Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2014; 4(11):1123-1132. doi:10.7150/thno.9627 This issue Cite
Research Paper
High-Purity Isolation and Recovery of Circulating Tumor Cells using Conducting Polymer-deposited Microfluidic Device
SeungHyun Jeon, WooYoung Hong, Eun Sook Lee, Youngnam Cho ![]()
New Experimental Therapeutic Branch, National Cancer Center, 111 Jungbalsan-ro, Ilsamdong-gu, Goyang, Gyeonggi-do 410-769, South Korea.
Received 2014-5-11; Accepted 2014-7-10; Published 2014-9-5
Abstract

We have developed a conductive nano-roughened microfluidic device and demonstrated its use as an electrically modulated capture and release system for studying rare circulating tumor cells (CTCs). The microchannel surfaces were covalently decorated with epithelial cancer-specific anti-EpCAM antibody by electrochemical deposition of biotin-doped polypyrrole (Ppy), followed by the assembly of streptavidin and biotinylated antibody. Our method utilizes the unique topographical features and excellent electrical activity of Ppy for i) surface-induced preferential recognition and release of CTCs, and ii) selective elimination of non-specifically immobilized white blood cells (WBCs), which are capable of high-purity isolation of CTCs. In addition, the direct incorporation of biotin molecules offers good flexibility, because it allows the modification of channel surfaces with diverse antibodies, in addition to anti-EpCAM, for enhanced detection of multiple types of CTCs. By engineering a series of electrical, chemical, and topographical cues, this simple yet efficient device provides a significant advantage to CTC detection technology as compared with other conventional methods.
Keywords: cancer, cancer diagnosis, circulating tumor cells, conducting polymer, electrical stimulation.
Introduction
In recent years, efforts to detect circulating tumor cells (CTCs) have increased significantly, providing clinically useful information for cancer biology. The metastatic spread of cancer is mostly initiated once malignant cells detach from the primary tumor, travel through the bloodstream, and attach in other parts of the body.[1,2] A comprehensive understanding of the identity and characteristics of CTCs can provide important insight into the biology of metastasis, and can facilitate use of CTCs as diagnostic and prognostic indicator in clinical treatment of cancer. [3,4] CTCs, referred to as “liquid biopsy”, show promise as a diagnostic and cancer management tool. Unlike conventional tissue biopsy taken from tumor masses, a real-time and non-invasive CTC analysis provides quick and easy accessibility in cancer monitoring. However, the extreme rarity of CTCs, in comparison with the large number of haematologic cells present in the blood, poses a major challenge in establishing a useful and reliable strategy for clinical application. Existing technologies include density-gradient centrifugation, size-based filtration (isolation by size of epithelial tumor cells (ISET)), reverse-transcription quantitative PCR (RT-PCR)-based methods, immunomagnetic-based approaches, and microfluidic devices.[5-15] Although recent technological improvements provide enhanced opportunities for identifying and enumerating CTCs, the development of more efficient isolation methods with high-volume throughput and high purity are still required for detailed molecular and downstream analysis. In recent studies, we have developed a biotin-doped polypyrrole (Ppy) platform capable of highly efficient capture of epithelial-cell adhesion molecule (EpCAM)-positive cancer cells and subsequent non-detrimental release of the captured cells in response to an applied electrical potential.[16] We have expanded this approach based on the flexibility and the versatility of the conductive Ppy by (i) incorporating a conventional PDMS device for developing a high-throughput cell recovery platform, (ii) applying a series of negative and positive voltages for high-purity isolation of CTCs, and (iii) including several different types of antibodies, beyond anti-EpCAM, that can recognize multiple cell types in the bloodstream. It is well-established that epithelial cells lose their typical epithelial phenotype and gain a mesenchymal signature during epithelia-to-mesenchymal transition (EMT), consequently showing phenotypic heterogeneity.[17,18] Herein, we propose a technology using a biotin-doped conductive polypyrrole-deposited microfluidic system (Biotin/Ppy-microfluidic) that can efficiently recognize and isolate multiple CTC types, and readily eliminate white blood cells (WBCs) in the bloodstream (Figure 1a).
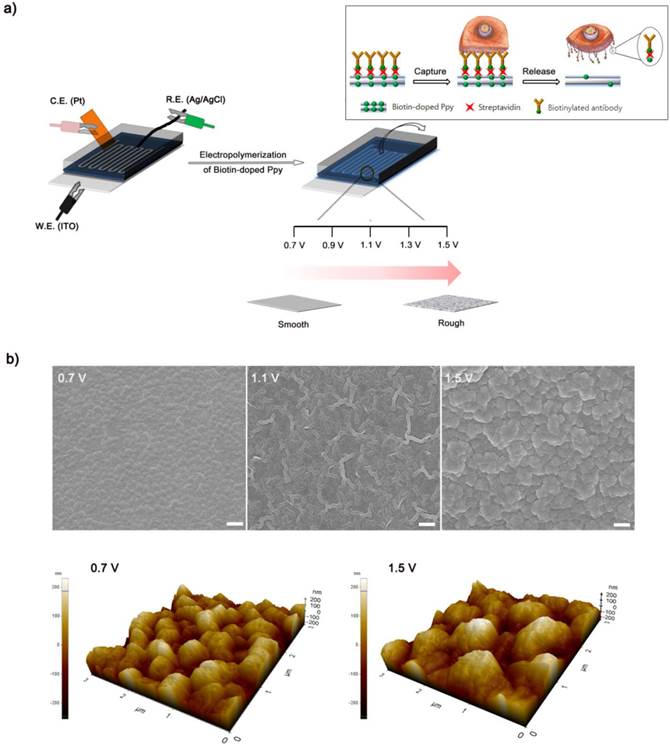
(a) Schematic representation of the process of electropolymerization of biotin-doped Ppy on the microfluidic device. The inset shows the specific capture and subsequent non-detrimental release of cancer cells using a biotin-doped, Ppy-deposited microfluidic device (Biotin/Ppy-microfluidic). A series of negative and positive voltages allows significantly enhanced isolation of CTCs by causing extensive damage to surface-immobilized WBCs. (b) SEM and AFM images of Ppy films electrodeposited on the microfluidic device as a function of applied voltage (Scale bars: 1 µm). The figures show the variation in the structural characteristics of Ppy depending on the electropolymerization conditions.
Current CTC-based methodologies are not sufficiently sensitive for reliable diagnosis or prognosis of cancer due to the co-existence of the very low numbers of isolated CTCs with a relatively large number of WBCs in the resulting cell suspension.[19] Therefore, we propose a novel strategy that efficiently enhances the capture yield of CTCs and preferentially eliminates WBCs using versatile electric fields. We assumed that the combined application of a series of negative and positive voltages may cause extensive damage to residual WBCs without decreasing the viability of CTCs. The negative electric voltages used here are designed to preferentially release captured CTCs from the surface, allowing the cancer cells to float free in the fluid. On the contrary, consecutive application of positive voltages is designed to incur electric field-induced damage to the cytoskeleton and nuclear membrane of surface-immobilized cells, especially WBCs.[20]
Results and Discussion
Preparation, Characterization, and Evaluation of Biotin/Ppy-microfluidic
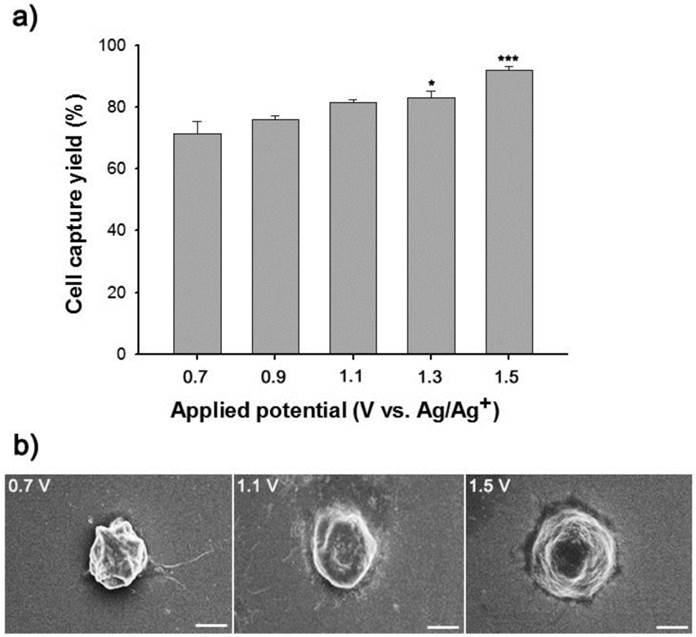
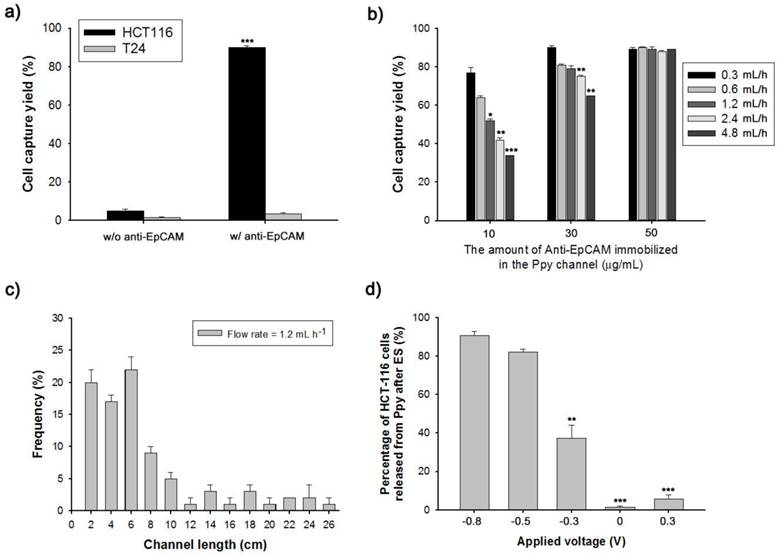
Conducting polymers such as Ppy, PANI, and PEDOT have been used as versatile platforms in wide range of applications because they can entrap a variety of biomolecules during electropolymerization process.[21-29] We expect that the application of electric fields coupled with a nanoroughened Ppy substrate will facilitate a synergistic improvement in the capture and release efficiency for CTCs, while minimizing non-specific capture of WBCs. Initially, to fabricate a conductive polymer-coated microfluidic device, electrochemical polymerization was performed in a solution containing pyrrole and biotin at a positive electrical potential by using a standard three-electrode system (Figure 1a). The biotin-doped Ppy was reacted with streptavidin, which allows for further conjugation with biotinylated antibodies. Biotin serves not only as a counter-anion for supporting the electrodeposition of Ppy, but also as an interface for subsequent assembly of biomolecules.[30] Furthermore, the surface roughness of a Ppy film deposited on a microfluidic channel can be finely controlled by varying the applied electrical voltage. Indeed, a subtle variation in the electro-polymerization conditions, ranging from +0.7 V to +1.5 V, significantly alters surface roughness from 25.5 ± 5 nm to 113.9 ± 8.3 nm. AFM and SEM images showed the morphological characteristics of synthesized Ppy as a function of the voltages applied during electrodeposition. The Ppy film appeared uniform with small grains when electrodeposited at +0.7 V, while high electric potentials accelerated the grain growth by coalescence, consequently permitting the growth of Ppy film with nanostructured features (Figure 1b). Indeed, the deposited Ppy was heavily dependent on the potentiostatic conditions employed in the electropolymerization to introduce nanoscale roughness. We then attempted to elucidate the impact of the nanoscale roughness of Ppy films on cell capture efficiency (Figure 2a). The biotin/Ppy-microfluidic device was first incubated with streptavidin and then conjugated with biotinylated anti-EpCAM as a model biomarker to evaluate CTC capture. As expected, cell capture densities for HCT116 cells (human colon cancer cells, EpCAM-positive) were strongly dependent on the Ppy synthesis conditions. The dramatic increase in cell capture efficiency, up to >90%, can be directly attributed to the macroscopic geometry of the Ppy. Because cells preferentially orient, adhere, and proliferate toward surfaces of increasing roughness, the Ppy film with nanoscale features facilitates local topographic interactions with nanoscale components of the cellular surface.[31, 32] SEM images provide additional detail on the topographic interaction between captured cells and Ppy platforms that differed in surface roughness. As seen in Figure 2b, cell morphology was strongly affected by surface topography. Indeed, the cells captured on the biotin/Ppy-microfluidic polymerized at 0.7 V showed no apparent spreading on the surface, and instead remained in a rounded state. In contrast, the nanoroughened Ppy surface strongly affected the strength of cell adhesion as well as cell shape. The cells captured on the rougher Ppy acquired a more stretched and elongated morphology, indicating that increased topographical features and roughness may be important characteristics for enhancing cell binding affinity. To explore the specificity of the cell recognition of the biotin/Ppy-microfluidic system, we compared its effect on cell capture yield using HCT116 and T24 (bladder cancer cell, EpCAM-negative) cell lines (Figure 3a). As can be readily observed, the presence of the antibody significantly enhanced the capture rate. Indeed, anti-EpCAM-coated biotin/Ppy-microfluidics are specially designed for the recognition of EpCAM-positive HCT116 cells; no apparent specific adhesion of T24 cells was observed regardless of the presence of the EpCAM antibody. Next, we observed cell capture efficiency using anti-EpCAM-coated biotin/Ppy-microfluidics, while varying two essential factors, fluid flow rate and the concentration of antibody immobilized on the channel (Figure 3b). Firstly, we focused on optimizing the flow rate at a concentration of 10-50 µg/mL anti-EpCAM, as flow rate is the primary influence on the frequency and duration of cell-surface contact. As expected, capture efficiency progressively decreased as flow rate increased. However, regardless of the flow rate, highly efficient capture of cancer cells, >90%, was achieved when 50 µg/mL antibody was used, eliciting a maximal affinity response with cancer-specific antigens. Next, the distribution of captured cells was analyzed at a flow rate of 1.2 mL/h (Figure 3c). 80% of the captured cells was located in the first 8 cm of the channel, indicating that a 24 cm-long microchannel would be appropriate to explore cell capture. In addition to cell recognition and capture performance, the feasibility of the proposed platform for controllable release of the captured cells was investigated. As seen in Figure 3d, the pattern of release of the captured cells was directly proportional to the applied negative potential, whereas positive electrical stimulation had no direct effect on cell release. Consistent with our previous work, >90% of the captured cells was rapidly released from microfluidics after exposure to -0.8 V for 15 s, that could be accounted for the reversible polymeric volume change during reduction-oxidation cycles.[33] The application of positive potentials (i.e., oxidative reaction) causes the film to expand, generating a free volume capable of incorporating large quantities of biotin moieties into the polymeric backbone. Adversely, upon application of negative potentials (i.e., reductive reaction), the Ppy backbone shrinks and simultaneously liberates conjugated biotin molecules and attached cells. The cells retrieved by on-demand and controlled releases were successfully cultivated in tissue culture dishes with multiple passages.
(a) Dependence of capture efficiency of EpCAM-positive HCT116 cells on the structural features of the Ppy microfluidic device depending on the electropolymerization conditions. (b) SEM images of captured HCT116 cells on anti-EpCAM-coated biotin/Ppy-microfluidics with varying surface roughness. Data are shown as mean ± s.d. (n = 5, ANOVA, ***p < 0.001, **p < 0.005, *p < 0.01).
(a) Evaluation of the cell capture yield of biotin/Ppy-microfluidics with or without subsequent anti-EpCAM conjugation using EpCAM-positive (HCT116) and EpCAM-negative (T24) cancer cells. (b) Dependence of HCT116 cell capture efficiency on the concentration of anti-EpCAM immobilized in the Ppy film and flow rate. (c) The distribution of the captured HCT116 cells in the microchannel at flow rates of 1.2 mL/h. (d) Controlled release of the captured cells as a function of the applied voltage. Data are shown as mean ± s.d. (n = 5, ANOVA, ***p < 0.001, **p < 0.005, *p < 0.01).
Performance Evaluation of Biotin/Ppy-microfluidic using Artificial Blood Samples
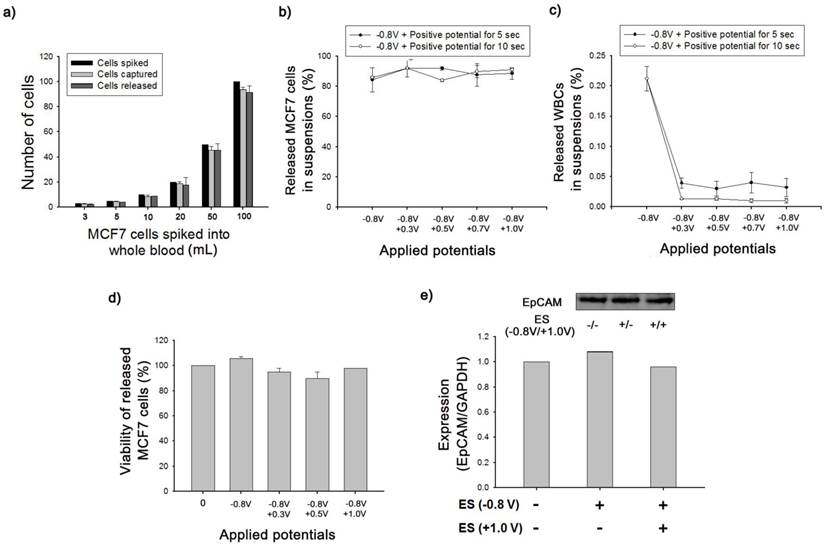
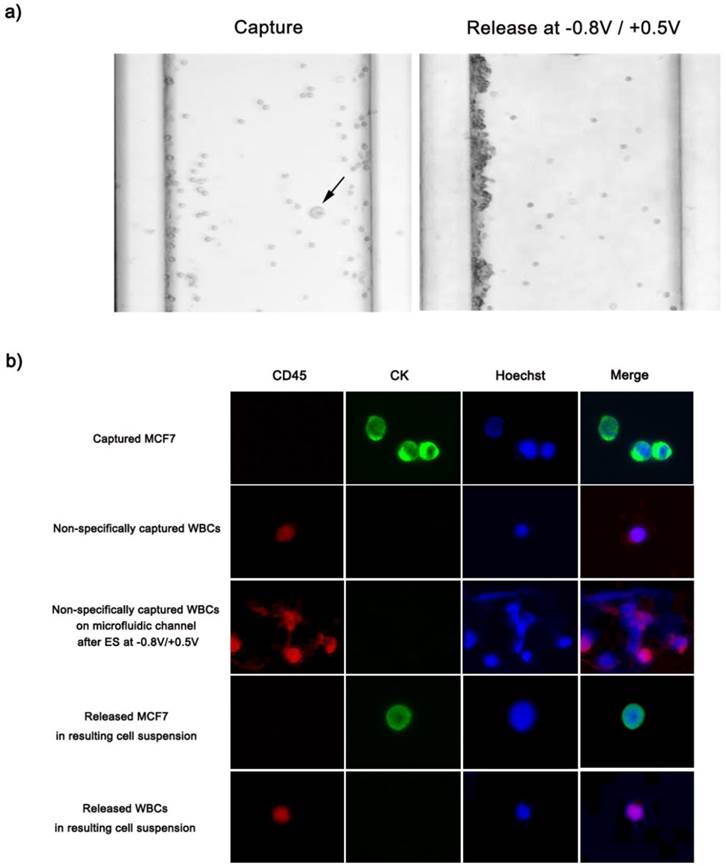
We further validated the clinical feasibility of anti-EpCAM-coated biotin/Ppy-microfluidics using artificial blood samples in which EpCAM-positive MCF7 cells (breast cancer cells) were spiked into healthy donor's blood in accordance with NCC Institutional Review Board Approval. As a proof-of-concept study, MCF7 cells were spiked into 1 mL of whole blood (containing approximately 2.5 × 106 WBCs/mL) at concentrations ranging from approximately 3 to 100 cells/mL and introduced into anti-EpCAM-coated biotin/Ppy-microfluidics at a flow rate of 1.2 mL/h. In all cases, the average CTC capture and release efficiency were >90% (Figure 4a). This approach showed particular sensitivity for low-frequency CTCs (3-5 cells) with capture and release rates of 97% and 95%, respectively. In addition, immediately following the exposure to -0.8 V, we applied various positive potentials, and observed the final numbers of CTCs and WBCs in the resulting cell suspension (Figure 4b-c). Control experiments were conducted at -0.8 V for 15 s but with no application of positive voltage; MCF7 capture efficiency was 85% and the rate of non-specific immobilization of WBCs was 0.21%. However, combined treatment with the two voltage sources greatly reduced the density of WBCs in the resulting cell suspensions to 0.01%, while maintaining a capture yield of MCF7 cells of >90%. In addition, we attempted to elucidate the effect of the positive potentials on the elimination of WBCs as a function of time. There appeared to be a distinct relationship between voltage application time and the number of WBCs in the resulting cell suspensions; however, the recovery rate of MCF7 cells was not influenced by exposure to additional positive potentials. Subsequently, the released cells were collected and re-cultured to evaluate their viability and phenotypic characteristics using quantitative western blot analysis. As shown in Figure 4d, the cell viability assay clearly showed that none of the potentials had a cytotoxic effect, even in combination with opposite potentials. In addition, the magnitude of EpCAM antigen expression of recovered cancer cells was verified using a Fujifilm Multigauge 3.0 (Figure 4e). A similar EpCAM protein band was observed for all HCT116 cells, even following application of combinatorial electrical potentials at -0.8 V for 15 s and +1.0 V for 5 s. As explained above, this key strategy to separate the captured cells is a result of the shrinkage of Ppy backbone, which subsequently untangles the biotin moieties and attached cells from the polymer but does not directly influence the structural and biological functions of the attached cells. Furthermore, we compared the cellular morphology of captured and released CTCs and WBCs following exposure to a series of electric potentials (Figure 5a). Optical microscopic images showed single cancer cells adhered to the channel, surrounded by large numbers of WBCs. Interestingly, the captured MCF7 cells were specifically released following application of electric fields at -0.8 V/+0.5 V, whereas most non-specifically immobilized WBCs remained adhered to the microchannel. In particular, these WBCs appeared to be agglomerated and thus formed large clusters along the wall of the microfluidic channel, resulting from electric field-induced breakdown of cell membrane integrity. Most clumps or agglomerates of WBCs remained in the channel, even after repetitive rinsing, indicating that this method will allow significant reduction of WBC contamination in the resulting cell suspensions. We next attempted to identify and enumerate the captured and released cells by visualizing them with cell-specific markers, such as anti-cytokeratin (anti-CK) for the identification of epithelial cells, Hoechst dye for nuclei, and anti-CD45 for haematologic cells (Figure 5b). MCF7 cells captured and subsequently released showed strong positive staining for anti-CK and Hoechst, but were negative for anti-CD45, with normal nuclear morphology. However, following exposure to electrical stimulation at -0.8 V/+0.5 V, WBCs on the microfluidic channel showed abnormalities in nuclear morphology and deterioration of plasma membrane structure with tiny and dispersed fragments. Hoechst-dye stained WBCs showed serious damage to the nucleus that appeared to be related to the electric field-induced injury. In contrast, non-specifically trapped WBCs without electrical stimulation showed typical regular, round morphologies with normal nuclei. Similarly, few of the WBCs released into the cell suspension following electrical stimulation were normal in shape and size.
(a) The number of MCF7 cells spiked into lysed blood at concentrations from 3 to 100 cells/mL, captured on anti-EpCAM-coated biotin/Ppy-microfluidics from spiked lysed blood, and subsequently released by applying an electric field at -0.8 V for 15 s. (b) The effect of applied electrical potentials and time on the release efficiency of spiked MCF7 cells. (c) The release efficiency of non-specifically immobilized WBCs under applied electrical potentials and time. (d) Viability of MCF7 cells released from microfluidic devices after the exposure to various electrical potentials. (e) Western blot analysis for EpCAM protein expression in MCF7 cells before and after electrical stimulation. The electrical stimulation (ES) was conducted at -0.8 V for 15 s and +1.0 V for 5 s.
(a) Optical microscopic images depicting the specific capture and subsequent electric field-mediated release of MCF7 cells spiked into artificial blood. EpCAM-positive MCF7 cells (black arrow) were captured on the center of the microchannel, whereas non-specifically trapped WBCs primarily formed large cell agglomerates or clumps against the wall of the microfluidic device. (b) Immunofluorescence images of captured and released cells. Cells were stained with Hoechst 33342, cytokeratin, and CD45. All studies were performed in quintuplicate.
Capture and Release of Cancer Cells with Different Levels of EpCAM Expression
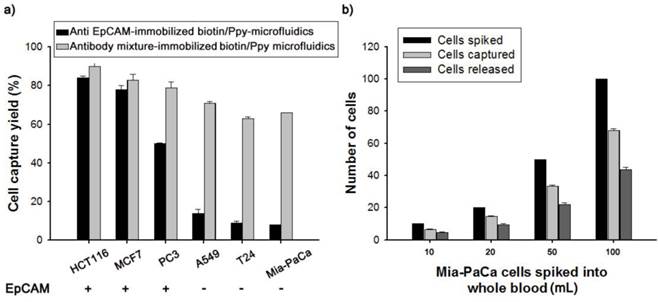
Current antibody-based technologies mostly rely on EpCAM, because of its overexpression on the surface of epithelial cells. However, loss of the EpCAM phenotype following EMT in the bloodstream has prompted exploration combining multiple antibodies (Figure 6). Thus, we incubated a biotin-doped Ppy-deposited microfluidic system with streptavidin to construct a versatile platform capable of reacting with a mixture of biotinylated antibodies, including anti-EpCAM, anti-TROP2, anti-EGFR, anti-N-Cadherin, and anti-vimentin. Subsequently, the feasibility and validity of this concept was demonstrated by comparison with an EpCAM- and antibody mixture-immobilized microfluidic platform, using cell lines that differed in EpCAM expression levels. Interestingly, when incubated with the antibody mixture-immobilized microfluidic platform, low EpCAM-expressing cell lines, such as A549, T24, and Mia-PaCa, showed a dramatic enhancement in cell capture efficiency, as high as 66-71% (Figure 6a). In distinct contrast, only 8% of Mia-PaCa cells were captured by the EpCAM-only microfluidic platform. Next, we evaluated the clinical potential of our capture assay using EpCAM-negative Mia-PaCa cells by spiking 10-100 cells into 1.0 mL of healthy donor's blood (Figure 6b). On average, the capture and release rates were greatly improved, at >65% and >70%, respectively. A biotin-doped Ppy-deposited microfluidic system integrated with the antibody cocktail can help detect previously unrecognized CTC subpopulations with improved recovery rates. The platform presented here is particularly well suited to identify and define various CTC subtypes beyond the EpCAM, providing important insights into the diagnostic and prognostic applications of CTCs.
(a) Cell capture yield of various cancer cell lines that differ in EpCAM expression, using anti EpCAM- and antibody mixture-immobilized biotin/Ppy-microfluidics. (b) The number of Mia-PaCa cells spiked into lysed blood at concentrations from 10 to 100 cells per mL, captured on antibody mixture-immobilized biotin/Ppy-microfluidics from spiked lysed blood, and subsequently released by applying an electric field at -0.8 V/+0.5 V.
Conclusions
In summary, we have developed a simple and effective conducting polymer-deposited microfluidic platform that allows for highly sensitive and specific capture and subsequent “on-demand” release of cancer cells without damage, using application of electrical potentials. The enhanced performance of our proposed platform is most likely owing to the synergistic effect of the electric field-mediated Ppy nanostructured surface, integration with an established PDMS device, and conjugation with multiple types of antibodies in addition to anti-EpCAM. In addition, this electric field-mediated approach damaged WBCs non-specifically trapped in the microchannel and forced WBCs to agglomerate on the microchannel; these effects are desirable for improving the purity of the resulting CTC suspensions. Overall, this approach is of great significance for both basic and clinical applications because it will facilitate sensitive, early detection of malignant cells, and permit sorting of cancer cells using simple and easy manipulation of electric fields, that ultimately may enable development of functional cell-based assays.
Experimental Section
Materials. Pyrrole, biotin, sodium dodecylbenzene sulfonate (NaDBS), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC), N-hydroxysuccinimide (NHS), and streptavidin were obtained from Aldrich. Bovine serum albumin (BSA) was obtained from Bovogen Biologicals Pty. Ltd. (Australia). Milli-Q water was used in the preparation of all water-based solutions. Sylgard 184 was purchased from Dow Corning. Human biotinylated antibodies (Anti-EpCAM, Vimentin, EGFR, N-Cadherin, and Trop-2) were purchased from R&D Systems.
Fabrication of a Biotin-doped Ppy-deposited Microfluidic Device. Polydimethylsiloxane (PDMS) microchannels were prepared as described previously.[1] A 10:1 (w/w) mixture of silicon elastomer base and silicon elastomer curing agent was initially degassed to remove bubbles, cast over SU-8 master molds patterned on an 8-inch silicon wafer, and cured in an oven at 60 °C for 2-3 h. A channel with dimensions 80 µm × 200 µm (height × width) was fabricated using a soft lithography technique. The inlet and outlet holes with 1 mm diameter were directly connected to a microchannel. PDMS microchannels were chemically activated by corona discharges and attached on a clean ITO to form a permanent bond. Ppy-deposited microfluidics were electrochemically prepared using a potentiostat/galvanostat (BioLogic SP-150), where a PDMS microchannel attached to an ITO surface, a platinum wire, and an Ag/AgCl reference were employed as the working, counter, and reference electrodes, respectively. For the preparation of the biotin-doped Ppy-deposited microfluidics, Ppy polymerization was performed in 0.01 M NaDBS and 0.1 M pyrrole aqueous solution containing 1 mM biotin into the microfluidic channels with the application of chronoamperometry (CA) at 0.7 V-1.5 V (versus Ag/AgCl) for 5 min. Then, the biotin/Ppy-microfluidic channel was coated with streptavidin (10 μg/mL), followed by anti-EpCAM (10 μg/mL in 1× PBS solution) at a flow rate of 1.2 mL/h. After washing with PBS, 1 wt% BSA in 1× PBS was added to microfluidics to block nonspecific binding of the antigen. Finally, the microchannel was washed with PBS several times.
Cell Culture. EpCAM-positive (HCT116, MCF7, and PC3) and EpCAM-negative (A549, T24, and Mia-PaCa2) cells were obtained from the American Type Culture Collection (ATCC) and cultured in Roswell Park Memorial Institute (RPMI)-1640 and Dulbecco's modified Eagle's medium(DMEM) supplemented with 10% fetal bovine serum (FBS; GenDepot).
Cell Capture. For the cell capture assay, cell suspensions were introduced into the devices using a connected syringe pump at constant flow rates of 0.3-4.8 mL/h. The resulting microfluidic channel was washed with normal media at a flow rate of 4.8 mL/h. The morphology of the captured cells was observed using FE-SEM. First, the captured cells were fixed with 2.5% formaraldehyde in PBS for 20 min at room temperature. Then sample dehydration was carried out through ethanol series (50%, 70%, 80%, 90% and 100%).
Electric Stimulation for Releasing the Captured Cells. The release profiles of cells captured in the microfluidic system were examined using a three-electrode system consisting of a reference electrode (Ag/AgCl), a counter electrode (Pt), and a working electrode (an ITO surface attached to the microfluidic channel) by applying a negative voltage for 15 s and a positive voltage for 5 ~ 10 s.
Western Blot. After washing with cold PBS, HCT116 cells were collected using an E-tube. Following lysis using a 0.4% RIPA buffer, media and PBS were removed by centrifugation. Protein samples (20 μg) were then separated on an 8% SDS-polyacrylamide gel and transferred to a nitrocellulose membrane (0.4 µm). The membranes were blocked with 5% nonfat dry milk and probed with antibodies against EpCAM (R&D Systems). An antibody against glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Santa Cruz Biotechnology) was used as an internal control. Positive reactions were visualized using an enhanced chemiluminescence detection system (Amersham Pharmacia).
Cell Viability Assay. Viability of the released cells was confirmed using a Cell Count Kit-8 (methylthiazole tetrazolium, Dojindo Molecular Technologies). Following 24 h of incubation in 96-well plates, absorbance was measured at 540 nm using a spectrophotometer (Molecular Devices, Emax).
Immunofluorescence. Cells were transferred to coverslips-in-plate, fixed, and permeabilized.[2] Then, cells were fixed with 3.7% paraformaldehyde and permeabilized using 0.3% Triton X-100 and 5% BSA, as described previously.[3] Antibodies against CK (cytokeratin) or CD45 were added for 2 h, and then an Alexa Fluor 488-conjugated (Invitrogen; green signal for CK) or Alexa Fluor 568-conjugated (Invitrogen; red signal for CD45) secondary antibody, respectively, was added for 40 min. Nuclear DNA (blue signal) was stained with 1 μg/mL Hoechst 33258 (Invitrogen). Labeled cells were examined under a Zeiss LSM 710 ConfoCor 3 fluorescence microscope.
Testing MCF7 Cell Capture and Release from Artificial Blood. Blood samples were collected from healthy volunteers in EDTA-Vacutainer tubes, where white blood cells (WBCs) were counted immediately after collection using a hemocytometer. The blood samples were prepared by spiking 10 µL RPMI media containing MCF7 cells at concentrations ranging from 3, 5, 10, 20, 50, and 100 cells into 1 mL of lysed blood. In addition, MCF7 cells were labeled with DiO green fluorescent dye prior to addition to the blood samples. After capturing, all of the immobilized cells were stained with three phenotypic markers: cytokeratin, Hoechst33258, and CD45.
Antibody Mixture-immobilized Microfluidic. For antibody mixture-immobilized biotin/Ppy-microfluidics, biotinylated microchannel was initially conjugated with streptavidin (10 μg/mL) and subsequently exposed to antibody mixture containing EpCAM, TROP-2, EGFR, vimentin, and N-cadherin with a concentration of 30 μg/mL, respectively. At a flow rate of 1.2 mL/h, EpCAM-positive cell lines (i.e., HCT116, MCF7, PC3) and EpCAM-negative cell lines (T24, A549, Mia-PaCa2) were used to evaluate their performance. The number of Mia-PaCa cells spiked into lysed blood were 10, 20, 50, 100 cell/mL and the capture and release experiments were conducted by sequentially applying the electrical stimulation at -0.8 V for 15 sec and at +0.5 V for 5 sec.
Acknowledgements
This research was supported by a National Cancer Center grant from the Republic of Korea (1310180).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Pantel K, Brakenhoff RH. Dissecting the metastatic cascade. Nat Rev Cancer. 2004;4:448-56
2. Klein CA. Cancer. The metastasis cascade. Science. 2008;321:1785-7
3. Hong B, Zu Y. Detecting circulating tumor cells: current challenges and new trends. Theranostics. 2013;3:377-94
4. Yu M, Stott S, Toner M, Mashswaran S, Haber DA. Circulating tumor cells: approaches to isolation and characterization. J. Cell Biol. 2011;192:373-82
5. Phillips JA, Xu Y, Xia Z, Fan ZH, Tan W. Enrichment of cancer cells using aptamers immobilized on a microfluidic channel. Anal. Chem. 2009;81:1033-9
6. Wang S, Liu K, Liu J, Yu Z TF, Xu X, Zhao L, Lee T, Lee EK, Reiss J, Lee YK, Chung L, W K, Huang J, Rettig M, Seligson D, Duraiswamy KN, Shen C KF, Tseng HR. Highly efficient capture of circulating tumor cells by using nanostructured silicon substrates with integrated chaotic micromixers. Angew. Chem. Int. 2011;50:3084-8
7. Galanzha EI, Shashkov EV, Keely T, Kim JW, Yang L, Zharov VP. In vivo magnetic enrichment and multiplex photoacoustic detection of circulating tumour cells. Nat. Nanotechnol. 2009;4:855-60
8. Hou HW, Warkiani ME, Khoo BL, Li ZR, Soo RA, Tan DS, Lim WT, Han J, Bhagat AS, Lim CT. Isolation and retrieval of circulating tumor cells using centrifugal forces. Sci. Rep. 2013;3:1259
9. Stott SL, Hsu CH, Tsukrov DI, Yu M, Miyamoto DT, Waltman BA, Rothenberg SM, Shah AM, Smas ME, Korir G K, Floyd J, Gilman AJ, Lord JB, Winokur A, Springer S, Irimia D, Nagrath S, Sequist LV, Lee RJ, Isselbacher KJ, Maheswaran S, Haber DA, Toner M. Isolation of circulating tumor cells using a microvortex-generating herringbone-chip. PNAS. 2010;107:18392-7
10. Nagrath S, Sequist LV, Maheswaran S, Bell DW, Irimia D, Ulkus L, Smith MR, Kwak EL, Digumarthy S, Muzikansky A, Ryan P, Balis UJ, Tompkins RG, Haber DA, Toner M. Isolation of rare circulating tumour cells in cancer patients by microchip technology. Nature. 2007;450:1235-9
11. Li P, Stratton ZS, Dao M, Ritz J, Huang TJ. Probing circulating tumor cells in microfluidics. Lab Chip. 2013;13:602-9
12. Yoon HJ, Kim TH, Zhang Z, Azizi E, Pham TM, Paoletti C, Lin J, Ramnath N, Wicha MS, Hayes DF, Simeone DM, Nagrath S. Sensitive capture of circulating tumour cells by functionalized graphene oxide nanosheets. Nat. Nanotechnol. 2013;8:735-41
13. Zhang N, Deng Y, Tai Q, Cheng B, Zhao L, Shen W, He R, Hong L, Liu W, Guo S, Liu K, Tseng HR, Xiong B, Zhao XZ. Electrospun TiO2 nanofiber-based cell capture assay for detecting circulating tumor cells from colorectal and gastric cancer patients. Adv. Mater. 2012;24:2756-60
14. Hughes AD, King MR. Nanobiotechnology for the capture and manipulation of circulating tumor cells. WIREs Nanomed Nanobiotechnol. 2012;4:291-9
15. Bourquin Y, Syed A, Reboud J, Ranford-Cartwright LC, Barrett MP, Cooper JM. Rare-Cell Enrichment by a Rapid, Label-Free, Ultrasonic Isopycnic Technique for Medical Diagnostics. Angew.Chem. Int. 2014;53:1
16. Jeon S, Moon JM, Lee ES, Kim YH, Cho Y. An Electroactive Biotin-Doped Polypyrrole Substrate That Immobilizes and Releases EpCAM-Positive Cancer Cells. Angew. Chem. Int. Ed. 2014;53:4897-2
17. Mikolajczyk SD, Millar LS, Tsinberg P, Coutts SM, Zomorrodi M, Pham T, Bischoff FZ, Pircher TJ. Detection of EpCAM-Negative and Cytokeratin-Negative Circulating Tumor Cells in Peripheral Blood. J. Oncol. 2011;2011:252361
18. Eisenwort G, Jurkin J, Yasmin N, Bauer T, Gesslbauer B, Strobl H. Identification of TROP2 (TACSTD2), an EpCAM-like molecule, as a specific marker for TGF-β1-dependent human epidermal Langerhans cells. J. invest. dermatol. 2011;131:5049-57
19. Zhao L, Lu YT, Li F, Wu K, Hou S, Yu J, Shen W, Wu D, song M, OuYang W, Luo Z, Lee T, Fang X, Shao C, Xu X, Garcia MA, Chung L. WK, Rettig M, Tseng HR, Posadas EM. High-Purity Prostate Circulating Tumor Cell Isolation by a Polymer Nanofiber-Embedded Microchip for Whole Exome Sequencing. Adv. Mater. 2013;25:2897-02
20. Stacey M, Fox P, Buescher S, Kolb J. Nanosecond pulsed electric field induced cytoskeleton, nuclear membrane and telomere damage adversely impact cell survival. Bioelectrochemistry. 2011;81:131-4
21. Geetha S, Chepuri RK, Rao M, Trivedi DC. Biosensing and drug delivery by polypyrrole. Anal. Chim. Acta. 2006;568:119-25
22. Wallace G, Spinks G. Conducting Polymers - Bridging the Bionic Interface. Soft Mater. 2007;3:665-71
23. Abidian M, Kim D, Martin D. Conducting-Polymer Nanotubes for Controlled Drug Release. Adv. Mater. 2006;18:405-9
24. Jeon G, Yang SY, Byun J, Kim JK. Electrically actuatable smart nanoporous membrane for pulsatile drug release. Nano lett. 2011;11:1284-8
25. Wadhwa R, Carl L, Xinyan C. Electrochemically controlled release of dexamethasone from conducting polymer polypyrrole coated electrode. J. Control. Release. 2006;110:531-41
26. Fredj HB, Helali S, Esseghaier C, Vonna L, Vidal L, Abdelghani A. Labeled magnetic nanoparticles assembly on polypyrrole film for biosensor applications. Tanlanta. 2007;75:740-7
27. Wallace G, Kane-Maguire L. Manipulating and monitoring biomolecular interactions with conducting electroactive polymers. Adv. Mater. 2002;14:953-60
28. Cho Y, Borens RB. The preparation of polypyrrole surfaces in the presence of mesoporous silica nanoparticles and their biomedical applications. Nanotechnology. 2010;21:275102
29. Pernaut JM, Reynolds JR. Use of Conducting Electroactive Polymers for Drug Delivery and Sensing of Bioactive Molecules. A Redox Chemistry Approach. J. Phys. Chem. B. 2000;104:4080-90
30. Cho Y, Borgens RB. Biotin-doped porous polypyrrole films for electrically controlled nanoparticle release. Langmuir. 2011;27:6316
31. Wang S, Wang H, Jiao J, Chen KJ, Owens GE, Kamei Ki, Sun J, Sherman DJ, Behrenbruch CP, Wu H, Tseng HR. Three-dimensional nanostructured substrates toward efficient capture of circulating tumor cells. Angew. Chem. Int. 2009;121:8970-3
32. Kim DH, Provenzano PP, Smith CL, Levchenko A. Matrix nanotopography as a regulator of cell function. J. Cell Biol. 2012;197:351-60
33. Otero TF, Martinez JG. Biomimetic intracellular matrix (ICM) materials, properties and functions. Full integration of actuators and sensors. J. Mater. Chem. B. 2013;1:26-38
Author contact
![]() Corresponding author: Email: ynchore.kr.
Corresponding author: Email: ynchore.kr.