Impact Factor
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials
Methods
Results
Discussion
Supplementary Material
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2015; 5(8):847-862. doi:10.7150/thno.10854 This issue Cite
Research Paper
Photodynamic Quenched Cathepsin Activity Based Probes for Cancer Detection and Macrophage Targeted Therapy
Yael Ben-Nun1, Emmanuelle Merquiol1, Alexander Brandis2, Boris Turk3,4,5, Avigdor Scherz2, Galia Blum1 ![]()
1. The Institute of Drug Research, The School of Pharmacy, The Faculty of Medicine, Campus Ein Karem, The Hebrew University, Jerusalem, Israel.
2. Department of Plant Sciences, Weizmann Institute of Science, Rehovot, Israel,
3. Department of Biochemistry and Molecular Biology, J. Stefan Institute, Ljubljana, Slovenia,
4. Faculty of Chemistry and Chemical Technology, University of Ljubljana, Slovenia,
5. Centre of Excellence for Integrated Approaches in Chemistry and Biology of Proteins, Ljubljana, Slovenia.
Received 2014-10-20; Accepted 2015-3-20; Published 2015-4-27
Abstract

Elevated cathepsins levels and activities are found in several types of human cancer, making them valuable biomarkers for detection and targeting therapeutics. We designed small molecule quenched activity-based probes (qABPs) that fluoresce upon activity-dependent covalent modification, yielding cell killing by Photodynamic Therapy (PDT). These novel molecules are highly selective theranostic probes that enable both detection and treatment of cancer with minimal side effects. Our qABPs carry a photosensitizer (PS), which is activated by light, resulting in oxidative stress and subsequent cell ablation, and a quencher that when removed by active cathepsins allow the PS to fluoresce and demonstrate PD properties. Our most powerful and stable PS-qABP, YBN14, consists of a selective cathepsin recognition sequence, a QC-1 quencher and a new bacteriochlorin derivative as a PS. YBN14 allowed rapid and selective non-invasive in vivo imaging of subcutaneous tumors and induced specific tumor macrophage apoptosis by light treatment, resulting in a substantial tumor shrinkage in an aggressive breast cancer mouse model. These results demonstrate for the first time that the PS-qABPs technology offers a functional theranostic tool, which can be applied to numerous tumor types and other inflammation-associated diseases.
Keywords: Cathepsins, Photodynamic Therapy, Activity-Based Probes, Macrophages, Non-Invasive Imaging
Introduction
Cathepsins are a subset of the cysteine proteases family, which share a conserved active site formed by a cysteine and a histidine residues. Besides their main function in the lysosomes as degrading proteases, cathepsins regulate numerous biological processes including immune response, prohormone processing, inflammation, epidermal homeostasis, angiogenesis and extracellular matrix (ECM) turnover. Therefore, it is not surprising that cathepsins are overexpressed in various pathologies such as arthritis, osteoporosis, atherosclerosis, cancer and other inflammation-associated diseases[1,2].
Out of the eleven cysteine cathepsins three cathepsins, B, L and S, were shown to have elevated expression and high levels of activity in different cancer types. Furthermore, cathepsins B and L were also found to be associated with poor prognosis and shorter survival periods of patients[2-5]. Cathepsins are thought to be involved in several processes of cancer progression, including angiogenesis initiation, tumor growth and invasiveness[6, 7]. They play a key role in metastasis spread by allowing cancer cells to migrate from the primary tumor mass by degrading ECM components such as laminin, fibronectin, and collagen IV. Moreover, pharmacological inhibition of cathepsins in several mouse models significantly delayed cancer progression[7-10], suggesting that cysteine cathepsins are valuable biomarkers and therapeutic targets for cancer.
Tumor-associated macrophages (TAMs) were reported to promote cancer progression by chemokine's and cytokine's paracrine loops that lead to angiogenesis, proliferation, migration, invasion and metastasis[11, 12]. Of particular significance is their role in helping the cancer cells to evade immune surveillance. Cathepsins were shown to be highly expressed and active in TAMs and play a critical role in several of their malignant activities[13]. In addition, the tumor mass can contain a substantial portion of macrophages, reaching up to 50-80% of the total tumor mass[14], suggesting that therapies targeting TAMs and TAM-related cathepsins should lead to improved cancer therapy.
Like other proteases, cathepsins are synthesized in the cell as inactive zymogens and their activity is regulated post-translationally by proteolytic activation and endogenous inhibitors[1, 15]. Therefore, the use of traditional tools such as antibodies to detect protease activity is often insufficient. Following their recent development, Activity-Based Probes (ABPs) have been extensively used to visualize and study proteases' activities both in normal and pathological conditions[16-21]. ABPs are small molecules that covalently bind to a target enzyme in an activity-dependent manner. Most ABPs have a short sequence that derives selectivity towards the target protease, an electrophile that facilitates covalent binding to the protease active site, and a tag, which enables the detection and characterization of the probe-labeled proteases. Quenched fluorescent ABPs (qABPs) include in addition a quencher moiety that diminishes the probes fluorescence as long as the probe is free in solution. Covalent modification of the qABP by an active protease releases the quencher and renders the probe-enzyme complex fluorescent, thus allowing for rapid detection of the active protease and biochemical analysis.
Several selective qABP for cathepsins have already been developed based on the acyloxymethyl ketone (AOMK) group as an electrophile. We further showed that the AOMK moiety is ideal for targeting cysteine cathepsins, because it can be used in the qABP strategy for real time imaging in living cells[21] and at the same time enables non-invasive in vivo imaging of cathepsin activity in animal cancer models[20].
Photodynamic therapy (PDT) is a method for cancer treatment that has been approved for clinical use in several indications. Cell ablation is induced by light irradiation of a photosensitive molecule in the presence of oxygen thus generating reactive oxygen species (ROS) such as singlet oxygen (1O2), which are lethal to their environment[22]. 1O2 and other ROS are cytotoxic since they oxidize proteins, lipid membranes and DNA, thereby impairing central cellular components[23-26]. The tissue destruction that PS causes after light exposure occurs only in the immediate vicinity of the PS since ROS are short-lived species. In general, most PSs fluoresce and possess a unique excitation spectrum different from most commercial fluorophores. Although some of the experimentally used PSs are conjugated to cell specific homing molecules the PD treatment of cancerous tissue is mainly confined by the light field. In several cases the lipophilic PS accumulates in skin causing light toxicity to the patients[27-29]. Targeting PSs to cancerous tissue and controlling its bio-distribution is therefore believed to significantly improve the efficacy of PDT and simultaneously diminish its side effects. In this work we therefore focused on a combination of two technologies, qABPs and PDT, to generate new theranostic agents, photosensitized quenched activity-based probes (PS-qABPs) that allow for combined tumor detection and macrophage-targeted treatment (see Scheme 1a).
General structure of PS-ABPs and PS-qABPs. (a) Design of PS-qABP. Covalent modification of the target active enzyme by the probe, leads to quencher release, thus the PS regain its fluorescence properties. Light activation of PS results in ROS generation leading to cell death. (b) Modified synthesis route of GB111-NH2, the precursor for all PS-ABP. (c) Activation of carboxylic acid residue to succinimide ester by TSTU. (d) General precursor structure of PS-qABP and PS-ABP and structure of QSY-21 coupled to hexdiamin linker. (e) Photosensitizers in use for generation of PS-qABP and PS-ABP and thier excitation wavelength. Fmoc, 9-fluorenylmethyloxycarbonyl; Boc, tert-butyloxycarbonyl; IBCF, isobutyl chloroformate; NMM, n-methylmorpholine; THF, tetrahydrofuran; DMF, dimethylformamide; TFA, trifluoro-acetic acid; DIEA, diisopropylethylamine; DCM, dichloromethane; DEA, diethylamine; Cbz, carboxybenzyl; DIC, diisopropylcarbodiimide; HOBt, 1-hydroxybenzotriazole; TSTU, O-(N-Succinimidyl)-1,1,3,3-tetramethyluronium tetrafluoroborate.
More specifically, development of a PS-labeled qABP based on cathepsin targeting should have the following advantages in vivo: (1) unlike traditional photosensitizers, these probes should be specifically targeted to cells in the tumor microenvironment with high cathepsin activity; (2) due to targeting tumor-associated cathepsins these PS-qABPs could be used for real-time imaging of tumors prior to light activation, in order to better localize the subsequent photodynamic anti-cancer treatment; (3) due to the quencher, the probes described here should have reduced fluorescent background and therefore should generate signal very rapidly without the need for clearance of unbound probe, and (4), unlike other probes, the PS-qABPs bind their targets covalently, so that the detection of the in vivo targets by the probe can be verified and quantified using biochemical analysis.
In this study we report on the development of a new class of qABPs that contain a PS moiety. These probes were generated based on the idea that the activity-based probe targeted to active cathepsins would drive the agent selectively to macrophages in the tumor-microenvironment. We have generated a library of PS-qABPs that were found to be quenched and to selectively bind cathepsins B, L and S. In addition, our leading probe induced TAM apoptosis in tumor-bearing mice upon light treatment. Thus, this new class of reagents has a potential to serve as an attractive cancer theranostic lead compound. A possible drawback of the PS-qABP strategy might be the lack of signal amplification at the tumor site, which can be obtained with the substrate-like probes[30]. However, the highly elevated cathepsin activity in the TAMs was found sufficient to overcome this problem and allow for a significant accumulation of PS at the tumor site leading to the desired theranostic effects.
Materials
Chemicals
General methods. Unless otherwise noted, all resins and reagents were purchased from commercial suppliers and used without further purification. Pheophorbide-a, Pyropheophorbide-a and Chlorine-e6 were purchased from Frontier Scientific, Inc (Logan, UT, USA). Visudyne was purchased from W&J PharmaChem, Inc. (Silver Spring, MD, USA). QSY-21 quencher was purchased from Life Technolegies (Grand Island, NY, USA). IRDye QC-1 quencher (QC-1) was purchased from Li-Core (Lincoln, NE, USA). The photosensitizer Bacteriochlorin, was generated as described for compound 11 in [31]. All water-sensitive reactions were performed in anhydrous solvents under positive pressure of nitrogen or argon. Reactions were analyzed by a Liquid Chromatography Mass Spectrometer (LC-MS) (Thermo Scientific MSQ-Plus), attached to an Accela UPLC system. Unless otherwise stated, all final compounds were purified by C18 reverse phase HPLC in acetonitrile/water gradient. Recombinant human cathepsins B, L and S were prepared as described[32-34]. The animal ethics committee of the Hebrew University approved all animal procedures. Male BALB/c mice (3-4 week-old) were used in all in vivo assays, 3-5 mice were used for each compound, repeating each experiment at least three times. A custom-built 760 nm light emitting diode array (LED array) (Roithner Lasertechnik GmbH, Vienna, Austria) was used for all light treatments.
Chemical Synthesis
Synthetic routes are presented in Scheme 1 and Supplementary Scheme 1.
Synthesis of Fmoc-Lys(Boc)-BMK, 1: as described in Kato et al 2005[35], in detail: Fmoc-Lys(Boc)-OH (1 eq., 3 gr, 6.4 mmol) was dissolved in anhydrous Tetrahydrofuran (THF) at 0oC in an inert atmosphere under Argon, N-methylmorpholine (NMM; 1.25 eq., 880 μl, 8.0 mmol) and isobutyl chloroformate (IBCF; 1.15 eq., 857 μl, 7.36 mmol) were sequentially added. The solution was stirred at 0oC for 10 minutes and additional two hours at -10oC. Then, excess of ethereal diazomethane (14 eq.), synthesis in situ, was added to the anhydride at -10oC. The reaction vessel was kept at -10oC for 30 minutes and then brought to room temperature during 2 hours while stirring. A solution of 1:1 acetic acid (AcOH): 37% HBr was added dropwise at 0oC while stirring for 2 minutes, until bubbling stopped. The reaction mixture was diluted by ethyl acetate (EA). The mixture was put in a separatory funnel, followed by the addition of water. The organic phase was separated and water phase was washed again with EA. The organic phases were pooled and washed with saturated NaHCO3 three times and twice with Brine. The organic fraction was dried over MgSO4 and evaporated to give yellow powder. The crude product, Fmoc-Lys(Boc)-BMK, was further dried by lyophilization to yield a bright yellow solid, 85% pure as judged by LC-MS and was used without further purification. Weight 4.49 gr, 8.24 mmol, M/z+1 545.
Synthesis of Fmoc-Lys(Boc)-AOMK, 2: based on the synthesis of asparagine AOMK described in Kato et al 2005[35] with modification, detailed synthesis is described here. Fmoc-Lys(Boc)-BMK (1 eq., 495 mg, 0.9 mmol) and 2,6-dimethylbenzoic acid (5 eq, 690 mg, 4.5 mmol) were dissolved in dry dimethylformamide (DMF) under argon. KF (10 eq., 534 mg, 9.2 mmol) was added and reaction was kept at room temperature for 2 hours while stirring in inert atmosphere. After work up as described above, the organic fraction was dried over MgSO4 and solvent was removed by vacuum to give yellow oil. The crude product, Fmoc-Lys(Boc)-AOMK, was found to be over 95% pure by LC-MS and was used without further purification. 365.2 mg, 65% yield, 0.594 mmol, M/z+1 615.
Synthesis of GB111-NH2, 3: Fmoc-Lys(Boc)-AOMK (365.2 mg, 0.594 mmol) was de-protected by 25% trifluoroacetic acid (TFA)/dichloromethane (DCM) (v/v) during 30 minutes followed by removal of solvent by co-evaporation with toluene in vacuum. 2-Chlorotrityl chloride resin (371.1 mg) was loaded by shaking of resin with de-protected Fmoc-Lysine(NH2)-AOMK (1 eq.), and diisopropylethylamine (DIEA) (3.5 eq, 360 μl) dissolved in anhydrous DCM for 1.5 hours. Methanol (1 ml/gr resin) was added, and the resin was shaken for 30 minutes followed by washing with DCM and DMF. The Fmoc protecting group was removed by two quick washes with 5% diethylamine (DEA)/DMF (v/v) followed by 5 minutes incubation with 5% DEA and then DCM and DMF washes. De-protection was verified by Kiser test. The peptide was elongated by addition of a solution of N-benzyloxycarbonyl-phenyalanine (3 eq., 226 mg), hydroxybenzotriazole (HOBT; 3 eq., 101.3 mg) and diisopropylcarbodiimide (DIC; 3 eq., 116 μl) in DMF for 2 hours. The resin was then washed with DCM and DMF and the final peptide product was cleaved from the resin by addition of 5% TFA/DCM (v/v) for 7 to 15 minutes. The cleaved solution was collected and solvent was removed by co-evaporation with toluene. The crude peptide was further dried in vacuum and purified by C18 reverse phase HPLC with an acetonitrile (ACN) / water gradient. GB111-NH2 eluted at 44% ACN to yield a white powder, 78.94 mg, 0.138 mmol, M/z+1 574, 56% yield relative to resin loading.
Synthesis of Cbz-Phe-Lys(Boc)-2,6 dimethyltherephthalic amide 6-aminohexane-QSY-21, 5 (Supplementary Scheme 1): synthesis of Cbz-Phe-Lys(Boc)-2,6 dimethyltherephthalic amide 6-aminohexane and coupling of commercial QSY-21 was described in Blum et al 2007[20] and shown in Supplementary Scheme 1.
Synthesis of Cbz-Phe-Lys(Boc)-2,6 dimethyltherephthalic amide 6-aminohexane-QC-1, 6 (Supplementary Scheme 1): commercial QC-1-NHS ester (1 eq., 1.6 mg, 2.21 μmol) was dissolved in DMSO and added to the free amine of Cbz-Phe-Lys(Boc)-2,6 dimethyltherephthalic amide 6-aminohexane (1 eq, 1.7 mg, 2.08 μmol) with DIEA (24 eq., 7 µl). The reaction was stirred in the dark for 2 hours. The resulting product, Cbz-Phe-Lys(Boc)-2,6 dimethyltherephthalic amide 6-aminohexane-QC-1, was purified from the crude reaction mixture by C4 reverse phase HPLC using a water /ACN gradient supplemented by 50 mM triethylammonium acetate (TEAA), pH 6.0. The product was eluted with 47.5-50% acetonitrile to obtain a blue-green solid, 2.26 mg, 1.20 μmol, 58% yield, M/z+1 1879.
Synthesis of PS-qABPs and PS-ABPs: the lysine's ε amine of each precursor 3 (GB111-NH2) (Scheme 1), or 5 or 6 (Supplementary Scheme 1) were Boc deprotected by 25-50% TFA/DCM followed by co-evaporation of solvent with toluene. Next, coupling of PSs to each one of the precursors was achieved by one of two synthesis routs: 1) activation of the PS carboxylic acid residue to succinimide ester (PS-NHS) by N,N'-Dicyclohexylcarbodiimide (DCC) and O-(N-Succinimidyl)-1,1,3,3-tetramethyluronium tetrafluoroborate (TSTU). After more than 90% of PS was converted to PS-NHS ester (detected by LC-MS as MW+97), the activated ester without further purification was coupled to the free amine precursors in basic conditions using 15 eq DIEA. 2) Reacting the free PS with the free amine using coupling reagent HOBT and DCC in basic conditions using 15 eq DIEA. In both methods, all reactions were stirred in the dark and then final products were purified by C18 reverse phase HPLC using ACN/water gradient. YBN14 was purified by C4 reverse phase HPLC using a water/ACN gradient supplemented by 50 mM TEAA. Supplementary Tables 1 and 2 summarize the method used for the synthesis of each probe, the precursor, ACN elution, color, yield and MW. A general structure of the probes is described in Scheme 1d, and a list of PSs attached to each of the probes is presented in Supplementary Table 3.
Methods
Biochemical evaluations
Quenching efficiency: Increasing concentrations of YBN1-8 in acetate buffer (50 mM acetate, 5 mM MgCl2, 2 mM DTT, pH 5.5) containing 1% DMSO were prepared in a 96-well plate. Fluorescence was imaged and quantified using an In Vivo Imaging System- Kinetic (IVIS-Kinetic) (Perkin Elmer, Waltham, MA, USA) equipped with an excitation filter of 430 nm and an emission filter of 690 nm for all compounds, or using SpectraMax M5 plate reader (Molecular Devices, Sunnyvale, CA, USA) with a suitable excitation/emission wavelength for each probe. Arbitrary fluorescent units were plotted versus probe concentrations, and the ratio between the slope of the non-quenched probe and the corresponding quenched probe was determined as the quenching efficiency. Similarly, quenching efficiency of the probes was evaluated in PBS. The quenching efficiency of YBN14 was evaluated in the same manner in 70% methanol. Fluorescence was imaged by a Typhoon FLA 9500 scanner (GE Healthcare Bio-Sciences AB, Uppsala, Sweden) with 785/810 nm excitation/emission filters, and quantified using ImageJ software.
Recombinant cathepsin labeling: Recombinant human Cathepsin L (0.6 µg), Cathepsin B (0.7 µg) or Cathepsin S (0.7 µg) were pretreated with 1 µM inhibitor GB111-NH2 or vehicle for 30 minutes (indicated samples) in reaction buffer (50 mM acetate, 2 mM DTT and 5 mM MgCl2, pH 5.5) at room temperature. Indicated concentrations of YBN1-8, 13 & 14 were added to samples for 90 minutes at 37oC. The reaction was stopped by addition of sample buffer x4 (40% glycerol, 0.2 M Tris/HCl pH 6.8, 20% beta-mercaptoethanol, 12% SDS and 0.4 mg/ml bromophenol blue). Samples were then boiled, separated on a 12.5% SDS gel and scanned for fluorescence by a Typhoon scanner FLA 9500 at excitation/emission wavelengths of 635/670 nm for YBN1-8 and 785/810 nm for YBN13 and YBN14, respectively.
Cell cultures: NIH-3T3 mouse fibroblast cells, Raw 264.7 mouse macrophage cells or 4T1 murine mammary gland epithelial cells were cultured in DMEM (Dulbecco's modified eagle's medium) supplemented with 10% fetal bovine serum (FBS), 1% penicillin and 1% streptomycin. Bone marrow derived macrophages (BMDM) were prepared by extracting bone marrow from BALB/C mice, followed by purification of mononuclear cells on a Ficoll gradient. Cells were then cultured in DMEM (as described above) supplemented with 10 µg/mL of MCSF (macrophage colony-stimulating factor, 1:200). Experiments were performed on either day 6 or day 7 after plating in culture. All cells were cultured in a humidified atmosphere of 95% air and 5% CO2 at 37oC.
Evaluation of probes permeability to intact cells, competition assay: NIH-3T3 cells (3x105 cells/well) were seeded in a six-well plate one day before treatment. Cells were treated with vehicle or 20 µM probes that were pre-dissolved in 0.1% DMSO, 0.9% ethanol in culture medium. After 4, 6 or 8 hours of probe incubation residual cathepsin activity was labeled with GB123[20] (5µM) (final DMSO concentration was maintained at 0.2% and 0.9% ethanol). Cells were washed with PBS and lysed by addition of sample buffer. Lysates were boiled for 5 minutes, centrifuged, and separated by 12.5% SDS-PAGE. Residual labeled proteases in cells were visualized by scanning the gel with an Odyssey Scanner, excitation/emission wavelengths of 680/700 nm.
Evaluation of probes in intact cells, direct labeling assay: Raw 264.7 cells or 4T1 cells (2.5x105 cells/well) were seeded in a six-well plate one day before treatment. Cells were treated with probes (20 µM, pre-dissolved in 0.9% ethanol, 0.1% DMSO, final concentrations) or with vehicle in culture medium at pH 7.4 for 4 hours. Cells were then washed with PBS and lysed by addition of sample buffer. Lysates were boiled for 5 minutes and centrifuged. 100 µg protein of RAW cell lysates or 150 µg of 4T1 cell lysates were then separated by 12.5% SDS-PAGE. Labeled proteases were visualized by scanning the gel with an Odyssey Scanner at excitation/emission wavelengths of 680/700 nm and 780/800 nm.
BMDM (3.8x105 cells/well) were seeded in a six-well plate four days before treatment. Cells were treated with YBN13 or YBN14 (20 µM) in culture medium for 4 hours (final DMSO concentration was 0.1% (v/v) throughout). Cells were then washed with PBS and lysed in RIPA buffer, pH 7.4 (1% Tergitol-type NP-40 (nonyl phenoxypolyethoxylethanol) 0.1% SDS, 0.5% sodium deoxycholate). Lysates were boiled for 10 minutes, centrifuged, and 20 μg lysates were separated by 12.5% SDS-PAGE. Labeled proteases were visualized by scanning the gel with an Odyssey Scanner at excitation/emission wavelengths of 780/800 nm.
Fluorescent imaging of protease activity in live cells: NIH-3T3 cells were seeded in an 8 well cover-slip chamber containing growth medium one day before treatment. Cells were either pretreated with inhibitor GB111-NH2 (5 μM) or with vehicle DMSO (0.1%) for 1 hour. Cells were labeled by addition of YBN7 or YBN8 (10 μM, 0.1% DMSO, 0.9% ethanol) to growth media (1%FBS) for 7 hours. Medium was then replaced with growth medium without phenol red. Fluorescent images were taken at 100X magnification using an Olympus inverted fluorescent microscope IX51 equipped with a Cy5 filter. Cells were either imaged directly or washed with growth medium for 16 hours and imaged in the presence of LysoTracker.
Stability of YBN14: Equal amount of YBN14 (83μM) in PBS (0.17% DMSO) was analyzed at different times after preparation by a LC-MS equipped with a C4 reverse phase column, acetonitrile/water gradient. Analysis spectrum of area under the curve was performed by Xcalibur Queal Browser software. Area under the curve was compared to time zero, determined as 100%.
In vivo imaging: 4T1 cells were grown to subconfluency, followed by detachment with trypsin, spin down and resuspention in 0.5% BSA in sterile PBS and 25% matrigel. Cells (1x106 per spot in a total volume of 20 μl) were injected subcutaneously at the indicated locations into 3-4 week-old male BALB/c mice under isoflurane anesthesia. Tumors were typically established 9-13 days after cells injections, the fur was removed from mice and compounds were injected intravenously via the tail vein as follows: YBN13 or YBN14 or bacteriochlorin (75 nmol) dissolved in 10% DMSO in PBS in a total volume of 100 μl. Mice anesthetized with isoflurane were then imaged at indicated time points after probe injection using the IVIS kinetic equipped with a 710/760 nm excitation/emission filter.
Therapeutic capability of YBN14 after light treatment in vivo: 4T1 cells (7.5x105 cells) were injected subcutaneously to the back of 3-4 week-old male BALB/c mice under isoflurane anesthesia. When tumors were established, the fur was removed from the mice. YBN14 or vehicle (10% DMSO) were injected intravenously via tail vein (as described above) on day 7, 9 and 11 post-subcutaneous cells injections. At 8 and 16 hours post each injection light treatment of 45 mW, 760 nm, 20 minutes was applied under anesthesia, light treatment was not applied to dark controls. Each experimental group contained five mice. Tumor sizes were measured daily by a digital caliper and volumes were calculated as width2 × length × 0.5. Mice were sacrificed at day 14.
Evaluation of the probes' ability to induce apoptosis in vivo after PDT: BALB/c mice bearing two tumor spots of 4T1 cells (see above for details) were injected via the tail vein with YBN14 or bacteriochlorin (75 nmol as described above). Light treatment (55 mW, 760 nm, 20 minutes) was applied at 8 and 16 hours to one tumor spot while the other tumor spot was kept in dark. 24 hours post YBN14 or bacteriochlorin injection, mice were anesthetized with isoflurane and sacrificed by cervical dislocation. Tumors, liver, kidneys and spleen were surgically excised and imaged ex vivo using the same IVIS kinetic as above; fluorescence intensity was calculated by dividing fluorescence signal intensity by area. Tissues were either frozen in liquid nitrogen and lysed using bead beater or dounced in RIPA buffer. Proteins were quantified by Bradford assay; total protein extracts (100 μg) were separated by 12.5% SDS-PAGE and visualized by scanning the gel with an Odyssey Scanner at excitation/emission wavelengths of 780/800nm, or tumors were incubated for 4 hours with 4% paraformaldehyde, followed by an overnight incubation with 30% sucrose at 4oC. Samples were then embedded in OCT and frozen at -80oC. Tissue was sectioned into 10 μm thick slices using a CM 1900 cryotome (Leica Microsystems, Wetzlar, Germany). Sample sections were stained overnight at 4oC with primary antibodies: F4/80 (Molecular Probes) and cleaved Caspase-3 (Cell Signaling) in Cas-Block (Invitrogen) and then with secondary antibodies coupled to Cy3 and Cy5 respectively. The samples were mounted with DAPI Fluoromount-G (Southren Biotech, AL, USA). Fluorescent pictures were taken with an Olympus FV10i confocal microscope (Olympus, Tokyo, Japan) equipped with Cy3, Cy5 and DAPI filters.
Results
Chemical synthesis
We set out to develop a library of quenched activity-based probes that carry photosensitizers (PS-qABPs). The library contains probes that are quenched and their corresponding non-quenched activity-based probes as controls. We based our design on the fluorescent qABPs that we developed previously [20, 21]. It contains an acyloxymethyl ketone electrophile that enables removal of the quencher upon enzyme binding. Our selection of PS moieties for attachment was based on two criteria: 1) high wavelength fluorescence in the near infrared range to enable non-invasive imaging, and 2) presence of an acid moiety enabling chemical binding to the core probe structure.
Synthesis of the probes was carried out using solid-phase peptide synthesis (SPPS) combined with solution synthesis. The precursor compound GB111-NH2[21], compound 3, was used for generating the non-quenched PS-ABP, its synthesis was modified from [21] as described in Scheme 1b. The commercial photosensitizer acid moiety was activated to an ester to enable attachment to the lysine ε free amine of compound 3 (Scheme 1c).
Compound 5 (Supplementary Scheme 1), a Boc protected amine of GB137 described in [20], which contains a quencher moiety was used as a precursor for the quenched PS-ABPs. Following Boc deprotection the activated PS moiety was coupled in the same manner as in compound 3. General structure of the probes is presented in Scheme 1d. We have generated a library of eight probes out of which four are quenched and four are non-quenched; the various PS structures and their corresponding excitation wavelengths are shown in Scheme 1e. Characterization of all compounds synthesized (structure, UV trace, mass spectrometry analysis of the major peak and maximal fluorescence absorption) is shown in Supplementary Material S4.
Biochemical evaluations of PS-qABPs and PS-ABPs
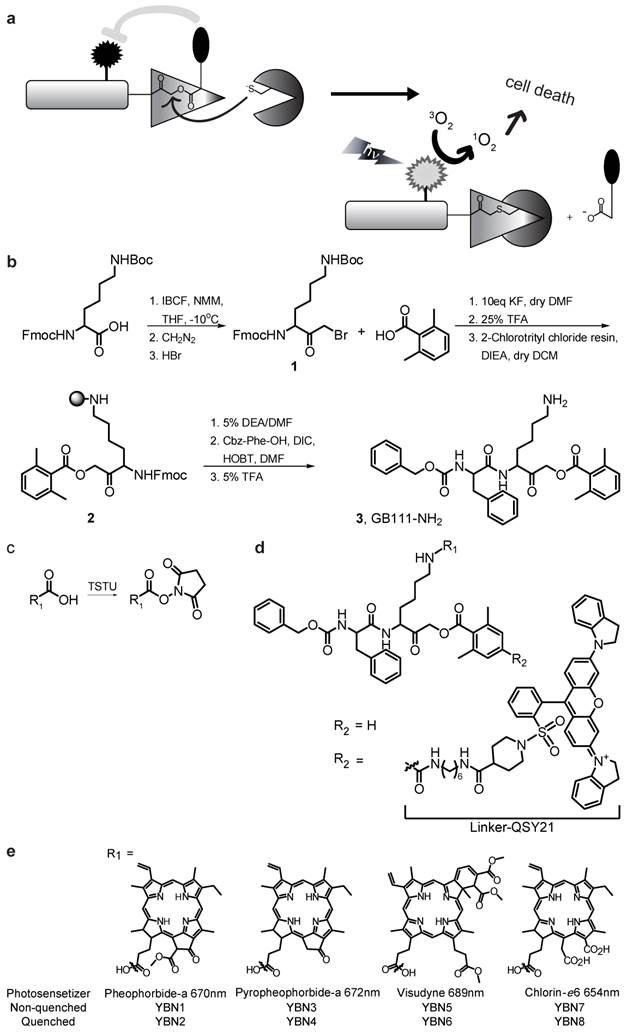
Taking in consideration the unique excitation and emission characteristics of the selected PSs, we set out to evaluate the fluorescent properties and quenching efficiency of the probes. We compared the fluorescence at increasing concentrations of the quenched probes relative to their non-quenched PS-ABPs counterpart at physiological or lysosomal pHs. For example Visudyne containing probes, YBN5 (non-quenched) and YBN6 (quenched), were excited at 430 nm and emission was measured at 690 nm. YBN6 was found to have more than 360-fold lower fluorescence in PBS (pH 7.5), when compared to YBN5 and more than 160-fold quenching in acetate buffer (Figure 1a). All of the PS-qABPs were excited at the wavelengths of their maximum absorption peak and were found significantly quenched (14 to 363-fold) in comparison to the corresponding non-quenched PS-ABPs (Supplementary Table 4). These results indicated that the quencher in our basic design allows absorption of energy emitted by the PS moiety as intended.
In vitro evaluation of first generation probes. (a) Quenching efficiency plot of YBN6. Increasing concentrations of YBN5 and YBN6 in acetate buffer (pH 5.5) were prepared in a 96-well plate. Fluorescence was imaged and quantified using an IVIS-Kinetic equipped with a 430/690nm excitation/emission filter. Fluorescent units were plotted versus probe concentrations; the ratio between the slopes was determined as quenching efficiency. (b) Labeling of recombinant cathepsins by PS-qABP and PS-ABP. Recombinant cathepsins B, L and S were treated with inhibitor GB111-NH2 or vehicle followed by incubation with indicated concentrations of YBN1-8. Samples were separated on SDS-PAGE and scanned for fluorescence by a Typhoon scanner at 635/670 nm. All probes show activity and dose dependent binding to the cathepsins. (c) Detection of cathepsin inhibition in intact cells by labeling of residual protease activities. NIH-3T3 cells were treated for 4 hours with probes (20 µM) or with DMSO in culture medium, followed by 2 hours labeling of residual enzyme activity by GB123 (5 µM). Samples were analyzed by SDS-PAGE, and scanned for fluorescence with an Odyssey Scanner at 680/700 nm. All PS-ABPs showed good cathepsin inhibition while their counterpart PS-qABPs showed poor cell permeability. (d) Average proteases inhibition by YBN1-8 quantified at 4 to 8 hour time points. (e) Direct labeling of endogenous cathepsins in intact RAW 264.7 and 4T1 cells. Cells were treated with YBN1-8 (20 µM) or with DMSO in culture medium for 4 hours, followed by cells lysis, SDS-PAGE separation and fluorescent scanning of the gel using an Odyssey Scanner at 680/700 nm.
Binding of ABPs to recombinant Cathepsin B, L and S
Next we evaluated the ability of the various PS-qABPs and PS-ABPs to bind to the target cathepsins by incubating them with recombinant human cathepsins B, L, or S. The free probes were separated from the enzyme-probe complex using SDS-PAGE. The detection of the probe-enzyme complex was done by a fluorescence scan of the gel. To assure that binding depends on protease activity, a control was performed in the presence of a potent active-site cathepsin inhibitor GB111-NH2.
All probes were found to bind the cathepsins in a dose-dependent manner. We assume that the different PSs have little influence on binding efficacy. The subtle fluorescence differences observed by the various probes were most likely derived from the discrepancy of the scanner and the unique excitation/emission wavelengths of each PS (Figure 1b).
ABP cell permeability and labeling of cathepsin in intact cells
After confirming that all the PS-qABPs and counterpart PS-ABPs label cathepsins B, L and S in vitro, we set out to determine the probes' cell permeability, specificity and their ability to target the endogenous cathepsins. We performed a competition study, in which NIH 3T3 cells were first treated with each one of the probes followed by labeling of the residual cathepsin activity with GB123 (our published Cy5 labeled probe that is easily detected)[20]. We found that all PS-ABPs (YBN# 1, 3, 5 and 7) showed good cell permeability and ability to bind cellular cathepsins. However, all the quenched probes (YBN# 2, 4, 6 and 8) showed lower cell permeability and thus lower endogenous cathepsins labeling (Figure 1c). Based on our previous results with qABPs carrying a similar quencher QSY-7 that showed reduced cells permeability[21], we increased the incubation time with the probes to eight hours, but without major improvement. A graph showing the average of cathepsins inhibition by the probes at 4-8 hours is presented in Figure 1d.
To detect direct labeling of the cellular cathepsins by the probes we investigated cathepsin labeling in cells that have increased cellular uptake such as Raw 264.7 macrophages and in cells that possess elevated cathepsin activity such as 4T1 breast cancer cells (Figure 1e). We detected direct labeling of endogenous cathepsins in these cells indicating the high specificity of most of our probes. The identity of the cathepsin labeled bands was determined by correlation to the cathepsins expression shown in Supplementary Figure S1. Non-specific bands of serum albumin at ~ 67kDa were detected similarly to our previously published cathepsin probes[20].
Fluorescence microscopy in intact cells
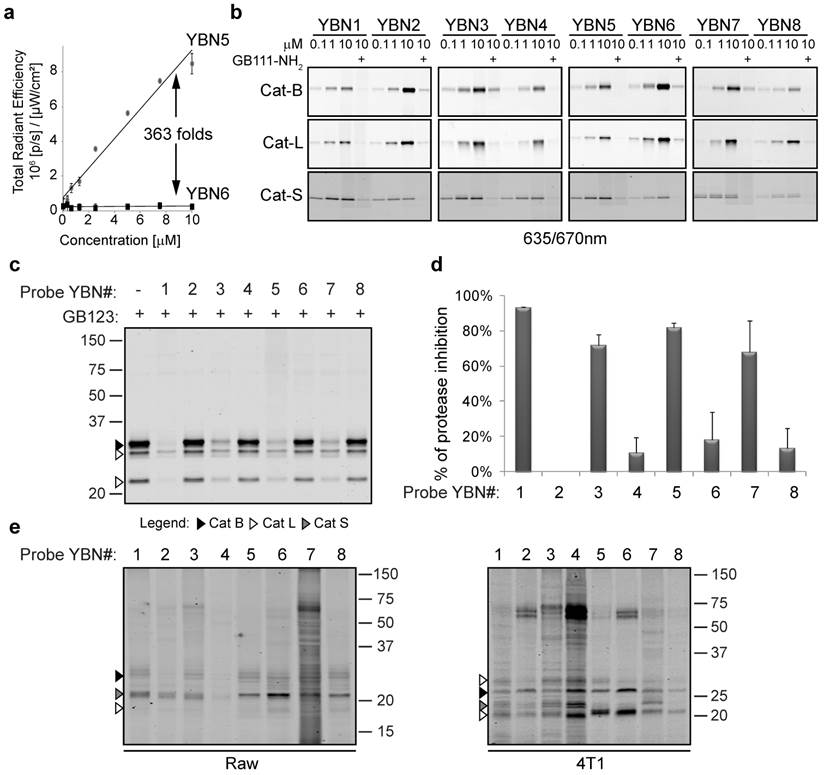
To verify the probe location and permeability in intact cells, we acquired fluorescence microscopic images of live NIH-3T3 cells treated with the probes. Images of PS-ABP YBN7 without washout show high accumulation and nonspecific staining of the entire cell as a result of free probe accumulation (Figure 2a). Such staining pattern of the non-quenched probe implied non-specific distribution throughout the cell without significant accumulation in any particular location. To confirm the probe specificity we next performed extensive washout of un-bound probe from cells, which resulted in punctate staining that overlapped with the signal of the lysosomal marker Lysotracker, and could be blocked by pretreatment of cells with a cathepsin inhibitor (Figure 2b). Similarly, imaging of cells that were incubated with the PS-qABP YBN8 also revealed a distinct punctate labeling pattern even without the washout step, as a consequence of efficient quenching of the probe. These results confirmed that the quenched YBN8 localized to the lysosomes where it interacted with active enzymes that removed the quencher. Moreover, the probe accumulated in cells in sufficient amounts to be detected by fluorescence microscopy.
Imaging protease activity in live cells. Cultures of NIH-3T3 cells were either pretreated with protease inhibitor GB111-NH2, or with control DMSO for 1 hour and then labeled by addition of YBN7 or YBN8 (10 µM) to growth medium for 7 hours. (a) Cells were imaged before washing by an inverted fluorescent microscope equipped with a Cy5 filter, bright field images are on the right. (b) Cells were imaged after a 16-hour wash with growth medium and after addition of lysosomal marker LysoTracker. Red fluorescence marks protease activity (PS-ABP or PS-qABP labeling); yellow fluorescence - lysosomal compartments; orange color - overlap of red and yellow signals.
Although the library of probes carrying the QSY-21 quencher allowed for adequate cathepsin detection, we were concerned that the low cell permeability will reduce the overall capabilities to induce sufficient PDT effect. Therefore, we decided to replace the quencher in order to improve probe cell permeability and thereby enhance its cell-killing properties required for efficient PDT in vivo.
We replaced the QSY-21 quencher with the commercial QC-1 quencher and used a new PS, a bacteriochlorin derivative[31], that is activated by a longer wavelength, approximately 750 nm, to better fit the quencher's absorbance energy. Longer wavelength probes are highly advantageous for in vivo applications since the light penetration is deeper than at lower wavelengths and auto-fluorescence is reduced.
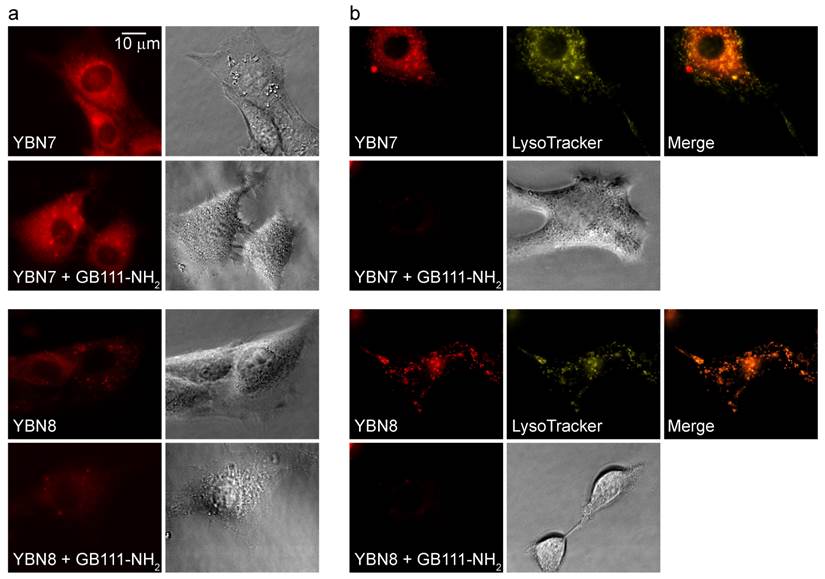
We synthesized the second generation of ABPs, the non-quenched YBN13 and the quenched YBN14, both possessing the bacteriochlorin photosensitizer (Figure 3a). A 10-fold quenching efficiency was achieved with the QC-1 quencher, when the signal of the YBN14 probe was compared with the YBN13 probe, most likely reflecting the QC-1 properties. However, based on the high expression levels of cathepsins in TAMs, such quenching should be sufficient for successful in vivo applications (Figure 3b). Similarly to the first-generation probes, the second-generation probes were found to bind recombinant cathepsins B, L and S in a dose- and activity-dependent manner (Figure 3c).
Second generation probes in vitro evaluation. (a) Structure of YBN13, PS-ABP and YBN14, PS-qABP. (b) Quenching efficiency of YBN14, a fluorescent image of a dilution series of YBN13 and YBN14 scanned by a Typhoon scanner at 785/810 nm. Fluorescence intensity was quantify by ImageJ software and plotted versus probe concentration. Ratio between slopes indicated a 10.76 fold quenching efficiency of YBN14. (c) Labeling of recombinant cathepsins. Recombinant cathepsins B, L and S were treated with YBN13 and YBN14 as described in Figure 1b. Samples were separated on SDS-PAGE and scanned for fluorescence by a Typhoon scanner at 785/810nm. Both YBN13 and YBN14 bind active cathepsins in a dose dependent manner. (d) Detection of cathepsin inhibition in intact cells by labeling of residual protease activities by GB123. NIH-3T3 cells were treated with YBN 13 and 14 as described in Figure 1c. (e) Direct labeling of endogenous cathepsins. Cells were treated with probes and analyzed similarly to as in (d), at 780/800nm detecting the PS bacteriochlorin.
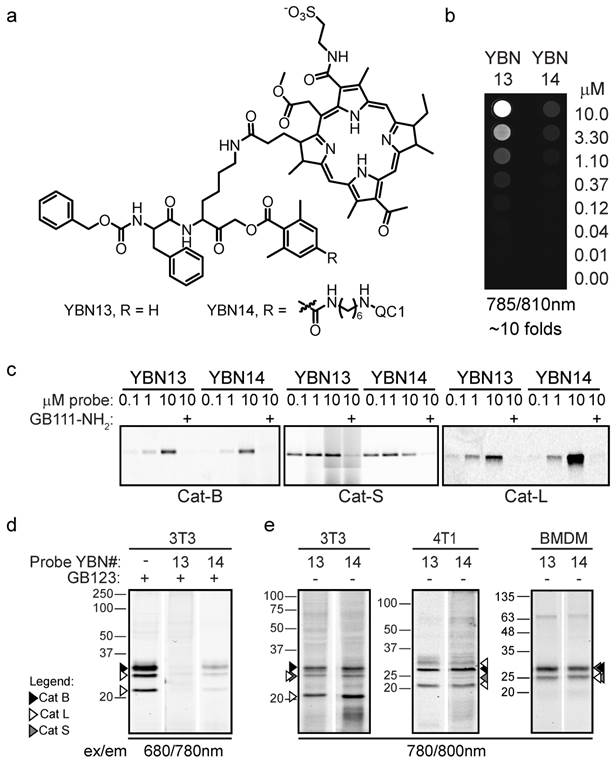
Importantly, both YBN13 and YBN14 probes were found to be highly cell permeable, as seen in the competition assay in Figure 3d. Detection of labeled cathepsins both in intact NIH-3T3 cells, 4T1 cells and in BMDM revealed high selectivity towards the intact cathepsins (Figure 3e).
In order to confirm that the quencher moiety and not the different PS is the reason for reduced cell permeability, a chimera PS-qABP carrying a bacteriochlorin analogue and the QSY-21 quencher was generated. While the chimera probe efficiently labeled recombinant cathepsins in an activity-dependent manner, it almost completely lacked cell permeability, (data not shown). These results suggested that improvement in cell permeability of YBN14 was largely achieved by using the new quenching moiety, QC-1. Moreover, YBN13 and YBN14 showed efficient cell killing in vitro after light treatment (Supplementary Figure S2). Based on these finding and the benefits of long wavelength PS for in vivo application, we advanced to evaluate these probes for theranostic capabilities in vivo.
Pharmacokinetic properties of probes in vivo
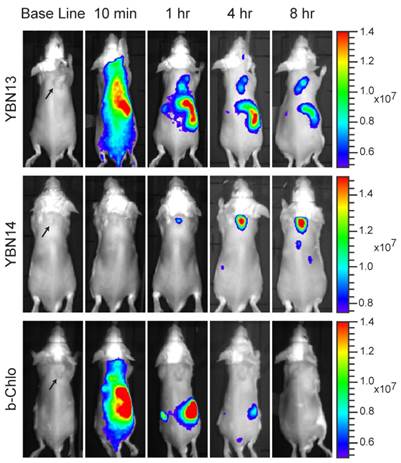
We started the in vivo studies by investigating the pharmacokinetics of YBN13 and YBN14 in tumor-bearing mice. This was done to establish the non-invasive imaging capabilities of these probes as well as the optimal time point for light treatment. At early time points the non-quenched probe, YBN13, exhibited high fluorescent signal in the whole mouse body (Figure 4). A clear signal from the tumor could be detected after 4 hours together with fluorescence of the probe distributed throughout the body. This non-specific fluorescence diminished over time with clearance of the un-bound probe from the body of the mouse, resulting in a tumor-specific signal. The quenched probe, YBN14, as expected, did not generate any background signal at early time points and a specific signal in tumor was visualized as early as one hour post probe injection (Figure 4). This specific signal increased over time and reached a maximum at 8 hours post probe injection. Due to the covalent nature of the probes a significant amount retained in the tumor up to 24 hours post injection (data not shown). We then evaluated the stability of YBN14 and found that the probe exhibited over 85% and 74% stability after 16 and 23 hours, respectively, demonstrating excellent correlation with the in vivo data. Based on these results, the optimal time for the light treatment was determined to be 8 and 16 hours after administration of the probe. Mice that were injected with the free PS, bacteriochlorin, showed rapid clearance with no fluorescence signal absorbed in the tumor, since this PS lacks the targeting moiety.
Non-invasive optical imaging of tumors in mice. 4T1cells were injected subcutaneously on the back of BALB/c mice. After tumors were established, YBN13, YBN14 or bacteriochlorin (b-Chlo) were injected intravenously (via tail vein). Mice anesthetized with isoflurane were imaged before and at indicated times after probe injections with an IVIS Kinetic equipped with 710/760nm excitation/emission filters, tumors are marked by arrows. Fluorescent scale bar is depicted on right of images in units of (p/sec/cm2/sr)/(μW/cm2). Fluorescent signal in tumor is detected with YBN13 and YBN14, but not with the free PS bacteriochlorin.
Photodynamic Therapy with YBN14
After establishing the optimal time point for light treatment, we evaluated the probe's ability to induce cell death after light treatment. We injected 4T1 tumor cells subcutaneously in two locations on the back of BALB/c mice. When tumors were established, YBN14 or bacteriochlorin were injected intravenously via the tail vein. Eight hours post probes injection a high fluorescent signal was detected in the tumors of YBN14 injected mice. At that time point one of the tumors was treated with light at 760 nm and the other tumor was covered, serving as a dark control. We repeated the light treatment of the same tumor at the 16 hour time point (Figure 5a). Mice were sacrificed 24 hours post probe injection, and then tumors and other major organs were excised and analyzed ex vivo. As suspected all organs of bacteriochlorin injected mice lacked detectable fluorescence. In contrast, mice injected with the quenched YBN14 probe showed significant fluorescence in organs with high cathepsin activity, i.e. kidney, liver and spleen. The tumors that were kept in dark showed significant fluorescence while the tumors that were light irradiated lacked fluorescence due to bleaching of the PS (Figure 5b). Tissue lysates were prepared and separated by SDS-PAGE, highly specific labeling of the cathepsins was detected by the fluorescent bands between 20-35 kDa, characteristic for these enzymes (Figure 5c).
PDT in tumor-bearing mice. (a) 4T1 cells were injected subcutaneously in two separate locations on the back of BALB/c mice. After tumors were established (marked by arrows), YBN14 or bacteriochlorin (b-Chlo) were injected intravenous (via tail vein). Mice were imaged at indicated time as described in Figure 4. After acquiring the image at 8 and 16 hours the right tumor was treated with light (indicated with a thunder cartoon) while the left tumor was kept in the dark. (b) Mice were sacrificed 24 hours after injections and tumor, spleen, kidneys, and liver were extracted and imaged ex vivo using the same IVIS Kinetic equipped with a 710/760nm excitation/emission filter. Right panel shows average of organ fluorescence in (p/sec/cm2/sr)/(μW/cm2) units. (c) Parts of the tumors and organs were lysed and equal protein samples were spread by SDS-PAGE, followed by fluorescent scanning of the gel using an Odyssey Scanner 780/800nm. Specific in vivo cathepsins labeling by YBN14 is detected on gel. (d) Mice bearing a single tumor were treated with YBN14 or vehicle at indicated times. Light treatments were applied at 8 and 16 hrs post probe injection. Growth curve presenting the fold increase in tumor volume relative to volume measured at day 6, * p<0.05. (e) Part of tumors from (a) were frozen in OCT, sectioned, and stained with cleaved Caspaese-3 and F4/80 antibodies labeled with Cy5 and Cy3 respectively. Representative fluorescent scan acquired with an Olympus FV10i confocal microscope are presented, green- F4/80, Red- cleaved Caspase-3, blue- DAPI. Apoptosis indicated by cleaved caspase 3 was detected only in F4/80 macrophages of YBN14 and light treated tumors.
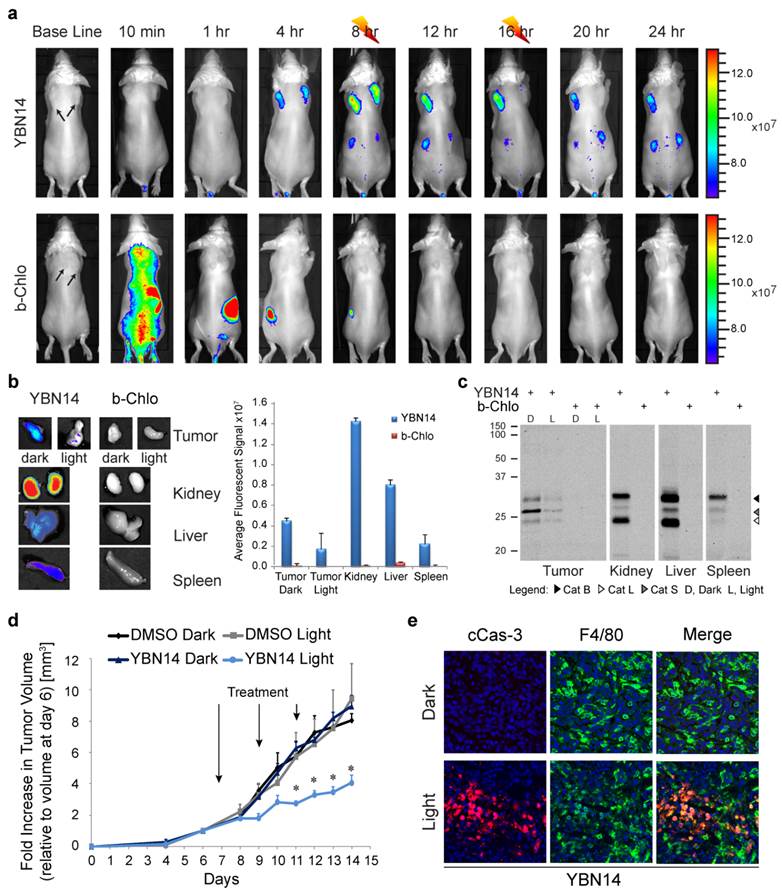
We then evaluated the ability of YBN14 to reduce tumor size after light treatment relative to controls (YBN14 - dark and vehicle - light). We injected YBN14 to tumor bearing mice on day 7, 9 and 11 and each injection was followed by two light treatments (at 8 and 16 hours post injection). We found a significant reduction in primary tumor size, as measured externally, in mice treated with YBN14 and light relative to controls. The reduction in tumor size was observed already on day 9 (two days after the first treatment) and became significant on day 11, reaching 50% on day 14, when mice were sacrificed (see tumor growth curve; Figure 5d). No skin toxicity was observed in the YBN14 treated mice.
To investigate if TAMs were affected by the PDT treatment, tumor sections were stained for cleaved caspase-3, an apoptosis marker, and F4/80, a macrophage marker. Significant signal for cleaved caspase-3 was detected in tumors treated with only a single dose of YBN14 and light, indicating that YBN14 triggered massive apoptosis in tumor tissue after light treatment. As expected, the apoptotic cell death was highly colocalized with the tumor macrophages known to exhibit high cathepsin activity (Figure 5e). Tumors that were kept in dark showed no apoptosis similar to the vehicle control. In addition, light treatment of tumors treated with the free photosensitizer bacteriochlorin at the eight-hour time point resulted in limited tumor apoptosis since the light treatment was performed several hours after its clearance (Supplementary Figure S3).
Discussion
Proteases are a large family of enzymes that are involved in several normal and pathological processes. Many key proteases serve as clinical drug targets, such as Angiotensin Converting Enzymes and HIV protease[36]. In this study we present a unique design of PS-qABP as theranostic agents, which target TAMs-expressed cysteine cathepsins B, L and S thereby allowing noninvasive optical visualization of their activity in vivo. Detection of elevated active cathepsins enables precise tumor imaging thereby enabling accuracy in localizing the light treatment that induces targeted cell death of macrophages within the tumor microenvironment. This design is expected to significantly reduce photosensitizers light toxicity to skin tissue since the free unbound probe is still attached to the quencher that absorbs the energy, preventing off-target ROS generation similar to the self-quenched probe L-SR15[37]. Importantly, the ABP scaffold was utilized here to direct and retain the PS specifically in cancerous tissue. The quencher/PS pair, QC-1/bacteriochlorin, substantially enhanced cell permeability, thereby targeting the high levels of intracellular cathepsins pool in TAMs, resulting in significantly enhanced macrophage-targeted treatment. Moreover, the function of QC-1/bacteriochlorin in high wavelength should enable utility in deeper tissue.
Furthermore, activation of the PS is dependent on two separate targeting mechanisms that ensure specific induction of cell death solely in the cancer microenvironment. Initially, the probes are targeted to the tissue that expresses high levels of active cysteine cathepsin proteases. In the next step, the probe's PS moiety is excited by localized light to induce defined tumor cell death. This is of particular importance since several tissues such as liver, spleen and kidneys express high levels of active cathepsins as seen also in Figure 5, suggesting that such confined light treatment would be highly beneficial for targeted therapy due to minimal toxicity to these organs. This is also a major advantage over most current PSs that do not have a targeting moiety, which directs and retains the PS specifically in the tumor.
Other self-quenched PS probes targeted to proteases were reported. One example is the smart substrate probe targeted to the cathepsin proteases consisting of a poly-L-lysine. The polymeric backbone is grafted with monomethoxy-polyethylene linked with several PSs residues in close proximity that create self-quenching of the PSs properties. Following proteolytic cleavage of the poly-lysine backbone, the released PS recovers its fluorescence properties and singlet oxygen generation upon light activation. These substrate probes showed good results in vivo in a mouse cancer model resulting in significant reduction in tumor size after light treatment[38]. In addition, other quenched PS protease substrate probes that target metalloproteases and caspases have been reported [39, 40]. These probes consist of a short core protease substrate sequence that is flanked by a PS and a quencher on opposite sides. Cleavage between the PS-quencher pair by an active protease leads to generation of the fluorescent signal and enables subsequent activation of the PS and generation of singlet oxygen. In vivo these probes generate a fluorescent signal that was detected non-invasively and were further exploited for PDT treatment [39, 40]. Although these PS protease substrate probes are highly valuable and show promise for combined cancer imaging and treatment they suffer from a major limitation of reduced retention of the free PS moiety in the targeted tissue after the substrate cleavage. In contrast, our PS-qABPs remains covalently bound to a single target protease molecule thereby overcoming rapid clearance. Although ABPs lack amplification we have shown previously that fluorescent ABPs produce higher and more specific signals than fluorescent polymeric substrate probes[19], most likely for two reasons: 1) in contrast to the protease substrate, ABPs covalently modify their enzyme targets, thus preventing clearance of the fluorophore leading to prolonged retention in the pathological tissue. 2) Our ABPs are small molecules that enter cells rapidly leading to attachment to both extracellular and a large pool of intracellular cathepsins while the polymeric substrates mostly target extracellular proteases.
A potential problem of the covalent ABPs is the lack of signal amplification since the target protease is inhibited upon binding to the probe. However, in TAMs that express high levels of cathepsins, this problem can be overcome, and they can still accumulate enough PS to induce significant tumor apoptosis, which is sufficient for efficient therapeutic applications (Figure 5).
TAMs serve as attractive targets since their abundance has recently been described to correlate with tumors having higher histopathological grade. In addition to promoting cancer, TAMs were found to alter chemotherapeutic agents efficacy[11,14]. Therefore, eliminating TAMs would be desired both as a monotherapy and as a combination treatment. Thus, the specific TAMs apoptosis achieved with our PS-qABP suggest that this methodology may be better used for aggressive tumors.
In conclusion, the novel quenched probe described here has theranostic capabilities and serve as a proof of concept for this design that can be further modified to other protease classes. One limitation of this technology is that tissue light penetration limits the ability to detect and treat tumors that are located deep in large subjects. However, this can be overcome by using PS functional at longer wavelengths (~750 nm) to increase light penetration as used here. In addition, several technical advancements have been developed to allow for improved optical imaging in deeper tissue such as fluorescent optical fibers, photo acoustic technologies, and molecular tomography technologies. These advancements and others that are still under development should enable translation of these types of probes into the clinics in future. Although targeting of PS to tumors have been reported in the literature[38, 41], here we describe a new concept of covalent attachment of the PS to highly expressed and active enzymes in the tumors. The PS quenched probes can thus potentially be tested as theranostic agents of other pathologies with high levels of cathepsin expression such as arthritis, atherosclerosis and other inflammation-associated diseases.
Supplementary Material
Supplementary Scheme 1, Supplementary Tables 1-4, Supplementary Figures S1-S3, Supplementary S4.
Abbreviations
1O2: Singlet oxygen
ABPs: Activity-based probes
ACN: Acetonitrile
AcOH: Acetic acid
AOMK: Acyloxymethyl ketone
b-Chlo: Bacteriochlorin
BMDM: Bone marrow derived macrophages
BMK: Bromo-methyl ketone
Boc: Tert-butyloxycarbonyl
Cbz: Carboxybenzyl
DAPI: 4',6-diamidino-2-phenylindole
DCC: N,N'-Dicyclohexylcarbodiimide
DCM: Dichloromethane
DEA: Diethylamine
DIC: Diisopropylcarbodiimide
DIEA: Diisopropylethylamine
DMEM: Dulbecco's Modified Eagle's medium
DMF: Dimethylformamide
DMSO: Dimethyl sulfoxide
DTT: Dithiothreitol
EA: Ethyl acetate
ECM: Extracellular matrix
FBS: Fetal bovine serum
Fmoc: 9-Fluorenylmethyloxycarbonyl
HOBT: Hydroxybenzotriazole
HPLC: High-pressure liquid chromatography
IBCF: Isobutyl chloroformate
IVIS: In Vivo Imaging System
LC-MS: Liquid chromatography mass spectrometer
LED: Light emitting diode
MCSF: Macrophage colony-stimulating factor
mM: Millimolar
MW: Molecular weight
mW: milliwatt
NHS: N-Hydroxysuccinimide
nm: Nanometer
NMM: N-methylmorpholine
NP-40: Nonyl phenoxypolyethoxylethanol
PAGE: Polyacrylamide gel electrophoresis
PBS: Phosphate buffer saline
PDT: Photodynamic Therapy
PS: Photosensitizer
PS-qABP: Photosensitizer- quenched activity-based probe
qABP: Quenched activity-based probe
ROS: Reactive oxygen species
SDS: Sodium dodecyl sulfate
SPPS: Solid-phase peptide synthesis
TAM: Tumor-associated macrophage
TEAA: Triethyl ammonium acetate
TFA: Trifluoroacetic acid
THF: Tetrahydrofuran
TSTU: O-(N-Succinimidyl)-1,1,3,3-tetramethyluronium tetrafluoroborate
Acknowledgements
This work was supported by the Israel Cancer Research Foundation (ICRF) (GB), the Binational US-Israel Science Foundation (BSF) (GB), the European Commission International Re-integration Grant (IRG) (GB) and the Israel Ministry of Health Chief Scientist Office (GB) Rostrees grant (UK) (GB), the Israel Cancer Association (GB), Israel's Council for Higher Education (GB) and by the grants from the Slovenian Research Agency (P1-0140 and J1-3602 to (BT)).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Turk V, Turk B, Turk D. Lysosomal cysteine proteases: facts and opportunities. EMBO J. 2001;20(17):4629-33
2. Jedeszko C, Sloane BF. Cysteine cathepsins in human cancer. Biol Chem. 2004;385(11):1017-27
3. Jagodic M, Vrhovec I, Borstnar S. et al. Prognostic and predictive value of cathepsins D and L in operable breast cancer patients. Neoplasma. 2005;52(1):1-9
4. Harbeck N, Alt U, Berger U. et al. Prognostic impact of proteolytic factors (urokinase-type plasminogen activator, plasminogen activator inhibitor 1, and cathepsins B, D, and L) in primary breast cancer reflects effects of adjuvant systemic therapy. Clin Cancer Res. 2001;7(9):2757-64
5. Lah TT, Cercek M, Blejec A. et al. Cathepsin B, a prognostic indicator in lymph node-negative breast carcinoma patients: comparison with cathepsin D, cathepsin L, and other clinical indicators. Clin Cancer Res. 2000;6(2):578-84
6. Gocheva V, Zeng W, Ke D. et al. Distinct roles for cysteine cathepsin genes in multistage tumorigenesis. Genes Dev. 2006;20(5):543-56
7. Joyce JA, Baruch A, Chehade K. et al. Cathepsin cysteine proteases are effectors of invasive growth and angiogenesis during multistage tumorigenesis. Cancer Cell. 2004;5(5):443-53
8. Mikhaylov G, Mikac U, Magaeva AA. et al. Ferri-liposomes as an MRI-visible drug-delivery system for targeting tumours and their microenvironment. Nat Nanotechnol. 2011;6(9):594-602
9. Shree T, Olson OC, Elie BT. et al. Macrophages and cathepsin proteases blunt chemotherapeutic response in breast cancer. Genes Dev. 2011;25(23):2465-79
10. Salpeter SJ, Pozniak Y, Merquiol E. et al. A novel cysteine cathepsin inhibitor yields macrophage cell death and mammary tumor regression. Oncogene. 2015;23:51
11. Mantovani A, Allavena P, Sica A. et al. Cancer-related inflammation. Nature. 2008;454(7203):436-44
12. Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer. 2004;4(1):71-8
13. Joyce JG, ter Meulen J. Pushing the envelope on HIV-1 neutralization. Nat Biotechnol. 2010;28(9):929-31
14. Zhang Y, Cheng S, Zhang M. et al. High-infiltration of tumor-associated macrophages predicts unfavorable clinical outcome for node-negative breast cancer. PLoS One. 2013;8(9):e76147
15. Kahlenberg JM, Dubyak GR. Mechanisms of caspase-1 activation by P2X7 receptor-mediated K+ release. Am J Physiol Cell Physiol. 2004;286(5):C1100-8
16. Edgington LE, Verdoes M, Ortega A. et al. Functional imaging of legumain in cancer using a new quenched activity-based probe. J Am Chem Soc. 2013;135(1):174-82
17. Hu M, Li L, Wu H. et al. Multicolor, one- and two-photon imaging of enzymatic activities in live cells with fluorescently Quenched Activity-Based Probes (qABPs). J Am Chem Soc. 2011;133(31):12009-20
18. Sellars JD, Landrum M, Congreve A. et al. Fluorescence quenched quinone methide based activity probes--a cautionary tale. Org Biomol Chem. 2010;8(7):1610-8
19. Blum G, Weimer RM, Edgington LE. et al. Comparative Assessment of Substrates and Activity Based Probes as Tools for Non-Invasive Optical Imaging of Cysteine Protease Activity. PLoS One. 2009;4(7):e6374
20. Blum G, von Degenfeld G, Merchant MJ. et al. Noninvasive optical imaging of cysteine protease activity using fluorescently quenched activity-based probes. Nat Chem Biol. 2007;3(10):668-77
21. Blum G, Mullins SR, Keren K. et al. Dynamic imaging of protease activity with fluorescently quenched activity-based probes. Nat Chem Biol. 2005;1(4):203-9
22. Dougherty TJ, Gomer CJ, Henderson BW. et al. Photodynamic Therapy. J Natl Cancer Inst. 1998;90(12):889-905
23. Klara S, Juan C, Gang Z. Killer Beacons for Combined Cancer Imaging and Therapy. Curr Med Chem. 2007;14(20):2110-25
24. Wiseman H, Halliwell B. Damage to DNA by reactive oxygen and nitrogen species: role in inflammatory disease and progression to cancer. Biochem J. 1996;313(Pt 1):17-29
25. Albert W G. New trends in photobiology: Photosensitized oxidation of cholesterol in biological systems: Reaction pathways, cytotoxic effects and defense mechanisms. J Photochem Photobiol B. 1992;13(2):105-18
26. Piette J. Mutagenic and genotoxic properties of singlet oxygen. J Photochem Photobiol B. 1990;4(3):335-9
27. Macdonald IJ, Dougherty TJ. Basic principles of photodynamic therapy. J Porphyr Phthalocyanines. 2001;5:105-29
28. Primbs GB, Casey R, Wamser K. et al. Photodynamic therapy for corneal neovascularization. Ophthalmic Surg Lasers. 1998;29(10):832-8
29. Mang TS, Allison R, Hewson G. et al. A phase II/III clinical study of tin ethyl etiopurpurin (Purlytin)-induced photodynamic therapy for the treatment of recurrent cutaneous metastatic breast cancer. Cancer J Sci Am. 1998;4(6):378-84
30. Hu HY, Vats D, Vizovisek M. et al. In vivo imaging of mouse tumors by a lipidated cathepsin S substrate. Angew Chem Int Ed Engl. 2014;53(29):7669-73
31. Brandis A, Mazor O, Neumark E. et al. Novel Water-soluble Bacteriochlorophyll Derivatives for Vascular-targeted Photodynamic Therapy: Synthesis, Solubility, Phototoxicity and the Effect of Serum Proteins. Photochem Photobiol. 2005;81(4):983-93
32. Rozman J, Stojan J, Kuhelj R. et al. Autocatalytic processing of recombinant human procathepsin B is a bimolecular process. FEBS Lett. 1999;459(3):358-62
33. Bromme D, Nallaseth FS, Turk B. Production and activation of recombinant papain-like cysteine proteases. Methods. 2004;32(2):199-206
34. Mihelic M, Dobersek A, Guncar G. et al. Inhibitory fragment from the p41 form of invariant chain can regulate activity of cysteine cathepsins in antigen presentation. J Biol Chem. 2008;283(21):14453-60
35. Kato D, Boatright KM, Berger AB. et al. Activity-based probes that target diverse cysteine protease families. Nat Chem Biol. 2005;1(1):33-8
36. Turk B. Targeting proteases: successes, failures and future prospects. Nat Rev Drug Discov. 2006;5(9):785-99
37. Shon SM, Choi Y, Kim JY. et al. Photodynamic therapy using a protease-mediated theranostic agent reduces cathepsin-B activity in mouse atheromata in vivo. Arterioscler Thromb Vasc Biol. 2013;33(6):1360-5
38. Choi Y, Weissleder R, Tung CH. Selective antitumor effect of novel protease-mediated photodynamic agent. Cancer Res. 2006;66(14):7225-9
39. Zheng G, Chen J, Stefflova K. et al. Photodynamic molecular beacon as an activatable photosensitizer based on protease-controlled singlet oxygen quenching and activation. Proc Natl Acad Sci U S A. 2007;104(21):8989-94
40. Stefflova K, Chen J, Li H. et al. Targeted photodynamic therapy agent with a built-in apoptosis sensor for in vivo near-infrared imaging of tumor apoptosis triggered by its photosensitization in situ. Mol Imaging. 2006;5(4):520-32
41. Goldshaid L, Rubinstein E, Brandis A. et al. Novel design principles enable specific targeting of imaging and therapeutic agents to necrotic domains in breast tumors. Breast Cancer Res. 2010;12(3):R29
Author contact
![]() Corresponding author: Galia Blum, Ph.D. Institute of Drug Research, The School of Pharmacy, The Faculty of Medicine The Hebrew University of Jerusalem, Jerusalem, Israel, 9112001. Phones: 972-2-675-8682; Fax: 972-2-675-7076; Email: galiablhuji.ac.il
Corresponding author: Galia Blum, Ph.D. Institute of Drug Research, The School of Pharmacy, The Faculty of Medicine The Hebrew University of Jerusalem, Jerusalem, Israel, 9112001. Phones: 972-2-675-8682; Fax: 972-2-675-7076; Email: galiablhuji.ac.il