Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and methods
Results and Discussion
Conclusion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2015; 5(8):890-904. doi:10.7150/thno.11821 This issue Cite
Research Paper
Site-Specific Drug-Releasing Polypeptide Nanocarriers Based on Dual-pH Response for Enhanced Therapeutic Efficacy against Drug-Resistant Tumors
Yaqiong Dong1,2,*, Jun Yang1,*, Hongmei Liu1,3, Tianyou Wang4, Suoqin Tang5, Jinchao Zhang2, ![]() , Xin Zhang1,
, Xin Zhang1, ![]()
1. National Key Laboratory of Biochemical Engineering, Institute of Process Engineering, Chinese Academy of Sciences, Beijing, 100190, China
2. College of Chemistry & Environmental Science, Chemical Biology Key Laboratory of Hebei Province, Key Laboratory of Medicinal Chemistry and Molecular Diagnosis of the Ministry of Education, Hebei University, Baoding, 071002, China
3. University of Chinese Academy of Sciences, Beijing, 100049, China
4. Capital Institute of Pediatrics, Beijing, 100020, China
5. Department of Pediatrics, The General Hospital of People's Liberation Army, Beijing, 100853, China
* These authors contributed equally to this work.
Received 2015-2-8; Accepted 2015-3-31; Published 2015-4-28
Abstract

To enhance effective drug accumulation in drug-resistant tumors, a site-specific drug-releasing polypeptide system (PEG-Phis/Pasp-DOX/CA4) was exploited in response to tumor extracellular and intracellular pH. This system could firstly release the embedded tumor vascular inhibitor (CA4) to transiently 'normalize' vasculature and facilitate drug internalization to tumors efficiently, and then initiate the secondary pH-response to set the conjugated active anticancer drug (DOX) free in tumor cells. The encapsulated system (PEG-Phis/DOX/CA4), both CA4 and DOX embedding in the nanoparticles, was used as a control. Comparing with PEG-Phis/DOX/CA4, PEG-Phis/Pasp-DOX/CA4 exhibited enhanced cytotoxicity against DOX-sensitive and DOX-resistant cells (MCF-7 and MCF-7/ADR). Moreover, PEG-Phis/Pasp-DOX/CA4 resulted in enhanced therapeutic efficacy in drug-resistant tumors with reduced toxicity. These results suggested that this site-specific drug-releasing system could be exploited as a promising treatment for cancers with repeated administration.
Keywords: polypeptide nanocarriers, site-specific releasing, combination cancer therapy, dual-pH response, drug resistance.
Introduction
Therapeutically effective concentration of chemotherapeutic agent in tumors plays a critical role in cancer chemotherapy. However, repeated administration leads to drug resistance of the tumor cells, which significantly hinders drug accumulation in tumor cells and finally results in failure in chemotherapy [1, 2]. Therefore, how to increase the drug internalization into drug-resistant tumors will be a great challenge.
Recently, many efforts have been directed towards developing drug efflux inhibitors, like P-glycoprotein (P-gp) inhibitors [3-5], to increase drug retention in drug-resistant tumor cells. Burt and coworkers, for example, found that combination delivery of paclitaxel (PTX)-loaded nanoparticles with a low-molecular-weight copolymer inhibitor of P-glycoprotein dramatically enhanced the cytotoxicity of PTX in multidrug-resistant P-gp-overexpressing cell lines [6]. However, due to the rapid growth, tumor microenvironment represents some abnormal phenomena such as high pressure, hypoxia, angiogenesis and acidic environment. The high pressure in the tumor interstitium directly results in the difficulty for delivering anticancer drugs into tumor tissues. The anoxic environment activates hypoxia-inducible factor (HIF-1) in making cancer cells resistant to the drugs. And the angiogenesis encourages thrombosis and impaired blood supply [7]. The tumor-specific abnormalities in the microenvironment sets up another hindrance for systems reaching the tumor tissues [8].
Therefore, researchers provide another strategy to enhance drug internalization into tumor tissues by combination delivery of tumor vascular inhibitors to transiently 'normalize' the tumor blood vessels [9]. This approach could indirectly normalize the tumor microenvironment by modifying the high-pressured and hypoxic environment and facilitate a large scale of drugs delivery to the tumor tissues [10]. Hence, combination delivery of tumor vascular inhibitor and chemotherapeutic agent could bring synergistic effect on drug internalization into cells and antitumor efficiency. For example, Wang and Ho reported a combinatorial drug delivery nanocapsule, which encapsulated a tumor vasculature inhibitor combretastatin A4 (CA4) into paclitaxel (PTX)-conjugated amphiphilic polyester. The inhibition effect of dual-drug loaded nanocapsule was 76.6%, which was higher than single CA4 (63.5%) and PTX (23.2%) against lewis lung carcinoma tumors [11]. This results indicated that combination of a conventional cytotoxic agent with an antiangiogenetic drug in optimal dosage could enhance the therapeutic efficacy. However, the ester linkage of the chemotherapeutic agents could not be selectively cleaved in tumor tissues, and the drugs could not specifically release in tumor cells. Moreover, the embedded tumor vascular inhibitor could not specifically release at the tumor vascular to regulate the tumor microenvironment and exert their synergistic effects well. Herein, the present approaches for combination delivery might result in insufficient drug accumulation in tumor cells, especially in drug-resistant tumors.
To address this challenge, it is crucial to fabricate a rational site-specific drug-releasing delivery system, which could not only unload the tumor vascular inhibitor specifically in the tumor vessels to temporarily 'normalize' the tumor vasculature to facilitate drug delivery, but also trigger in the tumor cells to increase effective drug concentration in drug-resistant tumor cells. With this concept, firstly, combretastatin A4 (CA4), one of the most potent antivascular agents [12], was used in our system. CA4 could rapidly inhibit the growth of vasculature in tumors by disrupting the cytoskeletal structures [12]. Doxorubicin (DOX), commonly used in the treatment of wide ranges of cancers, was chosen as anticancer drugs [13]. It could kill the tumor cells by intercalating into DNA. Hence, combination delivery of these two drugs should elicit synergistic effect in antitumor activity. Secondly, to realize the site-specific release, a smart dual-pH sensitive delivery system was designed to release CA4 and DOX in tumor vasculature and tumor cells, respectively. To construct our system, a biocompatible poly(ethylene glycol)-polyhistidine (PEG-Phis) polypeptide was chosen as the drug carrier (Figure 1A) since the imidazole groups of the polyhistidine block could be protonated at acidic condition [14]. The polyhistidine block could transform from hydrophobicity to hydrophilicity by protonation in response to the tumor intracellular pH. Then, DOX was conjugated with polyaspartate (Pasp) by a pH-triggered hydrazone bond to form an inert macromolecular prodrug (Pasp-DOX) [15]. The special linker could be broken at pH 5-6 [16], which was sensitive to the environment in tumor cells. The pH-triggered macromolecular prodrug Pasp-DOX and the small molecular inhibitor CA4 were encapsulated in the pH-sensitive PEG-Phis to construct the two-stage site-specific releasing system (Figure 1B).
(A) Illustration of the structures of PEG-Phis and Pasp-DOX, and the pH-sensitive hydrazone linker is shown in the black rectangle, which is marked in Pasp-DOX. (B) The preparation of PEG-Phis/Pasp-DOX/CA4 nanoparticles and the pH-responsive process: (1) self-assembly at pH 7.4; (2) protonation and swelling in tumor vessels at pH 7.0-6.0; (3) further swelling in tumor cells at pH 6.0-5.0; (4) demicellization. (C) Schematic interpretation of the system (PEG-Phis/Pasp-DOX/CA4) transporting from tumor vessels into tumor cells and the hypothetic drugs releasing process: (a) swelling in response to tumor extracellular pH and release of CA4; (b) cellular internalization; (c) further swelling in early endosomes; (d) demicellization and triggered the secondary pH response to release of DOX in late endosomes/lysosomes; (e) endosomal escape and release of DOX to nucleus.
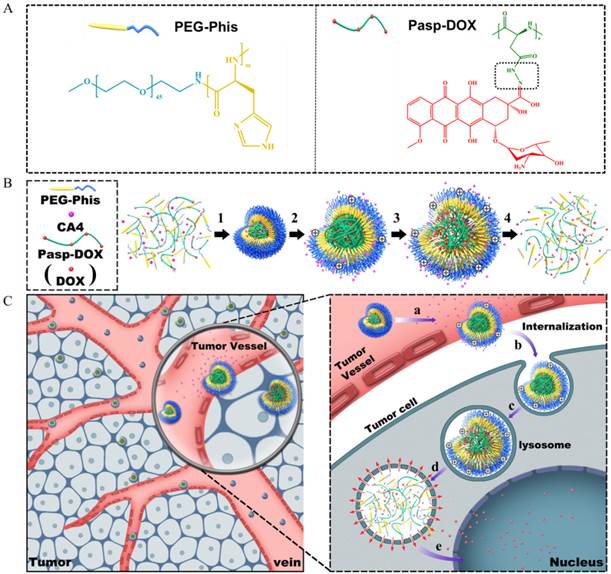
The suggested drug releasing process in the tumor tissues was hypothesized as follows (Figure 1B and 1C): (1) at the tumor vasculature, the polyhistidine block of PEG-Phis could protonate and swell in response to the tumor extracellular pH (pH 6-7) [17], releasing the encapsulated CA4 in tumor vasculature to transiently normalize tumor microenvironment; (2) after internalization into endosomes/lysosomes, the system further swelled until it disassembled, and the macromolecular prodrug Pasp-DOX triggered the secondary pH response (pH 5-6) in endosomes/lysosomes to release active anticancer drug DOX; (3) at the same time, the imidazole rings of the carriers could help to mediate endosomal escape through the proton sponge effect [15]; (4) the released DOX entered into nucleus. The potency of this delivery system was evaluated in DOX-resistant breast cancer cells (MCF-7/ADR cells) and MCF-7/ADR tumor-bearing mice. It was expected that this combination strategy could promote synergistic effect in drug-resistant tumors.
Materials and methods
Materials
N-Carbobenzyloxy-N-(2,4-dinitrophenyl)-2-histidine (N-CBZ-N-DNP-2-histidine) and β-benzyl-L-aspartate were purchased from GL Biochem (Shanghai, China) Ltd. Triphosgene was purchased from TCI (Japan). BisBenzimide H 33342 trihydrochloride (Hoechst 33342) and propidium iodide (PI) were purchased from Beyotime Biotechnology Ltd. (Shanghai, China). Poly(ethylene glycol 2000) methyl ether amine (CH3O-PEG2000-NH2), LysotrackerTM Green and (3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) were gotten from Sigma-Aldrich Shanghai Trading Co. Ltd. (Shanghai, China). Other reagents were bought from Alfa Aesar (China) Chemical Co. Ltd. (Shanghai, China). Doxorubicin hydrochloride (DOX·HCl) and Combretastatin A4 (CA4) were purchased from Dalian Meilun Biotech Co. Ltd. (Dalian, China). Doxorubicin hydrochloride was deprotonated with triethylamine in dimethylsulphoxide (DMSO) to obtain the hydrophobic DOX.
Synthesis and characterization of PEG-polyhistidine (PEG-Phis) conjugates
The PEG-Phis block polymers were prepared according to the previous report with some modifications (Supplementary Material: Figure S1) [18]. Briefly, thionyl chloride was added into the solution of N-Carbobenzyloxy-N-(2, 4-dinitrophenyl)-2-histidine (N-CBZ-N-DNP-2-histidine) in tetrahydrofuran to form N-DNP-L-histidine carboxyanhydride hydrochloride. Then, a predetermined amount of CH3O-PEG2000-NH2 was used to initiate N-DNP-L-histidine carboxyanhydride hydrochloride in N, N-dimethylformamide for ring-opening polymerization, and different molecular weights of PEG-poly((2, 4-dinitrophenyl) histidine) (PEG-Phis-DNP) were obtained. Finally, PEG-Phis copolymers were synthesized by deprotecting the DNP group of PEG-Phis-DNP with 2-mercaptoethanol. The structure of PEG-Phis was confirmed by using 1H nuclear magnetic resonance spectroscopy (1H-NMR, Supplementary Material: Figure S2) (400MHz, DMSO-d6). δ (ppm): 8.10 (imidazole-H2), 7.12 (imidazole-H5), 3.98 (-CH-(C=O)-), 3.51 (-OCH2CH2-), 3.25 (CH3-O-) and 3.15-3.01 (ImCH2-). The degree of polymerization (DP) of the PEG-Phis depended on the peak ratio between PEG2000 and histidine monomer was displayed in Supplementary Material: Table S1.
Synthesis and characterization of polyaspartate-DOX (Pasp-DOX)
The pH-sensitive drug poly(aspartate-hydrazone-doxorubicin) (Pasp-DOX) was prepared according to the previous report with some modifications (Supplementary Material: Figure S3) [19]. Briefly, β-benzyl-L-aspartate N-carboxyl-anhydride (BLA-NCA) was synthesized by reacting triphosgene with β-benzyl-L-aspartate in the presence of α-pinene. Then, isopropylamine was added to initiate the ring-opening polymerization of β-benzyl-L-aspartate N-carboxyl-anhydride (BLA-NCA) in tetrahydrofuran to synthesize poly-β-benzyl-L-aspartate (Poly-BLA). Hydrazine hydrate (NH2NH2·H2O) was added to Poly-BLA in tetrahydrofuran and refluxed to obtain poly(aspartate-hydrazine) (Pasp-hyd). Finally, Doxorubicin (DOX) was reacted with hydrazine groups of poly(aspartate-hydrazine) to get the pH-sensitive drug poly(aspartate-hydrazone-doxorubicin) (Pasp-DOX). The structure of Pasp-DOX was confirmed by 1H NMR (400 MHz, DMSO-d6, Supplementary Material: Figure S4). δ (ppm): 7.29 (Ph-H), 5.30 (DOX-H1), 3.88-4.03 (-PhOCH3, -(C=O)-CH-), 3.50(-(C=N)-CH2-OH), 2.62 (-(C=O)CH2-), 2.40 (-(C=O)CH2-), 2.00 (O-CH2-CH2-CH-NH2, HO-C-CH2-CH-O), 1.30-1.10 ((CH3)2CH- and DOX-H6).
Acid-base titration of PEG-polyhistidine (PEG-Phis)
The polymers and NaCl (control) were dissolved in deionized water and the solution was adjusted to pH 10 with 1 N NaOH [20]. The diluted solution was titrated by stepwise addition of 0.1 N HCl solution to obtain the titration profile.
Preparation and characterization of different formation of polymeric nanoparticles
PEG-Phis/Pasp-DOX/CA4 nanoparticles were prepared via a single-step nanoprecipitation method [21]. In brief, PEG-Phis (10 mg/mL DMSO), Pasp-DOX (10 mg/mL DMSO) and CA4 (10 mg/mL DMSO) were mixed together at the mass ratio of 5/5/1, and stirred for 1 h. The mixture was slowly dripped into deionized water under vigorous stirring conditions for 1 h, and followed by gently stirring for 3 h at room temperature. Then, the resulting mixture was transferred to an Amicon Ultra-4 centrifugal filter (MWCO: 3,500 Da, Billerica, MA) and dialyzed against deionized water for 3 d to remove residual free drugs. At the final time, the outer phase was replaced with fresh physiological saline and filtered with a Millipore filter (pore size: 0.45 μm).
The PEG-Phis/DOX/CA4 nanoparticles were prepared through the same method with PEG-Phis/Pasp-DOX/CA4. The solution was dialyzed against physiological saline and then filtered with a Millipore filter (pore size: 0.45 μm). The concentration of DOX in various formulations was measured by TU-1810 ultraviolet and visible spectrophotometer (Beijing, China). An amount of 0.5 mL PEG-Phis/DOX/CA4 nanoparticles were freeze-dried, then resolved with 1 mL DMSO, and measured. The loaded amount of DOX was obtained by quantifying the absorbance of DOX at 485 nm. The concentration of CA4 in various micelles was measured by reverse phase high performance liquid chromatography (HPLC) using a C18 column (4.6 mm×250 mm, Thermol, MA) with 65% acetonitrile (A) and 35% water (B) as eluents by wavelength monitoring at 295 nm. The injected volume is 20 μL and the flow rate is 1.0 mL/min. The encapsulation efficiency of DOX and CA4 were calculated using the following equation:
Drug encapsulation efficiency = Amount of loaded drug (mg)/Amount of added drug (mg) × 100%.
Characterization of nanoparticles and colloidal stability in vitro
Size distribution and zeta potential of PEG-Phis/Pasp-DOX/CA4 nanoparticles were measured using a dynamic light scatting (DLS) instrument (Malvern Nano ZS, UK). The morphology of PEG-Phis/Pasp-DOX/CA4 nanoparticles was performed by transmission electron microscopy (H-7650 TEM, Japan) with negative stain method. Before analysis, these nanoparticles were dripped onto 200 mesh copper grids coated with carbon and dyed with 2% (w/v) phosphotungstic acid for a moment and then air-dried prior to imaging. The colloidal stability was evaluated by incubating the PEG-Phis/Pasp-DOX/CA4 nanoparticles with Roswell Park Memorial Institute (RPMI) 1640 medium in the presence of 20% fetal bovine serum (FBS) at 37ºC. At each time point, the mean diameters of PEG-Phis/Pasp-DOX/CA4 nanoparticles were monitored using DLS.
pH-dependent structural change of nanoparticles
The PEG-Phis/Pasp-DOX/CA4 nanoparticles (200 μg/ml) were exposed to different pH values (pH = 7.4, 6.6, 5.8 and 5.0) at 37ºC for 0.5 h before measurement of the particle size and particle size distribution. And then the morphology was performed by TEM with negative stain method.
In vitro drug release
To determine the kinetics of DOX and CA4, 1 mL drug-loaded nanoparticles PEG-Phis/Pasp-DOX/CA4 were suspended in 1 mL PBS (pH = 7.4, 6.6, 5.8 and 5.0) and sealed in a dialysis bag (MWCO: 3,500 Da) [22]. The dialysis bags were incubated in 20 mL different pH buffer solution at 37ºC with gentle shaking. A 200 μL aliquot of sample was taken from the incubation medium outside of the dialysis bag at predetermined time intervals and stored frozen for analysis. The released DOX was examined by a microplate reader with excitation at 470 nm and emission at 590 nm. A calibration curve was prepared using different concentrations of free doxorubicin ranging from 0.1 to 10 mg/mL. Released CA4 was then quantified by HPLC. The injected volume is 20 μL and the flow rate is 1.0 mL/min.
Cell culture
The human umbilical vein endothelial cells (HUVECs), human breast adenocarcinoma cells (MCF-7) and human breast adenocarcinoma cells resistant to adriamycin (MCF-7/ADR), obtained from China Academy of Medical Sciences tumor cell bank (Beijing, China), were cultured in RPMI 1640 medium supplemented with 10% FBS and 1% penicillin/streptomycin at 37ºC and 5% CO2. In order to maintain drug resistance of MCF-7/ADR cells, DOX (0.8 μg/mL) was added in the medium.
In vitro non-contact co-culture studies
MCF-7 and MCF-7/ADR cells were seeded in 6-well plates and incubated for 24 h at 37ºC in 5% CO2. At a density of 20,000 cells/insert, HUVECs were cultured in transwell chambers (pore size: 0.4 μm) for 24 h and then placed in the wells above MCF-7 and MCF-7/ADR cells, respectively. After 24 h of incubation, the co-cultures were exposed to the drug treatments with DOX, DOX+CA4, Pasp-DOX, Pasp-DOX+CA4, PEG-Phis/DOX/CA4 and PEG-Phis/Pasp-DOX/CA4. The medium should be enough to exchange through the transwell. At the time point of 12, 24 and 36 h, the two layers were washed with cold PBS three times and fixed with 4% paraformaldehyde on ice for about 20 minutes. Then, the two layers were washed with cold PBS three times again. Tumor cells (MCF-7 and MCF-7/ADR) and HUVECs were stained with propidium iodide (PI, red signal) and Hoechst 33342 (blue signal), respectively. The cells were analyzed with a confocal lasers scanning microscope (CLSM) (Leica TCS SP5, German).
In vitro cytotoxicity
The cytotoxicity was evaluated by MTT assay against MCF-7 and MCF-7/ADR cells (6,000 cells/well), which were seeded on 96-well plates in 100 μL of RPMI 1640 medium and incubated at 37ºC in 5% CO2 for 24 h. The cells were treated with DOX, DOX+CA4, Pasp-DOX, Pasp-DOX+CA4, PEG-Phis/DOX/CA4, PEG-Phis/Pasp-DOX/CA4 and incubated for another 48 h and continued to incubate with fresh medium for additional 24 h. Subsequently, 10 μL MTT was added to each well and incubated for 4 h. Then the medium was removed, 150 μL DMSO was added to dissolve the crystals formed by living cells. The absorbance was measured using a microplate reader with excitation at 562 nm. The percentage of cell viability was determined by comparing cells treated with various drugs with the untreated control cells.
The subcellular localization
MCF-7 and MCF-7/ADR cells were seeded in a 35 mm glass bottom culture dish and incubated for 24 h at 37ºC in 5% CO2, and cultured with PEG-Phis/DOX/CA4, PEG-Phis/Pasp-DOX/CA4 in RPMI 1640 medium containing 10% FBS at 37ºC (5% CO2), respectively. After 1.5 h of incubation, the cells were washed with PBS three times, and fresh medium containing Hoechst 33342 and LysotrackerTM Green (Invitrogen, CA) was added for 0.5 h at 37ºC in dark. Then, the cells were washed with PBS for three times followed by observing with CLSM. In these images, the blue signals represented the nuclei, and the green signals represented the lysosomes/endosomes.
The mechanisms underlying the cellular internalization
The cellular internalizations of PEG-Phis/DOX/CA4 and PEG-Phis/Pasp-DOX/CA4 were performed in the presence of various endocytic inhibitors (chlorpromazine, genistein, mβCD, dynasore and wortmannin) [23, 24]. Energy-dependent endocytosis was completely blocked at 4ºC. MCF-7 and MCF-7/ADR cells were seeded in 12-well plates and incubated for 24 h at 37ºC in 5% CO2. Cells were cultured with chlorpromazine (10 μg/mL), genistein (100 μg/mL), mβCD (5 mM), dynasore (80 μM) and wortmannin (10 μg/mL) for 30 min prior to the addition of PEG-Phis/DOX/CA4 and PEG-Phis/Pasp-DOX/CA4, which was respectively presented in the cell culture medium throughout the uptake experiment at 37ºC for 2 h uptake. Then the cells were washed with cold PBS three times to remove the detached nanoparticles. Results were expressed as percentage uptake level of the control cells, which were incubated with PEG-Phis/DOX/CA4 and PEG-Phis/Pasp-DOX/CA4 without endocytic inhibitors at 37ºC for 2 h.
Drug accumulation
MCF-7 and MCF-7/ADR cells (50,000 cells/well) were seeded in 24-well plates and incubated for 24 h at 37ºC in 5% CO2. Cells were treated with DOX, DOX+CA4, Pasp-DOX, Pasp-DOX+CA4, PEG-Phis/DOX/CA4 and PEG-Phis/Pasp-DOX/CA4 in RPMI 1640 medium containing 10% FBS at pH 6.6 and pH 7.4 for 1, 2 and 4 h. The equivalent concentrations of DOX and CA4 were 2.5 μg/mL and 0.5 μg/mL, respectively. At predetermined time intervals, cells were washed three times with PBS, digested with trypsin solution, centrifuged and resuspended in 500 μL PBS to analysis the fluorescent intensity on a BD Calibur (BD Bio. Sciences, USA).
Blood pharmacokinetics
To determine pharmacokinetics, ICR mice were randomly divided into seven groups, and injected intravenously through the tail vein with DOX, CA4, DOX+CA4, Pasp-DOX, Pasp-DOX+CA4, PEG-Phis/DOX/CA4, and PEG-Phis/Pasp-DOX/CA4, respectively, at the equivalent dose of 2.5 mg DOX/kg and 0.5 mg CA4/kg. Approximately 100 μL of blood sample was collected in a heparinized tube from the orbital venous plexus at 5 min, 30 min, 1 h, 3 h, 8 h and 24 h post injection. To extract DOX and CA4, acetone and methanol was added respectively to the blood, vortexed, and then the solution was immediately centrifuged at 1,800 g for 10 min. The content of DOX and CA4 in supernatant was measured by fluorescence spectrophotometry and HPLC method, respectively.
In vivo antitumor effect
Female BALB/C nude mice, 6-8 weeks old, were purchased from the Academy of Military Medical Science of China. All procedures involving experimental animals were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee of Peking University, China. To develop the tumor xenograft, in vitro growing tumor cells were harvested by exposure to trypsin-EDTA, washed three times with PBS, and implanted subcutaneously into the right flanks of the mice (1×106 cells in 0.2 mL PBS). Antitumor effect of PEG-Phis/Pasp-DOX/CA4 was evaluated by monitoring the tumor volume in a double-blinded manner. After the tumor volume of bearing MCF-7/ADRmice reached 40 mm3, mice were randomly divided into eight groups (n=5). The mice were administrated intravenously with PBS, DOX, CA4, DOX+CA4, Pasp-DOX, Pasp-DOX+CA4, PEG-Phis/DOX/CA4 and PEG-Phis/Pasp-DOX/CA4 at the equivalent dose of 2.5 mg DOX/kg and 0.5 mg CA4/kg, respectively, every 2 days for 6 times. The tumor volume was measured to monitor the anticancer effect. Tumor volume was measured as follows: V=(a×b2)/2, a and b represented the major and minor axes of a tumor, respectively. The lengths of the axes were measured using a caliper. Tumor volume of each group on day 0 was normalized to 1 for all groups. Tumor volume in each group was compared by relative tumor volume: Relative tumor volume = tumor volume/initial tumor volume before treatment [25]. The tumor inhibition effect was calculated according to the following formula: Inhibition effect = 100% (mean relative tumor volume of control group - mean relative tumor volume of treatment group)/mean relative tumor volume of control group.
Immunohistology
At the 16th day after the first injection, the mice were sacrificed. Tumors tissues and normal organs were excised, weighted, photographed, fixed in 4% (w/v) paraformaldehyde. To determine the drug distributions of the formulations, DOX fluorescence in the tumors and normal organs was imaged by the Kodak In-Vivo Imaging System (USA) with excitation at 470 nm and emission at 600 nm. Then, the hearts, livers, and kidneys were fixed and embedded in paraffin. Thin sections were stained with haematoxylin-eosin and imaged. A Masson Trichrome kit (St. Louis, MO) was performed on paraffin sections of tumors. The slides were then stained according to the manufacturer's instructions. The tumor sections were further stained with CD31 primary antibody (Abcam, Cambridge, MA) followed by incubation with horseradish peroxidase-labeled secondary antibody. The sections were evaluated and digital images were acquired using a light microscope. Five randomly selected microscopic fields were quantitatively analyzed on ImageJ. Apoptosis in situ was then detected by TdT-dependent dUTP-biotin nick end labeling (TUNEL) assay using apoptosis detection kit (Millipore S7101, MA) according to the manufacturer's instructions.
Statistical analysis
Differences between two groups were evaluated by one-way ANOVA followed by Student's-test. P<0.05 was considered statistically significant. All values were expressed as mean±S.D. and each value was the mean of at least three repetitive experiments in each group.
Results and Discussion
Preparation and characterization of polypeptide nanoparticles
The PEG-Phis block polymers were prepared by ring-opening polymerization (Supplementary Material: Figure S1 and S2). To precisely tuning the pH-responsive range of the block polymer, a series of PEG-Phis (PEG-Phis20, PEG-Phis40, PEG-Phis60 and PEG-Phis80) was synthesized, and the degree of polymerization (DP) of the polyhistidine block was calculated by NMR (Supplementary Material: Table S1). The protonation of PEG-Phis was determined by using acid-base titration. Comparing with PEG-Phis20 and PEG-Phis80, PEG-Phis40 and PEG-Phis60 showed significant buffering capacity, in the pH range of 5.6-7.1 (Figure 2A), and responded to the tumor extracellular pH (pH 6-7) [26, 27]. The pH-responsive macromolecular prodrug Pasp-DOX was prepared by conjugating the ketone group of DOX with the hydrazine group of polyaspartate to form a hydrazone linker (Supplementary Material: Figure S3). The structure was confirmed by NMR spectra (Supplementary Material: Figure S4), and the DOX concentration was 65.9%, which was calculated by ultraviolet and visible spectrophotometer (UV) analysis.
(A) The pH-profile of PEG-Phis20, PEG-Phis40, PEG-Phis60, PEG-Phis80 copolymers and NaCl by acid-base titration. (B) TEM image of the PEG-Phis60/Pasp-DOX/CA4 nanoparticles prepared from the diluted solution (0.1 mg/mL) at pH 7.4. The scale bar represents 100 nm.
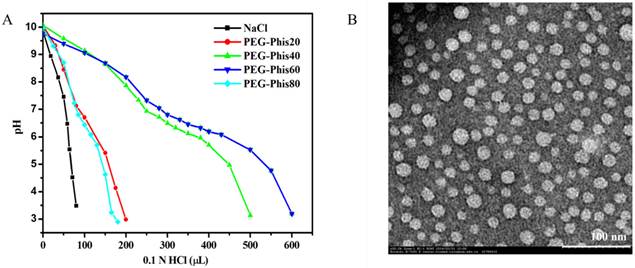
The dual-pH responsive system (PEG-Phis/Pasp-DOX/CA4) was prepared by assembling block polymer PEG-Phis, macromolecular prodrug Pasp-DOX and small molecular inhibitor CA4 via a single-step nanoprecipitation method. Comparing with PEG-Phis40, PEG-Phis60 could form relatively smaller and uniform nanoparticles (105 nm vs. 145 nm for PEG-Phis60 and PEG-Phis40, respectively, Supplementary Material: Figure S5 and Figure 2B), and exhibited much better stability when incubated with 20% FBS (Supplementary Material: Figure S6). Herein, PEG-Phis60/Pasp-DOX/CA4 was chosen as the optimal system in our following studies. The loading efficiency of DOX and CA4 in this system determined by UV and HPLC analyses was 96.1% and 98.1%, respectively, showing superior drug encapsulation.
In our hypothesis, PEG-Phis60/Pasp-DOX/CA4 nanoparticles should gradually swell until disassemble due to the nature properties of the polyhistidine block, and sequential release of encapsulated CA4 and conjugated DOX in the corresponding sites to finally enhance the drug concentration in tumors. To verify this, the sizes of the nanoparticles at acidic conditions were measured. The diameters of the nanoparticles sharply changed from 126.3 nm at pH 7.4 to 700.1 nm at pH 6.6 (Supplementary Material: Figure S7A), suggesting enormous volume swelling in respond to the acidic environment. The TEM also confirmed the morphology change as the sphere structures of the nanoparticles did not exist (Supplementary Material: Figure S7B).
In vitro drug release in response to pH
The drug release amount of PEG-Phis60/Pasp-DOX/CA4 nanoparticles at different pH buffers was tested by HPLC. As shown in Figure 3A, the release of CA4 was very slow at pH 7.4, whereas exhibited a rapid process when the pH changed to 6.6, with 8.7% vs. 64.1% for 12 h. This suggested that the nanoparticles was stable in physiological condition, but could quickly respond to the tumor extracellular pH to set the CA4 free. This was consistent with the pH buffering range of PEG-Phis60 and the size change of the nanoparticles, demonstrating that the system could initiate the primary pH response in tumor microenvironment. Seen from Figure 3B, the conjugated DOX had an obviously fast and continuous release when the pH changed from 7.4 to 5.8, 8.1% vs. 57.1% for two weeks, respectively. This demonstrated that the hydrazone bonds facilitated to break when the pH less than 5.8, which was in line with the endosomal pH.
The pH-dependent cumulative CA4 (A) and DOX (B) were released from the PEG-phis60/Pasp-DOX/CA4 nanoparticles at pH 7.4, 6.6, 5.8 and 5.0 with gentle shaking. The incubation temparature was 37 oC. Data are shown as the mean±S.D. of three independent experiments.
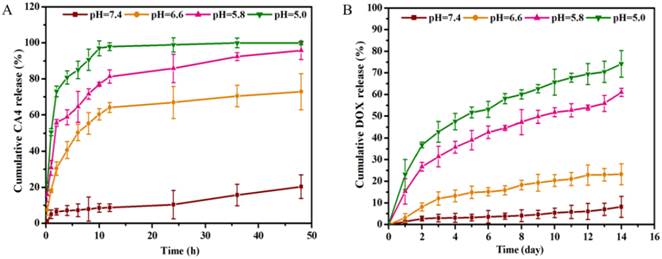
In vitro sequential release
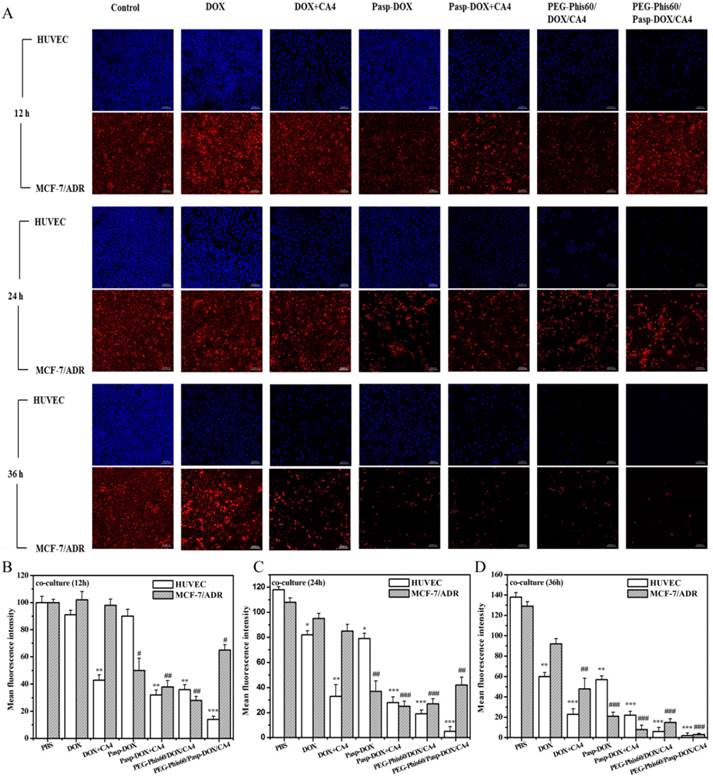
To evaluate if the drug system could sequentially induce cell apoptosis in tumor endothelial cells and tumor cells, an in vitro non-contact co-culture system was adopted (Supplementary Material: Figure S8). HUVECs was used as a model cell line of tumor endothelial cells [28], and MCF-7 and MCF-7/ADR cells as a model of tumor cells. They were applied in the assay to investigate antitumor efficiency in the drug-resistant cells. The encapsulated system PEG-Phis60/DOX/CA4, both DOX and CA4 embedded in the PEG-Phis60 with the same ratio as PEG-Phis60/Pasp-DOX/CA4, was used as a control. As given in Figure 4 and S9, DOX induced obvious apoptosis against MCF-7 after 12 h, whereas exhibited no significant effect on HUVECs for 12 h incubation and MCF-7/ADR cells even for 24 h, respectively. This indicated that DOX had no acute cytotoxity to HUVECs, and MCF-7/ADR cells showed drug resistant against DOX. However, the MCF-7/ADR cells showed significant inhibition when DOX was conjugated with polyaspartate. This suggested that conjugation with macromolecules could help free DOX bypass the drug efflux mechanism in drug-resistant cells. Comparing with DOX treated group, DOX+CA4 exhibited distinct inhibition to HUVECs and MCF-7 cells after 12 h incubation, demonstrating CA4 displayed cytotoxity to these two cells. In comparison with other treatments, the HUVECs with PEG-Phis60/Pasp-DOX/CA4 induced a significant apoptosis for 12 h incubation (mean fluorescence intensity (MFI) < 20%), whereas the MFI for MCF-7 and MCF-7/ADR cells were 63% and 65%, respectively. However, the HUVECs and tumor cells exhibited marked inhibition when treated with PEG-Phis60/DOX/CA4 for 12 h, 36% for HUVECs, 40% for MCF-7 and 28% for MCF-7/ADR cells, respectively. These results indicated that PEG-Phis60/Pasp-DOX/CA4 sequentially released CA4 and DOX to induce HUVECs and tumor cells apoptosis, respectively, whereas the encapsulated nanoparticles PEG-Phis60/DOX/CA4 simultaneously set the two drugs free to kill all the cells. The delayed release of DOX was due to the conjugation with polyaspartate. This phenomenon was consistent with the in vitro releasing assay.
In vitro non-contact co-culture studies. (A) The co-culture cells were treated with different formulations for 12, 24, and 36 h, respectively. After treatment, the HUVECs and tumor cells MCF-7/ADR were stained with Hoechst 33342 (blue) and propidium iodide (red), respectively. Quantitative analysis of mean fluorescence intensity of the co-culture cells after treated with different formulations for 12 h (B), 24 h (C) and 36 h (D). Results are mean±S.D. of three independent experiments. Statistical significance: *P<0.05, **P<0.005 and ***P<0.0005 versus the MFI of HUVEC cells after 12 h incubation. #P<0.05, ##P<0.005 and ###P<0.0005 versus the MFI of MCF-7/ADR cells after 12 h incubation. The scale bar was 100 μm.
In vitro antitumor activity
The in vitro antitumor activities of systems against MCF-7 and MCF-7/ADR cells were determined by MTT assay (Figure 5). The PEG-Phis60 polymer showed negligible cytotoxicity in tumor cells with the tested concentration. Free DOX and DOX+CA4 resulted in high cytotoxicity against MCF-7 cells, probably due to their toxicity and easy accumulation in cells. Free CA4 could induce apoptosis of MCF-7 cells, with 33.6% cell viability at the concentration of 20 μg/mL, which was in coincidence with the co-culture assay. PEG-Phis60/Pasp-DOX/CA4 and PEG-Phis60/DOX/CA4 displayed similar cytotoxicity as free drugs against MCF-7 cells. For MCF-7/ADR cells, the cytotoxicity of DOX and DOX+CA4 was sharply reduced, with 56.6% and 40.8% cell viability, respectively, even at the concentration of 40 μg/mL. Free CA4 induced similar cytotoxicity as it did against MCF-7 cells. However, PEG-Phis60/Pasp-DOX/CA4 still demonstrated relatively high cytotoxicity against the drug-resistant cell lines, with the IC50 value (the concentration inhibiting cell growth to 50% of control) 0.45 μg/mL. The encapsulated system PEG-Phis60/DOX/CA4 also exhibited better cytotoxicity than free drugs, but not as good as PEG-Phis60/Pasp-DOX/CA4, with IC50 value 2.81 μg/mL, which might be caused by the distinct cellular uptake of the nanoparticles.
Viability of MCF-7 cells (A) and MCF-7/ADR cells (B) after treatment with DOX, CA4, DOX+CA4, Pasp-DOX, Pasp-DOX+CA4, PEG-Phis60/DOX/CA4 and PEG-Phis60/Pasp-DOX/CA4 at different concentrations for 48 h. Data are shown as the mean±S.D. of three independent experiments.
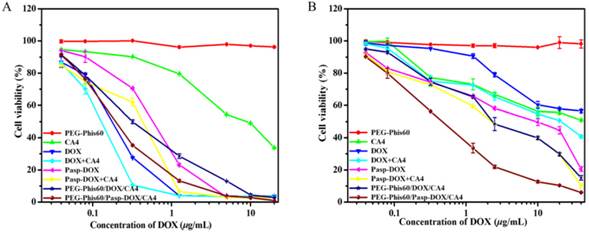
Cellular uptake and drug accumulation
The cellular uptake of systems was visualized with confocal laser scanning microscopy (CLSM). As shown in Supplementary Material: Figure S10 and S11, the yellow color in the overlapped images showed that DOX was mainly localized in lysosomes after 2 h incubation, suggesting that these two nanoparticles entered into cells by endocytosis. Especially, some of the internalized DOX entered into nucleus of MCF-7 cells after 2 h incubation, while none was in MCF-7/ADR cells. This might due to one of multidrug resistant (MDR) mechanisms to prevent the nuclear localization of DOX [29]. The internalization mechanism was further examined at lower temperature (4°C) or in the presence of various endocytic inhibitors to investigate the system-mediated cell penetration (Supplementary Material: Figure S12). PEG-Phis60/Pasp-DOX/CA4 entered into the MCF-7 cells mainly via energy-dependent endocytosis (4 °C), whereas got into MCF-7/ADR cells by caveolin-mediated (mβCD) and clathrin-mediated (dynasore) endocytosis. PEG-Phis60/DOX/CA4 was mainly depended on clathrin-mediated (chlorpromazine) and energy-dependent endocytosis (4°C) to get into MCF-7 cells, and entered into MCF-7/ADR cells by caveolin-mediated (mβCD) and clathrin-mediated (dynasore) endocytosis.
The accumulation of DOX in tumor cells was analyzed by the fluorescent intensity on a BD Calibur (Figure 6). DOX and DOX+CA4 exhibited higher accumulation than other formulations in DOX-sensitivecells (MCF-7) when the time extended to 4 h. This suggested that free drugs could quickly entered the cells by diffusion. However, in DOX-resistant cells, free DOX and DOX+CA4 were accumulated extremely low even when incubation for 4 h. This was probably due to the high expression of P-gp in MCF-7/ADR cells, which resulted in the exclusion of the DOX [29]. The accumulations significantly increased when treated with PEG-Phis60/Pasp-DOX/CA4 and PEG-Phis60/DOX/CA4, confirming that drug delivery by nanoparticles through endocytosis could significantly enhance cellular uptake in drug-resistant tumor cells. The cellular uptake of these formulations was also evaluated at pH 6.6, which was close to the acidic microenvironment of tumors (Supplementary Material: Figure S13). The trend of the DOX accumulation in MCF-7 and MCF-7/ADR cells was similar with the condition at pH 7.4, which suggested that the cellular uptake was not affected by the size change of the nanoparticles.
The accumulation of DOX in MCF-7 cells (A) and MCF-7/ADR cells (B) after incubation with PEG-Phis60/Pasp-DOX/CA4 at pH 7.4 for 1, 2 and 4 h. Data are shown as the mean±S.D. of three independent experiments. Statistical significance: *P<0.05, **P<0.005 and ***P<0.0005.
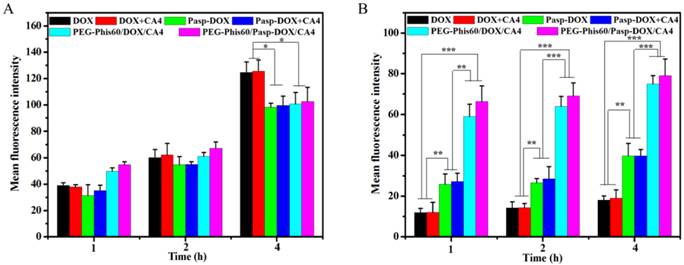
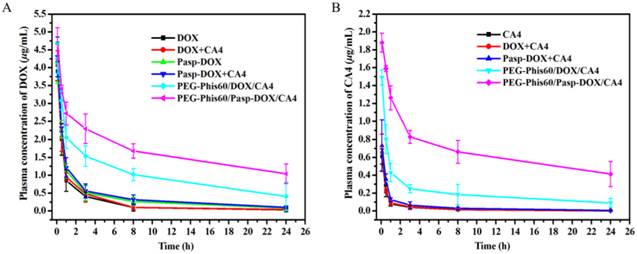
In vivo pharmacokinetics
Drug delivery in vivo is far more complicated since plasma is a concentrated, multi-protein solution [30]. Herein, the in vivo pharmacokinetics of all the formulations were investigated and calculated by non-compartment pharmacokinetic model [31]. As shown in Figure 7, free drugs, including DOX, CA4 and DOX+CA4, were quickly removed from the circulating system after intravenous administration, with the plasma half-time (t1/2) 5.1 h for free DOX and 6.2 h for free CA4, respectively. The plasma half-time of DOX and CA4 for PEG-Phis60/DOX/CA4 was 11.3 h and 14.7 h, which were 2-fold higher than that of free DOX and CA4. Among all the formulations, PEG-Phis60/Pasp-DOX/CA4 exhibited the longest circulation time, the plasma half-time of DOX and CA4 for PEG-Phis60/Pasp-DOX/CA4 was 19.4 h and 21.4 h, respectively, which was needed for passive delivery via enhanced permeability and retention effect (EPR) and better accumulation in tumors.
Plasma concentrationtime profiles of DOX (A) and CA4 (B) in mice after intravenous administration of various drug formulations at the equivalent dose of 2.5 mg DOX/kg and 0.5 mg CA4/kg. Data are shown as the mean±S.D. of three independent experiments.
In vivo antitumor studies against drug-resistant tumor
To evaluate the in vivo antitumor efficacy against drug-resistant tumor, MCF-7/ADR tumor xenograft model was used. The tumor inhibition effect was compared with the PBS control. As shown in Figure 8A and S14, except free DOX (with 8.3% inhibition effect), all the other formulations showed significant inhibition of tumor growth compared with PBS group after 16 days, probably due to the resistance of MCF-7/ADR tumor to DOX. Free CA4 also exhibited 60.8% inhibition of the tumor growth, in line with the previous report that CA4 could show cytotoxity against a variety of human cancer cells, including multi-drug resistant cell lines [32, 33]. The inhibition effects of PEG-Phis60/Pasp-DOX/CA4 and PEG-Phis60/DOX/CA4 were 91.8% and 54.8%, respectively. The statistical analysis showed that PEG-Phis60/Pasp-DOX/CA4 induced better inhibition of tumor growth than that of PEG-Phis60/DOX/CA4. Moreover, except free DOX and DOX+CA4, the body weight of the administrated groups did not show obvious loss weight (Figure 8B), demonstrating no severe side effects.
In vivo antitumor activity on MCF-7/ADR tumor-bearing BALB/C nude mice. (A) Relative tumor volume after intravenous injection with PBS, DOX, CA4, DOX+CA4, Pasp-DOX, Pasp-DOX+CA4, PEG-Phis60/DOX/CA4 and PEG-Phis60/Pasp-DOX/CA4 at the equivalent dose of 2.5 mg DOX/kg and 0.5 mg CA4/kg, respectively. The administration strategy was every 2 days for 6 times. (B) Body weight changes of MCF-7/ADR tumor-bearing mice after treatment with different formulations. Results are shown as mean±S.D. (n=5). Statistical significance: *P<0.05, **P<0.005 and ***P<0.0005. (C) DOX accumulation in tumor tissues after in vivo antitumor activity with different formulations at day 16. (D) Quantification of mean fluorescence intensity (MFI) of DOX was calculated. Results are expressed as mean±S.D. (n=5). Statistical significance: *P<0.05 and **P<0.005 versus the DOX treated group, #P<0.05.
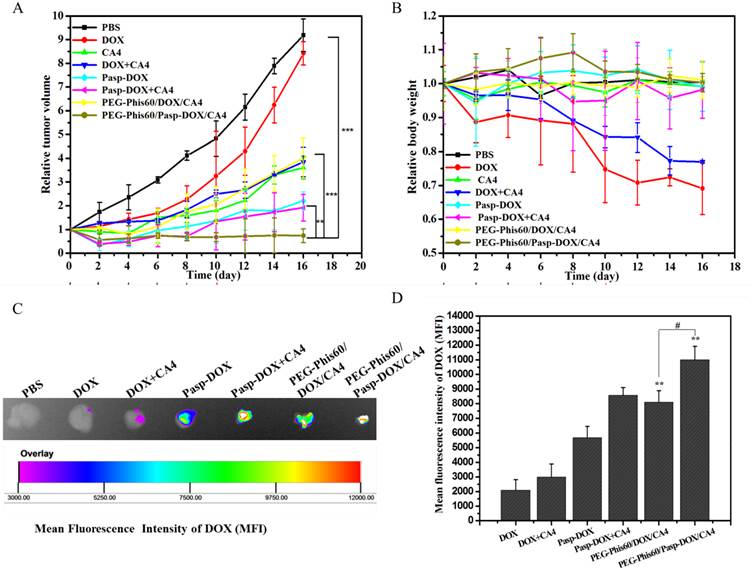
Due to the DOX self-fluorescene, the DOX distribution in tumor tissues of the treated mice was evaluated. As shown in Figure 8C and 8D, the strongest DOX fluorescence was observed in the tumor treated with PEG-Phis60/Pasp-DOX/CA4, indicating that more accumulation of PEG-Phis60/Pasp-DOX/CA4 than PEG-Phis60/DOX/CA4. Also, the biodistribution of normal organs was examined. As shown in Supplementary Material: Figure S15, free DOX and DOX+CA4 mainly perfused into kidney and liver. However, the DOX accumulation in these organs was significantly lower in the group treated with PEG-Phis60/Pasp-DOX/CA4. All these results might be ascribed to the smart dual-pH sensitive system, which could sequentially unload drugs in their corresponding sites and exhibit enhanced synergistic effects. The primary pH-response initiated CA4 release in tumor vasculature, which regulated the tumor microenvironment to facilitate drug delivery. The secondary pH-trigger guaranteed active drug DOX releasing in tumor cells and inducing cell apoptosis. These strategies ensured the high effective drug accumulation in tumor cells, and enhanced the chemotherapy in drug-resistant tumors.
To further reveal the toxicity of the different formulations, the white blood cell (WBC) counts were also observed, which was sensitive to the cytotoxic agents (Supplementary Material: Figure S16) [22, 34]. The results revealed that free DOX and DOX+CA4 exhibited the most serious decrease in WBC counts, whereas PEG-Phis60/Pasp-DOX/CA4 displayed no obvious toxicity. This was consistent with the above-mentioned results. Moreover, the histological examination suggested that PEG-Phis60/Pasp-DOX/CA4 showed less cytotoxicity to normal tissues, including heart, kidney and liver, suggesting the better biocompatibility of our system (Supplementary Material: Figure S17). However, treatments with other formulations induced different levels of damages to liver, such as degeneration, edema and inflammatory infiltration. This results were also in accordance with the biodistribution of these formulations in normal organs.
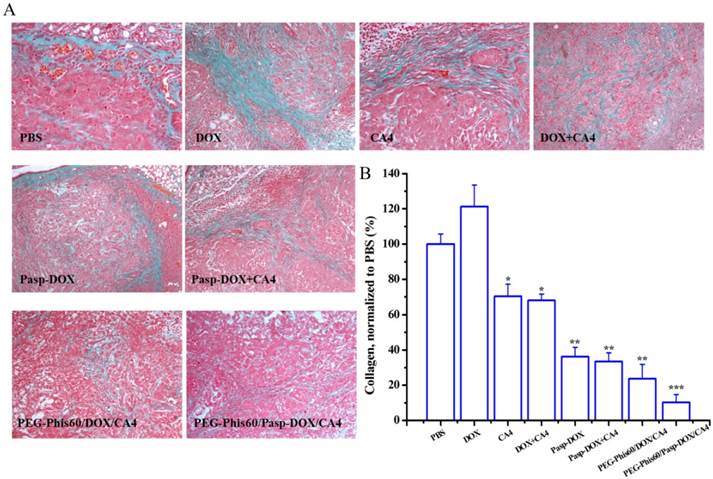
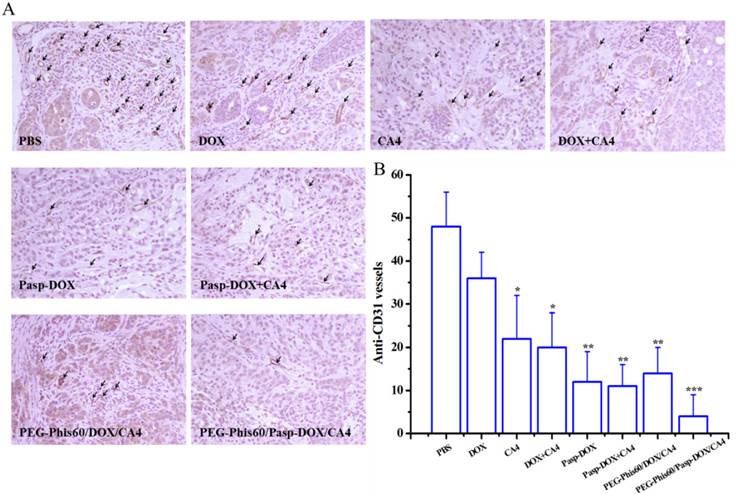
By temporarily normalizing the tumor microenvironment, our system exhibited enhanced drug accumulation in the tumor tissues. Herein, the relevant assays were exploited to examine the modification of the tumor microenvironment. Collagen, a major extracellular matrix component, was secreted by TAFs (tumor-associated fibroblasts) [35]. The collagen was stained using Masson Trichrome assay. As shown in Figure 9, PEG-Phis60/Pasp-DOX/CA4 significantly reduced the amount of collagen (blue color) compared to PBS control, suggesting the regulation of tumor microenvironment [35]. To further confirm the effect of the tumor vascular inhibitor to normalize the tumor microenvironment, blood vessels were stained by using an anti-CD31 antibody (Figure 10). Comparing with the control, PEG-Phis60/Pasp-DOX/CA4 significantly reduced the tumor angiogenesis, revealing that this system could regulate the balance between the pro-angiogenic and anti-angiogenic factors. It was in accordance with the Masson Trichrome assay results. These results revealed that PEG-Phis60/Pasp-DOX/CA4 could produce a superior anticancer efficacy against drug-resistant tumors, since they could release CA4 in tumor vessels to modify tumor microenvironment and induce more therapeutic drug accumulation in tumors.
(A) Tumor sections were stained with Masson Trichrome after treatment with different formulations. The blue color represented collagen content, and the cytoplasm was stained red. (B) The collagen content of five randomly selected microscopicfields after treatments was quantified using ImageJ. Results are shown as mean±S.D. (n=5). Statistical significance: *P<0.05, **P<0.005 and ***P<0.0005 versus control.
(A) Immunostaining with anti-CD31 tumor tissues was used to detect tumor vasculature (arrows represented) after treatment with different formulations. (B) Quantification of tumor vessel density after treatments. Five randomly selected microscopic fields after treatments were counted. Results are shown as mean±S.D. (n=5). Statistical significance: *P<0.05, **P<0.005 and ***P<0.0005 versus control.
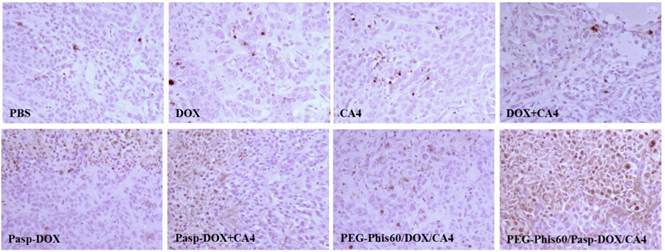
The cell apoptosis in the tumor tissues after treatments was also analyzed using TUNEL assay (Figure 11) and hematoxylin-eosin (H&E) staining (Supplementary Material: Figure S17). Compared with other formulations, PEG-Phis60/Pasp-DOX/CA4 markedly increased the number of TUNEL-positive tumor cells (brown color). H&E staining of the tumors also showed the same tendency. The tumor tissues treated with PEG-Phis60/Pasp-DOX/CA4 lost their normal structures, and the tumor cellularity significantly decreased. These results were consistent with the higher inhibition of the tumor growth induced by PEG-Phis60/Pasp-DOX/CA4.
Tumor sections were immunostained with TUNEL staining for tumor cell apoptosis after treatment with different formulations. Brown stains indicated apoptotic cells in TUNEL analysis.
Conclusion
In summary, a smart site-specific drug-releasing polypeptide system PEG-Phis60/Pasp-DOX/CA4 was contructed, which could sequentially release tumor vascular inhibitor CA4 and anticancer drug DOX in their corresponding sites by a two-stage pH response. This system firstly unloaded the CA4 specifically in the tumor vessels to temporarily 'normalize' the tumor vessels. This process indirectly regulated the tumor microenvironment and facilitated DOX delivery into drug-resistant tumor cells efficiently. Then the nanoparticles internalized into endosomes/lysosomes of the tumor cells by endocytosis, and initiated the secondary pH response to release DOX. This synergistic delivery exhibited enhanced effective drug concentration in drug-resistant tumor cells and superior antitumor effect in the drug-resistant breast tumor model with low systemic toxicity. Therefore, this programmed codelivery system hold great promise for chemotherapy to treat the drug-resistant tumors.
Supplementary Material
Supplementary Figures S1-S17, Table S1.
Acknowledgements
This work was financially supported by National Natural Science Foundation of China (Grants No. 21304099, 51203162, 51103159, 51373177 and 31470961), the National High Technology Research and Development Program (Grants No. 2014AA020708, 2012AA022703 and 2012AA020804), the Instrument Developing Project of the Chinese Academy of Sciences (Grant No. YZ201253 and YZ201313), the Open Funding Project of the National Key Laboratory of Biochemical Engineering (Grant No. Y22504A169), the “Strategic Priority Research Program” of the Chinese Academy of Sciences (Grant No.XDA09030301-3), the Hebei Province “Hundred Talents Program” (BR2-202), “Three Three Three” Talents Project of Hebei Province (A201401002), Key Basic Research Special Foundation of Science Technology Ministry of Hebei Province (14961302D), and Training Program for Innovative Research Team and Leading Talent in Hebei Province University (LJRC024).
Competing Interests
The authors have declared that no competing interests exists.
References
1. He QJ, Shi JL. MSN anti-cancer nanomedicines: chemotherapy enhancement, overcoming of drug resistance, and metastasis inhibition. Adv Mater. 2014;26:391-411
2. Szakacs G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov. 2006;5:219-34
3. Kuo WS, Ku YC, Sei HT, Cheng FY, Yeh CS. Paclitaxel-loaded stabilizer-free poly(D,L-lactide-co-glycolide) nanoparticles conjugated with quantum dots for reversion of anti-cancer drug resistance and cancer cellular imaging. J Chin Chem Soc-Taip. 2009;56:923-34
4. O'Connor R. The pharmacology of cancer resistance. Anticancer Res. 2007;27:1375
5. Binkhathlan Z, Lavasanifar A. P-glycoprotein inhibition as a therapeutic approach for overcoming multidrug resistance in cancer: current status and future perspectives. Curr Cancer Drug Tar. 2013;13:326-46
6. Wan CPL, Letchford K, Jackson JK, Burt HM. The combined use of paclitaxel-loaded nanoparticles with a low-molecular-weight copolymer inhibitor of P-glycoprotein to overcome drug resistance. Int J Nanomed. 2013;8:379-91
7. Danquah MK, Zhang XA, Mahato RI. Extravasation of polymeric nanomedicines across tumor vasculature. Adv Drug Deliver Rev. 2011;63:623-39
8. Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307:58-62
9. Zhang QB, Bindokas V, Shen JK, Fan HL, Hoffman RM, Xing HR. Time-course imaging of therapeutic functional tumor vascular normalization by antiangiogenic agents. Mol Cancer Ther. 2011;10:1173-84
10. Tong RT, Boucher Y, Kozin SV, Winkler F, Hicklin DJ, Jain RK. Vascular normalization by vascular endothelial growth factor receptor 2 blockade induces a pressure gradient across the vasculature and improves drug penetration in tumors. Cancer Res. 2004;64:3731-6
11. Wang Z, Ho PC. A nanocapsular combinatorial sequential drug delivery system for antiangiogenesis and anticancer activities. Biomaterials. 2010;31:7115-23
12. Tozer GM, Kanthou C, Parkins CS, Hill SA. The biology of the combretastatins as tumour vascular targeting agents. Int J Exp Pathol. 2002;83:21-38
13. Xu B, Shi PC, Fombon IS, Zhang YY, Huang F, Wang WG. et al. Disulfiram/copper complex activated JNK/c-jun pathway and sensitized cytotoxicity of doxorubicin in doxorubicin resistant leukemia HL60 cells. Blood Cell Mol Dis. 2011;47:264-9
14. Lee ES, Na K, Bae YH. Polymeric micelle for tumor pH and folate-mediated targeting. J Control Release. 2003;91:103-13
15. Bae Y, Fukushima S, Harada A, Kataoka K. Design of environment-sensitive supramolecular assemblies for intracellular drug delivery: polymeric micelles that are responsive to intracellular pH change. Angew Chem Int Edit. 2003;42:4640-3
16. Rihova B, Etrych T, Sirova M, Kovar L, Hovorka O, Kovar M. et al. Synergistic action of doxorubicin bound to the polymeric carrier based on N-(2-hydroxypropyl) methacrylamide copolymers through an amide or hydrazone bond. Mol Pharmaceut. 2010;7:1027-40
17. Lin W, Kim D. pH-sensitive micelles with cross-linked cores formed from polyaspartamide derivatives for drug delivery. Langmuir. 2011;27:12090-7
18. Lee ES, Shin HJ, Na K, Bae YH. Poly(L-histidine)-PEG block copolymer micelles and pH-induced destabilization. J Control Release. 2003;90:363-74
19. Bae Y, Nishiyama N, Fukushima S, Koyama H, Yasuhiro M, Kataoka K. Preparation and biological characterization of polymeric micelle drug carriers with intracellular pH-triggered drug release property: Tumor permeability, controlled subcellular drug distribution, and enhanced in vivo antitumor efficacy. Bioconjugate Chem. 2005;16:122-30
20. Urry DW, Peng SQ, Gowda DC, Parker TM, Harris RD. Comparison of Electrostatic-Induced and Hydrophobic-Induced Pka Shifts in Polypentapeptides - the Lysine Residue. Chem Phys Lett. 1994;225:97-103
21. Zhang LF, Chan JM, Gu FX, Rhee JW, Wang AZ, Radovic-Moreno AF. et al. Self-assembled lipid-polymer hybrid nanoparticles: a robust drug delivery platform. Acs Nano. 2008;2:1696-702
22. Sengupta S, Eavarone D, Capila I, Zhao GL, Watson N, Kiziltepe T. et al. Temporal targeting of tumour cells and neovasculature with a nanoscale delivery system. Nature. 2005;436:568-72
23. Tang HY, Yin LC, Lu H, Cheng JJ. Water-soluble poly(L-serine)s with elongated and charged side-chains: synthesis, conformations, and cell-penetrating properties. Biomacromolecules. 2012;13:2609-15
24. Yin LC, Song ZY, Kim KH, Zheng N, Gabrielson NP, Cheng JJ. Non-viral gene delivery via membrane-penetrating, mannose-targeting supramolecular self-assembled nanocomplexes. Adv Mater. 2013;25:3063-70
25. Cabral H, Matsumoto Y, Mizuno K, Chen Q, Murakami M, Kimura M. et al. Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size. Nat Nanotechnol. 2011;6:815-23
26. Helmlinger G, Yuan F, Dellian M, Jain RK. Interstitial pH and pO(2) gradients in solid tumors in vivo: high-resolution measurements reveal a lack of correlation. Nat Med. 1997;3:177-82
27. Zhao ZL, Meng HM, Wang NN, Donovan MJ, Fu T, You MX. et al. A controlled-release nanocarrier with extracellular pH value driven tumor targeting and translocation for drug delivery. Angew Chem Int Edit. 2013;52:7487-91
28. Wang YG, Yang TY, Wang X, Dai WB, Wang JC, Zhang XA. et al. Materializing sequential killing of tumor vasculature and tumor cells via targeted polymeric micelle system. J Control Release. 2011;149:299-306
29. Duan XP, Xiao JS, Yin Q, Zhang ZW, Yu HJ, Mao SR. et al. Smart pH-sensitive and temporal-controlled polymeric micelles for effective combination therapy of doxorubicin and disulfiram. Acs Nano. 2013;7:5858-69
30. Yuan YY, Mao CQ, Du XJ, Du JZ, Wang F, Wang J. Surface charge switchable nanoparticles based on zwitterionic polymer for enhanced drug delivery to tumor. Adv Mater. 2012;24:5476-80
31. Ma JS, Cai JZ, Lin GY, Chen HL, Wang XQ, Wang XC. et al. Development of LC-MS determination method and back-propagation ANN pharmacokinetic model of corynoxeine in rat. J Chromatogr B. 2014;959:10-5
32. Pettit GR, Singh SB, Hamel E, Lin CM, Alberts DS, Garciakendall D. Antineoplastic agents. 145. Isolation and structure of the strong cell-growth and tubulin inhibitor combretastatin-a-4. Experientia. 1989;45:209-11
33. Mcgown AT, Fox BW. Differential cytotoxicity of combretastatin-A1 and combretastatin-A4 in 2 daunorubicin-resistant P388 cell-lines. Cancer Chemoth Pharm. 1990;26:79-81
34. Wunderlich M, Mizukawa B, Chou FS, Sexton C, Shrestha M, Saunthararajah Y. et al. AML cells are differentially sensitive to chemotherapy treatment in a human xenograft model. Blood. 2013;121:E90-E7
35. Guo ST, Lin CM, Xu ZH, Miao L, Wang YH, Huang L. Co-delivery of cisplatin and rapamycin for enhanced anticancer therapy through synergistic effects and microenvironment modulation. Acs Nano. 2014;8:4996-5009
Author contact
![]() Corresponding authors: Xin Zhang, National Key Laboratory of Biochemical Engineering, Institute of Process Engineering, Chinese Academy of Sciences, No. 1 Beiertiao, Zhongguancun, Beijing, 100190, China. Tel.: +86 10 82544853; fax: +86 10 82544853. Email: xzhangac.cn. Jinchao Zhang, College of Chemistry & Environmental Science, Chemical Biology Key Laboratory of Hebei Province, Key Laboratory of Medicinal Chemistry and Molecular Diagnosis of the Ministry of Education, Hebei University, Baoding, 071002, China. Tel.: +86 312 5079525; fax: +86 312 5079386. Email: jczhang6970com.
Corresponding authors: Xin Zhang, National Key Laboratory of Biochemical Engineering, Institute of Process Engineering, Chinese Academy of Sciences, No. 1 Beiertiao, Zhongguancun, Beijing, 100190, China. Tel.: +86 10 82544853; fax: +86 10 82544853. Email: xzhangac.cn. Jinchao Zhang, College of Chemistry & Environmental Science, Chemical Biology Key Laboratory of Hebei Province, Key Laboratory of Medicinal Chemistry and Molecular Diagnosis of the Ministry of Education, Hebei University, Baoding, 071002, China. Tel.: +86 312 5079525; fax: +86 312 5079386. Email: jczhang6970com.