-responsive protein modification and its intracellular delivery for targeted cancer therapy. Angew Chem Int Ed Engl. 2014;53:13444-8
24. Hu Q, Katti PS, Gu Z. Enzyme-responsive nanomaterials for controlled drug delivery. Nanoscale. 2014;6:12273-86
25. Sun W, Jiang T, Lu Y, Reiff M, Mo R, Gu Z. Cocoon-like self-degradable DNA nanoclew for anticancer drug delivery. J Am Chem Soc. 2014;136:14722-5
26. Mo R, Jiang T, Di J, Tai W, Gu Z. Emerging micro- and nanotechnology based synthetic approaches for insulin delivery. Chem Soc Rev. 2014;43:3595-629
27. Zhang YQ, Yu JC, Shen QD, Gu Z. Glucose-Responsive Synthetic Closed-Loop Insulin Delivery Systems. Prog Chem. 2015;27:11-26
28. Hola K, Zhang Y, Wang Y, Giannelis EP, Zboril R, Rogach AL. Carbon dots—Emerging light emitters for bioimaging, cancer therapy and optoelectronics. Nano Today. 2014;9:590-603
29. Chen C, Peng J, Sun SR, Peng CW, Li Y, Pang DW. Tapping the potential of quantum dots for personalized oncology: current status and future perspectives. Nanomedicine (Lond). 2012;7:411-28
30. Cobley CM, Chen J, Cho EC, Wang LV, Xia Y. Gold nanostructures: a class of multifunctional materials for biomedical applications. Chem Soc Rev. 2011;40:44-56
31. Furukawa H, Cordova KE, O'Keeffe M, Yaghi OM. The chemistry and applications of metal-organic frameworks. Science. 2013;341:1230444
32. Sun B, Sun MJ, Gu Z, Shen QD, Jiang SJ, Xu Y. et al. Conjugated polymer fluorescence probe for intracellular imaging of magnetic nanoparticles. Macromolecules. 2010;43:10348-54
33. Traina CA, Bakus RC 2nd, Bazan GC. Design and synthesis of monofunctionalized, water-soluble conjugated polymers for biosensing and imaging applications. J Am Chem Soc. 2011;133:12600-7
34. Ahmed E, Morton SW, Hammond PT, Swager TM. Fluorescent multiblock pi-conjugated polymer nanoparticles for in vivo tumor targeting. Adv Mater. 2013;25:4504-10
35. Yuan Y, Liu J, Liu B. Conjugated-Polyelectrolyte-Based Polyprodrug: Targeted and Image-Guided Photodynamic and Chemotherapy with On-Demand Drug Release upon Irradiation with a Single Light Source. Angew Chem Int Ed Engl. 2014;126:7291-6
36. Jin GR, Mao D, Cai PQ, Liu RR, Tomczak N, Liu J. et al. Conjugated Polymer Nanodots as Ultrastable Long-Term Trackers to Understand Mesenchymal Stem Cell Therapy in Skin Regeneration. Adv Funct Mater. 2015;25:4263-73
37. Pu KY, Shuhendler AJ, Jokerst JV, Mei JG, Gambhir SS, Bao ZN. et al. Semiconducting polymer nanoparticles as photoacoustic molecular imaging probes in living mice. Nat Nanotechnol. 2014;9:233-9
38. Lassila JK, Zalatan JG, Herschlag D. Biological phosphoryl-transfer reactions: understanding mechanism and catalysis. Annu Rev Biochem. 2011;80:669-702
39. Zhang Z, Balogh D, Wang F, Willner I. Smart mesoporous SiO2 nanoparticles for the DNAzyme-induced multiplexed release of substrates. J Am Chem Soc. 2013;135:1934-40
40. Zhang Z, Balogh D, Wang F, Sung SY, Nechushtai R, Willner I. Biocatalytic release of an anticancer drug from nucleic-acids-capped mesoporous SiO2 Using DNA or molecular biomarkers as triggering stimuli. ACS nano. 2013;7:8455-68
41. Liao WC, Lu CH, Hartmann R, Wang F, Sohn YS, Parak WJ. et al. Adenosine Triphosphate-Triggered Release of Macromolecular and Nanoparticle Loads from Aptamer/DNA-Cross-Linked Microcapsules. ACS nano. 2015;9:9078-86
42. Mo R, Jiang T, DiSanto R, Tai W, Gu Z. ATP-triggered anticancer drug delivery. Nat Commun. 2014;5:3364
43. Gribble FM, Loussouarn G, Tucker SJ, Zhao C, Nichols CG, Ashcroft FM. A novel method for measurement of submembrane ATP concentration. J Biol Chem. 2000;275:30046-9
44. Gorman MW, Feigl EO, Buffington CW. Human plasma ATP concentration. Clin Chem. 2007;53:318-25
45. Leist M, Single B, Castoldi AF, Kühnle S, Nicotera P. Intracellular adenosine triphosphate (ATP) concentration: a switch in the decision between apoptosis and necrosis. J Exp Med. 1997;185:1481-6
46. Yu JC, Chen YL, Zhang YQ, Yao XK, Qian CG, Huang J. et al. pH-Responsive and near-infrared-emissive polymer nanoparticles for simultaneous delivery, release, and fluorescence tracking of doxorubicin in vivo. Chem Commun (Camb). 2014;50:4699-702
47. Martin AR, Vasseur JJ, Smietana M. Boron and nucleic acid chemistries: merging the best of both worlds. Chem Soc Rev. 2013;42:5684-713
48. Naito M, Ishii T, Matsumoto A, Miyata K, Miyahara Y, Kataoka K. A phenylboronate-functionalized polyion complex micelle for ATP-triggered release of siRNA. Angew Chem Int Ed Engl. 2012;51:10751-5
49. Zhu C, Liu L, Yang Q, Lv F, Wang S. Water-soluble conjugated polymers for imaging, diagnosis, and therapy. Chem Rev. 2012;112:4687-735
50. Wang X, Zhen X, Wang J, Zhang J, Wu W, Jiang X. Doxorubicin delivery to 3D multicellular spheroids and tumors based on boronic acid-rich chitosan nanoparticles. Biomaterials. 2013;34:4667-79
51. Bull C, Stoel MA, den Brok MH, Adema GJ. Sialic acids sweeten a tumor's life. Cancer Res. 2014;74:3199-204
52. Deshayes S, Cabral H, Ishii T, Miura Y, Kobayashi S, Yamashita T. et al. Phenylboronic acid-installed polymeric micelles for targeting sialylated epitopes in solid tumors. J Am Chem Soc. 2013;135:15501-7
53. Lee JY, Chung SJ, Cho HJ, Kim DD. Phenylboronic Acid-Decorated Chondroitin Sulfate A-Based Theranostic Nanoparticles for Enhanced Tumor Targeting and Penetration. Adv Funct Mater. 2015;25:3705-17
54. Tewey KM, Rowe TC, Yang L, Halligan BD, Liu LF. Adriamycin-induced DNA damage mediated by mammalian DNA topoisomerase II. Science. 1984;226:466-8
55. Qian CG, Zhu S, Feng PJ, Chen YL, Yu JC, Tang X. et al. Conjugated Polymer Nanoparticles for Fluorescence Imaging and Sensing of Neurotransmitter Dopamine in Living Cells and the Brains of Zebrafish Larvae. ACS Appl Mater Interfaces. 2015;7:18581-9
56. Borisov SM, Mayr T, Mistlberger G, Waich K, Koren K, Chojnacki P. et al. Precipitation as a simple and versatile method for preparation of optical nanochemosensors. Talanta. 2009;79:1322-30
57. Hu Q SW, Qian C, Wang C, Bomba HN, Gu Z. Anticancer Platelet-Mimicking Nanovehicles. Adv Mater. 2015;27:7043-50
58. Verrax J, Dejeans N, Sid B, Glorieux C, Calderon PB. Intracellular ATP levels determine cell death fate of cancer cells exposed to both standard and redox chemotherapeutic agents. Biochem Pharmacol. 2011;82:1540-8
59. Cai W, Chen X. Multimodality molecular imaging of tumor angiogenesis. J Nucl Med. 2008;49(Suppl 2):113S-28S
60. Zhang R, Fan Q, Yang M, Cheng K, Lu X, Zhang L. et al. Engineering Melanin Nanoparticles as an Efficient Drug-Delivery System for Imaging-Guided Chemotherapy. Adv Mater. 2015;27:5063-9
61. Chen H, Niu G, Wu H, Chen X. Clinical Application of Radiolabeled RGD Peptides for PET Imaging of Integrin alphavbeta3. Theranostics. 2016;6:78-92
Author contact
![]() Corresponding authors: E-mail: qdshenedu.cn, Phone: 86-25-89687807; zguunc.edu, zgu3edu, Phone: 1-919-515-7944.
Corresponding authors: E-mail: qdshenedu.cn, Phone: 86-25-89687807; zguunc.edu, zgu3edu, Phone: 1-919-515-7944.