Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2015; 5(2):110-123. doi:10.7150/thno.9717 This issue Cite
Research Paper
Chitosan/siRNA Nanoparticles Targeting Cyclooxygenase Type 2 Attenuate Unilateral Ureteral Obstruction-induced Kidney Injury in Mice
Chuanxu Yang2*, Line Nilsson1*, Muhammad Umar Cheema1, Yan Wang1, Jørgen Frøkiær1, Shan Gao2 ![]() , Jørgen Kjems2, Rikke Nørregaard1
, Jørgen Kjems2, Rikke Nørregaard1 ![]()
1. Department of Clinical Medicine, Aarhus University, Denmark;
2. Interdisciplinary Nanoscience Center, Department of Molecular Biology and Genetics, Aarhus University, Denmark.
*Chuanxu Yang and Line Nilsson contributed equally to this study.
Received 2014-5-22; Accepted 2014-9-22; Published 2015-1-1
Abstract

Cyclooxygenase type 2 (COX-2) plays a predominant role in the progression of kidney injury in obstructive nephropathy. The aim of this study was to test the efficacy of chitosan/small interfering RNA (siRNA) nanoparticles to knockdown COX-2 specifically in macrophages to prevent kidney injury induced by unilateral ureteral obstruction (UUO). Using optical imaging techniques and confocal microscopy, we demonstrated that chitosan/siRNA nanoparticles accumulated in macrophages in the obstructed kidney. Consistent with the imaging data, the obstructed kidney contained a higher amount of siRNA and macrophages. Chitosan-formulated siRNA against COX-2 was evaluated on RAW macrophages demonstrating reduced COX-2 expression and activity after LPS stimulation. Injection of COX-2 chitosan/siRNA nanoparticles in mice subjected to three-day UUO diminished the UUO-induced COX-2 expression. Likewise, macrophages in the obstructed kidney had reduced COX-2 immunoreactivity, and histological examination showed lesser tubular damage in COX-2 siRNA-treated UUO mice. Parenchymal inflammation, assessed by tumor necrosis factor-alpha (TNF-α) and interleukin 6 mRNA expression, was attenuated by COX-2 siRNA. Furthermore, treatment with COX-2 siRNA reduced heme oxygenase-1 and cleaved caspase-3 in UUO mice, indicating lesser oxidative stress and apoptosis. Our results demonstrate a novel strategy to prevent UUO-induced kidney damage by using chitosan/siRNA nanoparticles to knockdown COX-2 specifically in macrophages.
Keywords: Cyclooxygenase type 2, siRNA, chitosan, unilateral ureteral obstruction, mice.
Introduction
Ureteral obstruction is characterized by elevated intraluminal pressure, immediate macrophage infiltration, increased cytokine levels, and interstitial inflammation as well as oxidative stress [1-4], leading to marked induction of the inflammatory enzyme cyclooxygenase type 2 (COX-2). Previously, the importance of COX-2 in the inflammatory response to ureteral obstruction has been document [5-7]. Moreover, the elevated intraluminal pressure throughout the nephrons induces tubular apoptosis and leads to morphological changes and loss of the renal parenchyma [8]. In this study, we provided data demonstrating that targeting COX-2 in macrophages by i.p. injection of COX-2 chitosan/siRNA nanoparticles reduce renal tubular damage, inflammation, oxidative stress and apoptosis in mice subjected to three-day unilateral ureteral obstruction (UUO).
Cyclooxygenases are responsible for prostaglandin synthesis from arachidonic acid and are present in two isoforms in the kidney, COX-1 and COX-2. COX-1 is involved in the regulation of basic cellular functions, whereas COX-2 is a proinflammatory enzyme and is induced by inflammatory stimuli [9]. Specific COX-2 pharmacological inhibition has been suggested to prevent kidney damage and apoptosis in UUO models [10]. However, COX-2 inhibitors can cause side effects such as compromising the glomerular filtration rate, peripheral oedema, and hypertension, which could increase the risk of cardiovascular complications [11]. Based on these findings, treatments interfering with COX-2 expression could be considered as new therapeutic approaches to prevent or minimize the development of renal damage, apoptosis, inflammation, and oxidative stress in a UUO model.
Small interfering RNA (siRNA)-mediated knockdown of proinflammatory cytokines at the mRNA level, known as RNA interference, represents an alternative therapeutic strategy for inflammation. Therapies using siRNA have being developed for various clinical conditions, including acute kidney injury [12], diabetic nephropathy [13], and primary glomerular diseases [14], but they have primarily been used to treat cancer and viral infections [15, 16]. Chitosan, a naturally occurring cationic polysaccharide is an attractive drug delivery material due to its biocompatibility, low toxicity and low immunogenicity [17, 18]. Chitosan has been successfully used for mucosal delivery such as oral, nasal, ocular and pulmonary due to its mucoadhesive and mucosa permeation properties [19, 20]. More recently, chitosan has been developed as siRNA carriers due to its the protonatable amine groups capable of interacting with the negatively charged siRNA and facilitate efficient cellular uptake by promoting escape endosomes [21-23]. We have previously demonstrated that targeting TNF-α in macrophages by intraperitoneal (i.p.) injection of chitosan/Dicer-substrate siRNA (DsiRNA) nanoparticles prevents radiation-induced fibrosis and rheumatoid arthritis in mice without showing cytotoxic side effects [24]. Therefore, administration of chitosan/DsiRNA nanoparticles i.p. allows direct delivery into macrophages as a method to induce both local and systemic effects. Furthermore, because macrophages are recruited to local sites of the inflamed kidney and are critical during the UUO inflammatory response, they are an ideal target for RNAi-based COX-2 therapies.
This study was designed to investigate a novel treatment for UUO-induced renal damage using knockdown of COX-2 via chitosan/siRNA nanoparticles to reduce renal inflammation, apoptosis, and oxidative stress by targeting macrophages i.p.
Materials and Methods
Chemicals and siRNA
Chitosan was purchased from Heppe Medical Chitosan GmbH (150 kDa, 95% deacetylation; Frankfurt, Germany). Three candidate siRNA duplexes against murine COX-2 were synthesized from GenePharma (Shanghai, China) with the sequences: siRNA-1, sense 5′-GGAUUUGACCAGUAUAAGUTT-3′, antisense 5′-ACUUAUACUGGUCAAAUCCTG-3′; siRNA-2, sense 5′-AGACAGAUCAUAAGCGAGGTT-3′, antisense 5′-CCUCGCUUAUGAUCUGUCUTT-3′; siRNA-3, sense 5′-AACCUCGUCCAGAUGCUAUTT-3′, antisense 5′-AUAGCAUCUGGACGAGGUUTT-3′, negative control siRNA, sense 5′-GACGUAAACGGCCACAAGUTC-3′, antisense 5′-ACUUGUGGCCGUUUACGUCGC-3′. For in vivo fluorescence imaging and for immunofluorescent staining, Cy5-labelled siRNA (Ribotask, Odense, Denmark) was used, sense 5′-Cy5-GACGUAAACGGCCACAAGUTC-3′, antisense 5′-ACUUGUGGCCGUUUACGUCGC-3′. For in vivo knockdown experiment, LNA-modified siRNA (Ribotask) was used: siCOX-2, sense 5′-GGAUUUGACCAGUAUAAGUTT-3′, antisense 5′-ACUUAUACUGGUCAAAUCCTG-3′; negative control siEGFP sense 5′-GACGUAAACGGCCACAAGUTC-3′, antisense 5′-ACUUGUGGCCGUUUACGUCGC-3′. The LNA modification is indicated in bold.
Formation of chitosan/siRNA nanoparticles
Chitosan/siRNA nanoparticles were formulated as described in our earlier reports with some modifications [25]. Briefly, chitosan was dissolved in sodium acetate buffer (300 mM NaAc, pH 5.5) to obtain a 1 mg/mL solution. Twenty microliters of siRNA (100 μM) was added to 1 mL filtered chitosan solution while stirring for 1 h. The obtained nanoparticles were concentrated using an Amicon ultracentrifuge tube (10 kDa, MWCO).
COX-2 knockdown in murine macrophages in vitro
The murine macrophage cell line RAW 264.7 (ATCC, Manassas, VA, USA) was used to evaluate candidate siRNAs against COX-2. The cells were maintained in RPMI media supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin at 37°C in 5% CO2 and 100% humidity. One day before transfection, cells were seeded in a 12-well plate at 2 × 105 cells/well and incubated overnight. siRNAs were mixed with TransIT-TKO® (Mirius Bio, Madison, WI, USA) according to the commercial protocol and added at 50 nM final siRNA concentration per well. Following incubation for 24 hours, the medium was changed to stimulation medium containing 100 ng/mL LPS, and the cells were incubated for another 6 hours. COX-2 mRNA levels were quantified by real-time PCR, and protein levels were detected by western blotting. Meanwhile, the supernatants were harvested for PGE2 measurement using the Prostaglandin E2 EIA Monoclonal Kit (Cayman Chemical, Ann Arbor, MI, USA), according to the manufacturer protocol.
Experimental animals
All animal procedures were approved by the Animal Experiments Inspectorate, under the Danish Veterinary and Food Administration, license no. 2011/561-2007. Studies were performed on 10-week old adult male C57BL/6 mice. Animals had free access to a standard rodent diet (Altromin, Lage, Germany) and tap water. During the experiments, animals were kept in groups of three mice per cage, with a 12:12-hour light-dark cycle, at a temperature of 21 ± 2ºC, and at 55 ± 2% humidity. Animals were allowed to acclimate to the cages 7 days before surgery.
In vivo imaging of chitosan/siRNA nanoparticle accumulation in obstructed kidney
A UUO renal inflammatory model was performed by ligating left ureter as previously described [2]. Briefly, mice received sevoflurane anaesthesia and were placed on a heating pad to maintain body temperature. Through a midline abdominal incision, the left ureter was exposed and occluded with a 6-0 silk ligature. To confirm whether chitosan/siRNA nanoparticles would specifically accumulate in the left obstructed kidney, UUO mice were monitored by in vivo fluorescent imaging. At 3 days post-surgery, the mice were injected i.p. with nanoparticles containing Cy5-labelled siRNA at a dose of 0.5 mg/kg. After 1, 2, 4, and 20 hours post-injection, the mice were scanned using an IVIS® 200 imaging system (Xenogen, Caliper Life Sciences, Hopkinton, MA, USA) under anesthesia with 2.5% isoflurane. Cy5 excitation (λex = 640 nm) and emission (λem = 700 nm) filters were used. At 20 hours post-injection, the mice were sacrificed, and the individual organs were collected and scanned. Meanwhile, two control groups were imaged, including sham-operated mice that were injected with Cy5-labelled nanoparticles and UUO mice that were injected with buffer.
The fluorescent intensity from each kidney was quantified using the Living Image 4.0 software package (Caliper Life Sciences). The radiant efficiency of the kidney was measured (photons/sec/cm2/sr)/(μW/cm2), which presents radiance/illumination power density. Background fluorescence was subtracted prior to analysis.
Northern blotting analysis of siRNA distribution and integrity
Total RNA from each kidney was isolated by Trizol® reagent (Invitrogen, Copenhagen, Denmark) according to manufacturer's protocol. Four micrograms total RNA from kidneys were run on a 15% denatured polyacrylamide gel and transferred onto Hybond™-N+ membrane (Amershan Biosciences, Copenhagen, Denmark). Nanoparticles containing 2, 0.1, or 0.01 ng siRNA were also included as control samples. After UV-cross-linking, the membrane was probed with [γ-32P] ATP-labelled sense strand LNA-modified siRNA, according to the standard procedure of northern blotting [26]. The membrane was visualized through and analysed on the Typhoon scanner.
Isolation of primary peritoneal macrophages
Mice were subjected to 3 day UUO as previously described and injected i.p. with 200 µl chitosan/Cy-5-siRNA nanoparticles at a dose of 0.25 mg/kg or buffer solution 2 hour prior to termination. Primary peritoneal macrophages were harvested by injecting 10 ml PBS into the peritoneal cavity and the abdomen was gently agitated for 10 s. The PBS containing resident peritoneal cells was slowly withdraw, and the cell suspension centrifuged and the pellet were plated in RPMI 1640 cell culture medium (Gibco, Invitrogen) supplemented with 10% heat inactivated FCS containing 50 IU penicillin, 50µg streptomycin, and 2 mM glutamine (PSG) per ml (Gibco, Invitrogen). The macrophages were allowed to adhere for 2 hours before non-adherent cells were wash away with sterile PBS. Uptake of Cy-5 labeled siRNA was monitored in adhesive macrophages by a Zeiss semi-confocal epifluorescence microscope.
Chitosan/siRNA nanoparticle-mediated COX-2 knockdown in 3dUUO mice
Three-day UUO mice were randomly allocated into three groups receiving i.p. injections of chitosan/siCOX-2 nanoparticles or chitosan/siEGFP nanoparticles as a negative control. Three days prior to UUO surgery, chitosan/siRNA nanoparticles (COX-2 siRNA (n = 6) or control siRNA (n = 6)) were injected i.p. at a dose of 0.5 mg/kg (siRNA/body weight), and, hereafter, the mice were treated every second day throughout the study. UUO was induced for 3 days, a blood sample was taken from the left ventricle, mice were sacrificed, and the kidneys were collected for QPCR, immunoblotting, and immunofluorescence. Time-matched, sham-operated controls were prepared and observed in parallel (n = 6). Plasma osmolality as well as plasma creatinine, urea, potassium, and sodium were measured (Supplementary Material: Table S1) (Roche Cobas 6000 analyzer, Roche Diagnostic, Hvidovre, Denmark).
RNA isolation and quantitative PCR analysis
RNA was extracted from cells or tissues using Trizol® reagent (Invitrogen), according to manufacturer's protocol. Following the cDNA synthesis (Superscript® II Reverse Transcriptase, Invitrogen), the obtained cDNA served as the template for quantitative PCR (QPCR) using the SYBR® Green kit (Invitrogen) running on a LightCycler® 480 Real-Time PCR System (Roche). Primers used for QPCR amplification are specified in Supplementary Material (Table S2).
Semiquantitative immunoblotting
RAW 264.7 cells were collected and lysed using the M-PER Mammalian Protein Extraction Reagent (Thermo Scientific, Vedbaek, Denmark). Cell suspensions were centrifuged at 14,000 × g at room temperature for 10 min. The kidney was homogenized and centrifuged at 1,000 × g for 15 min at 4°C, and the supernatant was used for immunoblotting [6]. Samples were run on 12% polyacrylamide gels (Criterion™ TGX™ Precast Gel, Bio-Rad, Copenhagen, Denmark), electroeluted to nitrocellulose membranes, and subjected to immunolabelling.
Histology
Kidneys from UUO mice and sham-operated control mice were fixed by retrograde perfusion via the abdominal aorta with 4% paraformaldehyde in 0.01 M PBS buffer. Next, kidneys were fixed for an additional hour and washed 3 times (10 min) with 0.01 M PBS buffer. Fixed kidneys were then dehydrated, embedded in paraffin, and cut into 2-µm sections on a rotary microtome (Leica Microsystems A/S, Herlev, Denmark).
Paraffin-embedded sections were stained with hematoxylin and eosin to assess the grade of tubular damage. Under high magnification (20x), 10 non-overlapping fields from each section of the renal cortex were photographed. The tubular luminal area of each section was measured using the image analysis software ImageJ (http://rsbweb.nih.gov/ij/, National Institutes of Health, USA). All analyses were performed blind. Representative pictures of the renal cortex are shown in 40x magnification.
Fluorescence confocal microscopy
To evaluate siRNA localisation in the kidney, mice were subjected to 3 day UUO and injected i.p. with nanoparticles containing Cy5-labelled siRNA at a dose of 0.5 mg/kg 4 hours prior termination. For immunofluorescence, the kidneys were immersion-fixed in 4% paraformaldehyde, and samples were prepared, embedded in paraffin, and processed for immunofluorescence by using previously characterized antibodies. For Mac-2 labelling, sections were de-waxed, rehydrated, blocked with Protein Block (Dako, Glostrup, Denmark), and incubated with primary antibody diluted in 1% BSA in TBS for 1 h at room temperature. Hereafter, they were washed in TBST and subsequently incubated with secondary antibody. For COX-2 labelling, sections were de-waxed, rehydrated, blocked with 0.33% H2O2 diluted in methanol, and incubated with TEG-buffer for 10 min. at 100°C. After cooling, sections were quenched in 50 mM NH4Cl in PBS and preincubated with blocking solution (1% BSA in PBS). Sections were incubated overnight at 4°C with primary antibody diluted in 0.1% BSA in PBS plus 0.3% Triton X-100, subsequently washed, and incubated with secondary antibody. Sections were counterstained for TOPRO-3 (Life Technologies, San Fransisco, CA, USA) or DAPI (Life Technologies). Coverslips were mounted with SlowFade® Light Antifade Kit (Life Technologies). Fluorescent imaging was performed on a Leica DM IRE2 inverted confocal microscope using a Leica TCS SP2 laser mole and an HCX PC APO CS 63x/1.32 NA oil objective with 8-bit depth. Fluorescence signals were captured within the dynamic range of signal intensity. The fluorescence signals and background were corrected by using ImageJ (http://rsbweb.nih.gov/ij/, National Institutes of Health, USA).
Primary antibodies
COX-2 (Abcam, Cambridge, UK); SOD1 and HO-1 (Enzo, Farmingdale, NY, USA); SOD2 (Merck Millipore, Darmstadt, Germany); KIM-1 (R&D Systems, Abingdon, UK); caspase 3, cleaved caspase 3, and GAPDH (Cell Signaling, Danvers, MA, USA); and Mac-2 (Cederlane, Burlington, ON, Canada) were used for experiments.
Statistics
Two experimental groups were compared using Student's t-test for unpaired observations. For comparisons of more than two groups, one-way ANOVAs followed by post hoc t-tests were performed. p-values of <0.05 was considered significant.
Results
Accumulation of chitosan/siRNA nanoparticles in obstructed kidneys
Chitosan/siRNA nanoparticles have previously been shown to accumulate at an inflammatory site in vivo, presumably because of macrophage migration [27]. To evaluate the biodistribution of chitosan/siRNA nanoparticles in the UUO model, Cy5-labelled siRNA was formulated with chitosan and injected i.p. into UUO mice. Two control groups were included: Sham-operated mice that were injected with the same Cy5-labelled siRNA nanoparticles and UUO mice that were injected with PBS buffer. The mice were scanned at different time points using an IVIS® 200 imaging system. A strong fluorescent signal was observed in the region of the obstructed left kidney at 1 h after injection (Fig. 1A), whereas no clear signal was observed in the non-obstructed right kidney. More importantly, the Cy5 signal remained strong in the obstructed kidney at 2 and 4 h post-injection and persisted for 20 hours. In contrast, no significant fluorescent signals, compared to the auto-fluorescence of the body, were detected in the two control groups (Fig. 1A). These results suggest that chitosan/siRNA nanoparticles specifically accumulated in the inflamed kidney.
Optical fluorescence imaging of chitosan/Cy5 siRNA nanoparticles in a murine UUO model. (A) Mice subjected to sham operation or 20-hour UUO were administered chitosan/Cy5-labelled siRNA nanoparticles or buffer i.p. Fluorescent optical imaging was performed at distinct time points before and 1, 2, 4 and 20 hours post-injection of chitosan/Cy5-siRNA. (B) Ex vivo fluorescent imaging was performed on isolated organs, including the liver, lung, spleen, heart, and kidneys 20 h after injection of chitosan/Cy5-siRNA nanoparticles on mice subjected to sham operation or UUO and showed Cy5 signal restricted to the renal region. (C) Ex vivo fluorescent imaging of Cy5 signal from the obstructed and unobstructed kidneys 20 hours after injection of chitosan/Cy5-siRNA nanoparticles. (D) Quantification of fluorescence intensity in the obstructed (LK) and unobstructed kidney (RK) 20 h after injection of chitosan/Cy5-siRNA nanoparticles.
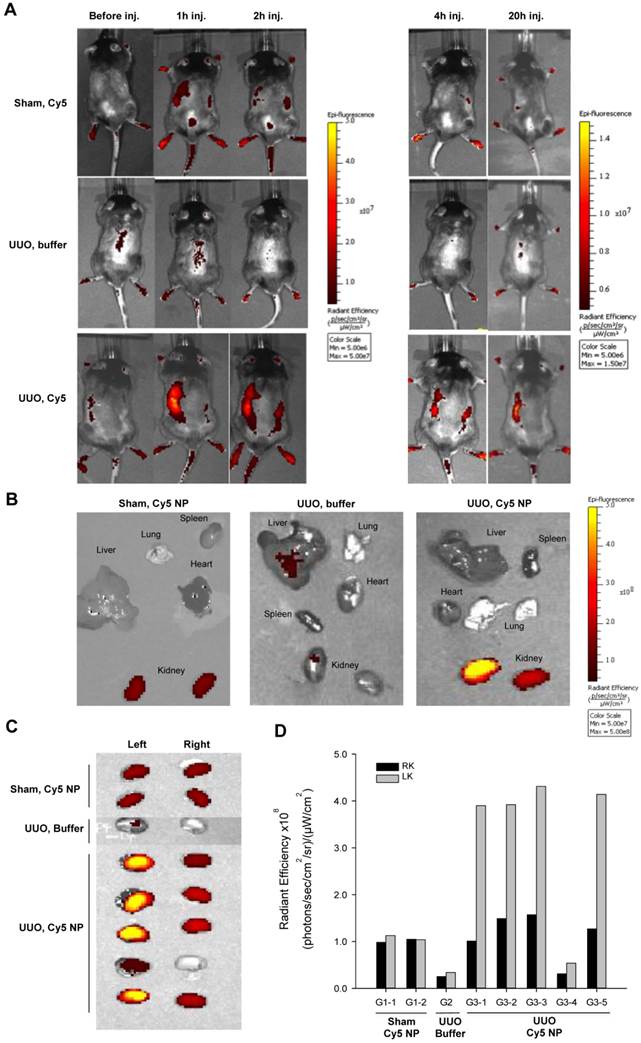
To study the biodistribution of chitosan/siRNA nanoparticles, we sacrificed all mice at 20 hours post-injection and isolated the organs for ex vivo fluorescent imaging. First, we compared the distribution of Cy5 signals in different organs, including the liver, spleen, kidney, and lung (Fig. 1B). A significantly higher Cy5 signal was emitted from the obstructed left kidney compared to the non-obstructed right kidney as well as other organs, confirming the specific Cy5-labelled siRNA accumulation in the obstructed kidney. In contrast, the sham-operated mice injected with the same amount of nanoparticles showed very weak signals from both kidneys, while the fluorescent signal from all other organs was at the level of the background. UUO mice injected with buffer also showed no or very low signal. To better compare the nanoparticle or siRNA concentrations, we scanned both kidneys from all mice in one frame. As shown in Figure 1C, the Cy5 signals from the obstructed left kidneys in the UUO group were significantly higher than those were from the right kidneys. The fluorescence intensity, defined as the radiant efficiency [(photons/sec/cm2/sr)/(μW/cm2)], was measured from each kidney using the Living Image 4.0 software package (Caliper Life Sciences, Hopkinton, MA, USA). The radiant efficiencies of the left kidneys in the UUO group were approximately 2.5-fold higher than those were of the right kidneys, which were comparable to kidneys from the sham-operated mice (Fig. 1D). It should be noted that the relatively low fluorescent signal from the kidney of mouse G3-4 was due to a poor i.p. injection, and this was also confirmed by quantifying the siRNA deposition by northern blot.
siRNA biodistribution in macrophages in the obstructed kidney
To investigate the integrity of the siRNA after accumulation in the obstructed kidney, RNA samples isolated from kidneys were analyzed by northern blotting. Three control samples containing a pre-determined amount of siRNA were included for quantification of the siRNA. The siRNA was predominately full length in all samples together with a minor component of 1-2 nucleotide shorter product (Fig. 2A). In accordance with our imaging data, there was a significantly higher amount of siRNA observed in the obstructed kidneys compared to both the non-obstructed kidneys and kidneys from sham-operated mice (Figs. 2A and B).
siRNA distribution and macrophage accumulation in response ureteral obstruction. Mice were subjected to 3-day UUO and treated with Cy5-labeled siRNA nanoparticles i.p. at a dose of 0.5 mg/kg as described in materials and methods. (A) Total RNA was isolated from whole kidney tissue by Trizol reagent, and Northern blot was performed to analyse siRNA distribution in the right (RK) and left (LK) kidney from both sham-operated and UUO mice. Nanoparticles containing 2, 0.1, or 0.01 ng siRNA were included as control samples, Ctrl1, Ctrl 2 and Ctrl 3 respectively. (B) Quantification of Cy5-labeled siRNA nanoparticle amount in the left kidney and right kidney. (C) QPCR analysis was performed to analyse the macrophage marker CD68 mRNA levels in left and right kidney from sham-operated and 3-day UUO mice. (D) Fluorescent micrograph showing chitosan siRNA nanoparticle uptake in macrophages extracted 2 hours after i.p. administration (0.25 mg/kg chitosan/Cy5-siRNA nanoparticles). Merge pictures shows co-localization of Cy5 and the M2 macrophage marker, Mac-2, combined with DAPI. (E) Mice were subjected to 3 days UUO and treated with Cy5-flourescent labeled siRNA (0.5 mg/kg) 4 hours prior termination. Sections were stained for the macrophage marker, Mac-2. Uptake of Cy5-fluorescent labeled siRNA nanoparticles in Mac-2 positive macrophages was investigated using fluorescent confocal microscopy in UUO mice. Cy5 (red), Mac-2 (green), counterstained for DAPI (blue). Arrows represent Cy5 in Mac-2 positive cell. Original magnification: ×63. Bars = 10 µm.
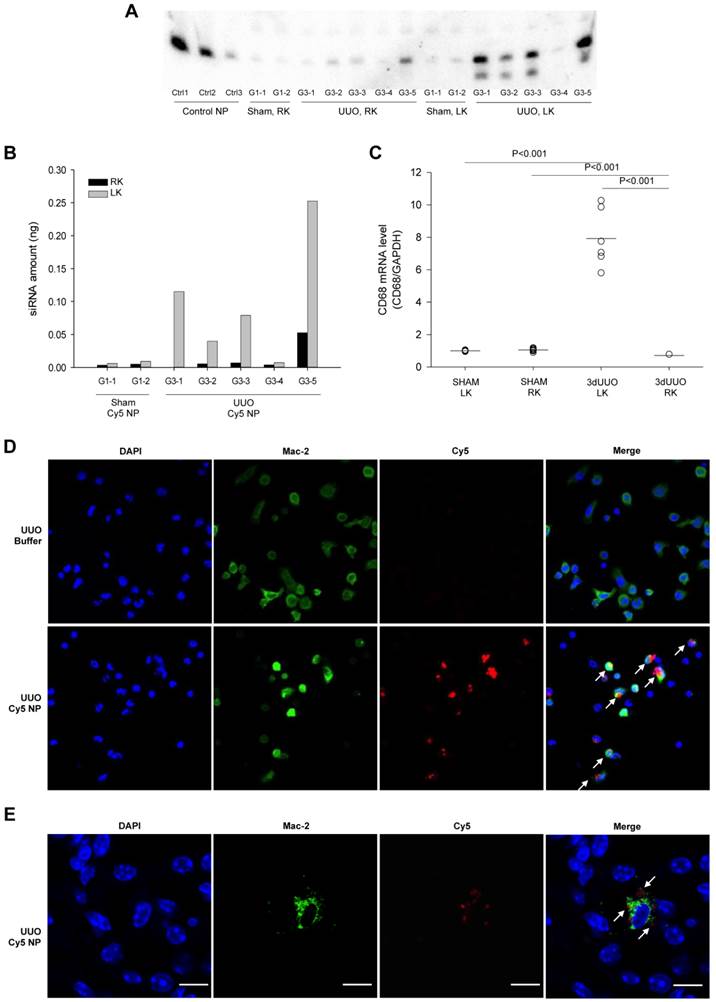
As previous findings showed that chitosan nanoparticles predominantly accumulate in macrophages [27], we analysed the population of macrophages in the obstructed kidney using macrophage-specific CD68 mRNA. A clear increase of CD68 mRNA expression was seen in the obstructed kidneys compared to the contralateral non-obstructed kidneys and kidneys from sham-operated mice, denoting the increased macrophage infiltration (Fig. 2C). The ability of macrophage uptake of chitosan/siRNA nanoparticles was demonstrated by i.p. administration of Cy5 labeled chitosan nanoparticles in mice subjected to 3dUUO. Nanoparticles containing Cy5 labeled siRNA were mainly present in peritoneal macrophages (Mac-2 positive cells) harvested 2 hours after injection (Fig. 2D).
To further assess the localization of the nanoparticles that migrated to the obstructed kidney, we performed fluorescent confocal microscopy using the M2 macrophage marker Mac-2. The Cy5-siRNA signal was detected in the cytoplasm of cells also positive for the Mac-2 marker, indicating that chitosan/Cy5-siRNA nanoparticles resided within the M2 macrophages in the obstructed kidney (Fig. 2E). No signal was observed in the contralateral kidneys or kidneys from sham-operated mice (data not shown).
In vitro knock down of COX-2 in macrophages
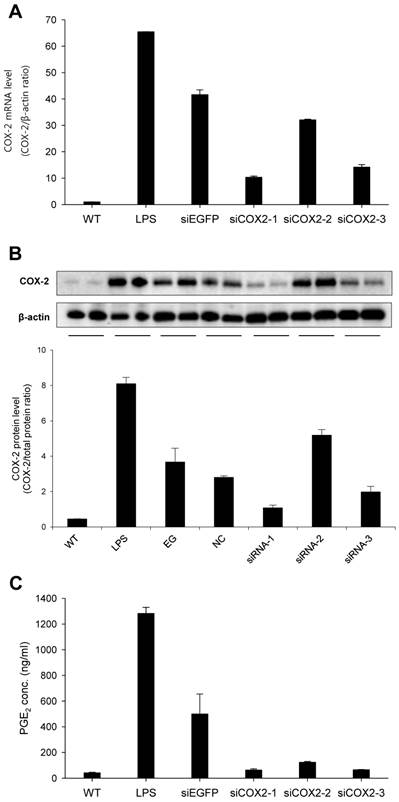
To obtain a highly potent siRNA against murine COX-2 (siCOX2), three sequences were designed and evaluated for COX-2 knockdown in RAW 264.7 macrophages. Following transfection, cells were stimulated with LPS before RNA and protein extraction. As shown in Figure 3, siCOX2-1 significantly reduced COX-2 production at both the mRNA (approximately 75%) (Fig. 3A) and protein levels (approximately 70%) (Fig. 3B) compared to siEGFP transfected RAW 264.7 cells exposed to LPS stimulation. Meanwhile, the supernatants of LPS-stimulated RAW 264.7 cells contained significantly reduced concentrations of prostaglandin E2 (PGE2), a crucial mediator of the inflammatory response during ureteral obstruction, when COX-2 was silenced (Fig. 3C). Because of its silencing efficiency, the siCOX2-1 sequence was chosen, and the 3′-terminally LNA-modified version of the siRNA was synthesized for subsequent in vivo treatments.
Transfection of murine macrophage RAW 264.7 cells. Three siRNA candidates against murine COX-2 were evaluated; siCOX2-1, siCOX2-2, and siCOX2-3. siRNAs were mixed with Trans-IT-TKO® and added at 50nM final siRNA concentration to cell cultures and incubated for 24 hours. Afterwards, transfected cells were stimulated by LPS (100 ng/mL) for 6 hours to induce COX-2. (A) RNA was extracted from cells followed by cDNA synthesis, which served as template for QPCR. COX-2 mRNA levels was investigated by QPCR. (B) Protein was extracted from cells and western blot analysis was performed to detect COX-2 protein levels. ß-actin was run in parallel to ensure equal protein loading. (C) PGE2 levels in cell culture media evaluated by ELISA. Results are means ± SEM. WT: wildtype, LPS: lipopolysaccharide, siEGFP: negative coding control siRNA, NC: non-coding control siRNA, siCOX2: siRNA targeting murine COX-2.
The effect of Chitosan/COX-2 siRNA in mice subjected to UUO in vivo
To investigate the nanoparticle-mediated in vivo silencing of COX-2, mice were subjected to 3-day UUO, and COX-2 knockdown was mediated by i.p. injection of chitosan-formulated COX-2 siRNA nanoparticles. As demonstrated in Figure 4A, the UUO COX-2 mRNA induction was attenuated by treatment with the COX-2 siRNA nanoparticles.
siCOX-2 induced regulation of renal COX-2 and inflammation in mice subjected to 3-day UUO. Mice were treated with chitosan/siCOX-2 nanoparticles or chitosan/siEGFP as negative control. Following three days obstruction, whole kidney tissue was purified for QPCR and western blotting analysis. (A) QPCR analysis of COX-2 mRNA levels. (B) QPCR analysis of TNF-α and (C) IL-6 mRNA levels in response to siCOX-2 treatment. (D) QPCR analysis of CD68 mRNA level in response to 3-day UUO and siCOX-2 treatment. Each graph of statistical dot plots shows the median per cent (black bars) and p value between experimental groups (n = 6).
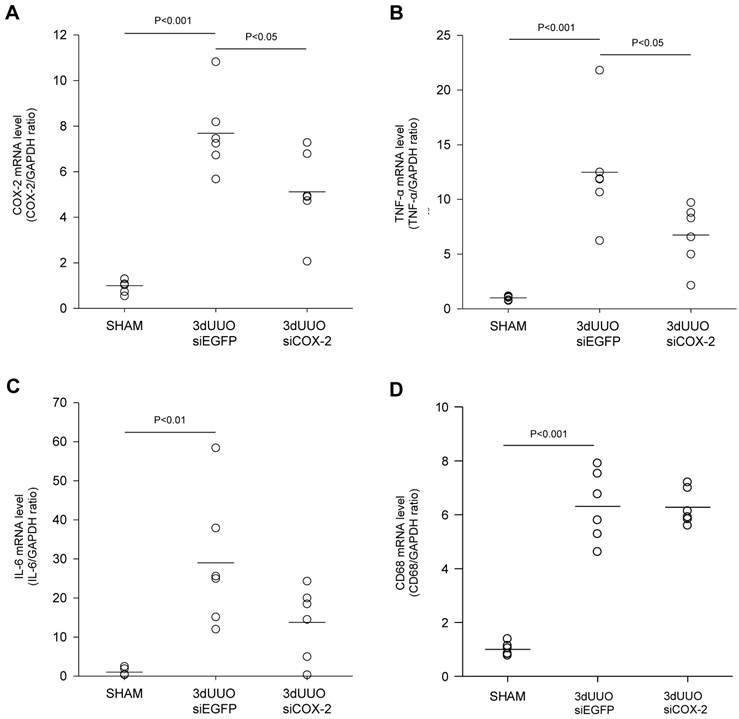
We analyzed the renal inflammatory state by measuring the mRNA expression of TNF-α and IL-6. UUO mice had increased TNF-α and IL-6 mRNA levels, and COX-2 siRNA significantly attenuated TNF-α mRNA levels, whereas IL-6 mRNA was only partially reduced (P=0.08) (Figs. 4B and C). No difference in the amount of macrophages between the COX-2 siRNA treated and untreated UUO mice was observed based on CD68 mRNA quantification (Fig. 4D).
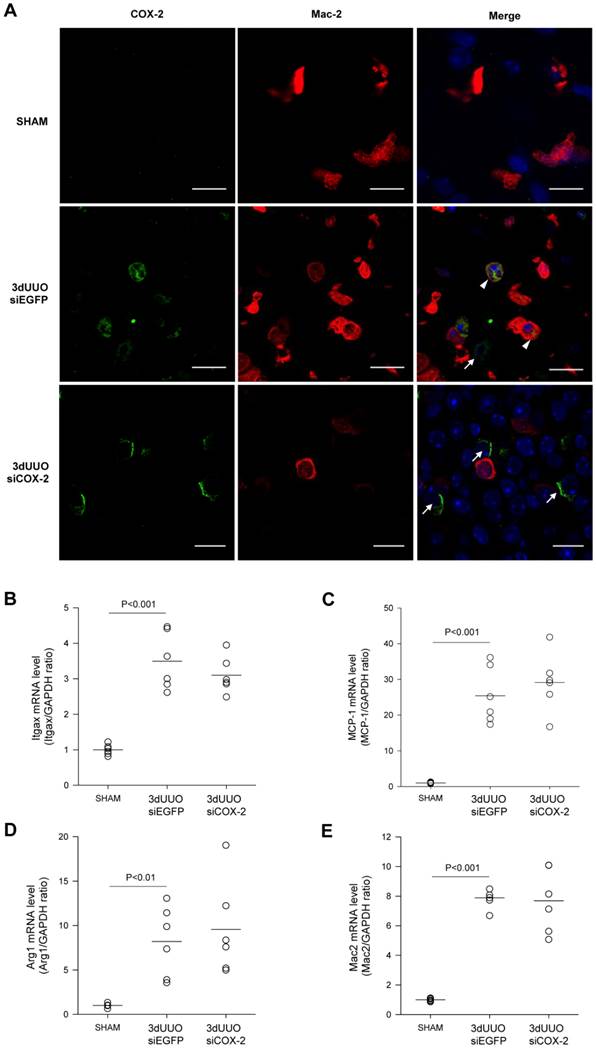
Chitosan nanoparticles containing COX-2 siRNA are supposed to mainly accumulate in the macrophages. To examine whether COX-2 was expressed in the macrophages of the obstructed kidney, we performed double immunofluorescent labelling with antibodies against COX-2 and the M2 macrophage-specific marker Mac-2. As shown in Figure 5A, COX-2 immunoreactivity localized to the macrophages in the obstructed kidney, and after COX-2 siRNA treatment, the number of COX-2 positive cells was diminished. Furthermore, to determine if COX-2 knockdown causes differentiation in macrophage phenotype, we examined distinct M1 and M2 macrophage markers. In response to 3-day UUO we saw a significant increase in the mRNA levels of the M1-specific markers, integrin alpha X (Itgax) and monocyte chemoattractant protein-1 (MCP-1) (Fig. 5B and C). Furthermore, the mRNA levels of the M2-specific markers, arginase 1 (Arg1) and galactose specific lectin 3 (Mac-2), were significantly increased in response to 3-day UUO (Figs. 5D and E). Neither M1, nor M2 markers were affected by COX-2 siRNA treatment. These findings suggest, that the effect of COX-2 siRNA treatment is not through the alteration of macrophage phenotypes.
COX-2 labeling of macrophages in renal inner medullary region and effect of siCOX-2 treatment on macrophage phenotype. (A) Double staining for COX-2 and macrophage marker, Mac-2, in the renal inner medullary region. Sham, 3-day UUO siEGFP, and 3-day UUO siCOX-2 kidney sections stained for Mac-2 (red), COX-2 (green), and counterstained for TOPRO-3 (blue). 'Merge' shows co-localization of COX-2 and Mac-2. Macrophages characterized by colocalization of Mac-2 and COX-2 (arrowheads); Renal interstitial cells characterized by COX-2 positive and Mac-2 negative cells (arrows). Original magnification: ×63. Bars = 10 µm. (B, C) mRNA expression of the M1 macrophage markers, Itgax and MCP-1. (D, E) mRNA expression of the M2 macrophage markers, Arg1 and Mac-2. Each graph of statistical dot plots shows the median per cent (black bars) and p value between experimental groups (n = 6).
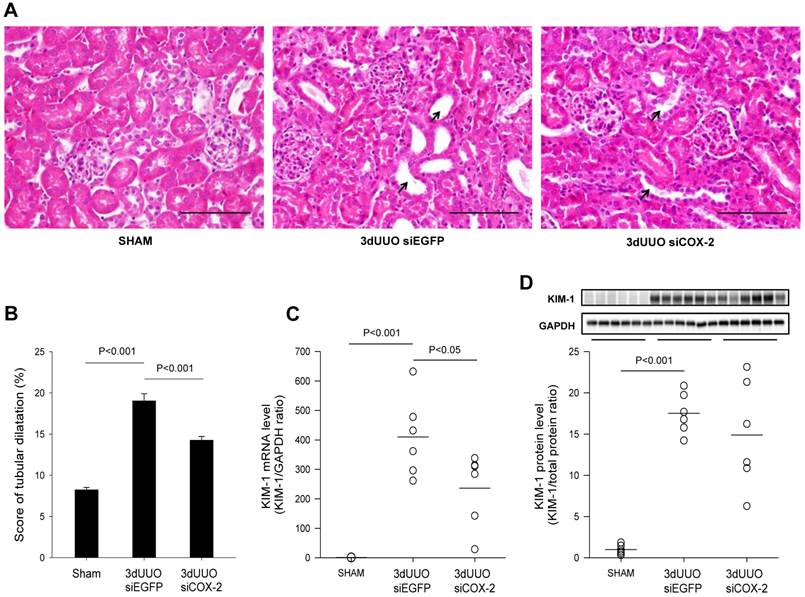
COX-2 siRNA administration minimizes tubular damage in 3-day UUO mice
To determine the effect of the COX-2 siRNA administration on tubular damage, we performed H&E staining on kidney sections. Mice subjected to UUO showed tubular dilatation and severe tubular damage in the obstructed kidneys, which was less pronounced in the COX-2 siRNA-treated mice (Figs. 6A and B). Kidney injury molecule-1 (KIM-1) is regarded as a marker of tubular injury based on its upregulation after injury in the proximal tubular segments of the nephron [28]. We observed a marked upregulation of KIM-1 mRNA and protein levels in response to 3-day UUO. In response to COX-2 treatment KIM-1 mRNA level was significantly attenuated compared with non-treated controls (Fig. 6C). However, COX-2 siRNA treatment did not significantly reduce KIM-1 protein expression although there was a trend (Fig. 6D). Collectively, these findings suggest that COX-2 siRNA administration reduces tubular damage in UUO mice.
Histological evaluation of the effect of COX-2 siRNA on tubular damage in response to 3-day UUO. Following three days ureteral obstruction, renal tissue were fixed in 4% paraformaldehyde, embedded in paraffin and subsequently cut into 2µm sections. To assess the grade of tubular damage sections were stained with hematoxylin and eosin (H&E). Arrows, examples of dilated tubules. (A) Representative images of H&E staining of cortical sections. Magnification: ×40. Bars = 100 µm. (B) H&E analysis of renal tubular damage following 3-day UUO by tubular dilatation (n=20). (C, D) mRNA and protein was obtained from whole kidney tissue for QPCR and western blotting analysis. Renal tubular injury marker KIM-1 mRNA and protein levels in response to 3-day UUO and siCOX-2 treatment. Each graph of statistical dot plots shows the median per cent (black bars) and p value between experimental groups (n = 6).
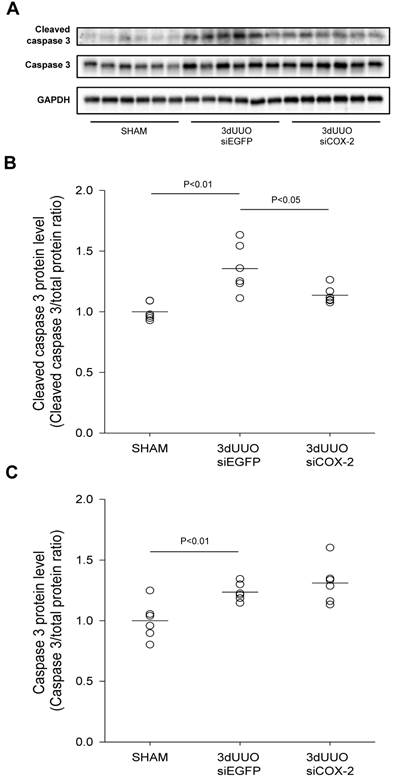
COX-2 siRNA administration attenuated apoptosis and oxidative stress in 3-day UUO mice
Caspase-dependent signalling plays a major role in the development of apoptosis [29]. We observed increased cleaved caspase-3 protein in the obstructed kidneys, demonstrating UUO-induced apoptosis (Figs. 7A, B, and C). COX-2 siRNA treatment significantly reduced the active cleaved caspase-3, indicating reduced apoptosis upon COX-2 knockdown (Fig. 7B).
Effect of COX-2 siRNA on apoptosis in response to 3-day UUO. Mice were subjected to 3-day UUO and treated with siCOX-2 or siEGFP for control. Protein was purified from whole kidney tissue and used for western blot analysis. (A) Immunoblots of caspase-3, and cleaved caspase-3 protein levels, with GAPDH to ensure equal protein loading. (B, C) Apoptosis markers, caspase-3 and cleaved caspase-3, protein levels in response to 3-day UUO and siCOX-2 treatment. Results are shown with total protein normalization. Each graph of statistical dot plots shows the median per cent (black bars) and p value between experimental groups (n = 6).
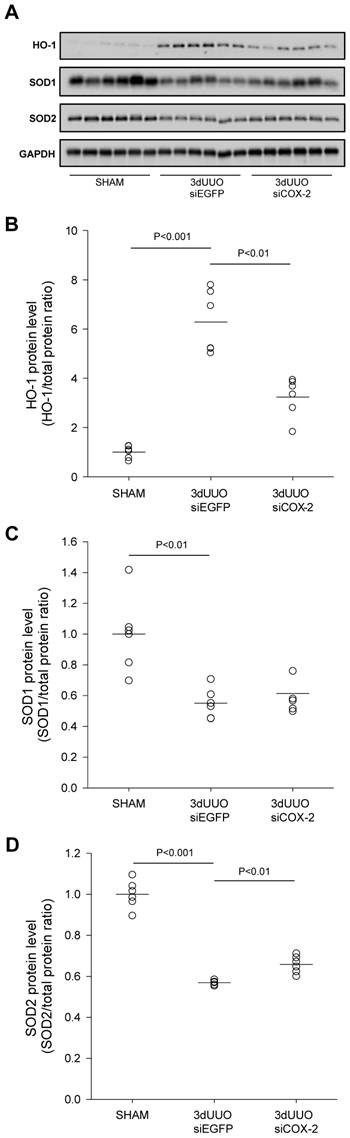
To evaluate the effectiveness of COX-2 siRNA treatment on oxidative stress, renal tissue was analysed for the oxidative stress marker HO-1 and the antioxidant enzymes SOD1 and SOD2. Our results confirmed increased oxidative stress in response to UUO, indicated by a significant increase in the oxidative stress marker HO-1 (Figs. 8A and B). In accordance with this, the level of both the SOD1 and SOD2 antioxidant enzymes decreased in response to UUO (Figs. 8C and D), which confirmed previous findings [30]. Notably, COX-2 siRNA-treated UUO mice had reduced HO-1 protein levels and attenuated the downregulation of SOD2 protein levels.
Effect of COX-2 siRNA on oxidative stress in response to 3-day UUO. Mice were subjected to 3-day UUO and treated with siCOX-2 or siEGFP for control. Protein was purified from whole kidney tissue and used for western blot analysis. (A) Immunoblots for HO-1, SOD1, and SOD2 protein levels, with GAPDH to ensure equal protein loading. (B) Protein analysis of the oxidative stress marker HO-1 protein levels in response to 3-day UUO and siCOX-2 treatment. (C, D) Regulation of antioxidant enzymes SOD1 and SOD2 protein levels in response to 3-day UUO and siCOX-2 treatment. Each graph of statistical dot plots shows the median per cent (black bars) and p value between experimental groups (n = 6).
Taken together, these findings suggest that COX-2 siRNA therapy reduces both oxidative stress and apoptosis in UUO mice.
Discussion
This study describes a novel treatment approach for UUO-induced damage using chitosan/siRNA nanoparticles to knockdown COX-2. COX-2 function has been targeted in many disorders by using COX-2 selective pharmacological inhibitors. Ureteral obstruction increases COX-2 expression significantly [6, 7, 31], and the selective COX-2 inhibitor etodolac has been previously shown to attenuate UUO-induced damage in mice [10]. However, the COX-2 independent side effects of these inhibitors have led to an increased demand for more potent and specific inhibitory agents of COX-2 activity. Here, we demonstrate that siRNA knockdown of COX-2 prevents or minimizes renal damage, inflammation, oxidative stress, and apoptosis in a UUO model, placing RNAi as a new potential therapeutic in COX-2 inhibition.
Efficient therapeutic use of siRNAs depends on their efficient delivery and ability to suppress the expression of target proteins that contribute to the progression of injury. We demonstrated that siRNA targeting COX-2 was effective at reducing UUO-induced COX-2 expression and resulting kidney injury. The utilization of chitosan nanoparticles for i.p. siRNA delivery in our study had three major advantages: (i) the ability to target macrophages through their phagocytic activity while avoiding the serum instability of polyplexes, (ii) the specific accumulation of siRNA into the injured kidney by nature of macrophage homing, and (iii) the efficacy of low-dose siRNA to achieve the therapeutic effect.
Previous studies have shown efficient chitosan/siRNA nanoparticle knockdown of TNF-α as a novel anti-inflammatory therapy for rheumatoid arthritis and radiation-induced fibrosis (RIF) [24, 25, 27]. Similar to the methods of this study, chitosan/siRNA nanoparticles were injected into the serum-free, macrophage-rich peritoneal cavity, where they were phagocytosed by macrophages.
The subsequent recruitment of macrophages to inflammatory tissues is a consequence of inflammatory signals and can lead to tissue repair and fibrosis formation [32]. For the UUO model used in this study, the recruitment of macrophages in the inflammatory kidney was confirmed by the increased expression of CD68, a general macrophage marker. We characterized the macrophages subtypes in this UUO model and demonstrated increased expression of both M1 and M2 related markers in the obstructed kidney. There was no difference in the expression of the M1 or M2 related genes after COX-2 siRNA administration, suggesting that knock-down of COX-2 did not regulate the macrophage phenotype. Furthermore, we also showed COX-2 immunoreactivity in Mac-2-positive macrophages in the obstructed kidney, indicating that COX-2 is expressed in the infiltrating M2 macrophages, which are recruited locally, e.g., from the intraperitoneal cavity to the obstructed kidney. Our approach takes advantage of this homing process to specifically silence COX-2 in the inflamed kidney.
To monitor the delivery process and study the biodistribution of siRNA, an optical imaging system was used to track the fluorescently labelled nanoparticles. As shown in figure 1, chitosan/Cy5-labelled siRNA nanoparticles were primarily present in the obstructed kidney, demonstrating successful targeting with this approach. In addition, the accumulation process occurred quickly, as it was observed at 1 hour post-administration and remained for at least 20 hours, consistent with the previously observations in the RIF model [27]. Furthermore, we showed the association of chitosan/Cy5-labelled siRNA nanoparticles with peritoneal macrophages in the obstructed kidney, indicating that the Cy5-labelled siRNA was carried by macrophages into the obstructed kidney. This targeting effect was further confirmed by northern blotting for the siRNA in the kidneys. These results suggest that this approach successfully delivered siRNA specifically to the obstructed kidney, which is very important to both avoid side effects and enhance the therapeutic effect.
Efficient siRNA delivery to the kidney is challenging due to the rapid clearance of naked siRNA. This normally requires an injection of high-dose siRNA to achieve the silencing effect [33, 34]. In the present study, a significant COX-2 expression knockdown was achieved by only a 10 µg dose of siRNA for each injection for three total injections, which is far less than other reported studies [33]. Furthermore, our strategy of targeting macrophages through intraperitoneal injection also avoids the tight junctions of the glomerular membrane barrier that make it difficult for nanoparticles to reach the targeting sites in the kidney. Effective delivery and therapeutic efficacy were accomplished without the need for either hydrodynamic injection into the tail vein [33] or intrarenal, local delivery via the renal artery [35], renal vein [36], or intrauretral administration in the renal pelvis [37].
The clinical relevance of i.p. administration has been documented by several studies [38, 39]. I.p. administration is easily performed and can, if necessary, be repeated by placing an i.p. catheter which will result in a higher peritoneal content of a certain drug compared to the plasma levels and thereby reducing the systemic toxicity [38]. Since, urinary tract obstruction is well known to stimulate migration of macrophages into the obstructed kidney as a part of the inflammatory response, the strategy of using macrophages to transport chitosan/siRNA may also be an attractive option in experimental treatment of diseased kidneys [2, 40-42]. In fact, a recent phase I clinical trial of i.p. administration of a novel nanoparticle formulation of the antimitotic paclitaxel (NTX) demonstrated that i.p. injection of NTX is well tolerated with minimal systemic exposure (Williamson SK. et al. J Clin Oncol 31, suppl; abstr 2558, 2013) indicating that i.p. administration using nanoparticles may be suitable in a clinical setting as well. Finally, i.p. administration may be an attractive option for patients with peritoneal carcinomatosis.
Conclusion
Several aspects of the data presented here augment the potential clinical application of siRNA for kidney-related disorders. First, the obstructed kidney is the primary site of siRNA distribution following i.p. injection. Second, the delivery of chitosan/siRNA nanoparticles via macrophages minimizes the exposure of other organs. Third, injections of very low doses of COX-2 siRNA attenuated the UUO-induced kidney injury by reducing tubular damage, inflammation, apoptosis, and oxidative stress.
This study demonstrates a novel treatment for UUO-induced kidney damage by using chitosan/siRNA nanoparticles to knockdown COX-2 in macrophages. This approach offers further insight into the development of RNAi-based therapy for inflammatory diseases.
Supplementary Material
Figure S1. Characterization of Chitosan/siRNA nanoparticles. Table S1. Effect of COX-2 siRNA (0.5 mg/kg) on weight, kidney weight and blood chemistry. Table S2. Primers used for QPCR amplification.
Acknowledgements
The authors thank Frederik Dagnaes-Hansen, Gitte Skou, Gitte Kall, and Line V. Nielsen for expert technical assistance. This study was supported by the Danish Research Council for Independent Research - Medical Sciences, the Lundbeck Foundation, the NOVO Nordisk Foundation, and the AP Moller Foundation.
Competing interests
None declared.
References
1. Li S, Mariappan N, Megyesi J. et al. Proximal tubule PPARalpha attenuates renal fibrosis and inflammation caused by unilateral ureteral obstruction. Am J Physiol Renal Physiol. 2013
2. Schreiner GF, Harris KP, Purkerson ML. et al. Immunological aspects of acute ureteral obstruction: immune cell infiltrate in the kidney. Kidney Int. 1988;34:487-493
3. Dendooven A, Ishola DA Jr, Nguyen TQ. et al. Oxidative stress in obstructive nephropathy. Int J Exp Pathol. 2011;92:202-210
4. Ostergaard M, Christensen M, Nilsson L. et al. ROS dependence of cyclooxygenase-2 induction in rats subjected to unilateral ureteral obstruction. Am J Physiol Renal Physiol. 2014;306:F259-70
5. Ricciotti E, FitzGerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol. 2011;31:986-1000
6. Nilsson L, Madsen K, Topcu SO. et al. Disruption of cyclooxygenase-2 prevents downregulation of cortical AQP2 and AQP3 in response to bilateral ureteral obstruction in the mouse. Am J Physiol Renal Physiol. 2012;302:F1430-9
7. Norregaard R, Jensen BL, Topcu SO. et al. Cyclooxygenase type 2 is increased in obstructed rat and human ureter and contributes to pelvic pressure increase after obstruction. Kidney Int. 2006;70:872-881
8. Gobe GC, Axelsen RA. Genesis of renal tubular atrophy in experimental hydronephrosis in the rat. Role of apoptosis. Lab Invest. 1987;56:273-281
9. Harris RC, Zhang MZ. Cyclooxygenase metabolites in the kidney. Compr Physiol. 2011;1:1729-1758
10. Miyajima A, Ito K, Asano T. et al. Does cyclooxygenase-2 inhibitor prevent renal tissue damage in unilateral ureteral obstruction? J Urol. 2001;166:1124-1129
11. Harris RC, Breyer MD. Update on cyclooxygenase-2 inhibitors. Clin J Am Soc Nephrol. 2006;1:236-245
12. Molitoris BA, Dagher PC, Sandoval RM. et al. siRNA targeted to p53 attenuates ischemic and cisplatin-induced acute kidney injury. J Am Soc Nephrol. 2009;20:1754-1764
13. Xie P, Sun L, Oates PJ. et al. Pathobiology of renal-specific oxidoreductase/myo-inositol oxygenase in diabetic nephropathy: its implications in tubulointerstitial fibrosis. Am J Physiol Renal Physiol. 2010;298:F1393-404
14. Takabatake Y, Isaka Y, Mizui M. et al. Chemically modified siRNA prolonged RNA interference in renal disease. Biochem Biophys Res Commun. 2007;363:432-437
15. Whitehead KA, Langer R, Anderson DG. Knocking down barriers: advances in siRNA delivery. Nat Rev Drug Discov. 2009;8:129-138
16. Akhtar S, Benter IF. Nonviral delivery of synthetic siRNAs in vivo. J Clin Invest. 2007;117:3623-3632
17. Dash M, Chiellini F, Ottenbrite RM. et al. Chitosan—A versatile semi-synthetic polymer in biomedical applications. Progress in Polymer Science. 2011;36:981-1014
18. Ragelle H, Vandermeulen G, Preat V. Chitosan-based siRNA delivery systems. J Control Release. 2013;172:207-218
19. de la Fuente M, Ravina M, Paolicelli P. et al. Chitosan-based nanostructures: a delivery platform for ocular therapeutics. Adv Drug Deliv Rev. 2010;62:100-117
20. Chen MC, Mi FL, Liao ZX. et al. Recent advances in chitosan-based nanoparticles for oral delivery of macromolecules. Adv Drug Deliv Rev. 2013;65:865-879
21. Howard KA, Rahbek UL, Liu X. et al. RNA interference in vitro and in vivo using a novel chitosan/siRNA nanoparticle system. Mol Ther. 2006;14:476-484
22. Ma Z, Yang C, Song W. et al. Chitosan Hydrogel as siRNA vector for prolonged gene silencing. J Nanobiotechnology. 2014;12:23_3155_12_23
23. Liu X, Howard KA, Dong M. et al. The influence of polymeric properties on chitosan/siRNA nanoparticle formulation and gene silencing. Biomaterials. 2007;28:1280-1288
24. Howard KA, Paludan SR, Behlke MA. et al. Chitosan/siRNA nanoparticle-mediated TNF-alpha knockdown in peritoneal macrophages for anti-inflammatory treatment in a murine arthritis model. Mol Ther. 2009;17:162-168
25. Nawroth I, Alsner J, Behlke MA. et al. Intraperitoneal administration of chitosan/DsiRNA nanoparticles targeting TNFalpha prevents radiation-induced fibrosis. Radiother Oncol. 2010;97:143-148
26. Gao S, Dagnaes-Hansen F, Nielsen EJ. et al. The effect of chemical modification and nanoparticle formulation on stability and biodistribution of siRNA in mice. Mol Ther. 2009;17:1225-1233
27. Nawroth I, Alsner J, Deleuran BW. et al. Peritoneal macrophages mediated delivery of chitosan/siRNA nanoparticle to the lesion site in a murine radiation-induced fibrosis model. Acta Oncol. 2013;52:1730-1738
28. Ichimura T, Bonventre JV, Bailly V. et al. Kidney injury molecule-1 (KIM-1), a putative epithelial cell adhesion molecule containing a novel immunoglobulin domain, is up-regulated in renal cells after injury. J Biol Chem. 1998;273:4135-4142
29. Favaloro B, Allocati N, Graziano V. et al. Role of apoptosis in disease. Aging (Albany NY). 2012;4:330-349
30. Cho MH, Jung KJ, Jang HS. et al. Orchiectomy attenuates kidney fibrosis after ureteral obstruction by reduction of oxidative stress in mice. Am J Nephrol. 2012;35:7-16
31. Norregaard R, Jensen BL, Topcu SO. et al. Urinary tract obstruction induces transient accumulation of COX-2-derived prostanoids in kidney tissue. Am J Physiol Regul Integr Comp Physiol. 2010;298:R1017-25
32. Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11:723-737
33. van de Water FM, Boerman OC, Wouterse AC. et al. Intravenously administered short interfering RNA accumulates in the kidney and selectively suppresses gene function in renal proximal tubules. Drug Metab Dispos. 2006;34:1393-1397
34. Wesche-Soldato DE, Chung CS, Lomas-Neira J. et al. In vivo delivery of caspase-8 or Fas siRNA improves the survival of septic mice. Blood. 2005;106:2295-2301
35. Takabatake Y, Isaka Y, Mizui M. et al. Exploring RNA interference as a therapeutic strategy for renal disease. Gene Ther. 2005;12:965-973
36. Hamar P, Song E, Kokeny G. et al. Small interfering RNA targeting Fas protects mice against renal ischemia-reperfusion injury. Proc Natl Acad Sci U S A. 2004;101:14883-14888
37. Xia Z, Abe K, Furusu A. et al. Suppression of renal tubulointerstitial fibrosis by small interfering RNA targeting heat shock protein 47. Am J Nephrol. 2008;28:34-46
38. Freedman RS, Lenzi R, Kudelka AP. et al. Intraperitoneal immunotherapy of peritoneal carcinomatosis. Cytokines Cell Mol Ther. 1998;4:121-140
39. De Smet L, Ceelen W, Remon JP. et al. Optimization of drug delivery systems for intraperitoneal therapy to extend the residence time of the chemotherapeutic agent. ScientificWorldJournal. 2013;2013:720858
40. Lefkowith JB, Okegawa T, DeSchryver-Kecskemeti K. et al. Macrophage-dependent arachidonate metabolism in hydronephrosis. Kidney Int. 1984;26:10-17
41. Klahr S. Interstitial macrophages. Semin Nephrol. 1993;13:488-495
42. Klahr S. Obstructive nephropathy. Intern Med. 2000;39:355-361
Author contact
![]() Corresponding authors: Rikke Norregaard, Department of Clinical Medicine, Aarhus University, Aarhus, Denmark. E-mail: rikke.norregaardau.dk or Jørgen Kjems, Interdisciplinary Nanoscience Center, Department of Molecular Biology and Genetics, Aarhus University, Aarhus, Denmark. E-mail: jkau.dk or Shan Gao, Interdisciplinary Nanoscience Center, Department of Molecular Biology and Genetics, Aarhus University, Aarhus, Denmark. E-mail: shgau.dk.
Corresponding authors: Rikke Norregaard, Department of Clinical Medicine, Aarhus University, Aarhus, Denmark. E-mail: rikke.norregaardau.dk or Jørgen Kjems, Interdisciplinary Nanoscience Center, Department of Molecular Biology and Genetics, Aarhus University, Aarhus, Denmark. E-mail: jkau.dk or Shan Gao, Interdisciplinary Nanoscience Center, Department of Molecular Biology and Genetics, Aarhus University, Aarhus, Denmark. E-mail: shgau.dk.