Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2015; 5(6):618-630. doi:10.7150/thno.11251 This issue Cite
Research Paper
Disclosing the CXCR4 Expression in Lymphoproliferative Diseases by Targeted Molecular Imaging
Hans Jürgen Wester1 ![]() , Ulrich Keller2, Margret Schottelius1, Ambros Beer3#, Kathrin Philipp-Abbrederis2, Frauke Hoffmann1, Jakub Šimeček1*, Carlos Gerngross3, Michael Lassmann4, Ken Herrmann4, Natalia Pellegata5, Martina Rudelius6, Horst Kessler7,8, Markus Schwaiger3
, Ulrich Keller2, Margret Schottelius1, Ambros Beer3#, Kathrin Philipp-Abbrederis2, Frauke Hoffmann1, Jakub Šimeček1*, Carlos Gerngross3, Michael Lassmann4, Ken Herrmann4, Natalia Pellegata5, Martina Rudelius6, Horst Kessler7,8, Markus Schwaiger3
1. Pharmaceutical Radiochemistry, Technische Universität München, Garching, Germany
2. III. Medical Department, Technische Universität München, Munich, Germany
3. Department of Nuclear Medicine, Technische Universität München, Munich, Germany
4. Department of Nuclear Medicine, University Hospital Würzburg, Würzburg, Germany
5. Institute of Pathology, Helmholtz Zentrum München, Munich, Germany
6. Institute of Pathology, Universität Würzburg, Comprehensive Cancer Center Mainfranken, Würzburg, Germany
7. Institute for Advanced Study, Department of Chemistry, Technische Universität München, Garching, Germany
8. Chemistry Department, Faculty of Science, King Abdulaziz University, Jeddah, Saudi Arabia
# Current address: Ulm University, Department of Nuclear Medicine; Ulm, Germany.
* Current address: Scintomics GmbH, Fuerstenfeldbruck, Germany.
Received 2014-12-6; Accepted 2015-2-6; Published 2015-3-1
Abstract

Chemokine ligand-receptor interactions play a pivotal role in cell attraction and cellular trafficking, both in normal tissue homeostasis and in disease. In cancer, chemokine receptor-4 (CXCR4) expression is an adverse prognostic factor. Early clinical studies suggest that targeting CXCR4 with suitable high-affinity antagonists might be a novel means for therapy. In addition to the preclinical evaluation of [68Ga]Pentixafor in mice bearing human lymphoma xenografts as an exemplary CXCR4-expressing tumor entity, we report on the first clinical applications of [68Ga]Pentixafor-Positron Emission Tomography as a powerful method for CXCR4 imaging in cancer patients. [68Ga]Pentixafor binds with high affinity and selectivity to human CXCR4 and exhibits a favorable dosimetry. [68Ga]Pentixafor-PET provides images with excellent specificity and contrast. This non-invasive imaging technology for quantitative assessment of CXCR4 expression allows to further elucidate the role of CXCR4/CXCL12 ligand interaction in the pathogenesis and treatment of cancer, cardiovascular diseases and autoimmune and inflammatory disorders.
Keywords: CXCR4, chemokine receptor, positron emission tomography, lymphoma, in vivo imaging
Introduction
Chemokine receptors form a large family of G-protein coupled receptors that mediate chemotaxis of cells towards a gradient of chemokines. The chemokine receptor subtype CXCR4 exerts its biological effect by binding its ligand CXCL12 (stromal cell-derived factor-1, SDF-1) which activates downstream pathways such as the MAP kinase and the PI3 kinase pathway, ultimately resulting in altered expression of adhesion molecules and cell homing. Physiologically, the CXCR4/CXCL12 interaction plays a pivotal role in a variety of processes that rely on the recruitment and homing of stem and progenitor cells or of immune cells, i.e. in embryogenesis, neoangiogenesis, hematopoiesis and in inflammation [1-3]. CXCR4 is normally expressed on T-lymphocytes, B-lymphocytes, monocytes, macrophages, neutrophils and eosinophils as well as hematopoietic stem and progenitor cells (HSPC) in the bone marrow [4]. Antagonizing the CXCR4-mediated retention of HSPC in these niches by anti-CXCR4 directed treatment with e.g. the cyclam-based antagonist AMD3100 (plerixafor) allows mobilization of HSPC for autografting upon myeloablative treatment [5]. Plerixafor treatment has also been shown to simultaneously mobilize various lymphocyte populations into the peripheral blood, highlighting the important role of CXCL12/CXCR4 for lymphocyte trafficking in vivo [6,7]. Pathological CXCR4 overexpression has been reported in more than 30 different types of cancer, including breast, pancreatic, ovarian, lung, prostate, colorectal and skin cancer, and in hematopoietic malignancies such as leukemia and lymphoma [8-12]. In tumors, CXCR4 overexpression and receptor activation by CXCL12 binding are key triggers for enhanced tumor growth and progression, tumor invasiveness and metastasis [3]. It is therefore not surprising that CXCR4 overexpression has been identified as an adverse prognostic factor in a subset of the above malignancies, e.g. in non-small cell lung cancer (NSCLC), in breast, ovarian, colorectal and pancreatic cancer as well as in AML [4,8,13-18]. In addition, clinical studies revealed that CXCR4 expression correlates with disease extent [15-24]. Hence, the CXCR4/CXCL12 axis represents a highly relevant molecular target of cancer biology and offers promising new approaches and techniques for targeted cancer therapy [25, 26].
During the last decade, the modern molecular imaging techniques have become valuable clinical tools in the assessment and quantification of biomarkers for early evaluation of therapy response, in particular in hematological cancers [27,28].
Consequently, a variety of strategies towards CXCR4 ligands suitable for molecular imaging in vivo have been investigated. Apart from 99mTc-labelled [29,30] and fluorescent [31,32] CXCL12 conjugates, several AMD derivatives have been investigated for 64Cu- [33-38], 18F- [39], and even 11C-labeling [40]. In addition, even smaller AMD analogues generated by molecular modelling approaches have been labeled with 18F-fluoride and successfully evaluated in first in vivo studies [41]. T140, a cyclic peptide comprising 14 amino acids [42], has been investigated as lead compound for potential 68Ga- [43,44], 64Cu- [45,46], 111In- [47], and 18F- [48,49] labeling. Furthermore, radiolabeled peptidomimetics [50], nanoparticles [51-53] and antibodies [54] have been evaluated preclinically. Excellent reviews on the development and assessment of these probes have recently been published [55-58].
Unfortunately, despite the fundamental role of CXCR4 in cancer biology and its putative significance as an attractive target for therapeutic approaches, a highly sensitive methodology for CXCR4-receptor quantification in men has been lacking so far.
To meet this clinical need, our group has started very early with the development and evaluation of cyclic pentapeptide structures [59-63]. We recently developed [68Ga]pentixafor ([68Ga]CPCR4.2), a high-affinity CXCR4-targeted nuclear probe for PET [61,62]. [68Ga]Pentixafor is a synthetic pentapeptide based on the cyclo(D-Tyr1-[NMe]-D-Orn2-Arg3-2-Nal4-Gly5) scaffold, which is conjugated at D-Orn2 with DOTA via 4-(aminomethyl) benzoic acid. With its high CXCR4-affinity, its excellent in vivo pharmacokinetics and high and specific accumulation in CXCR4-positive OH-1 human small cell lung cancer tumor xenografts [62], [68Ga]pentixafor-Positron Emission Tomography (PET) represents a promising method for the in vivo assessment of the CXCR4 expression status in cancer patients.
The particular focus of this work was to assess the potential of [68Ga]pentixafor-PET, a new CXCR4-targeted functional imaging technique, in the context of cancer research and treatment. For this proof-of-concept study hematological malignancies, in particular lymphoma, were selected as exemplary tumor entities due to the well documented CXCR4 expression in these cancers. Furthermore, especially in the case of lymphomas, there is an unmet clinical need for more specific functional imaging tools for the diagnosis, prognostic assessment and stratification of lymphoma patients. To date, only metabolic nuclear imaging, i.e. [18F]FDG-PET (2-[18F]Fluoro-2-desoxyglucose-PET), has been clinically integrated into treatment protocols for lymphomas - already with considerable impact on treatment algorithms [64-67]. Other PET-imaging probes such as the proliferation marker [18F]FLT are currently being investigated as alternative sensitive probes for early treatment response imaging [66,67].
The present investigation therefore aimed at demonstrating the potential of [68Ga]pentixafor-PET as a novel and powerful tool for sensitive in vivo quantification of CXCR4 expression in vivo using hematological malignancies as a representative example. This investigation can also be regarded as the first step towards a future theranostics concept, i.e. the combination of [68Ga]pentixafor-PET and subsequent peptide receptor radiotherapy using corresponding pentixafor derivatives labeled with suitable therapeutic radioiosotopes, such as β- or α-emitters.
Methods
Radiolabeling
Radiosynthesis of [68Ga]pentixafor
For in vitro and animal studies, [68Ga]pentixafor was prepared using 3.5 nmol of pentixafor in a Gallelut+ module (SCINTOMICS GmbH, Germany) similarly to a previously published procedure [61,62].
The syntheses of [68Ga]pentixafor for patient application were performed in a fully automated, GMP-compliant procedure using a GRP® module (SCINTOMICS GmbH, Germany) equipped with a disposable single-use cassette kit (ABX, Germany), using a standardized labeling sequence [70]. Briefly, a 68Ge/68Ga generator with SnO2 matrix (iTHEMBA LABS, South Africa) was eluted with 1 M HCl (5.5 mL), and the eluted 68Ga-activity was immobilized on a strong cationic exchanger cartridge (SCX - Chromafix, size M, Macherey-Nagel, Düren, Germany). Upon washing with water, 68Ga3+ was eluted into the reaction vessel (containing 20 µg pentixafor in 3 mL 1.5 M HEPES) using 1.7 mL 5 M NaCl [71]. The reaction mixture was heated to 140°C for 10 min and, upon cooling, transferred onto a C18 light cartridge (Waters, Eschborn, Germany). The cartridge was washed with water to remove unreacted 68Ga-activity, and [68Ga]pentixafor was eluted using 2 mL of EtOH. The eluate was diluted with 12 mL of PBS and passed through a sterile filter. The radiochemical purity of the ready-to-inject formulation (14 mL) was always >99% as confirmed by radio-HPLC and TLC, the specific activity was in the range of 30-65 GBq/µmol.
Radio-HPLC was carried out on a Shimadzu prominence HPLC system equipped with a CT0-20AC column oven (30 °C) and a SPD-20A UV/Vis detector. Radioactivity detection was performed using a Gabistar activity detector (raytest, Straubenhardt, Germany) connected to the outlet of the UV-detector. For HPLC quality control of [68Ga]pentixafor, a Chromolith Performance RP-18e column (100 × 4.6 mm; Merck Millipore, Darmstadt, Germany) was used. Elution was carried out at a constant flow of 3 mL/min using the following protocol: 0-1 min: 10% acetonitrile (0.1% TFA) in water (0.1% TFA), 1-6 min: 10-60% acetonitrile (0.1% TFA) in water (0.1% TFA).
Radio-TLC was carried out using two different systems: TLC1: silica gel 60 and 0.1 M aq. sodium citrate as a mobile phase; TLC2: Varian silica impregnated ITLC-chromatography paper and a 1:1 (v/v) mixture of 1 M aq. NH4OAc and MeOH as mobile phase. TLC-strips were analyzed using a miniGita TLC analyzer (raytest).
In vitro Evaluation
Cell culture
The human T lymphocyte cell line Jurkat was purchased from American Type Culture Collection (ATCC) and maintained in RPMI-1640 medium supplemented with 10% FCS, 2 mM L-glutamine, and 100 units/mL of penicillin/streptomycin. Daudi cells (human Burkitt lymphoma) were grown in RPMI-1640 medium supplemented with 10% FCS, 2 mM L-glutamine, 1% non-essential amino acids, 50 µM β-mercaptoethanol and 100 units/mL of penicillin/streptomycin, SU-DHL-8 (human large B-cell lymphoma) cells were maintained in RPMI-1640 medium supplemented with 20% FCS, 2 mM L-glutamine, 1% non-essential amino acids, 50 µM β-mercaptoethanol and 100 units/mL of penicillin/streptomycin. CHO-K1 cells (Chinese hamster ovary cells, DSMZ) were cultivated in RPMI-1640 medium supplemented with 10% FBS, 2 mM L-glutamine, and 100 units/mL of penicillin/streptomycin. All cell lines were maintained at 37°C in a humidified 5% CO2 atmosphere.
All media and supplements were obtained from Biochrom (Berlin, Germany) or Gibco (life technologies, Darmstadt, Germany). In the assay medium used for uptake and internalization studies, FCS was replaced by 5% bovine serum albumin (BSA; Sigma, St.Louis, USA).
For cell counting, a Countesse automated cell counter (Invitrogen, Carlsbad, USA) was used.
Transient transfection with CXCR4 and CXCR7
Transient transfection was accomplished using jetPRIME reagent (PEQLAB Biotechnology, Erlangen, Germany; #13-114) according to manufacturer's instructions. CHO-K1 cells were seeded at a density of 50000 cells/well in poly-L-lysine-coated 24-well plates, transfected with 0.5 µg/well of receptor-encoding constructs (kindly supplied by Prof. Dr. S. Schulz and Prof. Dr. R. Stumm, Institute of Pharmacology and Toxicology, Jena University Hospital, Germany) 16 h after plating, and used for assay 24 h after transfection.
Flow Cytometry
Receptor expression levels were quantified using flow cytometry. The human CXCR4-receptor was only available with an N-terminal haemagglutinin (HA)-tag. To demonstrate the independence of ligand binding of the presence or absence of a HA-tag on the receptor protein, a comparative evaluation of the murine receptors with and without the HA-tag was performed. For Flow Cytometry, adherent cells were harvested using Trypsin/EDTA (0.05% and 0.02%, respectively) in PBS (1 min at RT) and centrifuged. Suspension cultures were directly centrifuged. The cells (0.5-1.0 × 106 per sample) were washed once with PBS and incubated with antibody solution in PBS 1% BSA for 45 min on ice. For CXCR4-detection, the phycoerythrin (PE) conjugated antibodies or isotype controls anti-human CXCR4 (Becton Dickinson GmbH, Heidelberg, Germany, #555974, #555574) or anti-mouse CXCR4 (Becton Dickinson GmbH, Heidelberg, Germany, #551966, #553989) were used. For CXCR7-detection, the primary mouse anti-human CXCR7/RDC-1 antibody (clone 11G8) (# MAB42273; R&D Systems; Wiesbaden-Nordenstadt; Germany) was after several washes detected with the secondary FITC conjugated rat anti-mouse IgG antibody (#415-095-166 Dianova GmbH; Hamburg; Germany). Subsequently, cells were washed twice with PBS, resuspended in PBS containing 0.5% BSA and analyzed using a COULTER EPICS XL Flow Cytometer (Beckman Coulter GmbH, Krefeld, Germany).
Binding specificity
Transfection with hHACXCR4, mCXCR4, mHACXCR4, hCXCR7, mCXCR7 and mHACXCR7 was performed as described above. On the day of the experiment the transfection medium was removed and the cells were left to equilibrate in 200 µL of assay medium (RPMI + 5% BSA) at 37 °C for a minimum of 15 min before the experiment. Untransfected CHO cells (negative control) were treated identically. Then, 25 µL/well of either assay medium (Control) or of a 1 mM solution of AMD3100 in HBSS (determination of non-specific binding) were added (n = 3, respectively, for each receptor construct), followed by the addition of 25 µL of [68Ga]pentixafor in assay medium, leading to a final peptide concentration of 1 nM.
Upon incubation at 37 °C for 30 min, the supernatant was removed, and cells were rinsed twice with 200 µL of HBSS and lysed using 200 µL of 1 N NaOH. The lysate was transferred to vials and combined with 200 µL of HBSS used for rinsing the wells. Quantification of the amount of free and bound activity was performed in an Automatic Gamma Counter (WALLAC; 1480 WIZARDTM 3”).
Binding kinetics using lymphoma cell lines
In the case of the suspension cell lines (Jurkat, Daudi, SUDHL-8), samples containing 2 × 105 cells in assay medium were incubated with [68Ga]pentixafor (1 nM) at 37 °C for different time points up to 60 min in the presence (non-specific binding) or absence (control) of 100 µM AMD3100 (n=3 per concentration). The total sample volume was 250 µL. After incubation, the tubes were centrifuged (3 min, 1300 rcf, Megafuge 1.0, Heraeus Thermo Scientific) and the supernatant was carefully removed. After washing twice with 200 µL of cold HBSS, the amount of bound radioligand in the cell pellet was quantified using a γ-counter.
In vivo animal studies
Tumor models
For the induction of tumor growth, female CB-17 SCID mice (6-8 weeks, Charles River, Sulzfeld, Germany) were injected subcutaneously with app. 5×106 Daudi (right shoulder) or SU-DHL-8 (left shoulder) cells suspended in 100 µL of a 1:1 (v/v) mixture of serum free culture medium and Matrigel (BD Biosciences, Heidelberg, Germany). Within 10-21 days, solid palpable tumors had grown (100-800 mg), and the animals were used for the experiments. All animal studies were approved by the responsible regional authorities.
Biodistribution studies
For biodistribution studies, animals (n = 6) were injected intravenously with ≈ 5 MBq (≈ 0.05 nmol peptide per mouse) of [68Ga]pentixafor in PBS (100 µL) into the tail vein under isoflurane anaesthesia. The animals were sacrificed 90 min post injection (p.i.), and the organs of interest were dissected. The radioactivity was measured in weighted tissue samples using a γ-counter. Data are expressed in % ID/g tissue (mean ± SD).
Small animal PET imaging
All animal experiments were approved by local authorities and were in compliance with the institutions guidelines. For static small animal PET-studies using [68Ga]pentixafor, SCID mice were injected i.v. with the radioligand (4-10 MBq) as described and were then allowed to wake up from anaesthesia. After 75 min, mice were again anaesthesized with isoflurane and a CT scan was performed, followed by static PET imaging (90-110 min p.i.) using a SIEMENS Inveon small animal PET/CT. To demonstrate CXCR4-specificity of [68Ga]pentixafor uptake in tumors, competition studies (coinjection of 50 µg of AMD3100/mouse) were also performed.
For dynamic small animal PET-studies, animals were placed in the PET/CT-scanner under isoflurane anaesthesia on a heating mat and a CT-scan was performed. PET data acquisition was started immediately upon i.v. injection of [68Ga]pentixafor (10-15 MBq) using the following acquisition frames: 1 × 30, 4 × 60, 1 × 180, 4 × 300, 1 × 450, 5 × 600, 1 × 900 s, amounting to a total imaging time of 100 min.
For static [18F]FDG-PET imaging, animals were fasted for 2 hours before scanning, anaesthesized using isoflurane, and injected i.v. into the tail vein with 10-15 MBq of [18F]FDG in saline (100 µL). After 35 min, animals were placed in the PET/CT-scanner, and a CT-scan (5 min) was performed, followed by static PET imaging (45-60 min p.i.). During the entire time after tracer administration, animals were kept on a heating mat under isoflurane anaesthesia.
All images were reconstructed using 3D ordered-subsets expectation maximum (OSEM3D) algorithm without scanner and attenuation correction, and data analysis was carried out using the Inveon Research Workspace software.
Immunohistochemistry and immunofluorescence
Immunohistochemistry staining of SU-DHL-8 and Daudi xenografts was performed on an automated immunostainer (Ventana Medical Systems, Tucson, AZ, USA) as previously described following the manufacturer's protocols with minor modifications: a) for background reduction an avidin/biotin blocking step (0.1 mg/ml, 20 min) with ready-to-use reagents (Roche, Penzberg, Germany) was added, and b) the first and secondary antibody dilutions were pipetted manually onto the slides. The following antibodies were used: Ki-67 (Clone B56, Dako, Hamburg, Germany) (1:200); CXCR4 (clone UMB-2, kindly provided by Prof. Dr. S. Schulz, Institute of Pharmacology and Toxicology, Jena University Hospital, Germany) (1:10). Antibodies were diluted in Dako REALTM antibody diluent. The supersensitive detection system (BioGenex, Munich, Germany) was used and immunoreactions were developed in the DAB solution supplied by the kit (Vector lab, Burlingame, CA).
For double immunofluorescence, primary antibodies were detected by incubation with the following secondary antibodies: goat anti-rabbit conjugated with Alexa Fluor 555 (Invitrogen) and goat anti-mouse conjugated with FITC (Invitrogen). After incubation of slides with conjugated secondary antibody (1:100 for 60 min), they were counterstained and mounted with mounting medium (Vectashield, Vector laboratories; Bulingame, CA).
For immunohistochemistry of patient biopsy material the following antibodies were used: anti-CK20 mouse monoclonal antibody (Progen Biotechnik GmbH; HD, Germany (1:50)), anti-CD30 (DAKO, HB, Germany (1:20)), anti-CD138 mouse monoclonal antibody (Cell Marque, CA, USA (1:250)) and anti-CXCR4 rabbit polyclonal antibody (Abcam (clone UMB2), Cambridge, UK (1:100)).
After deparaffinization and rehydration, the slides were placed in a pressure cooker in 0.01 M citrate buffer (pH 6.0) and were heated for 7 min. Incubation with the different antibodies was carried out overnight at 4°C. Detection was performed with DAKO en vision system according to the manufactorer´s protocol.
For double immunofluorescence, primary antibodies were detected by incubation with the following secondary antibodies: donkey anti-rabbit conjugated with Dylight 488 (Jackson ImmunoResearch) and donkey anti-mouse conjugated with Cy5 (Jackson ImmunoResearch). After incubation of slides with conjugated secondary antibody (1:100 for 30 min), they were counterstained and mounted with mounting medium (Vectashield, Vector laboratories; Bulingame, CA).
Patient PET/CT studies
The PET investigations have been approved by the local authority (Reg. von Oberbayern) in accordance with the national regulations. Informed written consent was obtained from the patients prior to the investigation.
PET/CT acquisition was performed according to standard clinical protocols on a Siemens mCT PET/CT scanner (Siemens Medical Solutions, Erlangen, Germany). For [18F]FDG PET/CT, the patients fasted for 6 hours before scanning, and blood glucose levels were measured just before injection to ensure a value below 150 mg/dL. The patients were injected with 300-500 MBq of [18F]FDG and the acquisitions were started after 90 min p.i.. Acquisition time was 2 min per bed position (BP). For [68Ga]pentixafor PET/CT, the patients were injected with 200 MBq [68Ga]pentixafor, and the acquisition was started 50 min p.i.. Acquisition time was 3 minutes per bed position.
For attenuation correction in both scans, a low-dose CT (120 keV, 20 mAs) in shallow inspiration was performed. Emission data were corrected for randoms, dead time, scatter and attenuation. A 3D-OSEM (ordered-subset expectation maximization) iterative reconstruction algorithm was applied with 3 iterations and 21 subsets, 4 mm full-width at half-maximum Gaussian smoothing and zoom 1. Attenuation maps were obtained from the CT data by bilinear transformation, as implemented in the postprocessing software of the PET/CT scanner and used for attenuation correction of the PET/CT data, as previously described [72].
Radiation Dosimetry
Radiation dosimetry of [68Ga]Pentixafor will be described in detail elsewhere [72]. Briefly, patients were injected intravenous with [68Ga]Pentixafor (n=5; 134±25 MBq; peptide amounts <20µg/patient). Three dynamic whole-body scans were followed by four static scans (30 min, 60 min, 120 min and 240 min p.i.). Whole-body distribution and radiation dose estimates were calculated using OLINDA/EXM by means of venous blood samples and multi-exponential regression of organ region-of interest data normalized by administered activity [73].
Results
[68Ga]Pentixafor binds with high specificity and selectivity to human CXCR4
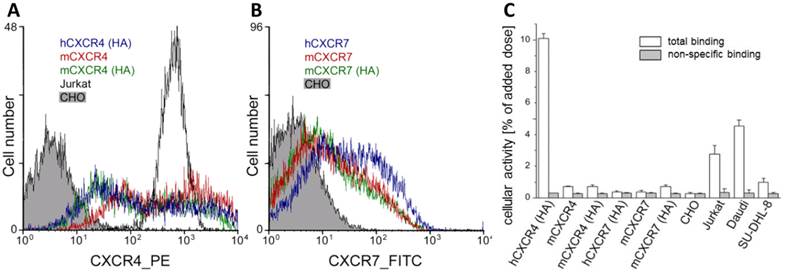
[natGa]Pentixafor has been shown to possess high affinity to hCXCR4 (Jurkat cells: IC50 = 5.0 ± 0.7 nM with radioiodinated FC-131 as the radioligand) [62]. In addition, and in contrast to alternative CXCR4 ligands such as e.g. AMD-analogs, we were able to demonstrate in this study using transiently transfected CHO cells that [68Ga]Pentixafor does not bind to mCXCR4, rCXCR4 or h-, m-CXCR7, an alternative receptor for CXCL12 (Fig. 1), and thus is a highly species selective PET-ligand for the human CXCR4.
CXCR4-expression on transfected CHO cells. (A) CXCR4-expression profile on wt-CHO cells, Jurkat cells and CHO cells transfected with HA-tagged human CXCR4, murine CXCR4 and HA-tagged murine CXCR4 was analyzed by flow cytometry demonstrate. Cells were incubated with the phycoerythrin conjugated anti-h/mCXCR4 Mab 2B11, 551966. The wt-CHO and Jurkat cells were used as control. (B) CXCR7-expression profile on wt-CHO cells and CHO cells transfected with human or murine CXCR7 and HA-tagged murine CXCR7 was analyzed by flow cytometry. Cells were incubated with the primary mouse anti-h/mCXCR7/RDC-1 Mab 11G8. Antibody binding was detected with a FITC-labeled secondary anti-mouse IgG antibody. The wt-CHO cells were used as a negative control. (C) Binding of [68Ga]pentixafor to human Jurkat, Daudi and SU-DHL-8 lymphoma cell lines and to parental CHO cells or CHO cells transiently transfected with human and murine chemokine receptors (30 min, 37°C) in the absence (white bars) and presence of competitor (100µM AMD3100, grey bars).
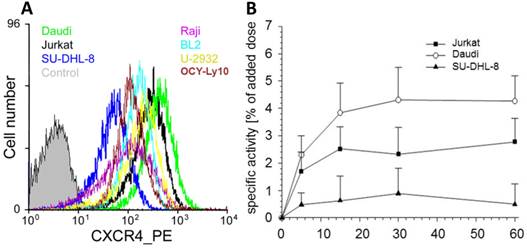
CXCR4 expression correlates with cellular uptake
Based on the pivotal role of the CXCL12/CXCR4 interaction in lymphocyte migration and homing and on the overexpression of CXCR4 in lymphoma [4,72], we decided to use this particular cancer entity for a first translational proof-of-concept investigation. From a series of lymphoid cancer cell lines with different surface CXCR4 expression levels (Fig. 2A; Daudi > Jurkat control > U-2932 > BL2 > OCI-Ly10 > Raji > SU-DHL-8), two cell lines were selected for the generation of mouse subcutaneous xenograft models of lymphoma with high (Daudi) and low (SU-DHL-8) CXCR4 expression. For Daudi, Jurkat and SU-DHL-8 cells, the extent of CXCR4-receptor mediated cellular accumulation of [68Ga]pentixafor within a 60 min incubation period (Daudi > Jurkat > SU-DHL-8, Fig. 2B) was found to directly correlate with the observed ranking order of cellular CXCR4-expression levels.
(A) CXCR4 is highly expressed in various lymphoma cell lines. CXCR4 expression level in the untransfected parental cell line CHO-K1 (control) and various frequently used human lymphoma cell lines (Burkitt's lymphoma cell lines: Daudi, Raji and BL2; diffuse large B-cell lymphoma cell lines: SU-DHL-8, OCI-Ly10, U-2932). Cells were incubated with the PE conjugated anti-hCXCR4 Mab 12G5. Jurkat cells were used as control. (B) Kinetics of CXCR4 mediated cellular accumulation of [68Ga]pentixafor in the depicted human lymphoma cell lines (30 min, 37°C).
Biodistribution and µPET/CT in a mouse tumor model
SU-DHL-8 (low CXCR4) and Daudi (high CXCR4) tumor models were subsequently used to successfully demonstrate the sensitivity of [68Ga]pentixafor for visualization of CXCR4 expression in lymphoma xenografts with high and low CXCR4 density by means of small animal PET imaging (Fig. 3A,B) and comparison with ex vivo biodistribution (Fig. 3C,D).
In vivo evaluation of [68Ga]pentixafor. (A,B) Coronal µPET/CT maximum intensity projections depict CXCR4-expressing Daudi (left tumor) and SU-DHL-8 lymphomas (right tumor) after injection of 4 MBq [68Ga]pentixafor (A). For competition (B), [68Ga]pentixafor (4 MBq) was coinjected with 50 µg AMD3100 (~2mg/kg). Bladder activity was blanked out. (C) Biodistribution of 5 MBq [68Ga]pentixafor 90 min p.i. in Daudi and SU-DHL-8 lymphoma bearing SCID mice, expressed as percent injected dose/g tissue (%ID/g), mean±sd, n=6. (D) Tumor (Daudi) / tissue uptake ratios as calculated from the biodistribution data (C).
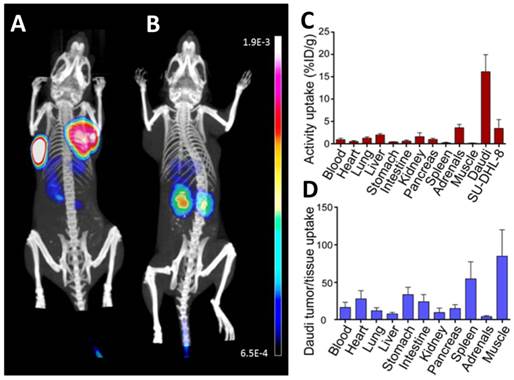
The µPET/CT studies revealed excellent imaging characteristics, even for the SU-DHL-8 tumor xenograft. Tumor uptake, tumor/muscle ratios and tumor/blood ratios of [68Ga]pentixafor 90 min post injection (p.i.) reached values of 16.2% ID/g, 85 and 16 for Daudi and 3.5% ID/g, 18.5 and 3.7 for SU-DHL-8 xenografts, respectively (Fig. 3C,D). Considering the selectivity of [68Ga]pentixafor for hCXCR4, these studies can be assumed to yield a realistic depiction of differential signal strength in human lymphomas.
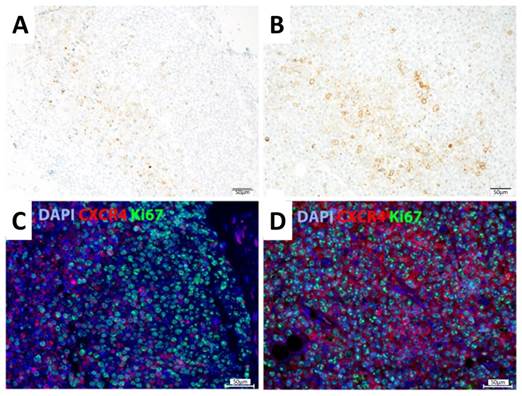
Immunohistochemical and immunofluorescent analysis of representative Daudi and SU-DHL-8 lymphoma xenograft samples showed colocalized CXCR4 and Ki67 expression patterns in the case of Daudi tumors, whereas CXCR4 expression and enhanced proliferation did not clearly correlate on a cellular level in the case of SU-DHL-8 tumors (Fig. 4).
Staining of SU-DHL-8 (A,C) and Daudi xenografts (B,D). (A,B) Immunohistochemical staining was performed with anti-CXCR4 antibody and counterstained with hematoxylin. (C,D) Immunofluorescent staining was performed with antibodies against CXCR4 (red) and Ki-67 (green). Nuclei were counterstained with DAPI (blue). Scale bars: 50 µm.
[68Ga]pentixafor-PET in humans
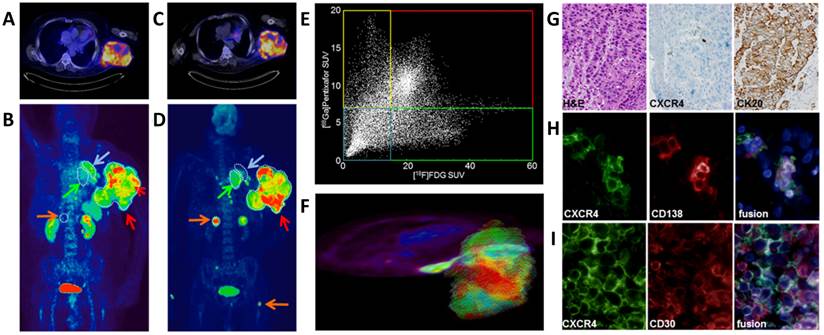
Based on these promising preclinical imaging studies, four patients were investigated by [68Ga]pentixafor-PET. In a patient with histologically proven CD30-positive aggressive T-cell lymphoma and a metachroneously (i.e. after relapse of lymphoma) diagnosed NSCLC, [68Ga]pentixafor- (Fig. 5A,B) and [18F]FDG-PET/CT (Fig. 5C,D) were performed on two consecutive days. [68Ga]pentixafor and [18F]FDG both showed intense tracer uptake in the lymphoma lesion (mean standardized uptake value (SUVmean)/maximum standardized uptake value (SUVmax) [68Ga]pentixafor: 9.91/16.95; SUVmean/SUVmax [18F]FDG: 29.82/50.91) (Fig. 5B,D; red arrow), whereas [68Ga]pentixafor showed substantially lower uptake in the biopsy-proven NSCLC lesion compared to [18F]FDG (SUVmean/SUVmax [68Ga]pentixafor: 2.70/3.86; [18F]FDG: 26.36/38.57) (Fig. 5B,D; green arrow). These findings were confirmed by immunohistochemistry of biopsies of both the NSCLC and the lymphoma, demonstrating relevant CXCR4 expression only in the lymphoma (Fig. 5G,I), whereas in the lung biopsy only few infiltrating plasma cells exhibited positivity for CXCR4 and the carcinoma remained negative. In accordance with these results, [68Ga]pentixafor also showed little to no uptake in several lesions, which were strongly [18F]FDG-avid and, according to their imaging features and distribution pattern, were most likely metastases from NSCLC and not lymphoma-associated lesions (Fig. 5B,D; orange arrows; right adrenal gland, several small bone lesions). Moreover, in lesions avid for both [68Ga]pentixafor- and [18F]FDG, the uptake of the two compared tracers was heterogeneous when analyzed on a voxel-by-voxel basis (Fig. 5E,F). The CXCR4/FDG PET imaging information obtained from this patient with two different cancer types may suggest that the two investigated PET tracers could provide complementary information on tumor dissemination and tumor biology. [68Ga]pentixafor showed excellent imaging properties with only little to moderate uptake in normal organs and background tissue (SUVmean/SUVmax blood pool 1.87/3.38, muscle 0.77/1.38, lung 0.75/1.20, liver 1.46/2.99, spleen 5.69/7.53, bone marrow (BM) 4.00/5.05, kidney 5.06/11.89, bladder 36.4/71.0). As expected, there was only little unspecific tracer uptake in tissues like muscle, lung and liver or in the blood pool, resulting in excellent lesion-to-background contrast for the CXCR4-positive lesions. Some physiological uptake in the BM was evident, as CXCR4 plays a crucial role in hematopoietic cell homing to the BM.
[18F]FDG- and [68Ga]pentixafor-PET/CT imaging of CXCR4 expression. [68Ga]pentixafor-PET/CT in a patient with a large T-cell lymphoma and metastatic non-small cell lung adenocarcinoma of the lung. A) transaxial [68Ga]pentixafor-PET/CT image demonstrate high and heterogeneous tracer uptake in the T-cell lymphoma. B) coronal [68Ga]pentixafor-PET MIP with heterogeneous tracer uptake in the lymphoma (red arrows), medium uptake in left upper lobe dystelectasis (blue arrow) and no uptake in the NSCLC primary tumor(green arrow) and its metastases (orange arrows). C) high and heterogeneous glucose utilization in the T-cell lymphoma, as demonstrated by the transaxial [18F]FDG-PET/CT image. D) coronal [18F]FDG-PET MIP with heterogeneous tracer uptake in the lymphoma (red arrow), no uptake in left upper lobe dystelectasis (blue arrow), but uptake in the NSCL cancer primary (green arrow) and its metastases (orange arrows). E) Heterogeneity of glucose utilization and CXCR4 expression disclosed by correlation of the SUVs of both tracers on a voxel-by-voxel basis. (red: high uptake of both tracers, blue: low uptake of both tracers, yellow: higher relative [68Ga]pentixafor uptake, green: higher relative [18F]FDG uptake. F) 3D model for the SUV based voxel-by-voxel comparison. G) Immunohistological assessment of CXCR4 expression; NSCLC section histologically stained with H&E (left) shows no anti-CXCR4 immunostaining (middle), but strong CK20 positive staining (right); H) CXCR4 (left) and CD138 (middle) immunofluorescent staining of a NSCLC section reveal only weak stromal CXCR4 expression co-localized with infiltrated CD138+ plasma cells (fusion, right); I) strong CXCR4 (left) and CD30 (middle) immunofluorescent staining of the T-cell lymphoma section shows strong CXCR4 expression of activated T-cells (fusion, right)
Further patients with lymphoproliferative cancers were then assessed. In a patient with relapsed diffuse large B-cell lymphoma (Fig. 6A) [68Ga]Pentixafor-PET/CT resulted in excellent tumor uptake (SUVmean/SUVmax: 15.93/23.32), and even clear delineation of the lesions within the brain (background SUVmean/SUVmax: 0.03/0.21, Fig. 6B) previously identified by MR imaging, and negligible uptake in non-tumor tissue (SUVmean/SUVmax blood pool 2.25/2.68, muscle 0.65/0.77, lung 0.45/0.62, liver 1.94/2.95, spleen 9.20/10.58, BM 3.59/4.32, kidney 3.93/5.91). In a patient with chronic lymphocytic leukemia and suspected transformation into aggressive B-cell lymphoma (Fig. 6C, D), all nodal lesions were visible with even higher tracer uptake as compared to [18F]FDG-PET ([68Ga]Pentixafor-PET SUVmean/SUVmax: 5.05/6.64; [18F]FDG-PET SUVmean/SUVmax: 3.66/5.94; [18F]FDG-PET not shown).
[68Ga]Pentixafor-PET/CT in patients with lymphoproliferative malignancies. A) [68Ga]Pentixafor- PET/CT in a patient with relapsed diffuse large B-cell lymphoma (PET MIP) with B) transaxial PET/CT image at the level of the brain. C) [68Ga]Pentixafor-PET/MR in a patient with chronic lymphocytic leukemia and suspected transformation into aggressive B-cell lymphoma (PET MIP) with D) transaxial PET/MR image at the level of the kidneys. E) [68Ga]Pentixafor PET/MR in a patient with multiple myeloma (PET MIP) with F) transaxial image at the level of the maxilla demonstrated no uptake in the maxilla (green arrow). G) Corresponding [18F]FDG PET/MR (PET MIP) of the patient depicted in E) and F) with corresponding H) [18F]FDG PET/MR transaxial image at the level of the maxilla (red arrow, same region as depicted in F) showed [18F]FDG uptake in the maxilla/ floor of the maxillary sinus, most likely caused by a dental infection.
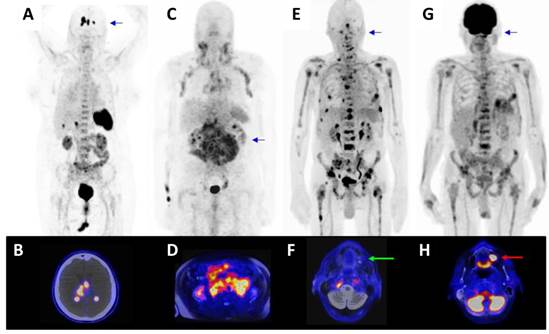
Superior imaging characteristics were also observed in a patient with multiple myeloma (Fig. 6E, G) with extensive bone involvement. [68Ga]Pentixafor-PET/MR exhibited a high contrast between the bone marrow and the lesions with a SUVmean/SUVmax: 18/25.87 of the hottest lesion in the 5th rib left compared to [18F]FDG uptake (SUVmean/SUVmax: 5.23/8.06). Moreover, [68Ga]Pentixafor-PET allowed to better differentiate the individual lesions from the surrounding tissue than the [18F]FDG-PET, especially with regard to the activity in the bone marrow (bone marrow (BM) SUVmean/SUVmax [68Ga]Pentixafor-PET: 2.69/4.19; [18F]FDG-PET 3.77/6.24). Several manifestations, which showed high uptake in [68Ga]Pentixafor-PET, were not seen in [18F]FDG-PET, particularly in the area of the thoracic and pelvic skeleton and the extremities. Because of the bone marrow activation due to chemotherapy, many lesions are masked in the [18F]FDG-PET and are visually and quantitatively not recognizable. In addition to a higher uptake of the CXCR4-targeted probe into numerous lesions, an [18F]FDG avid focus, most likely representing a dental infectious in the maxilla was found to be [68Ga]Pentixafor-PET negative (Fig. 6E).
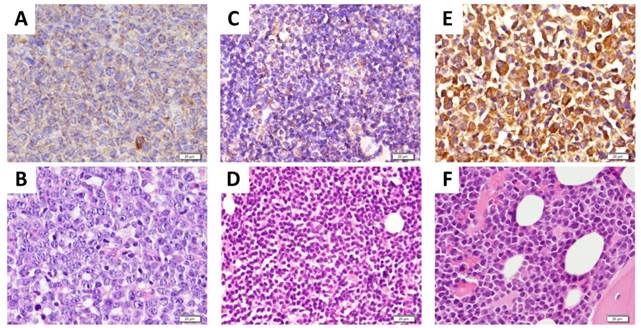
Importantly, previously obtained tumor biopsies from all patients depicted in Fig. 6 stained positive for CXCR4 as assessed by immunohistochemistry (Fig. 7).
CXCR4 expression in tumor bisopsies. Previously obtained representative biopsies (A, B: from DLBCL patient shown in Fig. 6A; C, D: from CLL patient with suspected transformation into DLBCL shown in Fig. 6C; E,F: from MM patient shown in Fig. 6E) were assessed for CXCR4 expression by immunohistochemistry using an anti-CXCR4 antibody and counterstaining with hematoxylin (A, C, E) or staining with eosin and hematoxylin (H&E) (B, D, F). Scale bars: 20 μm.
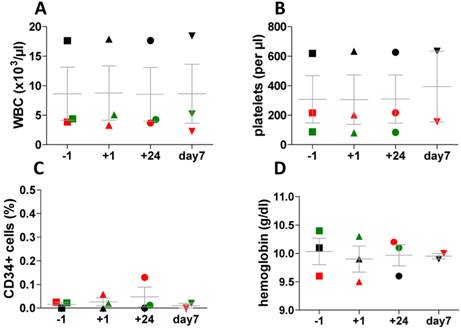
Application of [68Ga]pentixafor was well tolerated and there were no substantial changes in vital parameters (ECG, blood pressure, heart rate) before and at 3 hours and 24 hours after injection. Moreover, assessment of peripheral blood cell counts using pre- and post-imaging standard blood count assessment and standard CD34+ flow cytometry did not show any significant changes associated with tracer application (Fig. 8). In particular, the CD34+ HSPC percentage in the peripheral blood was in the normal range in all samples tested.
Assessment of peripheral blood cell counts and standard CD34 flow cytometry. Assessment was performed directly before (-1h) and after tracer injection (+1h, +24h, + 7days) in three patients with lymphoproliferative disease. In one patient CD34 value has not been measured at day 7, in another patients values of hemoglobin and platelets are missing on day 7. (A) white blood cell counts (WBC), (B) platelets, (C) hemoglobin and (D) percentage of CD34+ cells in the peripheral blood.
Radiation Dosimetry
Compared to the most often used sst-agonists [68Ga]DOTATOC and [68Ga]DOTATATE69, the values for organs with the highest absorbed doses after application of [68Ga]pentixafor, such as urinary bladder wall (15.06 mGy/185MBq), spleen (9.95 mGy/185MBq), or kidneys (6.48 mGy/185MBq), and the total effective dose are notably lower (total effective dose for [68Ga]Pentixafor, [ 68Ga]DOTATOC and [68Ga]DOTATOC: 2.89 mSv, 3.89 mSv and 3.89 mSv, respectively) (Table 1). Thus, based on the excellent pharmacokinetic profile and fast clearance kinetics, [68Ga]Pentixafor exhibits a favorable dosimetry. A detailed analysis of the biodistribution and radiation dosimetry of [68Ga]Pentixafor in humans is given elsewhere67.
Effective dose coefficients [mSv/MBq], total effective doses [mSv/185 MBq] and absorbed doses in selected organs [mGy/185 MBq] after administration of [68Ga]Pentixafor, compared to the corresponding values for [68Ga]DOTATOC# and [68Ga]DOTATATE#.
| [68Ga]Pentixafor | [68Ga]DOTATATE# | [68Ga]DOTATOC# | |
|---|---|---|---|
| effective dose coefficient [mSv/MBq] | 1.56E-02 | 2,10E-02 | 2,10E-02 |
| effective dose [mSv/185 MBq] | 2,89 | 3,89 | 3,89 |
| absorbed doses [mGy/185 MBq] | |||
| Urinary Bladder Wall | 15,06 | 18,13 | 22,02 |
| Spleen | 9,95 | 20,17 | 19,98 |
| Kidneys | 6,48 | 17,21 | 15,17 |
| Heart Wall | 4,91 | - | - |
| Osteogenic Cells | 3,40 | - | - |
| Liver | 3,31 | 9,25 | 7,59 |
| Gallbladder Wall | 2,70 | - | - |
| Red Marrow | 2,59 | 2,78 | 2,96 |
| Uterus | 2,52 | - | - |
| Pancreas | 2,37 | - | - |
| Ovaries | 2,33 | - | - |
| LLI Wall | 2,32 | - | - |
| Adrenals | 2,28 | 15,91 | 14,25 |
| Small Intestine | 2,28 | - | - |
| ULI Wall | 2,24 | - | - |
| Stomach Wall | 2,21 | - | - |
| Thymus | 2,10 | - | - |
| Lungs | 2,04 | 1,11 | 1,30 |
| Testes | 2,04 | - | - |
| Muscle | 2,02 | - | - |
| Thyroid | 2,00 | - | - |
| Brain | 1,85 | - | - |
| Breasts | 1,83 | - | - |
| Skin | 1,76 | - | - |
Discussion
Activation of the CXCR4/CXCL12 axis is a frequent event in a wide spectrum of pathologies, ranging from vascular diseases such as chronic heart failure, cardiac regeneration after acute myocardial infarction [35] and recovery after cerebral stroke [76] over autoimmune diseases [77] to tumorigenesis, cancer cell proliferation and metastasis [78]. These processes are often accompanied by CXCR4 overexpression in the diseased tissues, qualifying CXCR4 as a highly attractive biochemical target for non-invasive molecular imaging techniques using specific and sensitive CXCR4-targeted imaging probes.
Lymphoma is a frequent cancer with high CXCR4 expression and thus suitable for the evaluation of a specific imaging technique for planning and monitoring of potential future CXCR4 targeted therapies. To evaluate the potential of [68Ga]pentixafor-PET in this context, we assessed [68Ga]pentixafor as a probe for CXCR4 imaging in lymphoma cell lines and the corresponding xenograft models and in a first human proof-of-concept study in patients with lymphoproliferative malignancies.
[68Ga]pentixafor shows excellent affinity towards hCXCR4 (IC50 = 5.0 ± 0.7 nM) [62], and it is a highly specific ligand for hCXCR4, that shows no or negligible binding to either mCXCR4 or h- and mCXCR7 receptors (Fig. 1). This pronounced selectivity of [68Ga]pentixafor for hCXCR4 has the beneficiary effect of allowing unambiguous interpretation of [68Ga]pentixafor-PET images with respect to the observed signal. CXCR7 is oftentimes co-expressed with CXCR4, and its expression is also enhanced in a variety of human tumors and is highly associated with tumor associated vasculature [79].
[68Ga]pentixafor binding to hCXCR4-expressing cells in vitro (Fig. 2B) closely correlates with the respective receptor expression levels as determined by flow cytometry (Fig. 2A), and this finding is also reflected in the respective tumor accumulations of [68Ga]pentixafor in Daudi (high CXCR4) and SU-DHL-8 (low CXCR4) tumor xenografts (Fig. 3). With its excellent in vivo pharmacokinetic profile, [68Ga]pentixafor is clearly superior to previously reported CXCR4-targeted PET probes [56].
As demonstrated in this first proof-of-concept investigation in man, [68Ga]pentixafor-PET seems to represents a highly selective and specific method for the in vivo quantification of CXCR4 expression and thus can be of particular value for the pretherapeutic confirmation of CXCR4 expression density prior to novel CXCR4 targeted therapies.
The development and clinical evaluation of CXCR4 targeted therapeutics is a very active field of research. Initially, the major focus was directed towards their evaluation as effective anti-HIV agents and safe agents for the mobilization of HSPC for autologous transplantation in patients with hematological malignancies such as lymphoma and multiple myeloma. Plerixafor (AMD3100, Mozobil) is the only approved drug for this application so far [5], but other candidates such as the 14-mer peptidic antagonist BKT140 [80] also showed excellent results in patients with multiple myeloma and is now evaluated in clinical trials [81]. However, triggered by the recognition of the important role of CXCR4 in tumor growth and metastasis, the focus in the clinical evaluation of these compounds has shifted towards an additional assessment of their suitability and efficacy as anti-cancer and anti-metastatic drugs, both for their use in monotherapy as well as in combination therapy. Furthermore, other novel therapeutics such as the fully human IgG4 Mab BMS-936564/MDX-1338 have entered the arena. BMS-936564 demonstrates high efficacy as monotherapy in tumor-bearing mice [82] and is currently in phase I clinical trials for the treatment of various relapsed/refractory AML [81]. In the context of these actual and future therapeutic trials, [68Ga]pentixafor-PET could certainly be a powerful molecular diagnostic-based approach for patient selection.
The voxel-by-voxel analysis in one patient (Fig.5 E,F) and the mismatch of CXCR4 expression versus proliferation signal in the SU-DHL-8 model indicate a remarkable inter- and intralesion heterogeneity in the uptake of [68Ga]pentixafor and [18F]FDG. This demonstrates that the biological information provided by both tracers could prove supplementary even in lesions that show generally increased uptake of both tracers. Moreover, in some patients, [68Ga]pentixafor uptake was even higher compared to [18F]FDG. In these cases, [68Ga]pentixafor-PET could emerge as the imaging modality of choice.
Due to the high specificity of [68Ga]pentixafor uptake in CXCR4 expressing tumor, the resulting excellent target to non-target ratios and the first dosimetry data, corresponding analogues suitable for labeling with therapeutic β-- or α-emitting radionuclides could allow the realization of an endoradiotherapeutic approach e.g. in lymphoma therapy. A similar concept pairing a therapeutic agent with a corresponding imaging probe is represented by e.g. [177Lu/90Y]DOTATATE-therapy of somatostatin receptor expressing tumors after pre-therapeutic sst-receptor quantification using [68Ga]DOTATATE [83]. When applying the theranostics concept to CXCR4, especially in radiation sensitive hematological malignancies, we expect increased tissue selectivity compared to a ´conventional´ CXCR4-targeted pharmacological intervention and a more homogeneous treatment effect, as heterogeneity in target expression is expected to be compensated by the so-called cross-fire effect. The concept of CXCR4 targeted radiotherapy is currently under evaluation.
In summary, our data document the first methodology for clinical PET imaging of CXCR4 chemokine receptor expression. Having this powerful imaging technique at hand is highly interesting in several regards: a) it can provide detailed and quantitative information on the density, frequency and kinetics of CXCR4 expression in various diseases; b) it might provide a means for patient selection, treatment planning and monitoring in diseases where CXCR4-targeted treatment is now and in the future evaluated, and c) it opens a broad field of clinical investigations on the relevance of CXCR4 expression and regulation for a multitude of biological processes.
Abbreviations
SDF-1: Stromal-cell derived factor-1; HSPC: hematopoietic stem/progenitor cells; NSCLC: non-small cell lung cancer; AML: acute myeloid leukemia; PET: positron emission tomography; DOTA: 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid; [18F]FLT: [18F]Fluorothymidine; [18F]FDG: [18F]Fluorodeoxyglucose; HA-tag: haemagglutinin-tag; EDTA: ethylendiaminetetraacetic acid; CHO cells: chinese hamster ovary cells; DAB: diaminobenzidine; SUV: standardized uptake value.
Acknowledgements
We are grateful to Stephanie Schöffmann and Dr. Gloria Lutzny for assistance with flow cytometry and to Prof. Dr. Stephan Schulz and Prof. Dr. Ralf Stumm from Institute of Pharmacology and Toxicology, Jena University Hospital, for supplying the receptor-encoding constructs. We thank Dr. Misu Lee for performing the immunohistochemical stainings. Furthermore, we thank Simone Seifert, Simone Groß, Michael Schulze-Glück (members of the nuclear medicine PET team, Würzburg) and I. Grelle for their support and assistance.
H.J.W., U.K., A.B., N.S.P., M.S. are supported by the Deutsche Forschungsgemeinschaft (SFB824; subprojects Z1/4, A5/7/8, B5/8 and C3. U.K. is further supported by DFG grants KE 222/7-1 and DJCLS R11/18. H.K. thanks for financial support by the Center of Integrated Protein Science Munic (CIPSM) and by the King Abdulaziz University KAU (grant No. HiCi/25-3-1432).
Competing interests
Hans-J. Wester is founder, owner and CEO, of Scintomics GmbH, Fuerstenfeldbruck, Germany. Jakub Šimeček is research associate at Scintomics GmbH, Fuerstenfeldbruck, Germany. All other authors have declared that no competing interest exists.
References
1. Zlotnik A, Burkhardt A.M, Homey B. Homeostatic chemokine receptors and organ-specific metastasis. Nat Rev Immunol. 2011;11:597-606
2. Jacobson O, Weiss I.D. CXCR4 chemokine receptor overview: biology, pathology and applications in imaging and therapy. Theranostics. 2013;3:1-2
3. Domanska UM, Kruizinga RC, Nagengast WB. et al. A review on CXCR4/CXCL12 axis in oncology: no place to hide. Eur J Cancer. 2013;49:219-230
4. Teicher B.A, Fricker S.P. CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin Cancer Res. 2010;16:2927-2931
5. Brave M, Farrell A, Ching Lin S. et al. FDA review summary: Mozobil in combination with granulocyte colony-stimulating factor to mobilize hematopoietic stem cells to the peripheral blood for collection and subsequent autologous transplantation. Oncology. 2010;78:282-288
6. Kean LS, Sen S, Onabajo O. et al. Significant mobilization of both conventional and regulatory T cells with AMD3100. Blood. 2011;118:6580-6590
7. McDermott DH, Liu Q, Ulrick J. et al. The CXCR4 antagonist plerixafor corrects panleukopenia in patients with WHIM syndrome. Blood. 2011;118:4957-4962
8. Cojoc M, Peitzsch C, Trautmann F. et al. Emerging targets in cancer management: role of the CXCL12/CXCR4 axis. Onco Targets Ther. 2013;6:1347-1361
9. Burger J.A. & Peled, A. CXCR4 antagonists: targeting the microenvironment in leukemia and other cancers. Leukemia. 2009;23:43-52
10. Weilbaecher K.N, Guise T.A, McCauley. Et al. Cancer to bone: a fatal attraction. Nat Rev Cancer. 2011;11:411-425
11. Müller A, Homey B, Soto H. et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50-6
12. Burger JA, Kipps TJ CXCR4. a key receptor in the crosstalk between tumor cells and their microenvironment. Blood. 2006;107:1761-7
13. Tavernier-Tardy E1, Cornillon J, Campos L. et al. Prognostic value of CXCR4 and FAK expression in acute myelogenous leukemia. Leuk Res. 2009;33:764-8
14. Burger JA1, Bürkle A. The CXCR4 chemokine receptor in acute and chronic leukaemia: a marrow homing receptor and potential therapeutic target. Br J Haematol. 2007;137:288-96
15. Tavernier E, Aanei C, Solly F, Flandrin-Gresta P. et al. CXCR4: a new therapeutic target of the leukaemic cell? Role of the SDF-1/CXCR4 axis in acute myeloid leukaemia. Bull Cancer. 2014;101:593-604
16. Moreno MJ, Bosch R, Dieguez-Gonzalez R. et al. CXCR4 expression enhances diffuse large B cell lymphoma dissemination and decreases patient survival. J Pathol. 2015;235:445-55
17. Ko SY, Park CJ, Park SH. et al. High CXCR4 and low VLA-4 expression predicts poor survival in adults with acute lymphoblastic leukemia. Leuk Res. 2014;38:65-70
18. Ahn JY, Seo K, Weinberg OK, et a. The prognostic value of CXCR4 in acute myeloid leukemia. Appl Immunohistochem Mol Morphol. 2013;21:79-84
19. Spoo AC, Lubbert M, Wierda W. et al. CXCR4 is a prognostic marker in acute myelogenous leukemia. Blood. 2007;109:786-91
20. Hiller DJ, Meschonat C, Kim R. et al. Chemokine receptor CXCR4 level in primary tumors independently predicts outcome for patients with locally advanced breast cancer. Surgery. 2011;150:459-65
21. Ottaiano A, Franco R, Aiello Talamanca A. et al. Overexpression of both CXC chemokine receptor 4 and vascular endothelial growth factor proteins predicts early distant relapse in stage II-III colorectal cancer patients. Clin. Cancer. Res. 2006;12:2795-803
22. Spano JP, Andre F, Morat L. et al. Chemokine receptor CXCR4 and early-stage non-small cell lung cancer: pattern of expression and correlation with outcome. Ann Oncol. 2004;15:613-7
23. Jiang YP, Wu XH, Shi B. et al. Expression of chemokine CXCL12 and its receptor CXCR4 in human epithelial ovarian cancer: an independent prognostic factor for tumor progression. Gynecol Oncol. 2006;103:226-33
24. Maréchal R, Demetter P, Nagy N. et al. High expression of CXCR4 may predict poor survival in resected pancreatic adenocarcinoma. Br J Cancer. 2009;100:1444-51
25. Uy GL, Rettig MP, Motabi IH. et al. A phase 1/2 study of chemosensitization with the CXCR4 antagonist plerixafor in relapsed or refractory acute myeloid leukemia. Blood. 2012;119:3917-24
26. Kuhne MR, Mulvey T, Belanger B. et al. BMS-936564/MDX-1338: a fully human anti-CXCR4 antibody induces apoptosis in vitro and shows antitumor activity in vivo in hematologic malignancies. Clin Cancer Res. 2013;19:357-66
27. Groves AM, Win T. et al. Non-[18F]FDG PET in clinical oncology. Lancet Oncol. 2007;8:822-30
28. James ML, Gambhir SS. A molecular imaging primer: modalities, imaging agents, and applications. Physiol Rev. 2012;92:997-65
29. Kanki S, Segers VF, Wu W. et al. Stromal cell-derived factor-1 retention and cardioprotection for ischemic myocardium. Circ Heart Fail. 2011;4:509-18
30. Misra P, Lebeche D, Ly H. et al. Quantitation of CXCR4 expression in myocardial infarction using 99mTc-labeled SDF-1alpha. J Nucl Med. 2008;49:963-9
31. Zwier JM, Roux T, Cottet M. et al. A fluorescent ligand-binding alternative using Tag-lite® technology. J Biomol Screen. 2010;15:1248-59
32. Hatse S, Princen K, Liekens S. et al. Fluorescent CXCL12AF647 as a novel probe for nonradioactive CXCL12/CXCR4 cellular interaction studies. Cytometry A. 2004;61:178-88
33. Weiss ID, Jacobson O, Kiesewetter DO. et al. Positron emission tomography imaging of tumors expressing the human chemokine receptor CXCR4 in mice with the use of 64Cu-AMD3100. Mol Imaging Biol. 2012;14:106-14
34. Woodard LE, De Silva RA, Behnam Azad B. et al. Bridged cyclams as imaging agents for chemokine receptor 4 (CXCR4). Nucl Med Biol. 2014;41:552-61
35. De Silva RA, Peyre K, Pullambhatla M. et al. Imaging CXCR4 expression in human cancer xenografts: evaluation of monocyclam 64Cu-AMD3465. J Nucl Med. 2011;52:986-93
36. Weiss ID, Jacobson O, Kiesewetter DO. et al. Positron emission tomography imaging of tumors expressing the human chemokine receptor CXCR4 in mice with the use of 64Cu-AMD3100. Mol Imaging Biol. 2012;14:106-14
37. Nimmagadda S, Pullambhatla M, Stone K. et al. Molecular imaging of CXCR4 receptor expression in human cancer xenografts with [64Cu]AMD3100 positron emission tomography. Cancer Res. 2010;70:3935-44
38. Jacobson O, Weiss ID, Szajek L. et al. 64Cu-AMD3100—a novel imaging agent for targeting chemokine receptor CXCR4. Bioorg Med Chem. 2009;17:1486-93
39. Oltmanns D, Zitzmann-Kolbe S, Mueller A. et al. Zn(II)-bis(cyclen) complexes and the imaging of apoptosis/necrosis. Bioconjug Chem. 2011;22:2611-24
40. Hartimath SV, van Waarde A, Dierckx RA. et al. Evaluation of N-[(11)C]Methyl-AMD3465 as a PET Tracer for Imaging of CXCR4 Receptor Expression in a C6 Glioma Tumor Model. Mol Pharm. 2014;11:3810-7
41. Liang Z, Zhan W, Zhu A. et al. Development of a unique small molecule modulator of CXCR4. PLoS One. 2012;7:e34038
42. Tamamura H, Omagari A, Oishi S. et al. Pharmacophore identification of a specific CXCR4 inhibitor, T140, leads to development of effective anti-HIV agents with very high selectivity indexes. Bioorg Med Chem Lett. 2000;10:2633-7
43. George GP, Stevens E, Åberg O. et al. Preclinical evaluation of a CXCR4-specific (68)Ga-labelled TN14003 derivative for cancer PET imaging. Bioorg Med Chem. 2014;22:796-803
44. Hennrich U, Seyler L, Schäfer M. et al. Synthesis and in vitro evaluation of 68Ga-DOTA-4-FBn-TN14003, a novel tracer for the imaging of CXCR4 expression. Bioorg Med Chem. 2012;20:1502-10
45. Jacobson O, Weiss ID, Szajek LP. et al. Improvement of CXCR4 tracer specificity for PET imaging. J Control Release. 2012;157:216-23
46. Jacobson O, Weiss ID, Szajek LP. et al. PET imaging of CXCR4 using copper-64 labeled peptide antagonist. Theranostics. 2011;1:251-62
47. Hanaoka H, Mukai T, Tamamura H. et al. Development of a 111In-labeled peptide derivative targeting a chemokine receptor, CXCR4, for imaging tumors. Nucl Med Biol. 2006;33:489-94
48. Jacobson O, Weiss ID, Kiesewetter DO. et al. PET of tumor CXCR4 expression with 4-18F-T140. J Nucl Med. 2010;51:1796-804
49. Zhang XX, Sun Z, Guo J. et al. Comparison of (18)F-labeled CXCR4 antagonist peptides for PET imaging of CXCR4 expression. Mol Imaging Biol. 2013;15:758-67
50. Sato T, Iwase M, Miyama M. et al. Internalization of CCR4 and inhibition of chemotaxis by K777, a potent and selective CCR4 antagonist. Pharmacology. 2013;91:305-13
51. Bunschoten A, Buckle T, Kuil J. et al. Targeted non-covalent self-assembled nanoparticles based on human serum albumin. Biomaterials. 2012;33:867-75
52. Huang X, Zhang F, Wang Y. et al. Design considerations of iron-based nanoclusters for noninvasive tracking of mesenchymal stem cell homing. ACS Nano. 2014;8:4403-14
53. Lee Y, Chen Y, Tarasova NI. et al. The structure of monomeric components of self-assembling CXCR4 antagonists determines the architecture of resulting nanostructures. Nanotechnology. 2011;22:505101
54. Nimmagadda S, Pullambhatla M, Pomper MG. Immunoimaging of CXCR4 expression in brain tumor xenografts using SPECT/CT. J Nucl Med. 2009;50:1124-30
55. Chatterjee S, Behnam Azad B, Nimmagadda S. The intricate role of CXCR4 in cancer. Adv Cancer Res. 2014;124:31-82
56. Nayak TR, Hong H, Zhang Y, et al.Multimodality imaging of CXCR4 in cancer. current status towards clinical translation. Curr Mol Med. 2013;13:1538-48
57. Kuil J, Buckle T, van Leeuwen FWB. Imaging agents for the chemokine receptor 4 (CXCR4). Chem. Soc. Rev. 2012;41:5239-5261
58. Weiss ID, Jacobson O. Molecular Imaging of Chemokine Receptor CXCR4. Theranostics. 2013;3:76-84
59. Demmer O, Dijkgraaf I, Schottelius M. et al. Introduction of functional groups into peptides via N-alkylation. Org Lett. 2008;10:2015-8
60. Demmer O, Gourni E, Schumacher U. et al. PET imaging of CXCR4 receptors in cancer by a new optimized ligand. ChemMedChem. 2011;6:1789-91
61. Demmer O, Dijkgraaf I, Schumacher U. et al. Design, synthesis, and functionalization of dimeric peptides targeting chemokine receptor CXCR4. J Med Chem. 2011;54:7648-62
62. Gourni E, Demmer O, Schottelius M. et al. PET of CXCR4 expression by a (68)Ga-labeled highly specific targeted contrast agent. J Nucl Med. 2011;52:1803-10
63. Demmer O, Frank AO, Hagn F. et al. A conformationally frozen peptoid boosts CXCR4 affinity and anti-HIV activity. Angew Chem Int Ed Engl. 2012;51:8110-3
64. Cheson BD. Role of functional imaging in the management of lymphoma. J Clin Oncol. 2011;29:1844-54
65. Engert A, Haverkamp H, Kobe C. et al. Reduced-intensity chemotherapy and PET-guided radiotherapy in patients with advanced stage Hodgkin's lymphoma (HD15 trial): a randomised, open-label, phase 3 non-inferiority trial. Lancet. 2012;379:1791-1799
66. Moskowitz C. Interim PET-CT in the management of diffuse large B-cell lymphoma. Hematology Am Soc Hematol Educ Program. 2012;2012:397-401
67. Casasnovas RO, Meignan M, Berriolo-Riedinger A. et al. Early interim PET scans in diffuse large B-cell lymphoma: can there be consensus about standardized reporting, and can PET scans guide therapy choices? Curr Hematol Malig Rep. 2012;7:193-199
68. Graf N, Herrmann K, Numberger B. et al. [18F]FLT is superior to [18F]FDG for predicting early response to antiproliferative treatment in high-grade lymphoma in a dose-dependent manner. Eur. J. Nucl. Med. Mol. Imaging. 2013;40:34-43
69. Herrmann K, Buck AK, Schuster T. et al. Week one FLT-PET response predicts complete remission to R-CHOP and survival in DLBCL. Oncotarget. 2014;5:4050-9
70. Martin R, Juettler S, Mueller et al. Cationic eluate pretreatment for automated synthesis of [68Ga]CPCR4.2. Nucl Med Biol. 2014;41:84-9
71. Mueller D, Klette I, Baum RP. et al. Simplified NaCl based 68Ga concentration and labeling procedure for rapid synthesis of 68Ga radiopharmaceuticals in high radiochemical purity. Bioconjugate Chem. 2012;23:1712-7
72. Kinahan PE, Hasegawa BH, Beyer T. X-ray-based attenuation correction for positron emission tomography/computed tomography scanners. Semin Nucl Med. 2003;33:166-179
73. Herrmann K, Lapa C, Wester HJ. et al. Biodistribution and radiation dosimetry for the novel chemokine receptor CXCR4 targeting probe [68Ga]Pentixafor. J Nucl Med. 2015 accepted for publication
74. Pistoia V, Corcione A, Dallegri F. et al. Lymphoproliferative disorders and chemokines. Curr Drug Targets. 2006;7:81-90
75. Sandstrom M, Velikyan I, Garske-Roman U. et al. Comparative biodistribution and radiation dosimetry of 68Ga-DOTATOC and 68Ga-DOTATATE in patients with neuroendocrine tumors. J Nucl Med. 2013;54:1755-1759
76. Dong F, Harvey J, Finan A. et al. Myocardial CXCR4 expression is required for mesenchymal stem cell mediated repair following acute myocardial infarction. Circulation. 2012;126:314-24
77. Ruscher K, Kuric E, Liu Y. et al. Inhibition of CXCL12 signaling attenuates the postischemic immune response and improves functional recovery after stroke. Cereb Blood Flow Metab. 2013;33:1225-34
78. Hummel S, Van Aken H, Zarbock A. Inhibitors of CXC chemokine receptor type 4: putative therapeutic approaches in inflammatory diseases. Curr Opin Hematol. 2014;21:29-36
79. Sánchez-Martín L, Sánchez-Mateos P, Cabanas C. CXCR7 impact on CXCL12 biology and disease. Trends Mol. Med. 2013;19:12-23
80. Peled A, Abraham M, Avivi I. et al. The high-affinity CXCR4 antagonist BKT140 is safe and induces a robust mobilization of human CD34+ cells in patients with multiple myeloma. Clin. Cancer Res. 2014;20:469-79
81. U.S. National Institutes of Health. ClinicalTrials.gov is a registry and results database of publicly and privately supported clinical studies of human participants conducted around the world; identifier: NCT01120457 and NCT01359657. http://clinicaltrials.gov
82. Kuhne MR, Mulvey T, Belanger B. et al. BMS-936564/MDX-1338: a fully human anti-CXCR4 antibody induces apoptosis in vitro and shows antitumor activity in vivo in hematologic malignancies. Clin Cancer Res. 2013;19:357-66
83. van Essen M, Krenning EP, Kam BL. et al. Peptide-receptor radionuclide therapy for endocrine tumors. Nat Rev Endocrinol. 2009;5:382-93
Author contact
![]() Corresponding author: Hans-Jürgen Wester: phone +49-89-2891 2212; fax +49-89-2391 2204; E-mail: h.j.westerde
Corresponding author: Hans-Jürgen Wester: phone +49-89-2891 2212; fax +49-89-2391 2204; E-mail: h.j.westerde