Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2015; 5(8):834-846. doi:10.7150/thno.12040 This issue Cite
Research Paper
A Two-Step Pretargeted Nanotherapy for CD20 Crosslinking May Achieve Superior Anti-Lymphoma Efficacy to Rituximab
Te-Wei Chu1,†, Rui Zhang1,†, Jiyuan Yang1, Mark P. Chao2, Paul J. Shami1,3, Jindřich Kopeček1,4, ![]()
1. Department of Pharmaceutics and Pharmaceutical Chemistry, University of Utah, Salt Lake City, UT 84112, USA.
2. Institute for Stem Cell Biology and Regenerative Medicine, Stanford University, Stanford, CA 94305, USA.
3. Division of Hematology and Hematologic Malignancies and Huntsman Cancer Institute, University of Utah, Salt Lake City, UT 84112, USA.
4. Department of Bioengineering, University of Utah, Salt Lake City, UT 84112, USA.
† These authors contributed equally to this work.
Received 2015-3-4; Accepted 2015-3-25; Published 2015-4-26
Abstract

The use of rituximab, an anti-CD20 mAb, in combination with chemotherapy is the current standard for the treatment of B-cell lymphomas. However, because of a significant number of treatment failures, there is a demand for new, improved therapeutics. Here, we designed a nanomedicine that crosslinks CD20 and directly induces apoptosis of B-cells without the need for toxins or immune effector functions. The therapeutic system comprises a pretargeting component (anti-CD20 Fab' conjugated with an oligonucleotide1) and a crosslinking component (N-(2-hydroxypropyl)methacrylamide (HPMA) copolymer grafted with multiple complementary oligonucleotide2). Consecutive treatment with the two components resulted in CD20 clustering on the cell surface and effectively killed malignant B-cells in vivo. To enhance therapeutic efficacy, a two-step pretargeting approach was employed. We showed that the time lag between the two doses can be optimized based on pharmacokinetics and biodistribution of the Fab'-oligonucleotide1 conjugate. In a mouse model of human non-Hodgkin lymphoma (NHL), increasing the time lag from 1 h to 5 h resulted in dramatically improved tumor growth inhibition and animal survival. When the 5 h interval was used, the nanotherapy was more efficacious than rituximab and led to complete eradication of lymphoma cells with no signs of metastasis or disease recurrence. We further evaluated the nanomedicine using patient mantle cell lymphoma cells; the treatment demonstrated more potent apoptosis-inducing activity than rituximab hyper-crosslinked with secondary antibodies. In summary, our approach may constitute a novel treatment for NHL and other B-cell malignancies with significant advantages over conventional chemo-immunotherapy.
Keywords: B-cell lymphoma, CD20, apoptosis, HPMA copolymer, rituximab, morpholino
Introduction
Non-Hodgkin lymphoma (NHL) is a prevalent cancer with an around 72,000 projected new cases in 2015 in the United States [1]. Approximately 85% of NHLs arise from B-lymphocytes, and the rest are of T-cell origin. Rituximab, an anti-CD20 monoclonal antibody (mAb), was approved in 1997 for the treatment of B-cell NHL and remains one of the best-characterized antibodies in cancer immunotherapy [2]. The current standard for the treatment of most B-cell NHLs is rituximab in combination with chemotherapy. However, in spite of significant success with this approach, primary resistance and relapse remain a problem [3]. This has been attributed to the inability of immune effector cells to hyper-crosslink ligated mAbs (crosslinking of CD20-bound antibodies by effector cells via Fc receptor (FcR)/Fc interaction) [4, 5], and FcR-mediated endocytosis [6] or “trogocytosis” [7] of CD20 antigens. These clinical problems warrant the development of new, improved therapeutic strategies.
CD20 is one of the most reliable biomarkers of B-lymphocytes. It is a non-internalizing [8] or slowly internalizing [9, 10] receptor, highly expressed on the surfaces of most malignant B-cells, as well as normal B-cells. However, CD20 is not expressed on stem cells and mature plasma cells [11]. Consequently, therapeutics targeting CD20 are safe because normal B-cells can be restored after treatments. It is a well-established fact that crosslinking of CD20 at the surface of B-cells induces apoptosis [12]. One widely accepted model suggests that when CD20-bound antibodies are hyper-crosslinked by FcR-expressing immune effector cells (e.g., macrophages, natural killer cells), the clustered CD20 tend to redistribute at the cell surface and become localized to lipid rafts [13]. Such events mediate the interaction of CD20 with Src-family kinases that are also located in lipid rafts, and trigger apoptotic signaling [14]. Without hyper-crosslinking, apoptosis initiated by ligated mAbs is very limited [15].
Based on the above-mentioned mechanism, we designed a biomimetic therapeutic system (Fig. 1) that crosslinks CD20 and directly induces apoptosis of lymphoma B-cells without the need for chemotherapeutic agents, toxins or immune effector functions. The therapeutic system is composed of two macromolecular conjugates: 1) Fab'-MORF1: an anti-CD20 Fab' antibody fragment attached to a single-stranded morpholino oligonucleotide, MORF1, and 2) P-MORF2: a linear N-(2-hydroxypropyl)methacrylamide (HPMA) polymer (P) backbone grafted with multiple copies of the complementary oligonucleotide, MORF2. We have previously shown that exposure of the human NHL B-cell line Raji to the first conjugate (Fab'-MORF1) decorated the cell surface with MORF1 via CD20 binding; further treatment of the cells with the second conjugate (P-MORF2) resulted in MORF1/MORF2 hybridization at the cell surface, which mediated CD20 crosslinking and induced apoptosis in vitro and in vivo [16]. We named the designed platform “drug-free macromolecular therapeutics” because it does not contain small-molecule cytotoxic compounds (e.g., chemotherapeutics) and the individual components do not have apoptosis-inducing activity [17, 18]. This new therapeutic approach aims to selectively target B-cells for direct apoptosis induction. It has the potential to improve on outcomes currently obtained with conventional chemo-immunotherapy.
Drug-free macromolecular therapeutics by a two-step pretargeting approach. Two macromolecular conjugates, Fab'-MORF1 and P-MORF2, are administered consecutively as the pretargeting dose and the crosslinking dose to induce apoptosis of malignant B-cells.
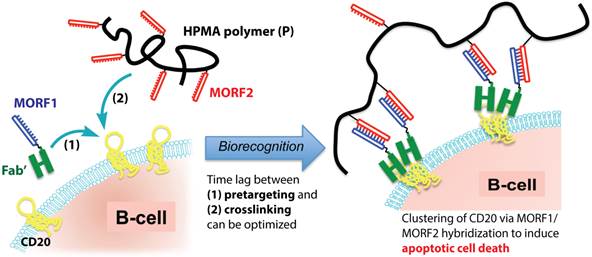
A significant advantage of the designed “two-step” therapeutics (i.e., consecutive administration of Fab'-MORF1 followed by P-MORF2) is the opportunity of pretargeting. Pretargeting is an approach used in cancer radio-immunotherapy, by which targeting functionality and therapeutic modalities are separated in order to achieve desirable pharmacokinetic (PK) goals and reduce adverse side reactions [19]. With the help of modern nanotechnology, the concept of pretargeting has been expanded in recent years and applied for such strategies as amplified therapeutic delivery [20] and universal targeting of different tumor ligands [21]. In the work presented here, Fab'-MORF1 and P-MORF2 are used as a pretargeting dose and a crosslinking dose (effector), respectively. We hypothesized that the time lag between the two doses could be optimized based on PK and biodistribution of the first dose (Fab'-MORF1), in order to achieve maximal tumor-to-tissue accumulation in individual patients. This approach would enable more efficient treatment and limit potential side effects associated with off-target binding.
In this report, we show the development and preclinical evaluation of drug-free macromolecular therapeutics for B-cell lymphomas. In vivo therapeutic efficacy was evaluated in mice using a luciferase-based imageable model of human B-cell NHL. The two-step pretargeting approach was employed where the time lag was optimized after determining the PK and biodistribution of Fab'-MORF1. The designed therapeutic system was compared with rituximab in mouse xenografts and against patient NHL cells in order to test its potential for clinical translation.
Materials and Methods
Preparation of conjugates Fab'-MORF1 and P-MORF2
A pair of 25 bp morpholino oligonucleotides (MORF1 and MORF2) with 3' primary amine modification was purchased from Gene Tools. MORF1 was modified with succinimidyl-4-(N-maleimidomethyl)cyclohexane-1-carboxylate (SMCC) and then conjugated to the Fab' fragment of the 1F5 mAb (IgG2a) via thiol-ene reaction (Supplementary Material: Fig. S1). The second conjugate was obtained in two steps: The polymeric precursor was first synthesized by reversible addition-fragmentation chain transfer (RAFT) copolymerization of HPMA and N-methacryloylglycylglycine thiazolidine-2-thione (MA-GG-TT). Then the complementary MORF2 was grafted to the copolymer backbone via amide linkage (Supplementary Material: Fig. S1). Both conjugates were purified by size exclusion chromatography [16]. A detailed description is provided in Supplementary Material: Supporting Methods.
Cell lines
Burkitt's B-lymphoma cell line Raji was purchased from the American Type Culture Collection (ATCC). ATCC confirmed this line tested positive for the presence of Epstein Barr viral DNA sequences via PCR. Luciferase-expressing Raji cell line (Raji-luc) was generated as previously described [22]. Raji-luc harbors a dual reporter gene L2G (Luc-2A-eGFP) containing a modified firefly luciferase gene joined to eGFP at the 3' end. The L2G construct was ligated into the pCDH-CMV-MCS lentiviral cDNA expression vector (System Biosciences).
Confocal fluorescence microscopy
Raji cells were incubated with rhodamine-labeled Fab'-MORF1 for 1 h and FITC-labeled P-MORF2 for another 1 h. Prior to imaging, cells were washed with PBS. For the CD20 pre-blocking control studies, all conditions were kept the same, except that the cells were pretreated for 1 h with excess amounts of a mouse anti-human CD20 mAb, 1F5 [23]. For detailed procedures, see Supplementary Material: Supporting Methods.
Pharmacokinetics study
Female C.B-17 SCID mice (6- to 8-week-old; 18-20 g; Charles River Laboratories) were used in all following animal experiments in this paper. Mice (n = 5) were intravenously injected with 125I-labeled Fab'-MORF1 (20 μCi per mouse; 1 nmol Fab' equivalent; 58 μg). At predetermined time intervals, 10 μL blood samples were collected from tail vein, and the radioactivity of each sample was measured with a Gamma Counter (Packard). The blood pharmacokinetic parameters were analyzed using a two-compartment model with WinNonlin 5.0.1 software (Pharsight).
Biodistribution study
Mice were intravenously injected with 4 × 106 Raji cells (in 200 μL PBS) via the tail vein. At day 1 or 7 post-inoculation, mice were i.v. administered with 125I-Fab'-MORF1 (20 μCi; 58 μg). Healthy mice (without tumor) were also given the same dose of 125I-Fab'-MORF1 as controls. At 1 h or 5 h post-administration of conjugates, mice (n = 4 per group) were sacrificed. Various organs and tissues were harvested, weighed, and counted for radioactivity with a Gamma Counter. Uptake of conjugates was calculated as the percentage of the injected dose per gram of organs or tissues (% ID/g).
Fluorescence molecular tomography (FMT) imaging
Raji cells were stained with 10 µM DiR (1,1'-dioctadecyl-3,3,3',3'-tetramethyl indotricarbocyanine iodide) (PerkinElmer) at 37 °C for 20 min. Following staining, cells were washed twice with cold PBS. Four million DiR-labeled Raji cells (in 200 μL PBS; used immediately after stained) were injected to mice (n = 4) via the tail vein. At 24 h post-inoculation, the mice were sacrificed, and various organs and tissues were harvested. The fluorescence signals of these organs and tissues were measured using an FMT camera (PerkinElmer) equipped with a 745 nm laser. Total signal intensities (count/energy) of each organ or tissue were quantified. Healthy mice (without tumor) were used as controls (n = 4).
In vivo anti-lymphoma efficacy study
Mice were injected via the tail vein with 4 × 106 Raji-luc cells. One week later, the inoculated mice were divided into groups (n = 6 or 7) and administered via the tail vein with three doses of different treatments (in 100 μL PBS) every other day. These treatments were: 1) PBS (100 μL), 2) Rituxan® (Genentech/Biogen Idec; 75 μg/20 g), 3) 1F5 mAb (75 μg/20 g), 4) Fab'-MORF1 (58 μg/20 g; 1 nmol MORF1) followed by P-MORF2 (119 μg/20 g; 5 nmol MORF2), 1 h interval, and 5) same as 4) but with 5 h interval. Mice were sacrificed at the onset of paralysis or when the body weight dropped below 80% of the initial; otherwise, the mice were maintained until 125 days and considered long-term survivors.
Bioluminescence and microcomputed tomography (microCT) imaging
Imaging of luciferase-expressing Raji lymphoma was performed in vivo and ex vivo, as previously described [22]. In vivo microCT imaging of mice was performed on week 10 after tumor implantation. For detailed procedures, see Supplementary Material: Supporting Methods. All above animal experiments were performed according to the protocol approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Utah.
Flow cytometry analysis of femoral bone marrow cells
After mice were sacrificed, fresh femurs (from both hind limbs) were purged with 1 mL PBS to obtain bone marrow cells. The suspension was passed through a 70-μm FalconTM cell strainer (BD Biosciences), followed by centrifugation (200 g, 5 min) and resuspension in 5 mL red blood cell lysis buffer. The suspension was incubated at room temperature for 5 min and washed/resuspended in cold PBS. Allophycocyanin (APC)-labeled mouse anti-human CD19 antibody (IgG1, κ isotype; BD Biosciences) (10 μL) was added to 100 μL single-cell suspension containing about 106 cells [24]. Cells were incubated for 45 min at 4 °C in the dark, and washed with 1.5 mL PBS prior to analysis. For flow cytometry, data of 5 × 105 cells were recorded.
Patient samples analysis and apoptosis assay
Leukocytes were isolated from four previously untreated mantle cell lymphoma (MCL) patients following ammonium chloride lysis. The isolated cells harbor the t(11;14) chromosomal translocation, a typical MCL phenotype. Specimens were collected after informed consent under a protocol approved by the University of Utah Institutional Review Board. For detailed procedures, see Supplementary Material: Supporting Methods.
Statistical analysis
Statistics was performed by Student's t test to compare between two groups, or one-way analysis of variance (ANOVA) to compare three or more groups (with p value < 0.05 indicating statistical significance). Animal survival analysis was performed with the log-rank test using the GraphPad Prism 5 software.
Results
Therapeutic conjugates Fab'-MORF1 and P-MORF2 were successfully synthesized
The morpholino oligonucleotide pair, MORF1 and MORF2 (Fig. 1), were 25 bp and both about 8.5 kDa. Their base sequences were designed to achieve optimal binding and solubility, and to avoid self-complementarity and off-target binding with human and murine mRNA [16]. The 3'-primary amine was used for conjugation to Fab' or the polymer backbone. The Fab' fragment was from a mouse anti-human CD20 mAb, 1F5 [23]; this antibody was selected as a proof-of-concept study. MORF1 was tethered to the terminal thiol of Fab' via a thioether bond (Supplementary Material: Fig. S1). This site-specific conjugation would prevent undesirable impact on antigen binding. The coupling reaction followed a 1:1 stoichiometry; molecular weight of the conjugate was 57.5 kDa. The hydrodynamic effective diameter of Fab'-MORF1 was 9 nm, as characterized by dynamic light scattering [16].
To prepare the multivalent P-MORF2 conjugate, we first synthesized a copolymer of HPMA with MA-GG-TT by RAFT polymerization [25]. The copolymer contained multiple side chains terminated in (amine-reactive) thiazolidine-2-thione groups and possessed a narrow distribution of molecular weights as determined by size exclusion chromatography (number average molecular weight, Mn = 136 kDa; polydispersity, Pd = 1.15). Then the MORF2 oligonucleotides (containing a 3'-primary amine) were attached to the HPMA copolymer side chains via amide bonds (Supplementary Material: Fig. S2). The valence (i.e., number of oligonucleotide grafts per polymer chain) of the P-MORF2 was 9. The hydrodynamic effective diameter of P-MORF2 was about 20 nm.
Fab'-MORF1 and P-MORF2 self-assembled on the surface of NHL B-cells
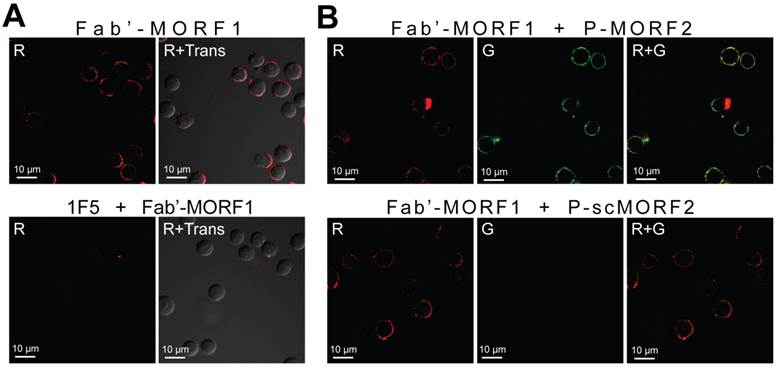
We have previously shown that the MORF1-MORF2 hybridization was fast (< 10 min) and stable (Tm = 59 °C) after conjugation to Fab' and the polymer [16]. When the two conjugates were mixed at equimolar MORF1/MORF2 concentration, a near 100% binding between Fab'-MORF1 and P-MORF2 was observed (Supplementary Material: Fig. S3). This indicated highly efficient in vitro self-assembly of the two conjugates. We further determined the cell surface biorecognition of the two conjugates by confocal microscopy (Fig. 2). In these experiments, Fab'-MORF1 was labeled with rhodamine (red), and P-MORF2 was labeled with FITC (green). Results showed that Fab'-MORF1 successfully decorated the surfaces of Raji cells (a human NHL B-cell line) via CD20 binding (Fig. 2A). Pre-blocking of the CD20 receptors at cell surfaces with an excess amount of 1F5 mAb resulted in no red signal decoration after the Fab'-MORF1 treatment. Furthermore, consecutive exposure of Raji cells to Fab'-MORF1 followed by P-MORF2 led to both the red and green signal decoration at the cell surface (Fig. 2B). This was mediated by the MORF1/MORF2 hybridization, as confirmed by controls using an HPMA copolymer labeled with FITC and grafted with multiple copies of a scrambled sequence of MORF2 (scMORF2). The polymer (P)-scMORF2 control conjugate, even in excess amounts, failed to bind to the surfaces of the MORF1-decorated B-cells, due to absence of the biorecognition pair. These data suggested that Fab'-MORF1 and P-MORF2 self-assemble on the surfaces of lymphoma B-cells with high functional specificity.
Biorecognition of Fab'-MORF1 and P-MORF2 at the surface of Raji B-cells. Fab'-MORF1 was labeled with rhodamine (red) and P-MORF2 with FITC (green). Trans: images under transmitted light, R: red channel, G: green channel. (A) Top panel, Fab'-MORF1 (1 μM) bound to the surfaces of B-cells. Bottom panel, pretreatment with 1F5 mAb (10 μM) followed by Fab'-MORF1 (1 μM). (B) Top panel, consecutive exposure to Fab'-MORF1 (1 μM) and P-MORF2 (1 μM; MORF eqv.). Bottom panel, polymer grafted with scrambled MORF2 (P-scMORF2; 5 μM MORF eqv.) failed to bind to the surfaces of the MORF1-decorated cells; scMORF2: 5'-GTATCCTTATTCCACGTTCATTTGT-3'. Note: P-MORF2 and P-scMORF2 used in this experiment only contained ~5 oligonucleotides per chain.
PK and biodistribution suggested an optimal time lag of 5 h for efficient tumor pretargeting
We planned to employ the two-step pretargeting strategy in the in vivo therapy experiments. Therefore, we analyzed the PK and biodistribution of the pretargeting dose, Fab'-MORF1, in order to determine the optimal time lag. The blood radioactivity versus time profile of the 125I-labeled Fab'-MORF1 conjugate in mice is illustrated in Fig. 3A, and the PK parameters are summarized in Fig. 3B. Results showed that Fab'-MORF1 had a terminal plasma half-life of about 5 h, which was longer than the half-lives of other Fab' fragments of IgG antibodies [26]. This was possibly due to the attachment of MORF1 since the PK of antibody fragments without Fc is molecular weight (MW)-dependent [27]. Importantly, at about 5 h after intravenous injection, Fab'-MORF1 reached a plateau or steady blood concentration, indicating a suitable timing for P-MORF2 administration. At that point, most Fab'-MORF1 were cleared from the blood or distributed to tumors and tissues. Consequently, at this time point there were minimal free conjugates in the blood (unbound to B-cells) that would interfere with the hybridization when P-MORF2 is administered. Based on this result, we hypothesized that, by using a time lag of 5 h, the pretargeting and second-step targeting can be more effective, and the therapeutic efficacy of drug-free macromolecular therapeutics can be improved over our previous experimental conditions that used a 1 h interval [16].
Optimization of tumor pretargeting time lag by pharmacokinetics and biodistribution. (A) The blood radioactivity-time profile of 125I-labeled Fab'-MORF1 in healthy SCID mice (n = 5). The closed circles represent the mean radioactivity, expressed as the percentage of the injected dose per gram of blood (% ID/g). (B) Two-compartment plasma pharmacokinetic (PK) parameters of Fab'-MORF1 in mice. T1/2, α, initial half-life; T1/2, β, terminal half-life; AUC, total area under the blood concentration versus time curve; CL, total body clearance; MRT, mean residence time; Vss, steady-state volume of distribution. (C) Biodistribution of Fab'-MORF1 in healthy SCID mice (no tumor) and SCID mice bearing human B-cell lymphoma xenografts (n = 4 per group). Raji cells were injected from the tail vein of mice 7 days prior to the study. Mice were sacrificed at 1 h or 5 h after i.v. injection of 125I-Fab'-MORF1. (D) Fluorescence molecular tomography (FMT) analysis of lymphoma B-cells dissemination in mice. Four million Raji cells were stained with the DiR dye and injected via the tail vein to SCID mice (n = 4). At 24 h post-tumor inoculation, organs were harvested for ex vivo imaging. L.N., lymph node. (E) Tumor-specific uptake of Fab'-MORF1. Mean radioactivity in each organ of healthy mice (no tumor; n = 4) is defined as the background. Tumor-specific uptake was analyzed by subtracting the background from the radioactivity of corresponding organs of the tumor-bearing mice (n = 4 per group). Negative values are shown as zero. All data are presented as mean ± SD. Statistics was performed with student's t test of unpaired samples (*: p < 0.05).
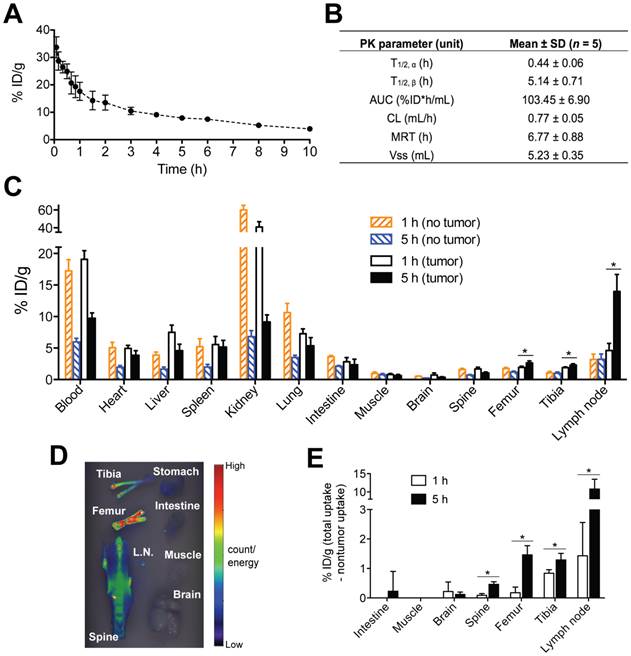
To further support this hypothesis, we compared the biodistribution of Fab'-MORF1 at 1 h and 5 h (Fig. 3C). The studies were performed in healthy SCID mice (no tumor), as well as SCID mice bearing systemically injected Raji B-cells (tumor). We first used fluorescence molecular tomography (FMT) imaging to determine the “hot spots” of Raji lymphoma dissemination (Fig. 3D); tumors were found abundantly in the tibiae, femora, spine, and lymph nodes, which is in agreement with previous reports [28]. Results of biodistribution showed that, 5 h after intravenous injection, Fab'-MORF1 indeed had a better tumor pretargeting efficiency when compared to 1 h post-injection. As shown in Fig. 3C, at 5 h, significantly more conjugates were found in the tibiae, femora, and lymph nodes of the tumor-engrafted mice, when compared to 1 h post-injection. These organs with substantially more uptake of Fab'-MORF1 matched with the Raji lymphoma tumor sites (as determined by FMT; Fig. 3D). Furthermore, in order to define tumor-specific uptake of Fab'-MORF1 (Fig. 3E), the mean radioactivity (125I) in each organ of healthy mice (no tumor; background) was subtracted from the radioactivity of corresponding organs of the tumor-bearing mice. The increase from the background indicated tumor-specific uptake of the conjugates. This analysis suggested significantly more Fab'-MORF1 tumor uptake at 5 h, when compared to 1 h. Differences were statistically significant (5 h vs. 1 h) for all lymphoma hot spots (tibia, femur, spine, and lymph node) (p < 0.05), but not for organs without tumors (e.g., brain, muscle, and intestine). These data confirmed that, in this animal model, an optimal time lag of 5 h was suitable for tumor pretargeting of Fab'-MORF1.
Interestingly, we observed less accumulation of Fab'-MORF1 at 5 h (vs. 1 h) in several major organs, including the liver, kidneys, and lungs (Fig. 3C), suggesting a lower chance of off-target binding after the administration of P-MORF2 (effector). This observation further strengthens the rationale of using 5 h as an optimal time lag. The biodistribution results were concordant with the PK data, which confirmed significant blood clearance of Fab'-MORF1 conjugates from 1 h (~18 % ID/g) to 5 h (~5 % ID/g for healthy mice). In addition, at 5 h, mice bearing tumors had more conjugates in the blood (~10 % ID/g) when compared to mice without tumors (~5 % ID/g). This phenomenon likely resulted from Fab'-MORF1 targeting to the circulating lymphoma B-cells in the blood. The biodistribution study was performed on day 7 post-tumor inoculation of mice. Similar trends were observed at day 1 post-tumor inoculation (Supplementary Material: Fig. S4). However, the tumor pretargeting efficiency was apparently better on day 7 (Fig. 3C) when compared to day 1, presumably due to more pronounced tumor dissemination and infiltration on day 7.
Pretargeted nanotherapeutics showed in vivo anti-lymphoma efficacy that is superior to rituximab
We used an imageable mouse model of systemically disseminated human B-cell lymphoma for preclinical evaluation of drug-free macromolecular therapeutics (Fig. 4). Luciferase-labeled Raji cells (Raji-luc) were engrafted intravenously into adult SCID mice, which have functional macrophages, natural killer cells and complement. Prior to tumor engraftment, we confirmed the CD20 expression of Raji-luc cells (Supplementary Material: Fig. S5). One week post-tumor inoculation, mice were administered three doses of different treatments every other day. Administration time and doses were based on a preliminary experiment (Supplementary Material: Fig. S6) and our previous studies [16, 29]. In this animal model, the hind-limb paralysis-free survival time reflects the therapeutic efficacy [28-30]. Luciferase imaging can be performed in vivo to determine tumor engraftment, dissemination, and growth/inhibition [22]. Here, we employed the two-step pretargeting strategy where Fab'-MORF1 and P-MORF2 were administered (via the tail vein) consecutively, using 1 h or 5 h as an interval. These two nanomedicine groups (Cons 1h, Cons 5h) were compared side by side with the immunotherapy control (1F5 mAb) and a clinically used anti-CD20 mAb, rituximab. The animal survival curve is shown in Fig. 4A. Data showed that mice in the placebo group (PBS) rapidly developed hind-limb paralysis within 4 weeks post-tumor injection; the median survival time was 25 days (0% survival). The designed nanomedicine using the 1 h interval (Cons 1h) substantially prolonged animal survival, resulting in a median survival of 55 days (14% survival; p = 0.003 compared to PBS). The therapeutic efficacy of Cons 1h was comparable to a Fab'-equivalent dose of rituximab, which had a median survival of 72 days (17% survival; p = 0.772 compared to Cons 1h). Importantly, when the 5 h interval (previously optimized time lag) was used, the nanomedicine achieved a significantly improved anticancer efficacy over the 1 h treatment (p = 0.008). This optimized treatment regimen (Cons 5h) produced an 83% animal survival, which was comparable to the immunotherapy control (1F5 mAb; p = 0.317) and was significantly better than rituximab (p = 0.009).
In vivo efficacy of drug-free macromolecular therapeutics against systemic B-cell lymphoma. SCID mice were injected with luciferase-expressing Raji cells (4 × 106) via the tail vein on day 0. Three doses of each treatment were administered on days 7, 9, and 11. PBS: mice injected with PBS (n = 6); Cons 1h: consecutive treatment of Fab'-MORF1 and P-MORF2, 1 h interval (n = 7); Cons 5h: consecutive treatment of Fab'-MORF1 and P-MORF2, 5 h interval (n = 6); Rituximab (n = 6); 1F5 mAb (n = 6). (A) Paralysis-free survival of mice presented in a Kaplan-Meier plot. Numbers of long-term survivors in each group are indicated. Statistics was performed with log-rank test (*: p < 0.05, **: p < 0.005, n.s.: no significant difference). (B) In vivo bioluminescence images at 25 days post-tumor injection. Mice were i.p. injected with 3 mg firefly D-luciferin 15 min prior to imaging. (C) Whole-body bioluminescence intensity of mice. Data are shown as mean ± SEM (n = 6 or 7). Statistics was performed by student's t test (**: p < 0.005). Black arrow: dose administration.
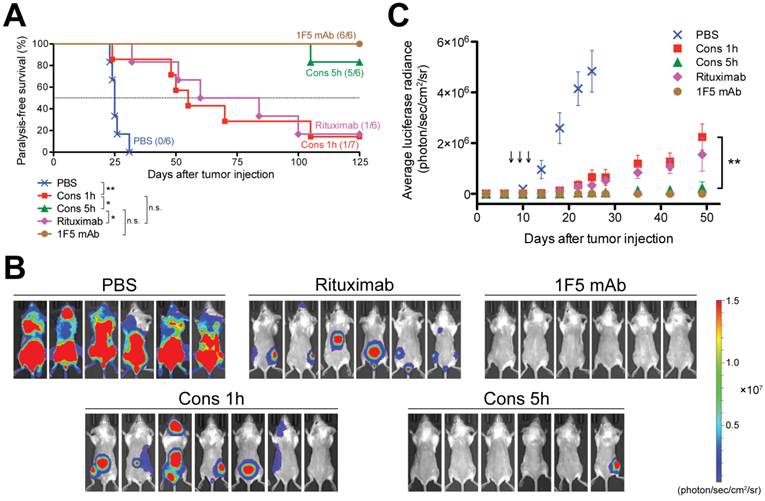
The in vivo tumor imaging studies (Fig. 4B and 4C) were in agreement with the animal survival. Bioluminescent images of mice (on day 25) are shown in Fig. 4B. These images demonstrate B-cell lymphoma dissemination in various organs, including the spine, femora and tibiae. Furthermore, whole-body bioluminescent intensities of mice were quantified to compare groups; the averaged tumor signal versus time profiles are illustrated in Fig. 4C. The placebo control mice (PBS) showed a rapid progression of tumor burden after 10 days. At day 25, the luciferase signals in the placebo group reached saturation. Our drug-free nanotherapeutic using the 1 h interval (Cons 1h) had a moderate effect of tumor inhibition, which was comparable to rituximab. The progression of lymphoma in these two groups (Cons 1h, Rituximab) was significantly delayed when compared to the placebo group. When the 5 h interval was used, the nanomedicine (Cons 5h) showed a substantial improvement over the 1 h treatment, leading to complete elimination of tumor load in 5 (out of 6) animals. At day 49, the average luciferase signal of Cons 5h was similar to the background and was significantly lower than Cons 1h (p = 0.004). Although there is no difference between Cons 5h and 1F5 mAb in tumor inhibition and animal survival, the anti-lymphoma efficacy of the designed nanotherapeutic, unlike mAbs, is independent of immune effector mechanisms, such as antibody-dependent cellular cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC) [31], and is a direct result of only apoptosis.
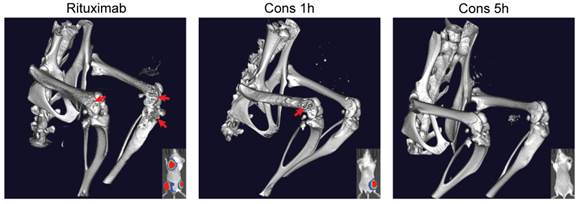
We also performed three-dimensional microcomputed tomography (3D microCT) imaging on the mice that bore Raji-luc lymphoma xenografts and underwent different treatments (Fig. 5). These images were focused on the lumbar spine and hind limbs (hot-spots of B-cell NHL metastasis). Results revealed extensive bone destruction in the rituximab-treated mice and moderate bone lesions in the mice administered drug-free nanotherapeutics using the 1 h interval (Cons 1h). Observed bone heterogeneity was extensive in the femur, and in some cases, the proximal tibia (close to the knee joint), indicating abnormal osteoclast activation (bone remodeling) stimulated by lymphoma metastases [32]. However, no signs of bone remodeling were found in mice treated with Cons 5h. Taken together, superior anti-lymphoma efficacy was achieved using the 5 h interval treatment, when compared to the 1 h treatment and rituximab. These results validate our hypothesis that 5 h is optimal for efficient pretargeting/2nd-step targeting in drug-free macromolecular therapeutics.
Three-dimensional microcomputed tomography (microCT) analysis. Mice were intravenously injected with Raji-luc cells and exposed to different treatments as in Fig. 4. Imaging was performed on day 70 and focused on the lumbar spine and hind limbs. Bone destruction, as a result of lymphoma metastasis, is indicated by the red arrow. Bioluminescence images (day 35) of the same mice are shown in the bottom right corner for comparison.
Optimized nanotherapy completely eradicates lymphoma B-cells in 83% of mice
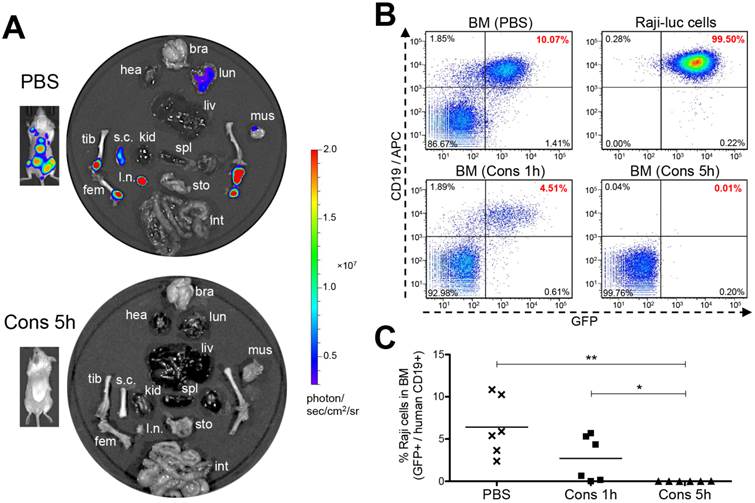
Ex vivo luciferase imaging was performed to examine tumors in multiple organs and tissues (Fig. 6A). Results were concordant with in vivo imaging and showed that Raji-luc lymphoma cells readily infiltrated in the femora, tibiae and lymph nodes of the negative control mice (PBS). Extensive infiltration of the spinal cord was also seen, likely accounting for the occurrence of hind-limb paralysis [28]. In some cases, we observed light to moderate tumor signals in the lung, liver or brain (Supplementary Material: Fig. S7) of control animals. However, most of the surviving mice (5 out 6) that were treated with the nanomedicine using a 5 h interval (Cons 5h) were completely tumor-free. Furthermore, we used flow cytometry to quantitatively analyze residual lymphoma B-cells in the femoral bone marrow (BM) of mice (Fig. 6B and 6C). Raji-luc cells harbor a dual reporter expressing both luciferase and green fluorescence protein (GFP) [22]. We additionally stained the cells with an anti-human CD19 antibody [24] and analyzed the percentage of GFP+ CD19+ cells (Supplementary Material: Fig. S8). Results showed that the negative control mice (PBS) harbored substantial amounts of Raji-luc cells in the bone marrow (average 6.4%). The mice treated with the nanomedicine using a 1 h interval (Cons 1h) demonstrated significant improvement (average 2.7%). In the group that was treated using optimized treatment conditions (Cons 5h), all mice, including five long-term survivors and one animal sacrificed on day 105 due to a large abdominal tumor, had 0% lymphoma B-cells in the bone marrow. Histology further confirmed that the long-term surviving mice were tumor-free (Supplementary Material: Fig. S9). Pathological examination suggested no acute or chronic toxicity caused by drug-free macromolecular therapeutics in any of the tissues evaluated, which corresponded to a stable body weight (Supplementary Material: Fig. S10). These results demonstrate excellent anti-NHL efficacy of the nanotherapeutics with the 2 components (Fab'-MORF1 and P-MORF2) administered at an optimal interval of 5 h. A low dose (58 μg × 3) of the pretargeting agent with a 5× excess of effectors was able to completely eradicate lymphoma B-cells in mice and was not toxic to normal tissues.
Eradication of lymphoma B-cells by drug-free macromolecular therapeutics. Mice i.v. injected with Raji-luc cells were exposed to different treatments as indicated in Fig. 4. (A) Representative ex vivo bioluminescent images of the control mice (PBS) and the mice treated consecutively with Fab'-MORF1 and P-MORF2 using 5 h as an interval (Cons 5h). In vivo images of the same mice are shown alongside. bra: brain, hea: heart, lun: lung, liv: liver, spl: spleen, kid: kidney, s.c.: spinal cord, l.n.: lymph node, tib: tibia, fem: femur, mus: muscle, sto: stomach, int: intestine. (B) Flow cytometry analysis of residual Raji-luc cells in the bone marrow (BM) of mice. BM cells isolated from mouse femur and Raji-luc cells (with GFP expression) from culture flasks were stained with an APC-labeled mouse anti-human CD19 antibody. (C) Comparison of % lymphoma cells in the bone marrow of control mice (PBS) and the nanomedicine-treated mice (Cons 1h and Cons 5h) as analyzed by flow cytometry. Each data point represents an individual mouse (n = 6 per group); mean % is indicated. Statistics was performed with Student's t test of unpaired samples (*: p < 0.05, **: p < 0.005).
Drug-free nanotherapeutic showed higher potency than rituximab in NHL patient specimens
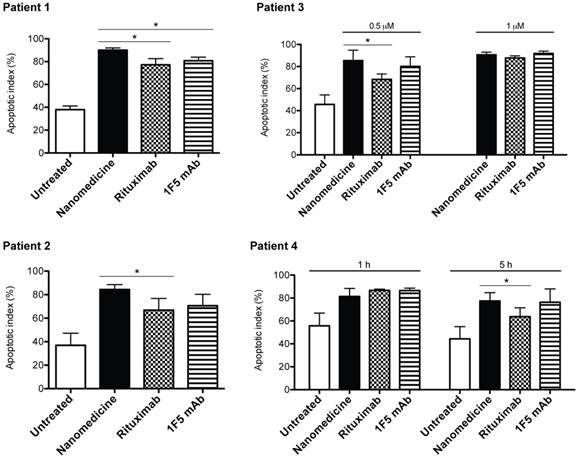
To further evaluate the clinical potential of the designed nanomedicine, primary cells were isolated from 4 mantle cell lymphoma (MCL) patients. MCL is an aggressive and incurable subtype of B-cell NHL. We treated the cells consecutively with Fab'-MORF1 and P-MORF2, as well as rituximab (or 1F5 mAb) hyper-crosslinked with secondary antibodies. The purpose of using secondary antibodies is to reproduce the function of FcR+ immune effector cells, which partly reflects the in vivo therapeutic efficacy of anti-CD20 mAbs [12]. Results showed that the designed nanomedicine effectively induced apoptosis of MCL cells from all 4 patients (Fig. 7). The percent of apoptotic cells was increased by about 2 fold when compared to the untreated cells. When compared to mAbs, the nanomedicine demonstrated superior apoptosis-inducing activity to rituximab in all 4 patient samples, and to 1F5 mAb in 1 sample (patient 1). In patient 3, we observed an increase of apoptotic index with increasing concentration for all compounds tested. The efficacy of 1F5 and rituximab seemed maximized at 1 μM, whereas the nanomedicine reached maximal apoptosis induction at a lower concentration (0.5 μM). In patient 4, we employed two different time intervals (1 h and 5 h) between the treatments. Data showed that, when the time lag was increased from 1 h to 5 h, there was no change in efficacy for the nanomedicine. However, for both mAbs, the apoptosis-inducing activity dropped when the longer time interval was used. This phenomenon can be attributed to Fc-mediated endocytosis of the mAb/CD20 complex, which was observed elsewhere by other investigators [6]. These results highlight the potential of drug-free macromolecular therapeutics as a novel and potent treatment against B-cell lymphomas.
Drug-free nanotherapeutic induces apoptosis of mantle cell lymphoma (MCL) cells from patients. Two samples were from lymph node biopsies (patients 1 and 2) and two from the peripheral blood (patients 3 and 4). Percent apoptotic cells was analyzed by annexin V binding and quantified with flow cytometry. Nanomedicine, Fab'-MORF1 followed by P-MORF2; Rituximab, rituximab followed by goat anti-mouse secondary Ab; 1F5 mAb, 1F5 followed by goat anti-mouse secondary Ab. Time interval between treatments was 1 h (unless otherwise indicated). Incubation time was 24 h. Fab' equivalent concentrations of treatments were 0.5 μM (for patients 1, 2 and 3) and 1 μM (for patients 3 and 4). Data are presented as mean ± SD (n = 2 or 3). Statistics was performed by student's t test (*: p < 0.05). All treatment groups had significantly higher apoptotic indices when compared to the untreated cells.
Discussion
Data presented here validated the proposed concept and showed that the anti-lymphoma efficacy of the designed nanomedicine was significantly better than rituximab, the most frequently used anti-CD20 mAb in the clinic, while comparable to 1F5. In mice, the better efficacy of 1F5 (mouse mAb) when compared to rituximab (human/mouse hybrid mAb) was likely due to stronger binding of the Fc region of 1F5 by (mouse) immune effector cells [33]. It will be interesting to compare our approach with type II anti-CD20 mAbs (e.g., obinutuzumab) that can also induce direct apoptosis [34]. It is worth noting that drug-free macromolecular therapeutics depleted tumors by a single, well-defined mechanism (apoptosis) whereas the efficacy of mAbs is related to apoptosis as well as other effector mechanisms, e.g., ADCC and CDC [31]. We believe that the superior efficacy of our therapeutic results from the multivalent effect, i.e., the P-MORF2 conjugate has multimeric interactions with targets, in contrast to mAbs with only two binding sites. Our laboratory has previously shown that the multivalency of anti-CD20 constructs can magnify binding affinity and apoptosis induction by several folds, when compared to their monovalent or divalent counterparts [35-37]. We have also reported that, in addition to valence, the polymer MW had a positive influence on CD20 clustering and apoptosis [37]. Thus, it is possible to further improve the efficacy by using P-MORF2 with a higher valence and/or a larger polymer backbone, together with a higher dose of the pretargeting agent. See Supplementary Material: Supporting Discussion for further comparison with other multivalent CD20 targeting platforms.
This research offers a unique new strategy in lymphoma treatment by direct apoptosis induction; no small-molecule toxic drug or immune activation is involved. This approach avoids the severe side effects associated with conventional chemo- and radiotherapy. It has potential to treat or sensitize chemoresistant malignancies [38]. Compared to immunotherapies using anti-CD20 mAbs, the designed nanomedicine kills B-cells without the need for effector cells. This allows to target diseases that are not responsive to immunotherapy, which constitute about half of all NHL patients [3]. In the clinic, a large population of rituximab nonresponders harbors polymorphism in the IgG Fc receptor (FcR) gene [4, 5], resulting in failure of effector cells to hyper-crosslink ligated mAbs (via Fc-FcR binding). Many other cases of mAb nonresponsiveness or resistance are due to CD20 downregulation [5-7]. For instance, repeated treatment of mAbs may lead to CD20 internalization, which is mediated by the inhibitory FcR expressed on the target B-cells themselves [6]. In contrast, once the Fc region was removed, Fab' of the same antibody was found to be non-internalizing [9]. More recently, resistance to rituximab has been attributed to “trogocytosis” (shaving of CD20 antigens from the surfaces of B-cells by macrophages via the regulatory FcR) [7]. It is noteworthy that all these mechanisms accounting for the nonresponsiveness/resistance are mediated by Fc-FcR recognition between mAbs and immune effector cells. Since our design does not contain Fc fragments, it may circumvent these mechanisms and benefit patients who do not respond to immunotherapy. Furthermore, our nano-sized therapeutic conjugates likely have better mobility and more rapid tumor penetration when compared to immune effector cells, which is advantageous to target solid tumors that are commonly seen in lymphoma patients.
The designed nanotherapeutic platform offers two types of treatment possibilities: (1) consecutive administration of Fab'-MORF1 and P-MORF2, and (2) premixture of the two conjugates. We have previously tested the in vivo anticancer efficacy of the premixture [16]. Such premixed treatment was able to extend the animal survival with an efficacy similar to that of the consecutive treatment. Although there is no significant difference in therapeutic outcomes, the proposed two-step (consecutive) treatment offers the opportunity of pretargeting. This is an advantage over the premixed treatment in that the timing of administration can be optimized based on PK and biodistribution of the pretargeting dose (Fab'-MORF1), in order to achieve maximal tumor-to-tissue accumulation and enable more efficient treatment. This concept has been successfully proven in the presented study. The optimized pretargeting time lag was 5 h after i.v. administration (in female SCID mice); at this time, most Fab'-MORF1 was cleared from the blood and reached a steady plasma concentration (Fig. 3A) and was efficiently distributed to the tumors (Fig. 3E). This approach, in combination with imaging methods or with predetermined clinical characteristics of individual patients, can potentially be applied for personalized therapy to enhance therapeutic efficacy. Studies have shown that the pharmacokinetic profile of rituximab differs between responders and nonresponders [39]. Therefore, a two-step pretargeted platform, which offers flexibility to individualize the PK of therapeutic effectors, may benefit patients with inadequate response to mAbs treatment by overcoming the pharmacokinetic limitations. For clinical application, the pretargeting parameters can be more precisely determined with the aid of mathematical modeling [40]. Our approach may constitute a novel personalized nanotherapy for lymphomas and potentially other cancers.
Our approach will ultimately require validation and confirmation in properly conducted clinical trials. Nonetheless, the selection of CD20 as a pharmacological target is validated by extensive prior experience. Our therapeutic effector P-MORF2 will likely result in prolonged B-cell depletion compared to mAbs, due to a long blood circulation time of the HPMA polymer backbone [41, 42]. The plasma half-life of P-MORF2 can be further increased using multiblock backbone degradable HPMA copolymers [25, 43, 44]. Before translation to the clinic, one major issue to be addressed is potential immunogenicity of the conjugates. Here, the preclinical evaluation was performed in SCID mice, which are severely immunocompromised. It will be important to conduct immunogenicity studies using immunocompetent animals. The HPMA polymer is non-immunogenic [45, 46]. Its safety has been proven in clinical trials [47]. Moreover, conjugation of immunogens to HPMA copolymers reduces the immunogenicity [45]. Therefore, we anticipate that the P-MORF2 conjugate will have a favorable safety profile. Indeed, in the animal experiment performed here, mice were administered 3 doses of the designed therapeutic containing a total of about 18 mg/kg P-MORF2, and no toxicity has been observed. In the reported system, we used Fab' from a mouse 1F5 mAb [23]; it is likely that when tested in humans, this system will trigger immune responses due to foreign, murine-derived protein fragments (e.g., production of anti-mouse antibodies and the associated allergic or hypersensitivity reactions). Such concerns can be addressed by switching to a humanized anti-CD20 mAb, e.g., ofatumumab, veltuzumab. Alternatively, we shall develop a humanized 1F5 mAb to address this issue.
Besides lymphomas, the therapeutic conjugates developed in this study can also be used for autoimmune diseases, such as rheumatoid arthritis, multiple sclerosis, and systemic lupus erythematosus, as well as chronic lymphocytic leukemia (CLL). All these diseases have been treated by anti-CD20 mAbs [48, 49]. Recently, we evaluated the designed therapeutics in patient CLL cells, including high-risk patients with the 17p13 deletion [50, 51]. Results showed that our treatment effectively induced apoptosis of CLL B-cells and led to subsequent cell death, and was more potent than the 1F5 mAb. In summary, we have developed a novel two-step pretargeted nanotherapy for treatments of B-cell malignancies. This approach is more direct and effective than type I anti-CD20 mAbs and possesses significant advantages over conventional chemo-, radio-, and immunotherapy.
Supplementary Material
Supporting Methods, Supporting Discussion, and Figures S1 - S10.
Acknowledgements
This work was supported in part by NIH grant GM95606 (to J.K.) from the National Institute of General Medical Sciences and the University of Utah Research Foundation. We thank Ken M. Kosak for assisting with patient samples analysis, Dr. Lawrence D. McGill for histopathological examination, Dr. Ruozhen Hu for assisting with flow cytometry analysis, and Dr. Pavla Kopečková for helpful discussion.
Competing Interests
J.K., J.Y. and T.-W.C. are inventors on a pending US patent application (PCT/US2014/023784; assigned to the University of Utah) related to this work. J.K. is Chief Scientific Advisor, J.Y. Scientific Advisor, and P.J.S. Chief Medical Advisor for Bastion Biologics. Otherwise, the authors declare no competing financial interests.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5-29
2. Maloney DG, Grillo-López AJ, White CA, Bodkin D, Schilder RJ, Neidhart JA. et al. IDEC-C2B8 (Rituximab) anti-CD20 monoclonal antibody therapy in patients with relapsed low-grade non-Hodgkin's lymphoma. Blood. 1997;90:2188-95
3. Molina A. A decade of rituximab: Improving survival outcomes in non-Hodgkin's lymphoma. Annu Rev Med. 2008;59:237-50
4. Cartron G, Dacheux L, Salles G, Solala-Celigny P, Bardos P, Colombat P. et al. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcγRIIIa gene. Blood. 2002;99:754-58
5. Smith MR. Rituximab (monoclonal anti-CD20 antibody): mechanisms of action and resistance. Oncogene. 2003;22:7359-68
6. Dransfield I. Inhibitory FcγRIIb and CD20 internalization. Blood. 2014;123:606-7
7. Pham T, Mero P, Booth JW. Dynamics of macrophage trogocytosis of rituximab-coated B cells. PloS One. 2011;6:e14498
8. Stashenko P, Nadler LM, Hardy R, Schlossman SF. Characterization of a human B lymphocyte-specific antigen. J Immunol. 1980;125:1678-85
9. Michel RB, Mattes MJ. Intracellular accumulation of the anti-CD20 antibody 1F5 in B-lymphoma cells. Clin Cancer Res. 2002;8:2701-13
10. Press OW, Farr AG, Borroz KI, Anderson SK, Martin PJ. Endocytosis and degradation of monoclonal antibodies targeting human B-cell malignancies. Cancer Res. 1989;49:4906-12
11. Anderson KC, Bates MP, Slaughenhoupt BL, Pinkus GS, Schlossman SF, Nadler LM. Expression of human B cell-associated antigens on leukemias and lymphomas: a model of human B cell differentiation. Blood. 1984;63:1424-33
12. Shan D, Ledbetter JA, Press OW. Apoptosis of malignant human B cells by ligation of CD20 with monoclonal antibodies. Blood. 1998;91:1644-52
13. Deans JP, Li H, Polyak MJ. CD20-mediated apoptosis: signalling through lipid rafts. Immunology. 2002;107:176-82
14. Hofmeister JK, Cooney D, Coggeshall KM. Clustered CD20 induced apoptosis: src-family kinase, the proximal regulator of tyrosine phosphorylation, calcium influx, and caspase 3-dependent apoptosis. Blood Cells Mol Dis. 2000;26:133-43
15. Ghetie MA, Bright H, Vitetta ES. Homodimers but not monomers of Rituxan (chimeric anti-CD20) induce apoptosis in human B-lymphoma cells and synergize with a chemotherapeutic agent and an immunotoxin. Blood. 2001;97:1392-8
16. Chu T-W, Yang J, Zhang R, Sima M, Kopeček J. Cell surface self-assembly of hybrid nanoconjugates via oligonucleotide hybridization induces apoptosis. ACS Nano. 2014;8:719-30
17. Wu K, Liu J, Johnson RN, Yang J, Kopeček J. Drug-free macromolecular therapeutics: induction of apoptosis by coiled-coil-mediated cross-linking of antigens on the cell surface. Angew Chem Int Ed. 2010;49:1451-5
18. Chu T-W, Kopeček J. Drug-free macromolecular therapeutics - a new paradigm in polymeric nanomedicines. Biomater Sci. 2015 doi: 10.1039/c4bm00442f; [Epub ahead of print]
19. Goodwin DA, Meares CF. Advances in pretargeting technology. Biotechnol Adv. 2001;19:435-50
20. Mulvey JJ, Villa CH, McDevitt MR, Escorcia FE, Casey E, Scheinberg DA. Self-assembly of carbon nanotubes and antibodies on tumours for targeted amplified delivery. Nat Nanotechnol. 2013;8:763-71
21. Gunn J, Park SI, Veiseh O, Press OW, Zhang M. A pretargeted nanoparticle system for tumor cell labeling. Mol Biosyst. 2011;7:742-8
22. Chao MP, Alizadeh AA, Tang C, Myklebust JH, Varghese B, Gill S. et al. Anti-CD47 antibody synergizes with rituximab to promote phagocytosis and eradicate non-Hodgkin lymphoma. Cell. 2010;142:699-713
23. Press OW, Appelbaum F, Ledbetter JA, Martin PJ, Zarling J, Kidd P. et al. Monoclonal antibody 1F5 (anti-CD20) serotherapy of human B cell lymphomas. Blood. 1987;69:584-91
24. Chen WC, Completo GC, Sigal DS, Crocker PR, Saven A, Paulson JC. In vivo targeting of B-cell lymphoma with glycan ligands of CD22. Blood. 2010;115:4778-86
25. Pan H, Yang J, Kopečková P, Kopeček J. Backbone degradable multiblock N-(2-hydroxypropyl)methacrylamide copolymer conjugates via reversible addition-fragmentation chain transfer polymerization and thiol-ene coupling reaction. Biomacromolecules. 2011;12:247-52
26. Covell DG, Barbet J, Holton OD, Black CD, Parker RJ, Weinstein JN. Pharmacokinetics of monoclonal immunoglobulin G1, F(ab')2, and Fab' in mice. Cancer Res. 1986;46:3969-78
27. Bazin-Redureau MI, Renard CB, Scherrmann JM. Pharmacokinetics of heterologous and homologous immunoglobulin G, F(ab')2 and Fab after intravenous administration in the rat. J Pharm Pharmacol. 1997;49:277-81
28. Ghetie MA, Richardson J, Tucker T, Jones D, Uhr JW, Vitetta ES. Disseminated or localized growth of a human B-cell tumor (Daudi) in SCID mice. Int J Cancer. 1990;45:481-5
29. Wu K, Yang J, Liu J, Kopeček J. Coiled-coil based drug-free macromolecular therapeutics: in vivo efficacy. J Control Release. 2012;157:126-31
30. Griffiths GL, Mattes MJ, Stein R, Govindan SV, Horak ID, Hansen HJ. et al. Cure of SCID mice bearing human B-lymphoma xenografts by an anti-CD74 antibody-anthracycline drug conjugate. Clin Cancer Res. 2003;9:6567-71
31. Okroj M, Österborg A, Blom AM. Effector mechanisms of anti-CD20 monoclonal antibodies in B cell malignancies. Cancer Treat Rev. 2013;39:632-9
32. Roodman GD. Biology of osteoclast activation in cancer. J Clin Oncol. 2001;19:3562-71
33. Cragg MS, Glennie MJ. Antibody specificity controls in vivo effector mechanisms of anti-CD20 reagents. Blood. 2004;103:2738-43
34. Herter S, Herting F, Mundigl O, Waldhauer I, Weinzierl T, Fauti T. et al. Preclinical activity of the type II CD20 antibody GA101 (obinutuzumab) compared with rituximab and ofatumumab in vitro and in xenograft models. Mol Cancer Ther. 2013;12:2031-42
35. Johnson RN, Kopečková P, Kopeček J. Synthesis and evaluation of multivalent branched HPMA copolymer-Fab' conjugates targeted to the B-cell antigen CD20. Bioconjug Chem. 2009;20:129-37
36. Johnson RN, Kopečková P, Kopeček J. Biological activity of anti-CD20 multivalent HPMA copolymer-Fab' conjugates. Biomacromolecules. 2012;13:727-35
37. Chu T-W, Yang J, Kopeček J. Anti-CD20 multivalent HPMA copolymer-Fab' conjugates for the direct induction of apoptosis. Biomaterials. 2012;33:7174-81
38. van der Kolk LE, Evers LM, Omene C, Lens SMA, Lederman S, van Lier RAW. et al. CD20-induced B cell death can bypass mitochondria and caspase activation. Leukemia. 2002;16:1735-44
39. Cartron G, Watier H, Golay J, Solal-Celigny P. From the bench to the bedside: ways to improve rituximab efficacy. Blood. 2004;104:2635-42
40. Liu G, Hnatowich DJ. A semiempirical model of tumor pretargeting. Bioconjug Chem. 2008;19:2095-104
41. Ulbrich K, Šubr V. Structural and chemical aspects of HPMA copolymers as drug carriers. Adv Drug Deliv Rev. 2010;62:150-66
42. Zalevsky J, Chamberlain AK, Horton HM, Karki S, Leung IW, Sproule TJ. et al. Enhanced antibody half-life improves in vivo activity. Nat Biotechnol. 2010;28:157-9
43. Yang J, Luo K, Pan H, Kopečková P, Kopeček J. Synthesis of biodegradable multiblock copolymers by click coupling of RAFT-generated heterotelechelic polyHPMA conjugates. React Funct Polym. 2011;71:294-302
44. Zhang R, Yang J, Sima M, Zhou Y, Kopeček J. Sequential combination therapy of ovarian cancer with degradable N-(2-hydroxypropyl)methacrylamide copolymer paclitaxel and gemcitabine conjugates. Proc Natl Acad Sci USA. 2014;111:12181-6
45. Říhová B, Kovář M. Immunogenicity and immunomodulatory properties of HPMA-based polymers. Adv Drug Deliv Rev. 2010;62:184-91
46. Kverka M, Hartley JM, Chu T-W, Yang J, Heidchen R, Kopeček J. Immunogenicity of coiled-coil based drug-free macromolecular therapeutics. Biomaterials. 2014;35:5886-96
47. Vasey PA, Kaye SB, Morrison R, Twelves C, Wilson P, Duncan R. et al. Phase I clinical and pharmacokinetic study of PK1 [N-(2-hydroxypropyl)methacrylamide copolymer doxorubicin]: first member of a new class of chemotherapeutic agents - drug-polymer conjugates. Clin Cancer Res. 1999;5:83-94
48. Dierickx D, Delannoy A, Saja K, Verhoef G, Provan D. Anti-CD20 monoclonal antibodies and their use in adult autoimmune hematological disorders. Am J Hematol. 2011;86:278-91
49. Jain P, O'Brien S. Anti-CD20 monoclonal antibodies in chronic lymphocytic leukemia. Expert Opin Biol Ther. 2013;13:169-82
50. Chu T-W, Kosak KM, Shami PJ, Kopeček J. Drug-free macromolecular therapeutics induce apoptosis of patient chronic lymphocytic leukemia cells. Drug Deliv Transl Res. 2014;4:389-94
51. Chu T-W, Kopeček J. Drug-free macromolecular therapeutics - a new paradigm in polymeric nanomedicines. Biomater Sci. 2015 doi: 10.1039/c4bm00442f
Author contact
![]() Corresponding author: J. Kopeček, University of Utah, Center for Controlled Chemical Delivery, 2030 East 20 South, Biopolymers Research Building, Room 205B, Salt Lake City, Utah 84112-9452, USA. Phone: +1 (801) 581-7211; Fax: +1 (801) 581-7848; E-mail: jindrich.kopecekedu
Corresponding author: J. Kopeček, University of Utah, Center for Controlled Chemical Delivery, 2030 East 20 South, Biopolymers Research Building, Room 205B, Salt Lake City, Utah 84112-9452, USA. Phone: +1 (801) 581-7211; Fax: +1 (801) 581-7848; E-mail: jindrich.kopecekedu