Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results and Discussion
Conclusions
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2015; 5(7):746-754. doi:10.7150/thno.10732 This issue Cite
Research Paper
Effect of PEG Pairing on the Efficiency of Cancer-Targeting Liposomes
Phei Er Saw1,2, Jinho Park1, Eunbeol Lee1, Sukyung Ahn1, Jinju Lee3, Hyungjun Kim1, Jinjoo Kim1, Minsuk Choi1, Omid C. Farokhzad2, Sangyong Jon1, ![]()
1. KAIST Institute for the BioCentury, Department of Biological Sciences, Korea Advanced Institute of Science and technology (KAIST), 291 Daehak-ro, Daejeon 305-701, Republic of Korea.
2. Laboratory of Nanomedicine and Biomaterials, Department of Anesthesiology, Brigham and Women's Hospital Harvard Medical School, Boston, MA 02115, U.S.A.
3. School of Life Sciences, Gwangju Institute of Science and Technology, 123-Cheomdangwagi-ro, Gwangju 500-712, Republic of Korea.
Received 2014-10-5; Accepted 2015-3-13; Published 2015-4-5
Abstract

Standardized poly(ethylene glycol)-modified (PEGylated) liposomes, which have been widely used in research as well as in pre-clinical and clinical studies, are typically constructed using PEG with a molecular weight of 2000 Da (PEG2000). Targeting ligands are also generally conjugated using various functionalized PEG2000. However, although standardized protocols have routinely used PEG2000, it is not because this molecular weight PEG has been optimized to enhance tumor uptake of nanoparticles. Herein, we investigated the effect of various PEG lipid pairings—that is, PEG lipids for targeting-ligand conjugation and PEG lipids for achieving 'stealth' function—on in vitro cancer cell- and in vivo tumor-targeting efficacy. A class of high-affinity peptides (aptides) specific to extra domain B of fibronectin (APTEDB) was used as a representative model for a cancer-targeting ligand. We synthesized a set of aptide-conjugated PEGylated phospholipids (APTEDB‑PEG2000‑DSPE and APTEDB‑PEG1000‑DSPE) and then paired them with methoxy-capped PEGylated phospholipids with diverse molecular weights (PEG2000, PEG1000, PEG550, and PEG350) to construct various aptide-conjugated PEGylated liposomes. The liposomes with APTEDB‑PEG2000/PEG1000 and APTEDB‑PEG1000/PEG550 pairings had the highest uptake in EDB-positive cancer cells. Furthermore, in a U87MG xenograft model, APTEDB‑PEG2000/PEG1000 liposomes retarded tumor growth to the greatest extent, followed closely by APTEDB‑PEG1000/PEG550 liposomes. Among the PEGylated liposomes tested, pairs in which the methoxy-capped PEG length was about half that of the targeting ligand-displaying PEG exhibited the best performance, suggesting that PEG pairing is a key consideration in the design of drug-delivery vehicles.
Keywords: aptides · liposomes · cancer therapy · poly(ethylene glycol) · drug delivery · extra domain B of fibronectin
Introduction
Liposomes are an attractive candidate as pharmaceutical nanocarriers that have already been used clinically.[1, 2] Liposomes are a good option for efficient drug-delivery because of their desirable attributes, including reasonable stability, extended circulation in the bloodstream, increased tumor accumulation, and reduced systemic toxicity associated with their use.[3-7] For in vivo applications, PEGylation of the liposomal surface is necessary to achieve a 'stealth' function.[8, 9] In most cases, PEG2000 (5 mol%) is standard in the preparation of PEGylated liposomes.[2, 4, 7, 9-14] In addition, the functionalization of PEG2000, for example with maleimide or amine groups, is used to introduce targeting ligands on the outermost surface of the PEGylated liposomes.[5, 15, 16] PEG2000 thus seems to be the 'gold standard' for liposomal formulation. However, its use as a stealth coating is more of a traditional standard rather than a choice based on functional optimization. Therefore, there is a need for further optimization of liposomal drug-delivery vehicles to achieve better outcomes in vivo.[17-19]
Recently, Stefanick and colleagues reported that the length of PEG used in conjugating targeting ligands and background stealth coating affects the cellular uptake efficiency of liposomes in vitro.[20] In their experimental setting, the liposome uptake efficiency was significantly higher when a targeting ligand displayed by PEG550 was paired with a background coating of PEG350 (peptide-PEG550/PEG350 liposome) compared with other combinations of PEG, including peptide-PEG2000/PEG2000 liposomes and peptide-PEG350/PEG2000 liposomes.[20] These results raise interesting questions regarding the importance of specific combinations of the targeting ligand-displaying PEG lipids and the methoxy-capped stealth PEG lipid. However, these original results were obtained in an in vitro cell-based system; thus, it is not clear whether these optimized PEG lipid parameters are feasible for in vivo cancer models.
Fibronectin extra domain B (EDB), which is specifically expressed in tumor-associated blood vessels and extra cellular matrix—and even on cancer cells—is recognized as a novel cancer-associated biomarker.[21, 22] Very recently, we reported an EDB-targeting liposome for anti-glioblastoma therapy in which an aptide specific to EDB (ATPEDB) that binds with high-affinity was conjugated to PEGylated liposomes containing anti-cancer drugs. Here, we report the results of the systematic optimization of PEG lipid pairs for use as a targeting ligand-conjugated PEG lipid and a methoxy-capped PEG lipid. To construct a series of EDB-targeting PEGylated liposomes, we prepared a set of aptide-conjugated PEGylated phospholipids with two different PEG lipid lengths (APTEDB‑PEG2000-DSPE and APTEDB-PEG1000-DSPE) and paired them with methoxy-capped PEG lipids of various lengths (PEG350, PEG550, PEG1000, and PEG2000). The cellular uptake of liposomes with the different PEG lipid pairings was then assessed in EDB-overexpressing cancer cells. Moreover, to determine the optimal combination of targeting ligand-displaying PEG lipid and methoxy-capped PEG lipid in vivo, we assessed and compared the cancer-targeting ability and therapeutic efficacy in EDB-positive tumor-bearing mice.
Methods
Materials
1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1'-rac-glycerol) (POPG), polyethylene glycol(2000)-DSPE (ammonium salt) (PEG2000-DSPE), polyethylene glycol(1000)-DSPE (ammonium salt) (PEG1000-DSPE), polyethylene glycol(750)-DSPE (ammonium salt) (PEG750-DSPE), polyethylene glycol(550)-DSPE (ammonium salt) (PEG550-DSPE), polyethylene glycol(350)-DSPE (ammonium salt) (PEG350-DSPE), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (ammonium salt) (Rh-DSPE), plant cholesterol (Chol), and a mini handheld extruder set were purchased from Avanti Polar Lipids (Alabaster, AL, USA). N-maleimide-PEG2000-DSPE (ammonium salt) (Mal-PEG2000-DSPE) and N-maleimide-PEG1000-DSPE (Mal-PEG1000-DSPE) were purchased from NanoCS (NY, USA). An aptide specific for the EDB domain of fibronectin (APTEDB) with an additional cysteine in the β-hairpin constant loop region, N'-CSSPIQGSWTWENGK(C)WTWGIIRLEQ-C', was custom-synthesized by Anygen Corp (Jangsung, JeonNam, South Korea). The hydrophilic anti-cancer drug, doxorubicin hydrochloride (Dox), was purchased from Boryung Pharmaceuticals Co. Ltd (Seoul, South Korea). A Sepharose CL-4B column was purchased from Sigma-Aldrich (St. Louis, MO, USA). Mounting solution was purchased from Dako (Glostrup, Denmark). An EZ-Cytox Cell Viability Assay Kit was purchased from Daeil Lab (Seoul, South Korea). All other chemicals were of reagent grade and were used as received. All animal experiments were performed according to the rules and regulations of animal care and handling established by the Animal Care Committee of Korea Advanced Institute of Science and Technology (KAIST).
Preparation of liposomes with various PEG length pairings
An anionic lipid film of POPC:Chol:POPG (4:3:3 molar ratio) was prepared as previously described [23]. All lipids were purchased from Avanti Polar Lipids unless stated otherwise and received in chloroform solution. Briefly, all components stated above were mixed in a glass vial accordingly. During the preparation of the lipid film, APTEDB-PEG2000-DSPE or APTEDB-PEG1000-DSPE was added to the original liposome formulation at a concentration of 2.5 wt%, respectively. The conjugation scheme of APTEDB-PEG1000/2000-DSPE is described in detail in the Supplementary Material (Fig. S1a). The methoxy-capped PEG was added to the respective liposome formulation at a concentration of 2.5 wt%. After all the components were added, the lipid film was allowed to dry in a vacuum and further lyophilized overnight to remove all excess chloroform. For cellular uptake experiments, 0.5% rhodamine-labeled DSPE was added to each formulation. This yielded seven different liposomes: APTEDB-PEG2000 LS containing PEG350, PEG550, PEG1000 or PEG2000; and APTEDB-PEG1000 LS containing PEG350, PEG550 or PEG1000. During rehydration, 1 mL of HEPES-buffered 5% glucose (HBG) was added to the lipid film to yield a liposome solution with a final concentration of 2 mg/mL. After brief sonication, the solution was extruded through a 100-nm polycarbonate membrane with a mini handheld extruder. At least 10 extrusions were done for each sample to ensure the formation of uniform-sized liposomes.
Cellular uptake of liposomes
The cellular uptake efficiency of liposomes was evaluated in U87MG human glioblastoma, PC3 human prostate cancer and SCC-7 murine oral squamous cell carcinoma cell lines. U87MG cells were cultured in EMEM medium, while PC3 and SCC-7 cells were cultured in RPMI medium. All media were supplied with 10% FBS and a 1% penicillin and streptomycin antibiotic solution. Cellular uptake of each liposome was determined by treating EDB-positive U87MG and SCC-7 cells and EDB-negative PC3 cells with each liposome formulation. Prior to this uptake experiment, EDB positive and negative cells were identified by real-time qRT-PCR and immunocytochemistry. Approximately, 50,000 cells were grown to confluency on a sterilized coverslip, which was placed in a 12-well plate. After 24 h, the cells were incubated for 1 h at 37 °C with each liposome group in serum containing medium at a concentration of 100 µg/mL. Cells were then washed with phosphate-buffered saline (PBS), fixed with 4% paraformaldehyde (PFA), counter-stained with the nuclear dye 4',6-diamidino-2-phenylindole (DAPI), mounted on glass slides, and examined by confocal microscopy.
In vitro cytotoxicity of liposomes in U87MG and SCC-7 cells
For Dox loading, doxorubicin hydrochloride, dissolved in HEPES Buffered Glucose (HBG) buffer, was added to liposomes at a final concentration of 50 μg/mL during the rehydration process. A standard curve for Dox was first plotted as described in the supporting information (Supplementary Material: Fig. S2). Dox-loaded liposomes were then purified by size-exclusion chromatography (CL-4B column). All liposome fractions were eluted out at fractions 3-5 while the smaller free Dox was eluted out at fractions 10-12.[24] After column purification, the encapsulation efficacy was determined with a spectrofluorometer. Cells were then treated with each liposomal formulation at various concentrations: 0.1-2 µM for U87MG cells and 0.5-5 µM for SCC-7 cells. After 72 h of co-incubating the cells with liposomes in serum containing media, cells were washed with PBS and incubated with EZ-Cytox assay reagent. The uptake of EZ-Cytox reagent was determined by spectrophotometry reading with an absorbance wavelength of 470 nm. The IC50 value for each formulation was determined by ProBit analysis using 10 replicates (n = 10) per condition.
In vivo uptake of liposomes in U87MG xenograft and SCC-7 allograft models
U87MG or SCC-7 cells were injected subcutaneously into Balb/c female nude mice at 5 × 106 cells per mouse. After 3 weeks, tumor growth was recorded and determined to be around 80-120 mm3. At this time, 200 µg of each rhodamine-labeled liposome were injected via the tail vein into a mouse. After 6 h, the mice were sacrificed, and the tumors were excised. The uptake of liposomes into tumors was visualized with the IVIS® imaging system.
In vivo anti-tumor efficacy of liposomes in a U87MG xenograft model
U87MG cells were injected into Balb/c female nude mice at 5 × 106 cells per mouse, and tumors were allowed to grow until reaching a volume of ~80-120 mm3. Mice were then randomly divided into seven groups (n = 5/group): (1) Control (saline), (2) free Dox (2 mg/kg), (3) non-targeting LS (Dox, 2 mg/kg), (4) APTEDB-PEG2000/PEG2000 LS (Dox, 2 mg/kg), (5) APTEDB-PEG2000/PEG1000 LS (Dox, 2 mg/kg), (6) APTEDB-PEG1000/PEG1000 LS (Dox, 2 mg/kg), and (7) APTEDB-PEG1000/PEG550 LS (Dox, 2 mg/kg). Mice from each group received samples every three days with a total of four intravenous injections. After the treatment was completed, monitoring of the mice continued every three days until the tumors in the control group had reached about 2,000 mm3. The mice were then euthanized, and the tumors were excised. The tumor size (1/2 x length x width x height) and weight were recorded.
Results and Discussion
Synthesis of seven pairs of EDB-targeting PEGylated liposomes
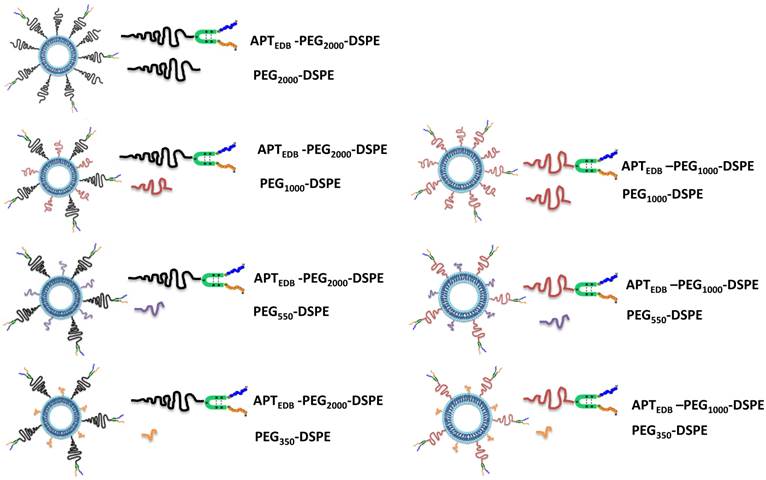
To examine the effect of PEG pairing on the cellular and tumoral uptake of liposomes in vitro and in vivo, respectively, we prepared a series of aptide-displaying liposomes paired with methoxy-capped PEG lipids of varying lengths. One consideration when pairing targeting ligand-displaying PEG lipid and methoxy-capped PEG lipid is that the former must be at least as long as the latter enabling its exposure for target binding. An EDB-specific aptide with an additional cysteine (Cys-APTEDB) was reacted with a maleimide-functionalized PEGylated phospholipid (either PEG2000-DSPE or PEG1000-DSPE) to yield APTEDB-PEG2000-DSPE or APTEDB-PEG1000-DSPE (Fig. S1a). Next, to construct a series of EDB-targeting PEGylated liposomes, APTEDB-PEG2000-DSPE was then paired with the methoxy-capped PEGylated phospholipids PEG2000-DSPE, PEG1000-DSPE, PEG550-DSPE and PEG350-DSPE (Fig. S1b) yielding the following four sets of liposomes (LS): APTEDB‑PEG2000/PEG2000 LS, APTEDB-PEG2000/PEG1000 LS, APTEDB-PEG2000/PEG550 LS and APTEDB-PEG2000/PEG350 LS. Three sets of liposomes were similarly prepared from APTEDB-PEG1000-DSPE, yielding APTEDB-PEG1000/PEG1000 LS, APTEDB-PEG1000/PEG550 LS and APTEDB-PEG1000/PEG350 LS. Because of the length criteria noted above, we did not prepare an APTEDB-PEG1000/PEG2000 LS in which the targeting ligand might be shielded by a longer methoxy-capped PEG lipid. Each aptide-lipid conjugate was first purified by HPLC and then subjected to MALDI-TOF (matrix-assisted laser desorption ionization time-of-flight) analysis to verify successful conjugation before incorporating into liposomes (Supplementary Material: Fig. S3). Fig. 1 shows a schematic representation of the seven pairs of liposomes with different combinations of aptide-displaying PEG lipids and methoxy-capped PEG lipids. The size and morphology of the aptide-conjugated PEGylated liposomes were characterized by dynamic light scattering (DLS) and transmission electron microscopy (TEM) (Supplementary Material: Fig. S4). Anionic liposomes were used in this experiment based on previous results that showed anionic liposomes have lower non-specific binding in cells.[24] In our previous paper, we observed that by conjugating an EDB specific aptide onto liposomes, the in vitro and in vivo tumor uptake significantly increased while that for non-targeting anionic liposomes only showed minimal uptake.[24, 25] Likewise, all the liposomes showed a layered membrane structure in the TEM images and had hydrodynamic sizes ranging from 100 to 130 nm in diameter, with slight differences in the size and morphology among the groups. (Supplementary Material: Fig. S4)
Schematic representation of all combinations of the APTEDB-PEG2000-DSPE (targeting ligand conjugated PEG) group paired with PEG2000, PEG1000, PEG550 and PEG350 (methoxy-capped PEG lipids) and the APTEDB‑PEG1000-DSPE group paired with PEG1000, PEG550 and PEG350 (methoxy-capped PEG lipids). Left column represents a set of liposomes prepared using the targeting ligand conjugated PEG2000-phospholipid as a key component: APTEDB-PEG2000/PEG2000 LS, APTEDB-PEG2000/PEG1000 LS, APTEDB-PEG2000/PEG550 LS, APTEDB-PEG2000/PEG350 LS. Right column represents a set of liposomes prepared using the targeting ligand conjugated PEG2000-phospholipid as a key component: APTEDB-PEG1000/PEG1000 LS, APTEDB-PEG1000/PEG550 LS, APTEDB-PEG1000/PEG350 LS.
Comparison of cellular uptake between the seven pairs of EDB-targeting PEGylated liposomes
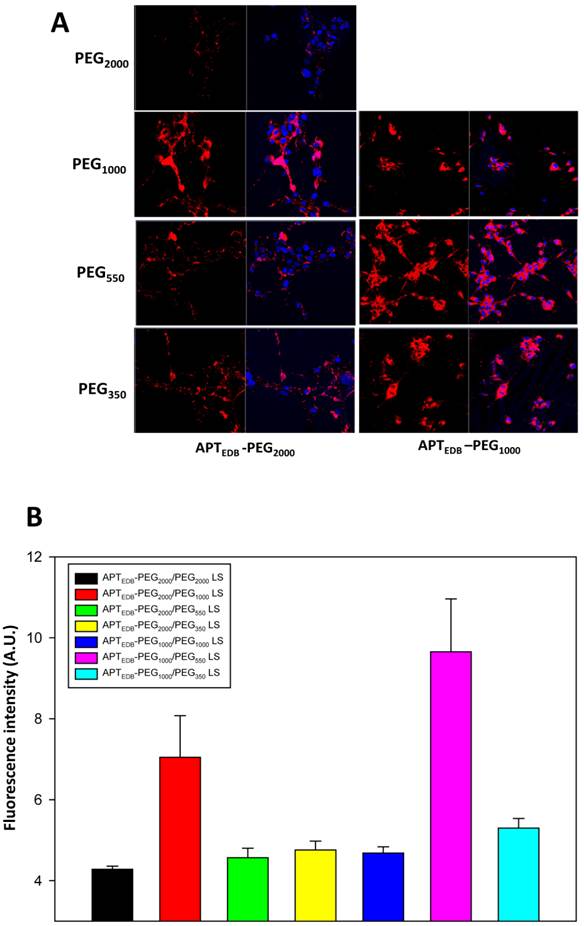
Cellular uptake for all the prepared liposomal formulations was assessed in U87MG cells, a human glioblastoma cell line known to overexpress EDB.[24] The expression levels of EDB in these cell lines studied were confirmed by conventional reverse transcription-polymerase chain reaction (RT-PCR), quantitative RT-PCR (qRT-PCR), and immunocytohistochemistry (Fig. S5). To qualitatively evaluate the uptake efficiency, we fluorescently labeled all the liposomes by incorporating 0.5% rhodamine-modified phospholipid during the liposome preparation. As shown in Fig. 2a, the uptake of APTEDB-PEG2000/PEG1000 LS was higher than that of APTEDB-PEG2000/PEG2000 LS as well as APTEDB-PEG2000/PEG550 LS and APTEDB-PEG2000/PEG350 LS. As for the right column, the APTEDB-PEG1000/PEG550 LS had the highest uptake compared with both APTEDB-PEG1000/PEG1000 LS and APTEDB-PEG1000/PEG350 LS. Quantification of these images with Image J further confirmed that the increase in uptake of APTEDB-PEG2000/PEG1000 LS and APTEDB-PEG1000/PEG550 LS were statistically significant compared to the other groups (Fig. 2b). These results indicate that liposomes containing a methoxy-capped PEG lipid shorter than the targeting ligand-displaying PEG lipid have a higher cellular uptake than PEG lipid pairs of equal length. Interestingly, both APTEDB-PEG2000/PEG1000 LS and APTEDB-PEG1000/PEG550 LS in which the length of the methoxy-capped PEG lipids was nearly half that of the ligand-displaying PEG lipid had a much higher cellular uptake than that of the other combinations.
In vitro cellular uptake of all liposomal systems in U87MG cells. (a) Uptake of each liposomal formulation in U87MG cells in vitro as determined by confocal microscopy. U87MG cells were co-incubated with a respective liposomal system for 1 h in serum containing medium, washed and fixed prior to confocal imaging. APTEDB-PEG2000/PEG1000 LS showed the highest cellular uptake among formulations in the APTEDB-PEG2000-DSPE set (left column), and APTEDB-PEG1000/PEG550 LS showed the highest cellular uptake among formulations in the APTEDB-PEG1000-DSPE set (right column). (b) Image J quantitative analysis on uptake efficiency of each liposomal system. Five confocal images were taken from each liposomal formulation and the mean value of rhodamine intensity was calculated for the respective liposome system. Statistical significance was calculated using Student's T test; with standard deviation (S.D.) being used in calculation.
Based on the in vitro cellular uptake results, we narrowed our subsequent investigations, including the in vivo animal experiments, to four pairs of formulations: APTEDB-PEG2000/PEG2000 LS, APTEDB-PEG2000/PEG1000 LS, APTEDB-PEG1000/PEG1000 LS, and APTEDB-PEG1000/PEG550 LS. To determine whether the pattern of cellular uptake shown in Fig. 2 is a general phenomenon, the cellular uptake efficiency was also assessed for these four liposomal systems in SCC-7 and PC-3 cells, which are EDB-positive and EDB-negative (control) cell lines, respectively. As expected, the cellular uptake of each liposomal system in the SCC-7 cells followed a trend similar to that found in the U87MG cells. In contrast, minimal cellular uptake was observed for all four liposomal systems in the EDB-negative PC3 cells, suggesting that cellular uptake of the aptide-conjugated PEGylated liposomes was mediated by specific binding of the aptide to EDB in both U87MG and SCC-7 cells (Supplementary Material: Fig. S6). We also observed minimal cellular uptake with the scrambled aptide (ATPscr)-conjugated liposomes and non-targeting liposomes in the cells tested (SCC-7, U87MG and PC-3) (Fig S7).
Next, we examined the cellular uptake pathway for each liposomal system using three intracellular trackers: ER, mitochondria, and lysosome. Interestingly, all four liposomal systems showed similar co-localization patterns in the three intracellular compartments of both U87MG and SCC-7 cells (Supplementary Material: Fig. S8), suggesting that there is little difference in the cellular uptake pathway of the liposomes. Therefore, we speculated that the enhanced cellular uptake with both APTEDB-PEG2000/PEG1000 LS and APTEDB-PEG1000/PEG550 LS is not caused by a difference in the cellular uptake mechanism but presumably due to the enhanced interactions between the targeting ligand and its target in both liposomal systems.
Effect of PEG lipid pairing on the anticancer activity of EDB-targeting, PEGylated liposomes encapsulating Dox
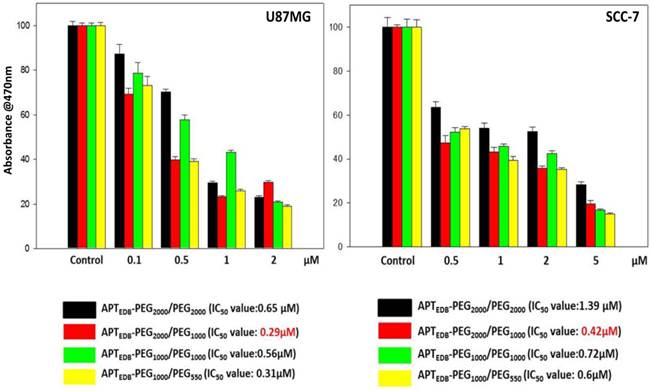
To investigate the functional implications of the PEG-pairing effect, we prepared four EDB-targeting, PEGylated liposomes encapsulating Dox, APTEDB-PEG2000/PEG2000 LS (Dox), APTEDB-PEG2000/PEG1000 LS (Dox), APTEDB-PEG1000/PEG1000 LS (Dox) and APTEDB-PEG1000/PEG550 LS (Dox), and compared their relative cytotoxicity in cancer cells. About ~7wt% of Dox was loaded into the liposomes and quantified by spectrofluorometry analysis. All liposomal formulations had similar loading efficiencies with minimal variation among the groups (Supplementary Material: Fig. S2 and Table S1). The 50% inhibitory concentration (IC50) value of each liposomal system was obtained in both U87MG and SCC-7 cells after treatment for 24 h in serum containing media. As shown in Fig. 3, in both cells tested, the lowest IC50 value was observed for APTEDB-PEG2000/PEG1000 LS (Dox), followed by APTEDB-PEG1000/PEG550 LS (Dox), whereas both APTEDB-PEG2000/PEG2000 LS (Dox) and APTEDB-PEG1000/PEG1000 LS (Dox) had significantly higher IC50 values. These anticancer activity results are in good agreement with the cellular uptake results observed for the same liposomal systems without encapsulated drugs. Taken together, these cellular uptake and cytotoxicity results clearly indicate that the pairing a targeting ligand-displaying PEG lipid with a half-length methoxy-capped PEG lipid yields a better performing liposome in terms of the efficiency for target-specific drug delivery.
Cell viability of U87MG (left) and SCC-7 (right) after treatment with Dox-encapsulated APTEDB-PEG2000/PEG2000 LS, APTEDB-PEG2000/PEG1000 LS, APTEDB-PEG1000/PEG1000 LS and APTEDB-PEG1000/PEG550 LS. After treatment of each respective liposomal system in serum containing media for 72 h, cell viability was assessed by EZ Cytox Kit (Cell Viability, Proliferation & Cytotoxicity Assay Kit). Absorbance at 470 nm is directly proportional to the amount of live cells. In both cell lines tested, the lowest IC50 values were identified from the APTEDB-PEG2000/PEG1000 LS (Dox) group: 0.29 μM for U87MG and 0.42 μM for SCC-7, respectively.
Effect of PEG lipid pairing on cancer targeting and therapy in vivo
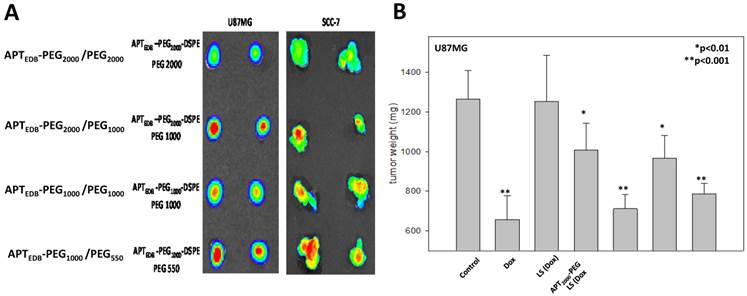
To examine whether the PEG lipid pairing effects observed in vitro were viable in an in vivo context, we first performed optical imaging experiments in a tumor xenograft and allograft model, created by using EDB-overexpressing U87MG and SCC-7 cells, respectively. Each liposomal system was fluorescently labeled with 0.5% rhodamine-DSPE during liposome preparation and then was intravenously injected into the tail vein of the tumor-bearing mice. After 6 h, tumor tissues were excised, and their fluorescence images were recorded. As shown in Fig. 4a, the fluorescence intensity was significantly higher for APTEDB-PEG2000/PEG1000 LS and APTEDB-PEG1000/PEG550 LS than for APTEDB-PEG2000/PEG2000 LS and APTEDB-PEG1000/PEG1000 LS in both U87MG and SCC-7 tumors, confirming that the PEG-pairing effect observed in vitro is viable for in vivo systems. The fact that this phenomenon was seen in both EDB-positive cancer models suggests that the PEG-pairing effect can be generalized to different targeted versions of the liposomal system.
In vivo tumor uptake of four liposomes in U87MG xenograft and SCC-7 allograft models. (a) Rhodamine-labeled liposomes were intravenously injected into two U87MG xenograft and SCC-7 allograft tumor-bearing mice via tail veins. After 24 h, mice were euthanized and tumors were excised and viewed with IVIS imaging system. The highest uptake was seen in both APTEDB-PEG2000/PEG1000 LS and APTEDB-PEG1000/PEG550 LS treated groups. (n=2 for each group for each cell line, s.e.) (b) In vivo anti-tumor efficacy was assessed in U87MG xenograft model. After treatment of Dox-encapsulated APTEDB-PEG2000/PEG2000 LS, APTEDB-PEG2000/PEG1000 LS, APTEDB-PEG1000/PEG1000 LS and APTEDB-PEG1000/PEG550 LS at a dose of 2 mg Dox/kg, tumor was excised; and the tumor size and weight were measured. The greatest suppression of tumor growth was achieved in APTEDB-PEG2000/PEG1000 LS (Dox) (~90%), followed by APTEDB-PEG1000/PEG550 LS (Dox) (~80%), while non-targeting LS and free Dox showed modest anti-tumor efficacy of ~28% and ~65%, respectively. Statistical analysis was carried out with Students' T Test, with *p value < 0.01 for APTEDB-PEG2000/PEG2000 LS and APTEDB-PEG1000/PEG1000 LS; **p value < 0.001 for APTEDB-PEG2000/PEG1000 LS, APTEDB-PEG1000/PEG550 LS as compared to control group.
Next, we did a biodistribution experiment with the U87MG xenograft mice. As shown in Supplementary Material: Fig. S9, the fluorescence intensity in the tumor areas of both APTEDB-PEG2000/PEG1000 LS and APTEDB-PEG1000/PEG550 LS treated mice was significantly higher than that of APTEDB-PEG2000/PEG2000 LS, APTEDB-PEG1000/PEG1000 LS, and non-targeted LS, while biodistribution in other vital organs including the liver, kidney, lung, heart and spleen was not markedly different from one another.
Encouraged by these in vivo cancer-targeting results, we next did in vivo anti-tumor efficacy experiments with the four different PEG-paired liposomal systems loaded with Dox. Saline, free Dox, and a non-targeting liposomal system (PEG2000/PEG2000 LS) lacking only the EDB-specific aptide were used for comparisons. After three intravenous injections on alternate days and with continuous observation for 30 days, the tumor sizes and weights were determined and recorded. As shown in Fig. 4b, the tumor weights were clearly different among the treatment groups: APTEDB-PEG2000/PEG1000 LS (Dox) retarded tumor growth to the greatest extent (~90%), followed closely by APTEDB-PEG1000/PEG550 LS (Dox) (~80%). Both APTEDB-PEG2000/PEG2000 LS (Dox) and APTEDB-PEG1000/PEG1000 LS (Dox) also significantly suppressed tumor growth compared to non-targeting liposomes, albeit to a lesser degree (~50% and 60%, respectively). Taken together, these results clearly indicate that simple differential combination and pairing of PEG lipids in a targeted liposomal system results in significantly different outcomes in terms of cancer targeting and therapeutic efficacy.
Conclusions
In this report, we tested the hypothesis that specific PEG lipid pairs in a targeting ligand-conjugated PEGylated liposomal system affect cancer targeting and therapeutic efficiency. Cellular and in vivo targeting experiments using EDB-targeting liposomes with four different combinations of PEG lipid pairing revealed an interesting trend: (i) both APTEDB-PEG2000/PEG1000 LS and APTEDB-PEG1000/PEG550 LS had much higher cellular and tumor uptake than that of APTEDB-PEG2000/PEG2000 LS and APTEDB-PEG1000/PEG1000 LS, respectively, and (ii) there was a slight difference in the efficacy between APTEDB-PEG2000/PEG1000 LS and APTEDB-PEG1000/PEG550 LS. Our interpretation of these observations is that, when the length or height of the methoxy-capped PEG lipid is similar to that of the targeting-ligand-displaying PEG lipid, the methoxy-capped PEG lipid might not only act as a shield but also produce a steric hindrance, thereby blocking interactions of the targeting ligand with its target. Because most targeting ligands are very small, they could easily be blocked by a continuously moving methoxy-capped PEG lipid. When the length of the methoxy-capped PEG lipid is decreased to the appropriate extent, it may serve as a backbone that strengthens the stem of the targeting-ligand-displaying PEG lipid by blocking the bending or folding of it on the liposomal surface, and thereby allowing the ligand to be stretched out. We speculate that these structural dynamics could be the basis for the enhanced uptake of APTEDB-PEG2000/PEG1000 LS and APTEDB-PEG1000/PEG550 LS.
Taken together, our results show the importance of the PEG lipid-pairing effect in the design of nanomaterials, highlighting the need to match PEG lipids of the appropriate length. In view of this, we conclude that ligand-mediated-targeting PEGylated liposome systems should be carefully optimized. By incorporating various PEG lipid pairs in our aptide-liposome system, we established the following: (i) PEG2000-lipid is not necessarily the best-length PEG lipid to use as a stealth function, and (ii) PEG lipid pairs in which the length of the methoxy-capped PEG lipid is approximately half that of the targeting ligand-displaying PEG lipid appears to be the best combination for enhancing cellular uptake in vitro and tumor uptake in vivo.
Supplementary Material
Synthetic procedure and characterization of aptide-conjugated phospholipids, TEM analysis of liposomes, qRT-PCR and immunocytochemistry for EDB expression, and in vitro cellular uptake study of liposomes.
Acknowledgements
This work was supported by grants from the Global Research Laboratory (grant no. 2014044002), the Radiation Technology R&D program (grant no. 2014033231), and the Intelligent Synthetic Biology Center (grant no. 2013M3A6A8073557) through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning.
Conflict of Interest
The authors declare no competing financial interest.
References
1. Barenholz Y. Doxil(R)--the first FDA-approved nano-drug: lessons learned. J Control Release. 2012;160:117-34
2. Luk BT, Fang RH, Zhang L. Lipid- and polymer-based nanostructures for cancer theranostics. Theranostics. 2012;2:1117-26
3. Malam Y, Loizidou M, Seifalian AM. Liposomes and nanoparticles: nanosized vehicles for drug delivery in cancer. Trends Pharmacol Sci. 2009;30:592-9
4. Allen TM, Cheng WW, Hare JI, Laginha KM. Pharmacokinetics and pharmacodynamics of lipidic nano-particles in cancer. Anticancer Agents Med Chem. 2006;6:513-23
5. Sapra P, Tyagi P, Allen TM. Ligand-targeted liposomes for cancer treatment. Curr Drug Deliv. 2005;2:369-81
6. Immordino ML, Dosio F, Cattel L. Stealth liposomes: review of the basic science, rationale, and clinical applications, existing and potential. Int J Nanomedicine. 2006;1:297-315
7. Allen TM. Liposomal drug formulations - Rationale for development and what we can expect for the future. Drugs. 1998;56:747-56
8. Lasic DD, Martin FJ, Gabizon A, Huang SK, Papahadjopoulos D. Sterically stabilized liposomes: a hypothesis on the molecular origin of the extended circulation times. Biochim Biophys Acta. 1991;1070:187-92
9. Gabizon AA. Stealth liposomes and tumor targeting: one step further in the quest for the magic bullet. Clin Cancer Res. 2001;7:223-5
10. Allen TM, Hansen C, Martin F, Redemann C, Yauyoung A. Liposomes Containing Synthetic Lipid Derivatives of Poly(Ethylene Glycol) Show Prolonged Circulation Half-Lives Invivo. Biochim Biophys Acta. 1991;1066:29-36
11. Gref R, Luck M, Quellec P, Marchand M, Dellacherie E, Harnisch S. et al. 'Stealth' corona-core nanoparticles surface modified by polyethylene glycol (PEG): influences of the corona (PEG chain length and surface density) and of the core composition on phagocytic uptake and plasma protein adsorption. Colloids Surf B Biointerfaces. 2000;18:301-13
12. Iyer AK, Khaled G, Fang J, Maeda H. Exploiting the enhanced permeability and retention effect for tumor targeting. Drug Discov Today. 2006;11:812-8
13. Nie Y, Ji L, Ding H, Xie L, Li L, He B. et al. Cholesterol derivatives based charged liposomes for doxorubicin delivery: preparation, in vitro and in vivo characterization. Theranostics. 2012;2:1092-103
14. Qhattal HS, Hye T, Alali A, Liu X. Hyaluronan Polymer Length, Grafting Density, and Surface Poly(ethylene glycol) Coating Influence in Vivo Circulation and Tumor Targeting of Hyaluronan-Grafted Liposomes. ACS Nano. 2014;8:5423-40
15. Kirpotin D, Park JW, Hong K, Zalipsky S, Li WL, Carter P. et al. Sterically stabilized Anti-HER2 immunoliposomes: Design and targeting to human breast cancer cells in vitro. Biochemistry. 1997;36:66-75
16. Yu MK, Park J, Jon S. Targeting strategies for multifunctional nanoparticles in cancer imaging and therapy. Theranostics. 2012;2:3-44
17. Sinha R, Kim GJ, Nie SM, Shin DM. Nanotechnology in cancer therapeutics: bioconjugated nanoparticles for drug delivery. Mol Cancer Ther. 2006;5:1909-17
18. Ferrari M. Cancer nanotechnology: Opportunities and challenges. Nat Rev Cancer. 2005;5:161-71
19. Farokhzad OC, Langer R. Impact of nanotechnology on drug delivery. ACS Nano. 2009;3:16-20
20. Stefanick JF, Ashley JD, Kiziltepe T, Bilgicer B. A systematic analysis of peptide linker length and liposomal polyethylene glycol coating on cellular uptake of peptide-targeted liposomes. ACS Nano. 2013;7:2935-47
21. Khan ZA, Chan BM, Uniyal S, Barbin YP, Farhangkhoee H, Chen S. et al. EDB fibronectin and angiogenesis -- a novel mechanistic pathway. Angiogenesis. 2005;8:183-96
22. Chen S, Chakrabarti R, Keats EC, Chen M, Chakrabarti S, Khan ZA. Regulation of vascular endothelial growth factor expression by extra domain B segment of fibronectin in endothelial cells. Invest Ophthalmol Vis Sci. 2012;53:8333-43
23. Saw PE, Ko YT, Jon S. Efficient Liposomal Nanocarrier-mediated Oligodeoxynucleotide Delivery Involving Dual Use of a Cell-Penetrating Peptide as a Packaging and Intracellular Delivery Agent. Macromol Rapid Commun. 2010;31:1155-62
24. Saw PE, Kim S, Lee IH, Park J, Yu M, Lee J. et al. Aptide-conjugated liposome targeting tumor-associated fibronectin for glioma therapy. J Mater Chem B. 2013;1:4723-6
25. Park J, Kim S, Saw PE, Lee IH, Yu MK, Kim M. et al. Fibronectin extra domain B-specific aptide conjugated nanoparticles for targeted cancer imaging. J Control Release. 2012;163:111-8
Author contact
![]() Corresponding author: syjonac.kr.
Corresponding author: syjonac.kr.