Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Conclusion and Discussion
Supplementary Material
Acknowledgements
Abbreviations
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2014; 4(11):1096-1111. doi:10.7150/thno.9423 This issue Cite
Research Paper
Co-delivery of Doxorubicin and Bmi1 siRNA by Folate Receptor Targeted Liposomes Exhibits Enhanced Anti-Tumor Effects in vitro and in vivo
Tan Yang1, Bin Li1, Shibo Qi1, Yong Liu1, Yongkang Gai1, Peng Ye1, Guang Yang1, Wendian Zhang1, Peng Zhang2, Xingxing He3, Weijie Li4, Zhiping Zhang1, Guangya Xiang1 ![]() , Chuanrui Xu1
, Chuanrui Xu1 ![]()
1. School of Pharmacy, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, P. R. China, 430030;
2. Department of Oncology of Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology;
3. Department of Hepatology of Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology;
4. Department of Pharmacy of Tongji Hospital; Tongji Medical College, Huazhong University of Science and Technology, Wuhan, P. R. China, 430030.
Received 2014-4-17; Accepted 2014-7-29; Published 2014-8-24
Abstract
Bmi1 gene overexpression is found in various human tumors and has been shown as a potential target for gene treatment. However, siRNA-based treatments targeting Bmi1 gene have been restricted to limited delivery, low bioavailability and hence relatively reduced efficacy. To overcome these barriers, we developed a folate receptor targeted co-delivery system folate-doxorubicin/Bmi1 siRNA liposome (FA-DOX/siRNA-L). The FA-DOX/siRNA-L was prepared through electrostatic interaction between folate doxorubicin liposome (FA-DOX-L) and Bmi1 siRNA. In vitro and in vivo studies showed that FA-DOX/siRNA-L inhibited tumor growth by combinatory role of Bmi1 siRNA and doxorubicin (DOX). Co-delivery of Bmi1 siRNA and DOX by FA-DOX/siRNA-L showed significantly higher efficacy than sole delivery of either DOX or Bmi1 siRNA. Real-time PCR and western blot analysis showed that FA-DOX/siRNA-L silenced the expression of Bmi1 gene. In addition, higher accumulation of the siRNA and DOX in tumor cells indicated that folate ligand displayed tumor targeting effect. These results suggest that Bmi1 is an effective therapeutic target for siRNA based cancer treatment that can be further improved by co-delivery of DOX through targeted liposomes.
Keywords: liposome, Bmi1 siRNA, doxorubicin, folate receptor, tumor targeting.
Introduction
Small interfering RNAs (siRNAs) have attracted considerable research attention for their potential therapeutic applications. Notably, a number of studies have explored the use of siRNAs to silence the expression of oncogenes and specific targets that promote the proliferation of tumor cells as an anticancer strategy [1]. Bmi1, a member of the mammalian polycomb group of multimeric transcriptional repressors, is involved in the regulation of development, stem cell self-renewal, cell cycle, and senescence [2, 3]. Deregulation of Bmi1 expression has been reported in various tumor types, including non-small-cell lung carcinoma, colon carcinoma, medulloblastoma, metastatic melanoma, and nasopharyngeal carcinoma [4-8]. In our past study, we found that downregulation of Bmi1 by lenti-virus mediated shRNA inhibits the growth of hepatocellular carcinoma both in vitro and in vivo, which suggests again that Bmi1 is a potential target for cancer therapy [9, 10]. However, due to lack of an effective and safe delivery strategy, the feasibility of Bmi1 targeted therapy for clinical translation has not been extensively investigated up to date.
Combination therapy with siRNAs and chemotherapeutics has been investigated as an alternative strategy for achieving enhanced anticancer activity. Several groups have designed delivery systems based on liposomes or nanoparticles for co-delivery of anticancer chemotherapeutics and siRNAs. Some of the reported studies include: cationic liposomes for co-delivery of doxorubicin (DOX) and siRNA targeting multi-drug resistance (MDR) protein to enhance anticancer efficacy in lung cancer cells [11]; mesoporous silica nanoparticles to deliver DOX and Bcl-2 siRNA for effective treatment of cancer cells [12], and micellar nanoparticles carrying paclitaxel (PTX) and siRNA specific for polo-like kinase 1 to induce a synergistic tumor-suppressive effect [13]. Among the various types of delivery systems, liposome has attracted considerable attention. It is one of the only two families of therapeutic nanocarriers that have been approved in clinical practice [14]. Compared with free chemical drugs, liposome can help prolong systemic circulation time, enhance tumor localization and overcome MDR [15, 16]. Liposome can also reduce the adverse effects of chemotherapeutic drugs, such as the cardiac toxicity of DOX [17-20]. For two decades, studies have been focused on the development of novel liposomal therapeutic agents with high encapsulation efficiency and enhanced passive targeting efficiency.
DOX, a potent anticancer drug, is effective against a wide range of human neoplasms. It has been widely applied as a chemotherapeutic agent for cancer treatment. However, the clinical uses of DOX are restricted largely due to limited tissue specificity and serious cardiotoxic effects resulted from the generation of free radicals and lipid peroxidation. To reduce the toxicity of DOX, liposomal formulation was applied in clinics. However, the efficacy of DOX treatment was not enhanced greatly by liposomal delivery. To further improve the efficacy of liposomal DOX, targeting liposomes was employed to promote the tumor targeting effect and hence improve the efficacy. In recent years, many studies have been focused on targeting delivery systems, such as folate receptor targeting systems [21], transferrin receptor targeting system [22], RGD conjugating delivery system etc [23].
Folate receptor (FR), a glycosyl phosphatidinositol (GPI)-anchored membrane protein, is overexpressed in 90% ovarian carcinomas and many types of other epithelial cancers [24-28]. The expression levels of FR in normal tissues are much lower than in tumor tissues. FR is an ideal target for drug delivery for its distinct expression between normal tissues and malignant tissues. Folate, the natural ligand of FR, has been extensively investigated for chemotherapeutic nanoparticle delivery considering its inherent high affinity, small size and non-toxicity [29-36]. Lee and Low synthesized the folate-conjugated liposomes by incorporating 0.1 mol% of folate polyethylene glycol distearoylphosphatidylethanolamine (FA-PEG-DSPE) construct into the lipid bilayer. The uptake of FA-PEG-DSPE liposomal DOX by KB cells was significantly increased and the cytotoxicity was enhanced as well [37]. Since then, various lipophilic folate derivatives have been evaluated to improve the targeting efficiency and stability of liposomes [21, 38, 39].
Despite the critical role of Bmi1 in cancer cell regulation, Bmi1 has not been sufficiently investigated as a new therapeutic target for siRNA, either as a monotherapy or combination therapy. In particular, no reported studies have explored the co-delivery of Bmi1 siRNA in combination with chemotherapy by an in vivo feasible targeted delivery system. In this study, we formulated for the first time an FR targeting cationic liposome encapsulating DOX and Bmi1 siRNA (FA-DOX/siRNA-L). After the preparation of FA-DOX/siRNA-L, characterization, FR dependent cellular uptake, gene silencing and cytotoxicity, in vivo pharmacokinetics and distribution, liver and kidney toxicity, anti-tumor efficacy, and the preliminary mechanisms were investigated to determine the efficacy and safety of co-delivery of Bmi1 siRNA and DOX.
Materials and Methods
Chemicals for preparation of liposomes
Doxorubicin hydrochloride (DOX·HCl), folate, dicyclohexylcarbodiimide (DCC), N-hydroxysuccinimide (NHS), cholesterol, DMSO, (NH4)2SO4, CHCl3, Sepharose CL-4B chromatography media and folate-free RPMI 1640 cell culture medium were purchased from Sigma-Aldrich Chemical Co (St. Louis, MO, USA). DMEM high glucose medium was purchased from the Hyclone of Thermo Scientific (IL, USA). Trypsin was purchased from Beyotime Institute of Biotechnology (Beijing, China). 1, 2-Dioleoyl-3-Trimethylammonium-Propane (Chloride Salt) (DOTAP), monomethoxy polyethylene glycol 2000-distearoyl phosphatidylethanolamine (mPEG-DSPE) and PEG-bis-amine (3350 Da) were purchased from Avanti Polar Lipid (Alabaster, AL, USA). PD-10 desalting columns were purchased from GE Healthcare Biosciences (PA, USA). All reagents and solvents were of analytical or HPLC grade and were used without further purification. FAM or Cy5-labeled Bmi1 siRNA, and Chol-modified Bmi1 siRNA (sense strand: 5'-CCA GAC CAC UAC UGA AUA UAA-3'; antisense strand: 5'-UUA UAU UCA GUA GUG GUC UGG UU-3') were synthesized by Ribobio Co., Ltd (Guangzhou, China).
Cell culture
HeLa, KB, Hep3B, A549, Huh7, MCF-7 and LO2 cells are all from the China Center for Type Culture Collection at Wuhan University (Wuhan, China). The cells were cultured with folate free RPMI 1640 or high glucose DMEM medium supplemented with penicillin, streptomycin, and 10% FBS in 37°C and 5% CO2 incubators. M-plasmocin (San Diego, CA, USA) at a concentration of 2.5 μg/mL was used to prevent the possible mycoplasma infections.
Preparation of FA-DOX-L and DOX-L
Folate-PEG-bis-amine and cholesterol-OTs (Chol-OTs) were synthesized by methods described in literatures [40, 41]. Briefly, for the synthesis of folate-PEG-bis-amine, folate (26.5 mg, 60 µmol) was dissolved in DMSO, and then PEG-bis-amine (167.5 mg, 50 µmol), DCC (15.5 mg, 75µmol), NHS (8.63 mg, 75 µmol) and triethylamine (34.8 µL, 250 µmol) were added into the solution and then reacted at 30°C overnight. The reaction mixture was then centrifuged for 7 min at 7,000 rpm, and then the supernatant was transferred into a new centrifuge tube. Three mL of Na2CO3 (50 mM) was added and the mixture was centrifuged for 15 min at 4,000 rpm. The upper layers were purified by Sephadex G-25 gel-filtration chromatography and downy yellow solid was obtained by lyophilization. For the synthesis of Chol-OTs, pyridine, cholesterol (5 g, 12.93 mmol) and 4-toluene sulfonyl chloride (2.71 g, 14.22 mmol) were mixed and stirred at 25°C for 12 h. The reactant was mixed with 25 mL of purified water and extracted 3 times with ethyl ether. The organic phases were combined and dried for 1 h with Na2SO4. The residue was then recrystallized by petroleum ether. Finally, for the synthesis of FA-CONH-PEG-NH-Chol (FA-PEG-Chol), folate-PEG-bis-amine (128 mg, 33.9 µmol), Chol-OTs (22.3 mg, 42.3 µmol) and triethylamine (11.8 µL, 84.6 µmol) were dissolved in CHCl3, and then reacted for 48 h under stirring at 25°C. The solvent (CHCl3) was then removed by rotary evaporation and the residue was treated by 8 mL Na2CO3 (50 mM), the mixture was ultra sounded for 10 min and further centrifuged at 4,000 rpm for 20 min. Supernatant was then dialyzed with dialysis membrane (MWCO 14 kDa). The product FA-PEG-Chol was then obtained by lyophilization, which yielded a yellow powder product (115 mg) with 81.6% yield. The molecular weight of the FA-PEG-Chol was confirmed by matrix-assisted laser desorption/ionization time-of-flight mass spectroscopy (MALDI-TOF-MS, Bruker Daltonics Inc., Fremont, CA) (Supplementary Material: Figure S1).
The cationic liposomes were prepared by thin film hydration and polycarbonate membrane extrusion method according to the literatures [31, 42]. The lipid compositions of the FR-targeted liposomes and non-targeted liposomes were DOTAP/Chol/mPEG-DSPE/FA-PEG-Chol at a molar ratio of 40:55:4.5:0.5 and DOTAP/Chol/mPEG-DSPE at a molar ratio of 40:55:5 respectively. DOX was remotely loaded into the liposomes by a transmembrane pH gradient. Briefly, the total lipids (85 mg) was dissolved in CHCl3 and dried to a thin film by rotary evaporation followed by further drying under vacuum at 40°C. The lipid film was then hydrated with 2 mL (NH4)2SO4 (250 mM) for 30 min at 60°C. The resultant of multilamellar vesicles were extruded 5 times through 0.2 µm pore-size polycarbonate membranes and 5 times through 0.1 µm pore-size polycarbonate membranes using a Lipex Extruder (Northern Lipids Inc., Canada) driven by pressurized nitrogen at 60°C to produce homogeneous unilamellar vesicles. The residual (NH4)2SO4 was removed with phosphate-buffered saline (PBS, pH 7.4) by size-exclusion chromatography on a PD-10 column. DOX·HCl dissolved in deionized H2O (10 mg/mL) was added to the liposomes at a DOX-to-lipid ratio of 1:20 (w/w), followed by a 30 min incubation at 65°C. Free DOX was separated from liposomes by size exclusion chromatography on a Sepharose CL-4B column. DOX concentration in the liposomes and drug encapsulation efficiency of the collected fraction were determined by measuring absorption at 480 nm on a UV-Vis spectrophotometer (756 PC, Shanghai Spectrum Instruments, Shanghai, China) following liposome lysis in ethanol.
Preparation of FA-DOX/siRNA-L and DOX/siRNA-L
The FA-DOX/siRNA-L and DOX/siRNA-L were prepared at a w/w (liposome/siRNA) ratio of 200:1 in RNase free H2O by adding stock solution of FA-DOX-L or DOX-L into a prepared siRNA solution. The samples were vortexed, and the solutions were then incubated at room temperature for 30 min to ensure siRNA loading efficiently. The loading efficiency of siRNA by DOX-L was determined by ethidium bromide (EB) dye displacement assay in the agarose gel. Free or unconjugated siRNA will migrate in the gels, but the conjugated siRNA will be stuck in the loading wells with liposomes.
Size and zeta potential determination
Particle size and zeta potential of the liposomes were measured by Zeta PALS (Zeta Potential Analyzer, Brookhaven Instruments Corporation, Austin, TX) according to the manufacturer's instructions. All the measurements were carried out at room temperature. Each parameter was measured three times, and average values and standard deviations were calculated.
Agarose gel electrophoresis
Agarose gel electrophoresis was used to evaluate the siRNA loading in liposomes. The siRNA or liposome (the dose of siRNA was 120 pmol) was applied to a 2% (w/v) agarose gel in TAE buffer containing Goldview staining reagent (Solarbio, China). The free siRNA or unconjugated siRNA will migrate in the gels but the conjugated siRNA will be stuck in the loading wells with liposomes. Images were obtained using a UV transilluminator and a digital imaging system (Life Science Technologies, USA).
Cellular uptake of liposomes
HeLa and KB cells grown in a monolayer were washed once with fresh folate free RPMI 1640 medium and then were incubated with FA-DOX-L, DOX-L, FA-siRNA-L (Cy5 labeled siRNA), siRNA-L (Cy5 labeled siRNA), FA-DOX/siRNA-L (FAM labeled siRNA) and DOX/siRNA-L (FAM labeled siRNA) for 1 h at 37°C. For folate blocking group, 1 mM free folate was added to the incubation media prior to the addition of FA-DOX/siRNA-L. After the transfection for 1 h, the cells were rinsed with PBS for 3 times and fixed with 4% paraformaldehyde solution (Thermo Scientific, USA) for 15 min. The solution of 4,6-Diamidino-2-phenylindole (DAPI, Thermo Fisher Scientific, USA) was employed for the staining of nucleus. Samples were observed with a fluorescence microscope (Olympus, Japan).
Real-time PCR
Total RNA was extracted from the cells or tissues by using Trizol reagent following the protocol suggested by the manufacturer. Then the cDNA was synthesized using the Reverse Transcription System (Promega, Madison, USA). Real-time PCR was performed with SYBR Green by using the Mx3005P QPCR instrument (Agilent Technologies, USA). The rRNA was applied as the input reference. The primers used in this study are listed as follows: Bmi1, F: TGG ACT GAC AAA TGC TGG AGA-3' R: GAA GAT TGG TGG TGG TTA CCG CTG-3'; rRNA, F: CGG CTA CCA CAT CCA AGG AA-3', R: GCT GGA ATT ACC GCG GCT-3'; p14 (Arf ), F: GTT CTT GGT GAC CCT CCG GAT T-3', R: ATC AGC ACG AGG GCC ACA G-3'; p16 (Ink4A), F: GCC CAA CGC ACC GAA TAG TT-3', R: GGG CAG TTG TGG CCC TGT AG-3'; p21 (Cip1), F: CCT GTC ACT GTC TTG TAC CCT-3', R: GCG TTT GGA GTG GTA GAA ATC T-3'.
Western blot analysis
Cells were lysed in lysis buffer CelLytic M Cell Lysis Reagent (Sigma-Aldrich, MA, USA) for 30 min on ice, and the supernatant was collected after centrifugation at 12,000 rpm (Eppendorf 5415D). Cell lysate was separated on a 10% acrylamide gel and transferred to a PVDF membrane. Membranes were blocked in 5% skim milk for 1 h and then incubated with monoclonal antibody against Bmi1 (1:1000, Millipore, USA) or β-actin (1:5000, Millipore, USA) overnight. Membranes were then washed in PBST (PBS with 0.1% Tween-20) 3 times and then incubated for 1 h with secondary antibody. Then the membranes were washed 4 times and developed by an enhanced chemiluminescence system according to the manufacturer's instructions (Perkin Elmer, Waltham, MA).
Cytotoxicity
The cytotoxic effect of liposomes against the KB, HeLa and Hep3B cells was measured by Cell Counting Kit-8 kit (CCK-8 kit, Dojindo laboratories, Kumamoto, Japan). KB, HeLa and Hep3B cells were seeded onto 96-well plates at a density of 1 × 104 cells/well. After 24 h, cells were treated with FA-DOX/siRNA-L, FA-DOX-L, FA-siRNA-L, DOX-L and free DOX. Cells were incubated for a further 24 h and the proportion of viable cells measured colorimetrically according to the user's manual. Cell viability within each group was expressed as a percentage of the viability of control cells.
Microscopy
Microscopic examination of cells or tissue section slides was performed with an Olympus SZX12 fluorescence microscope equipped with digital camera and connected to a PC running MagnaFire 2.0 camera software (Optronics, Goleta, CA). Pictures were taken at equal exposure time for each sample.
Stability test of the loading siRNA
The FA-DOX/siRNA-L was added into the mouse serum and then kept in 37ºC for 1 h, 3 h and 6 h respectively. Then the liposome solutions with serum were added into the 6-well KB cell culture and incubated for 1 h in the 37ºC CO2 incubator. After 1 h incubation, the cells were rinsed with PBS for 3 times and fixed with 4% paraformaldehyde solution (Thermo Scientific, USA) for 15 min. DAPI solution was employed for the staining of nucleus. Samples were observed with a fluorescence microscope (Olympus, Japan) and pictures were taken at same exposure time.
Pharmacokinetic analysis
Female Kunming mice (body weight ~25 g, 6 - 8 weeks old) were obtained from Laboratory Animal Center of the Huazhong University of Science and Technology. Liposomes or free drug was injected as a single intravenous bolus via the lateral tail vein at a dose of 5 mg/kg DOX, 4 mice for each group. At 10 min, 30 min, 1 h, 2 h, 4 h, 8 h, 12 h, and 24 h after the injection, 500 µL blood was collected in heparin-treated tubes and then centrifuged at room temperature (Eppendorf 5415D, 5,000 rpm, 5 min) to obtain plasma. Plasma aliquots of 100 µL were added to 1 mL ethanol and 100 µL deionized water, followed by addition of 25 µL 20% sodium dodecyl sulfate (SDS). The samples were then vortexed for 30 s and centrifuged at 4ºC (12000 g, 10 min) to extract the DOX from the plasma protein. DOX concentration in plasma was measured by its fluorescence intensity at 495 nm by fluorescence spectrophotometer (Hitachi F-2700, software FL solution, Japan). The concentration of DOX in each sample was calculated by using a calibration curve determined by a series of dilution of DOX solutions. Pharmacokinetic variables including AUC, Cmax, t1/2 and clearance were calculated by using pharmacological software Drug And Statistics (Hefei, China, version 2.0).
Tissue distribution, tumor tissue uptake and anti-tumor efficacy study in vivo
Female Balb/C nude mice (6 - 8 weeks of age, 16 - 18 g) were obtained from Huafukang technology corporation (Beijing, China), and kept in filter-topped cages with standard rodent chow and water available ad libitum, with a 12 h light/dark cycle. The experiment protocol was approved by the ethical committee of animal experiment of Huazhong University of Science and Technology. For the tissue distribution assay, subcutaneous KB tumors were seeded by inoculation of 2.5 × 106 KB cells in the front armpit of the mice. At the tumor volume of 500 mm3, liposomes or free drug was injected via the lateral tail vein at a dose of 170 μg/kg siRNA and 5 mg/kg DOX. After the injection of 3 h, 500 µL blood was collected in heparin-treated tubes and then centrifuged (5,000 rpm, 5 min) at room temperature to obtain plasma. Plasma aliquots of 100 µL were added with 1 mL ethanol, 100 µL deionized water, and 25 µL 20% sodium dodecyl sulfate (SDS), and then were vortexed 30 s and centrifuged (12000 g, 10 min) at 4ºC to extract the DOX from the plasma protein. Lung, heart, liver, spleen, kidney and tumor tissues were also collected at 3 h respectively after injection. Tissues of 0.1 mg were homogenized with the buffer (300 µL EtOH, 15 µL 37% HCl, 185 µL H2O) and then centrifuged (Eppendorf 5415D, 12,000 rpm, 15 min) at 4ºC to collect the supernatant. The concentration of DOX was determined by fluorescence spectrophotometer. DOX in plasma was measured by its fluorescence intensity at 495 nm by fluorescence spectrophotometer (Hitachi F-2700, software FL solution, Japan). For the efficacy study, 2.5 × 106 KB cells were injected in the front armpit of the nude mice. At the tumor volume of ~50 mm3, liposomes or free drug was administrated by a single dose tail vein injection (170 μg/kg siRNA, 8 mg/kg DOX). The tumor sizes were measured every other day and any death of the mice was recorded. Tumor tissue uptake were assayed using the tumor tissue removed from mice for distribution study. Apoptosis assay (TUNEL) and target gene expression analysis (qRT-PCR) were performed by using the resected tumor tissue from mice for efficacy study after the mice were sacrificed. The tumor tissue sections were examined by fluorescence microscope (Olympus SZX12, Japan).
Liver and kidney toxicity
To check the potential liver and kidney toxicity of FA-DOX/siRNA-L, mice were administrated with a single dose tail vein injection of saline, FA-L, and FA-DOX/siRNA-L at a liposome concentration of 310 mg/kg (n = 3). Blood samples were collected at 24 h after the injections and then centrifuged (5000 rpm for 5 min) at room temperature to remove the cells. Then the plasma was used to determine the ALT, AST, BUN and CR levels by automatic biochemical analyzer (Roche ISE900, Switzerland).
Statistical analysis
Significance analysis of the data was performed using Student's t-test (SPSS Software, Chicago, IL). A value of P < 0.05 was considered significant and P < 0.01 was considered highly significant when compared with the corresponding control. Multiple groups were compared by one-way ANOVA with Dunnett's t-test. A value of P < 0.05 was considered significant and P < 0.01 was considered highly significant.
Results
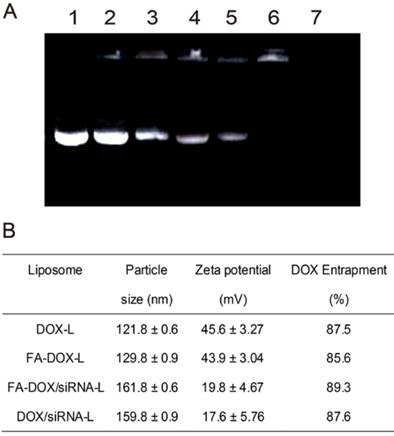
FA-DOX/siRNA-L preparation and characterization
DOX-L or FA-DOX/-L was prepared according to the methods to encapsulate DOX inside to achieve minimal premature drug release. Then the Bmi1 siRNA was loaded by DOX-L or FA-DOX/-L through electrostatic adherence (Fig.1). Then the liposomes were characterized to ensure the liposomes were prepared correctly. By dynamic light scattering detection, the average particle sizes of DOX-L and FA-DOX-L were 121.8 and 129.8 nm respectively (Fig.2B). To find an appropriate preparation composition, we mixed the liposomes and siRNA at the ratio 40:1, 80:1, 120:1, 160:1 and 200:1 (w/w) and performed the agarose gel electrophoresis. The result showed that when the liposomes and siRNA were combined at a ratio of 200:1 (w/w), the siRNA was fully entrapped in the loading wells with liposomes (Fig.2A). This suggests that the siRNA can be fully loaded by liposomes at the ratio of 200:1. After loading with Bmi1 siRNA at the lipid/siRNA ratio of 200:1 (w/w), the mean particle sizes of DOX/siRNA-L and FA-DOX/siRNA-L were 159.8 and 161.8 nm respectively (Fig.2B). This indicated that siRNA binding increased the particle size of liposomes about 30-40 nm. The zeta potential of DOX-L and FA-DOX-L were 45.6 and 43.9 mV respectively (Fig.2B). After loading with Bmi1 siRNA, the zeta potential of DOX/siRNA-L and FA-DOX/siRNA-L were 17.6 and 19.8 mV respectively (Fig.2B). This is due to that the positive zeta potential from DOTAP was neutralized partly by the siRNA with negative potential. The entrapments of DOX in DOX-L, FA-DOX-L, DOX/siRNA-L and FA-DOX/siRNA-L were 87.5, 85.6, 87.6 and 89.3% respectively (Fig.2B).
The structure and anti-tumor mechanisms of FA-DOX/siRNA-L. First, long circulating FA-DOX/siRNA-L is bound to and internalized into tumor cells via folate receptors. Then FA-DOX/siRNA-L is transported through the endosome and lysosome. DOX and siRNA are released to the cytoplasm. Finally, the Bmi1 siRNA binds to Bmi1 mRNA and the DOX binds to chromosomal DNA.
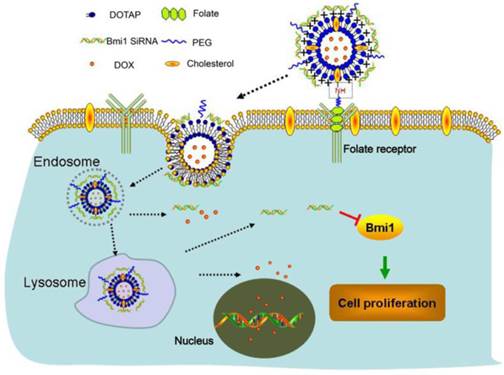
Complexation of siRNA with folate DOX liposomes (FA-DOX-L) and characterization of liposomes. A. A representative agarose gel electrophoresis image illustrating siRNA loading efficiency at the following w/w ratio (FA-DOX-L/siRNA): (1) 0:1 (siRNA only, control); (2) 40:1; (3) 80:1; (4) 120:1; (5) 160:1; (6) 200:1, and (7) FA-DOX-L only. SiRNA combined to FA-DOX-L has limited migration in the agarose gel, near the loading wells. B. Key parameters of DOX-L, FA-DOX-L, DOX/siRNA-L and FA-DOX/siRNA-L liposomes. Data are expressed as mean ± SD of three independent samples.
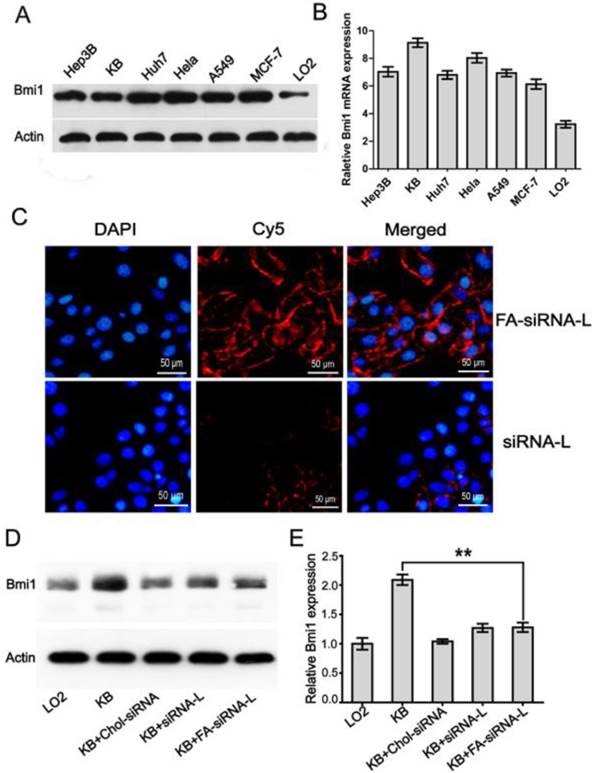
FA-DOX/siRNA-L delivers the Bmi1 siRNA and DOX efficiently and simultaneously in vitro
After the preparation and characterization, the efficiency of folate liposome to deliver DOX and siRNA was detected. We first assayed the expression of Bmi1 by western blotting and qRT-PCR in 6 cancer cell lines Hep3B, KB, Huh7, HeLa, A549, MCF-7, and 1 normal fetal liver cell line LO2. Results showed that all six cancer cell lines showed high level Bmi1 expression (Fig.3A). Quantitative RT-PCR detection showed identical results with western blotting, with the highest expression level in KB and HeLa cells (Fig.3B). Thus, we chose KB, HeLa, and Hep3B (negative for FR expression) cells for the successive studies and the capability of folate liposome to deliver Bmi1 siRNA was checked in these three cell lines. Fluorescence microscopy showed clearly that upon 1 h treatment with Bmi1 siRNA-L, the siRNA was aggregated at the membrane of KB cells but not internalized (Fig.3C). However, Bmi1 siRNA delivered by FA-siRNA-L was noticeably uptaken and released to the cytoplasm of KB cells after 1 h culture (Fig.3C). The fluorescence of siRNA was much greater in KB cells treated with FA-siRNA-L than that treated with siRNA-L (Fig.3C). This suggests that FA-siRNA-L can deliver the siRNA into the cells more efficiently than the siRNA-L (Fig.3C). Western blotting also showed that the Bmi1 expression in KB cells was inhibited by FA-siRNA-L although the effect was identical to siRNA-L and Chol-siRNA after 48 h treatments (Fig.3 D and E).
The Bmi1 siRNA delivery efficiency by FA-siRNA-L and repression effects on Bmi1 expression in KB cells. For western blotting and qRT-PCR assay, cells cultured in DMEM medium were harvested and protein or mRNA was extracted for the determinations. For the uptake study, KB and LO2 cells grown in a monolayer were incubated with FA-siRNA-L, siRNA-L, and Chol-siRNA for 1 h at 37°C and were then used for the next assays. A. The Bmi1 protein expression in cancer or normal cell lines detected by western blot. B. QRT-PCR quantitative analysis of the expression of Bmi1 in cancer cells in mRNA level. C. Cellular uptake and intracellular distribution of Bmi1 siRNA in the KB cells. D. Western blotting of Bmi1 expression in the cells treated with Bmi1 siRNA complex or liposomes. LO2 cells were used as the normal cell control and β-actin was used as the loading control. E. Quantitative analysis of the western blotting bands. Data are expressed as mean ± SD of 3 independent samples. **: P < 0.01.
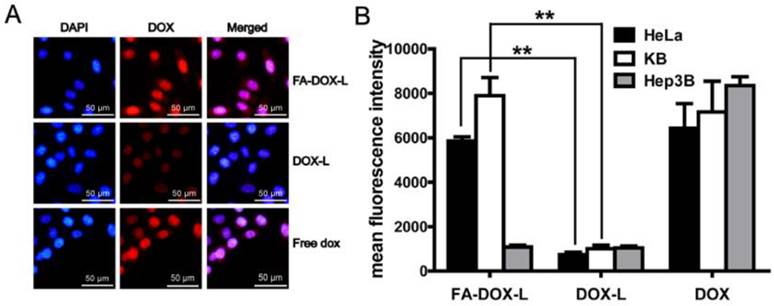
The efficiency of folate liposme to deliver DOX was also determined in KB and HeLa cells, with Hep3B cell as the negative control. KB cells treated with free DOX showed clear fluorescence in nucleus, which suggests the excellent permeability and uptake of DOX (Fig.4A). When entrapped by DOX-L, the uptake of DOX showed marked reduction (Fig.4A). However, FA-DOX-L can significantly improve the uptake of DOX compared with DOX-L. Quantitative analysis by fluorescence microplate reader also showed that the FA-DOX-L increased the uptake of DOX 6.8 and 8.2 fold in HeLa and KB cells than the DOX-L respectively (Fig.4B). But FA-DOX-L cannot increase the uptake of DOX in Hep3B cells compared with DOX-L (Fig.4B). These data suggest the FA-DOX-L can deliver the DOX efficiently and this effect is highly dependent on the folate ligands.
Cellular uptake and distribution of DOX in KB and HeLa cells. KB, HeLa and Hep3B cells (FR negative) grown in a monolayer were incubated with FA-DOX-L, DOX-L, and free DOX for 1 h at 37°C. A. Fluorescence detection and localization of DOX in the KB cells. B. Cellular uptake of DOX delivered by DOX-L and FA-DOX-L in KB, HeLa and Hep3B cells. Hep3B cell was used as a negative control. Data are expressed as mean ± SD of 3 independent samples. **: P < 0.01.
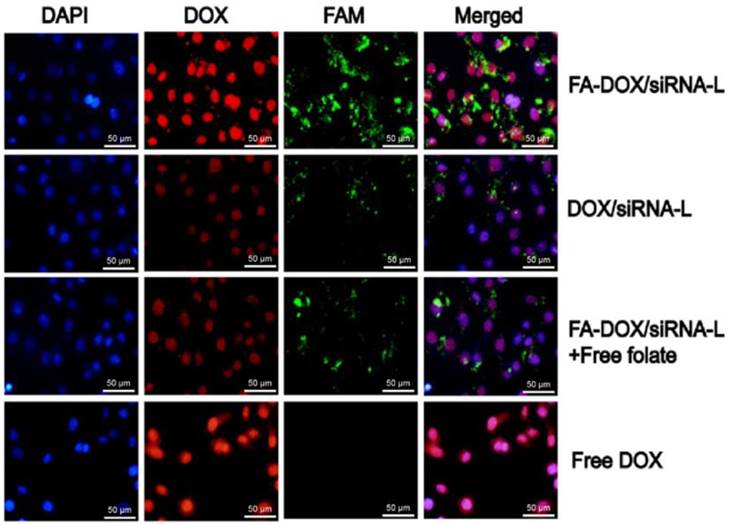
We then checked the co-delivery and localization of DOX and Bmi1 siRNA by FA-DOX/siRNA-L and DOX/siRNA-L. DOX/siRNA-L can deliver both the DOX (red) and siRNA (FAM labeled, green) to the KB cells, but the efficiency was not so high (Fig.5, second row). Compared with DOX/siRNA-L, FA-DOX/siRNA-L can significantly improve the uptake of DOX and siRNA simultaneously (Fig.5, first row) and this effect can be blocked by addition of 1 mM free folate (Fig.5, third row). Importantly, the distribution of DOX in nucleus and Bmi1 siRNA in cytoplasm were not affected each other when delivered by FA-DOX/siRNA-L. Similar result was observed in HeLa cells as in KB cells (Supplementary Material: Fig. S2). These data illustrated that FA-DOX/siRNA-L can deliver both DOX and siRNA efficiently and simultaneously into cells and ensure the release of the drugs.
Co-localization of Bmi1 siRNA and DOX in KB cells. KB cells grown in a monolayer were incubated with free DOX, DOX/siRNA-L, FA-DOX/siRNA-L and for 1 h at 37°C. For folate blocking group, 1 mM free folate was added to the incubation media prior to the addition of FA-DOX/siRNA-L. Bmi1 siRNA was labeled with FAM (Green) and DOX emits red fluorescence by itself. The nucleus was counterstained with DAPI (Blue).
FA-DOX/siRNA-L showed improved cytotoxic effects of Bmi1 siRNA and DOX
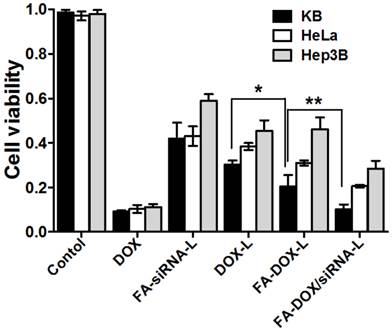
To verify the therapeutic effect of FA-DOX/siRNA-L, the cytotoxic effects of various Bmi1 and DOX formulations were evaluated in KB, HeLa and Hep3B cells. Compared with saline treated cells, FA-siRNA-L treated cells showed significant cell growth inhibition or cell death (Fig.6). After 24 h treatments, almost half of the KB and HeLa cells were diminished by FA-siRNA-L treatment, whereas the Hep3B cells survived well (Fig.6). After 24 h FA-siRNA-L treatment, 57% KB and 58% HeLa cells were diminished, while only 41% Hep3B cells were killed (Fig.6). Similarly, FA-DOX-L killed 81% KB cells, 73% HeLa cells and 53% Hep3B cells (Fig.6). As expected, FA-DOX/siRNA-L killed 90.5% KB cells, 82% HeLa cells and 68% Hep3B cells, much higher than FA-siRNA-L and FA-DOX-L (Fig.6). Together, these data showed that FA-DOX/siRNA-L displayed higher cytotoxicity than both FA-DOX-L and FA-siRNA-L. These data suggest clearly that Bmi1 siRNA and DOX delivered by FA-DOX/siRNA-L showed enhanced cytotoxic effects in vitro.
Cytotoxic effects of FA-DOX/siRNA-L in KB, HeLa and Hep3B cells. Cells were treated with liposomes with the final siRNA concentration of 100 nM and DOX concentration of 1 mg/mL. The Hep3B cells were used as a negative control. The multiple groups were compared by one-way ANOVA with Dunnett's t-test. Data are expressed as mean ± SD (n = 3). *: P < 0.05; **: P < 0.01.
Pharmacokinetic analysis
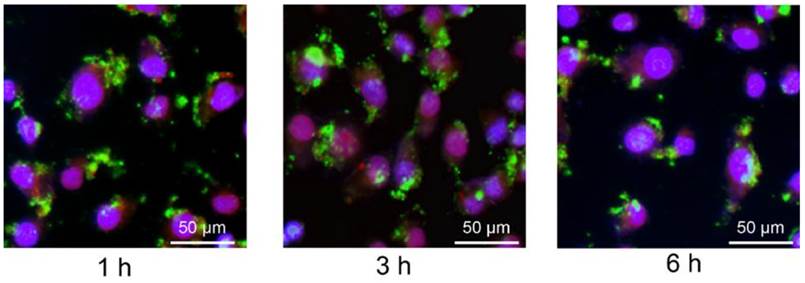
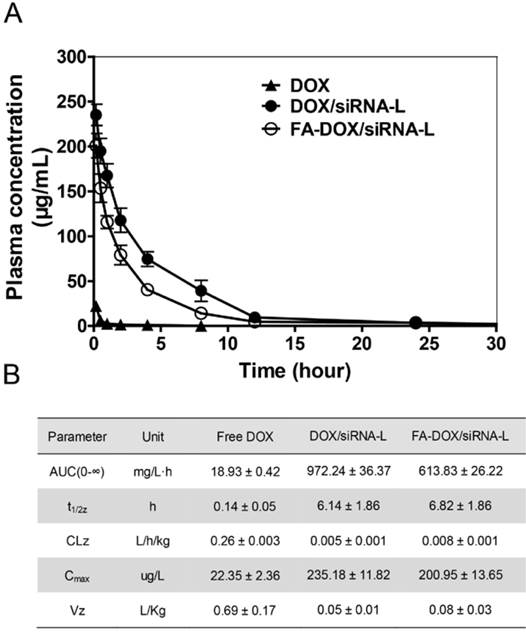
We first assayed the stability of siRNA conjugated with liposome in the serum. After 6 h culture with the mouse serum, the FA-DOX/siRNA-L still can be uptaken by KB cells in cell culture. In addition, the amount of siRNA absorbed by KB cells had no significant reduction after 6 h culture compared with 1 and 3 h culture with mouse serum. This suggests that the siRNA conjugated with FA-DOX/siRNA-L is stable in the blood circulation for at least 6 h (Fig.7). We then determined the pharmacokinetics of both FA-DOX/siRNA-L, DOX/siRNA-L and free DOX. Plasma clearance kinetics of liposomal DOX is shown in Figure 8A and pharmacokinetic parameters are summarized in Figure 8B. As expected, free DOX showed rapid clearance from the plasma and its concentration was almost undetectable after 30 min (Fig.8A). Meanwhile, DOX/siRNA-L showed a much longer circulation time, and its half-life increased to 6.14 h (Fig.8B). FA-DOX/siRNA-L showed similar t1/2 with DOX/siRNA-L, 6.82 h (Fig.8B). In addition, both FA-DOX/siRNA-L and DOX/siRNA-L showed much larger AUC than free DOX.
Stability test of the siRNA of the FA-DOX/siRNA-L. The FA-DOX/siRNA-L liposomes were cultured with mouse serum for 1, 3 and 6 h at 37 ºC. Then the liposomes solutions with serum were added to KB cell culture for 1 h under 37 ºC. The siRNA was labeled with FAM (green) and the nucleus was counterstained with DAPI (blue). Note the color of the nucleus was merged from blue (DAPI) and red (DOX).
The pharmacokinetic analysis of DOX in mouse plasma. A. Pharmacokinetic curves of DOX, DOX/siRNA-L, FA-DOX/siRNA-L in the mouse plasma. The mice were injected intravenously by a single dose of drugs at a DOX concentration of 5 mg/kg in female Kunming mice and the blood samples were collected at 8 time points (n = 4). B. Pharmacokinetic parameters of DOX calculated from the same experiment by a non-compartment model (n = 4 animals for each group). AUC: area under curve; CLz: clearance; t1/2z: plasma half-life; Vz: distribution volume during elimination phase; Cmax: maximum plasma drug concentration.
DOX distribution and Bmi1 repression by FA-DOX/siRNA-L delivery in vivo
Tissue distributions of DOX in normal and tumor tissues were observed in tumor-bearing nude mice after 3 h tail vein injection. Both DOX/siRNA-L and FA-DOX/siRNA-L injections showed evident DOX accumulation in livers with reference to other organs compared with free DOX (Fig.9A). However, the distribution of DOX delivered by DOX/siRNA-L and FA-DOX/siRNA-L in kidneys and hearts were similar to free DOX (Fig.9A). In tumor tissues, DOX/siRNA-L and FA-DOX/siRNA-L both increased the concentration of DOX greatly compared with free DOX (Fig.9B). In addition, FA-DOX/siRNA-L showed even higher DOX uptake than DOX/siRNA-L in tumor tissues (Fig.9B). These results showed that FA-DOX/siRNA-L efficiently and specifically delivered DOX to the tumor tissues.
DOX distribution and Bmi1 expression in normal and tumor tissues in xenograft nude mice. For panel A and B, the mice were injected via tail vein at a single dose of saline, free DOX (5 mg/kg), DOX/siRNA-L (170 μg/kg siRNA, 5 mg/kg DOX) and FA-DOX/siRNA-L (170 μg/kg siRNA, 5 mg/kg DOX) (n = 3) when the tumors grew to about 500 mm3. The tissues were collected at the 3 h after the injections. A. Quantitative analysis DOX distribution by the microplate reader in the normal tissues of xenograft nude mice. B. DOX distribution by fluorescence detection in tumor tissues of mice. C. Relative Bmi1 mRNA levels in tumor tissues treated with saline, free DOX, DOX-L, DOX/siRNA-L, FA-DOX-L and FA-DOX/siRNA-L by a single dose tail vein injection (170 μg/kg siRNA, 5 mg/kg DOX) (n = 3). Total mRNA was extracted from the isolated tumor tissues 24 h after the drug injections. Data are expressed as mean ± SD. *: P < 0.05.
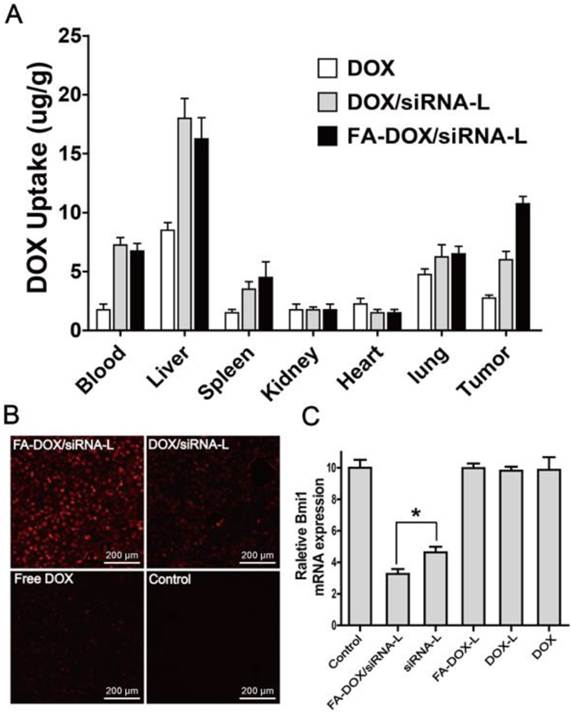
To minimize the degradation of siRNA during the tissue processing and fluorescence quenching, we used qRT-PCR to investigate the distribution of Bmi1 siRNA in the tumors tissues. We collected the tumor tissues 24 h after the drug administration and harvested the total mRNA to perform qRT-PCR assay. Similar to saline, the formulation of FA-DOX-L, DOX-L and DOX had no influence on Bmi1 expression (Fig.9C). However, siRNA-L and FA-DOX/siRNA-L administration significantly reduced Bmi1 expression in the tumors (Fig.9C). Moreover, FA-DOX/siRNA-L showed even greater repression effects than the siRNA-L (Fig.9C). These data suggest that through FA-DOX/siRNA-L delivery, Bmi1 siRNA can inhibit Bmi1 expression both in vitro and in vivo.
Evaluation of in vivo tumor inhibition ability
Anti-tumor therapeutic efficacy of various formulations composed of DOX and/or Bmi1 siRNA were further evaluated using a KB xenograft tumor mouse model. A single dose of DOX and/or Bmi1 siRNA formulation was administrated by tail vein injection upon the subcutaneous tumor grew to ~50 mm3. At the 25th day after the injection, the mice were sacrificed and tumors were removed for volume determination and histology examination. Compared with the saline group (2319 mm3), free DOX group (1287 mm3) and DOX-L group (1099 mm3) showed smaller tumors at 25 days (Fig.10A and B). Tumors of FA-DOX-L group (540 mm3) and FA-siRNA-L group (855 mm3) were even smaller than DOX and DOX-L treated tumors (Fig.10A and B). Notably, FA-DOX/siRNA-L treated tumors had the smallest sizes (248 mm3) among these formulations treatment groups (Fig.10A and B). The growth curve of tumors showed that the differences among these groups were due to continuous growth inhibition induced by these formulations (Fig.10B). Statistical analysis between FA-DOX/siRNA-L and FA-DOX -L group indicted a P value of 0.032 (Fig.10B). These data suggest that DOX and Bmi1 siRNA co-delivery by FA-DOX/siRNA-L can enhance the tumor inhibition capability and provide improved therapeutic effect.
Anti-tumor effects of FA-DOX/siRNA-L in xenograft mouse model. A. Excised tumors at the end point of the experiment (day 24 after the drug injection). B. Growth curves of xenograft tumors treated with saline, free DOX, DOX-L, FA-siRNA-L, FA-DOX-L and FA-DOX/siRNA-L by a single dose tail vein injection (170 μg/kg siRNA, 8 mg/kg DOX). The curves present the changes of tumor sizes from the day of injection (day 0). The comparisons were performed versus the FA-DOX-L group by the Student's test. Data are expressed as mean ± SD (n = 3). *: P < 0.05. C. Body weight changes of mice treated with saline, free DOX, DOX-L, FA-siRNA-L, FA-DOX-L and FA-DOX/siRNA-L. Saline injection was used as the control. Data are expressed as mean ± SD (n = 3).
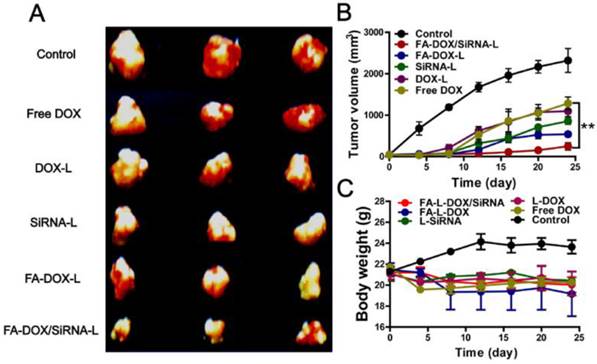
To evaluate the systemic toxicity of various formulations, the body weight changes in mice were examined every 4 days after injections. At the fourth day, the DOX injected mice showed sudden weight loss compared with other group mice (Fig.10C). The body weights of FA-DOX-L treated mice showed slight decrease, while other group mice kept slowly growing or unchanged. These data showed that co-delivery of DOX and Bmi1 siRNA by FA-DOX/siRNA-L did not increase systemic toxicity compared with FA-DOX-L or FA-siRNA-L (Fig.10C).
The enhanced apoptosis induction by FA-DOX/siRNA-L
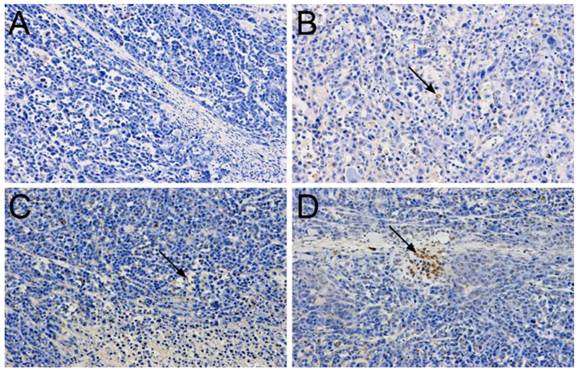
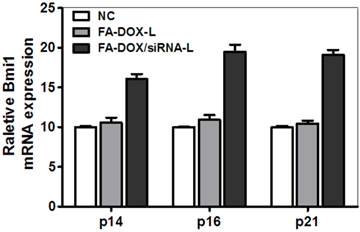
To determine whether there are synergetic anti-tumor effects of DOX and Bmi1 siRNA resulted from the enhanced apoptosis, we checked the apoptosis by TUNEL assay in tumor samples of treated mice. In saline injected mice, no TUNEL staining was observed (Fig.11A). In FA-siRNA-L and FA-DOX-L treated mice, widespread TUNEL staining clearly showed the existence of apoptosis (Fig.11B and C). In FA-DOX/siRNA-L treated mice, both the strength and amount of the signal were stronger than that of FA-siRNA-L or FA-DOX-L treated mice (Fig.11D). To determine whether the cellular apoptosis or growth inhibition was resulted from the Bmi1 expression suppression, we checked the expression of its downstream targets, p14 (Arf), p16 (Ink4A) and p21 (Cip1). The qRT-PCR results showed that Bmi1 knockdown upregulated the expression of p14, p16 and p21 to 1.61, 1.95 and 1.91 fold respectively (Fig.12). Considering the role of p14 and p21 inducing cellular apoptosis and p16 mediating cell cycle arrest, these data suggest that Bmi1 knockdown contributes as least partly to the cancer cell apoptosis and growth inhibition. Together, these data suggest that the enhanced treatment efficacy of Bmi1 siRNA and DOX co-delivery by FA-DOX/siRNA-L was resulted from the integrated apoptosis or cell cycle arrest induced by DOX and Bmi1 siRNA.
TUNEL staining in biopsies of KB tumors treated with a single dose injection of siRNA and/or DOX liposomes (170 μg/kg siRNA, 8 mg/kg DOX). The tumors were removed from the mice after the efficacy study (day 24 after the drug injection) and biopsies were made after 4% PFA fixation. TUNEL signal was colored by diaminobenzidine (DAB, brown color) and cell nucleus was counterstained by hematoxylin. A. Saline; B. FA-siRNA-L; C. FA-DOX-L; D. FA-DOX/siRNA-L.
The expression of Bmi1 target genes in tumor tissues when Bmi1 was knockdown. Tumor tissues were resected from the mice treated with a single dose injection of FA-DOX-L, FA-DOX/siRNA-L liposomes (170 μg/kg siRNA, 8 mg/kg DOX) and saline (NC group) after the efficacy study (day 24 after the drug injection). Total mRNA was extracted from the frozen tumor tissue samples (n = 3).
Liver and kidney toxicity of FA-DOX/siRNA-L
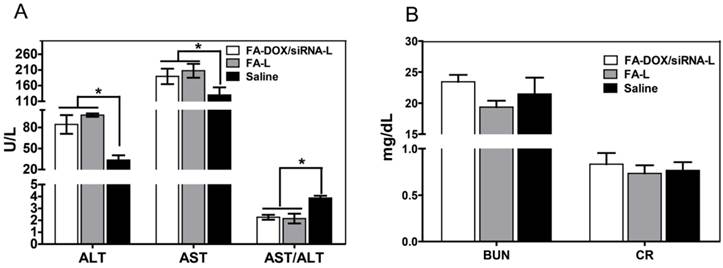
Besides the efficacy of liposomal chemotherapeutic drugs, another concern about the druggability of liposomal formulation is its safety. Considering a high concentration of DOX was accumulated in the liver, which might lead to potential liver toxicity, we assayed the liver and kidney toxicity of FA-DOX/siRNA-L in mice. It was found that 24 h after FA-L injection, the ALT and AST level rose to 97.7 ± 4.4 and 207.3 ± 12.9 U/L in comparison with 33.7 ± 3.8 and 133.3 ± 13.9 U/L of saline injection group. After FA-DOX/siRNA-L injection, the ALT and AST level rose to 84.3 ± 7.7 and 189.3 ± 14.2 U/L (Fig.13A). We also compared the AST/ALT ratio among the saline, FA-L and FA-DOX/siRNA-L injection groups. The AST/ALT ratios of FA-L and FA-DOX/siRNA-L groups were 2.1 and 2.3, whereas the ratio of saline group was 3.9, which hints the toxicity was a mild and short time effect (Fig.13A) [43, 44]. We also observed the BUN and CR level after the liposomes injection. After injection of FA-L or FA-DOX/siRNA-L, the BUN and CR levels in the mouse blood did not show significant increase compared with saline injections (Fig.13B). This was consistent with the observation that the distribution of FA-DOX/siRNA-L was relatively low in the kidney (Fig.9A).
Determination of the liver and kidney toxicity of FA-DOX/siRNA-L. Female Kunming mice were injected intravenously with a single dose of saline, FA-L, and FA-DOX/siRNA-L at a blank liposome concentration of 310 mg/kg (n = 3). Blood samples were collected 24 h after drug injections. A. Liver toxicity determined by AST, ALT level and AST/ALT ratio. B. Kidney toxicity determination by BUN and CR level in plasma. *: P < 0.05.
Conclusion and Discussion
Bmi1 gene has been extensively investigated in cancer biology, with growing evidences supporting its functions in tumor formation and disease relapse in a number of cancer types. Overexpression and activation of Bmi1 induce various forms of cancer, and are required for the onset and maintenance in various human tumors. Despite its roles in carcinogenesis, its therapeutic potential has not been adequately evaluated under a translational research setting. A possible circumstance is that expression of the Bmi1 is also essential for the self-renew and proliferation of many types of stem cells besides the cancer cells [45-48], which demands a targeted delivery system to minimize the undesired side effects to these tissues. To overcome this barrier, this study was aimed at designing a targeted liposomal formulation that can specifically deliver Bmi1 siRNA and DOX into the tumor site, down regulate Bmi1 expression in the tumor and achieve therapeutic cures.
To our knowledge, the current study demonstrated for the first time that inhibition of Bmi1 protein expression by siRNA could induce apoptosis in tumors together with DOX, and significantly suppress tumor growth of cancer cells in nude mice. Through co-delivery of DOX and siRNA by folate liposome, we have demonstrated such a feasibility using a model system to deliver siRNA and chemotherapeutics both in vitro and in vivo. We envisioned that this co-delivery system could be generalized to other anticancer drugs and other siRNAs.
In this study, contradictory to the hypothesis that the FR targeted FA-siRNA-L shows better repression effects than the non-targeted siRNA-L, the gene silencing effects between these two formulations were actually similar in vitro (Fig.3D). The possible reasons are listed as follows: first, the total siRNA delivered by various liposomal formulations was added into the cell culture in the same amount, which resulted the cells absorbed the same amount of siRNA after 48 h culture; second, the cationic liposome itself has a very high transfection capability even without the targeting ligand; third and most importantly, western blotting was assayed at 48 h post transfection, when the inhibitory effects of Bmi1 siRNA may have reached the plateau phase.
We also noticed in our study that both the FA-DOX/siRNA-L and DOX/siRNA-L liposomes showed higher accumulation in livers. This observation is consistent with studies from other groups that the cationic liposomes are preferentially uptaken by livers. A possible reason is that the macrophages or Kupffer cells in liver facilitated the uptake and hence retained the liposomes in livers [49]. The second possible reason is that the widespread blood vessel or capillary in the livers promoted the uptake of liposomes by the hepatocytes. Although the toxicity study suggests that the FA-DOX/siRNA-L injection induces acute liver toxicity, the toxicity of FA-DOX/siRNA-L is actually derived from the FA-L liposome itself instead of the DOX and siRNA since the toxicity of FA-DOX/siRNA-L was similar with FA-L (Fig.13). In addition, the decreased AST/ALT hints this toxicity is possibly mild and reversible since the AST/ALT value generally increase significantly in severe liver damages [43, 44]. On the other hand, this phenomenon may prompt that this delivery system is possibly appropriate for the treatment of liver cancers.
For targeted nanomedicines, conjugated targeting ligand can significantly improve the therapeutic effectiveness of conventional chemotherapeutics or gene therapy in cancer. Our data showed that the FA-DOX/siRNA-L could efficiently bind to KB and HeLa cells overexpressing FR and increase the uptake of DOX and Bmi1 siRNA, suggesting the effectiveness of FR targeting. On the other hand, the targeting activity of folate conjugated liposome is highly dependent on the folate receptor, which may limit the application of FR targeting liposome delivery. We also noticed that although Hep3B cells express very low level folate receptor, the FA-DOX/siRNA-L still showed relative high cytotoxicity on them in vitro. This effect was resulted from the passive targeting effects of liposome itself. That is, although the folate ligand dose not promote the uptake of FA conjugated liposomes by receptor mediated active targeting pathway, the FR negative Hep3B cell still can uptake the liposomes by regular endocytosis. This role for liposomes has been confirmed by commercial use as a delivery vector in vitro. For example, Lipofectamine 2000® has long been used a transfection reagent.
Importantly, our data validated the rational of co-delivery of DOX and Bmi1 siRNA since the selection of chemotherapeutics and gene drugs is critically important for combined therapy. Both in vitro and in vivo results illustrated that these two drugs showed additive or enhanced effects. As an oncogene, Bmi1 is not overexpressed in normal tissue cells. Co-delivery of Bmi1 siRNA will not increase the toxicity of chemotherapeutic drugs to normal tissues. Importantly, unlike TP53 or BCL-2 gene, Bmi1 mutations are seldom found in the cancer samples. Bmi1 functions as an oncogene mostly by amplification in copies, and this specific feature makes it an ideal target for siRNA based treatments since the mutations in the coding sequence may make the siRNA totally invalid. In terms of mechanisms, Bmi1 exerts its oncogenic function in many types of cancers mainly by silencing the Ink4a/Arf tumor suppressor locus encoding the p16 (Ink4A) and p14 (Arf) proteins. Whereas p16 inhibits cell cycle progression by disrupting the cyclin D/CDK4/6 complex, p14 suppresses Mdm2 function resulting in p53 stabilization and subsequent apoptosis [50]. Our data illustrated that Bmi1 knock down significantly resulted in the up-regulation of p14, p16 and p21 in tumor tissues. This suggests that Bmi1 siRNA may induce apoptosis by activating p14 derived and p21 dependent apoptotic pathway and in turn sensitize the cancer cells to DOX chemotherapy. In addition, we observed that the DOX delivered by the folate liposome was primarily localized in perinuclear region upon internalization, while the Bmi1 siRNAs were presented in the cytoplasm. This distinct intracellular distribution model perfectly matches their acting sites in the cells. Taken together, the co-delivery strategy of a broad spectrum chemotherapy with Bmi1 represents a promising approach to overcome existing barriers with cancer therapy that warrants further studies.
Finally, the synergistic or combined effects by co-delivering anti-cancer drugs and siRNAs have also been confirmed by other studies using different delivery system or targeting different genes. Chen et al designed a CD13 targeting (NGR peptide) PEGylated LPD (liposome-polycation-DNA) nanoparticle (LPD-PEG-NGR) to deliver c-MYC siRNA and DOX, and they observed the enhanced therapeutic effects in tumor mouse model [51]. Co-delivery of paclitaxel and Bcl-2 siRNA by cationic core-shell nanoparticles was also showed increased cytotoxicity in human breast cancer cells [52]. Co-delivery of EGFRvIII siRNA and erlotinib in glioblastoma cells was also found to significantly inhibit cell proliferation and induce apoptosis compared with the individual treatments [53]. Some studies focused on suppressing the drug-resistant capability of multiple drug resistant cells by co-delivering chemical drugs and siRNA targeting P-gp [11, 54] or other genes [11, 12]. These studies demonstrated that co-delivery of a chemotherapeutic agent and a P-gp or other siRNA to a drug-resistant cancer cell line could accomplish the cell killing in an additive or synergistic fashion. Some research teams also fabricated or applied novel material to deliver chemical drugs and siRNA, such as mesoporous silica nanoparticles (MSNP) [12, 55], nanostructured lipid carriers (NLCs) [56] and cationic core-shell nanoparticles (MDS-co-CES) [52] et al. When these materials were used for co-delivery anti-cancer drugs and siRNA, synergistic or enhanced anti-cancer effects were achieved as the liposome based drug delivery system dose. These studies, together with our study, collectively suggest that the synergistic or combined effect by co-delivering chemotherapeutic drugs and siRNA to cancer cells is a universal phenomenon and hold promise for the future personalized cancer treatments.
Supplementary Material
Fig.S1 - Fig.S2.
Acknowledgements
This work was financially supported by National Basic Research Program of China (973 Program, 2012CB932501), the National Natural Science Foundation of China (No.81370058, 81101824 and 81202095), and Wuhan Young Talents Program (No 2013070104010042).
Abbreviations
AST: aspartate amino-transferase
ALT: alanine amino-transferase
BUN: blood urea nitrogen
Chol: cholesterol
CR: creatine
DSPE: distearoylphosphatidylethanolamine
DOX: doxorubicin
DCC: dicyclohexylcarbodiimide
DOTAP: dioleoyl-3-trimethylammonium-propane
DSPE: distearoyl phosphatidylethanolamine
DMSO: dimethyl sulfoxide
DMEM: Dulbecco's modified Eagle medium
FAM: carboxyfluorescein
FBS: fetal bovine serum
FR: folate receptor
GPI: glycosyl phosphatidinositol
HPLC: high-performance liquid chromatography
MDR: multi-drug resistance
NHS: N-hydroxysuccinimide
PEG: polyethylene glycol
PBS: phosphate buffered saline
PCR: polymerase chain reaction
PVDF: polyvinylidene fluoride
PFA: paraformaldehyde
Competing Interests
The authors have declared that no competing interest exists.
References
1. Oh YK, Park TG. siRNA delivery systems for cancer treatment. Adv Drug Deliv Rev. 2009;61:850-62 doi:S0169-409X(09)00149-5 [pii] 10.1016/j.addr.2009.04.018
2. Park IK, Morrison SJ, Clarke MF. Bmi1, stem cells, and senescence regulation. Journal of Clinical Investigation. 2004;113:175-9 doi:Doi 10.1172/Jci200420800
3. Lobo NA, Shimono Y, Qian D, Clarke MF. The biology of cancer stem cells. Annual Review of Cell and Developmental Biology. 2007;23:675-99 doi:DOI 10.1146/annurev.cellbio.22.010305.104154
4. Mihic-Probst D, Kuster A, Kilgus S, Bode-Lesniewska B, Ingold-Heppner B, Leung C. et al. Consistent expression of the stem cell renewal factor BMI-1 in primary and metastatic melanoma. International Journal of Cancer. 2007;121:1764-70 doi:Doi 10.1002/Ijc.22891
5. Leung C, Lingbeek M, Shakhova O, Liu J, Tanger E, Saremaslani P. et al. Bmi1 is essential for cerebellar development and is overexpressed in human medulloblastomas. Nature. 2004;428:337-41 doi:Doi 10.1038/Nature02385
6. Vonlanthen S, Heighway J, Altermatt HJ, Gugger M, Kappeler A, Borner MM. et al. The bmi-1 oncoprotein is differentially expressed in non-small cell lung cancer and correlates with INK4A-ARF locus expression. British Journal of Cancer. 2001;84:1372-6 doi:DOI 10.1054/bjoc.2001.1791
7. Kim JH, Yoon SY, Kim CN, Joo JH, Moon SK, Choe IS. et al. The Bmi-1 oncoprotein is overexpressed in human colorectal cancer and correlates with the reduced p16INK4a/p14ARF proteins. Cancer Letters. 2004;203:217-24 doi:DOI 10.1016/j.canlet.2003.07.009
8. Sacchi S, Federico M, Dastoli G, Fiorani C, Vinci G, Clo V. et al. Treatment of B-cell non-Hodgkin's lymphoma with anti CD 20 monoclonal antibody Rituximab. Critical Reviews in Oncology Hematology. 2001;37:13-25 doi:Doi 10.1016/S1040-8428(00)00069-X
9. Xu CR, Lee S, Ho C, Bommi P, Huang SA, Cheung ST. et al. Bmi1 functions as an oncogene independent of Ink4A/Arf repression in hepatic carcinogenesis. Mol Cancer Res. 2009;7:1937-45 doi:1541-7786.MCR-09-0333 [pii] 10.1158/1541-7786.MCR-09-0333
10. Fan L, Xu C, Wang C, Tao J, Ho C, Jiang L. et al. Bmi1 is required for hepatic progenitor cell expansion and liver tumor development. PLoS One. 2012;7:e46472. doi:10.1371/journal.pone.0046472 PONE-D-11-24788 [pii]
11. Saad M, Garbuzenko OB, Minko T. Co-delivery of siRNA and an anticancer drug for treatment of multidrug-resistant cancer. Nanomedicine (Lond). 2008;3:761-76 doi:10.2217/17435889.3.6.761
12. Chen AM, Zhang M, Wei D, Stueber D, Taratula O, Minko T. et al. Co-delivery of doxorubicin and Bcl-2 siRNA by mesoporous silica nanoparticles enhances the efficacy of chemotherapy in multidrug-resistant cancer cells. Small. 2009;5:2673-7 doi:10.1002/smll.200900621
13. Sun TM, Du JZ, Yao YD, Mao CQ, Dou S, Huang SY. et al. Simultaneous delivery of siRNA and paclitaxel via a "two-in-one" micelleplex promotes synergistic tumor suppression. ACS Nano. 2011;5:1483-94 doi:10.1021/nn103349h
14. Sanhai WR, Sakamoto JH, Canady R, Ferrari M. Seven challenges for nanomedicine. Nat Nanotechnol. 2008;3:242-4 doi:nnano.2008.114 [pii] 10.1038/nnano.2008.114
15. Rahman A, Husain SR, Siddiqui J, Verma M, Agresti M, Center M. et al. Liposome-mediated modulation of multidrug resistance in human HL-60 leukemia cells. J Natl Cancer Inst. 1992;84:1909-15
16. Gabizon A, Shmeeda H, Horowitz AT, Zalipsky S. Tumor cell targeting of liposome-entrapped drugs with phospholipid-anchored folic acid-PEG conjugates. Adv Drug Deliv Rev. 2004;56:1177-92 doi:10.1016/j.addr.2004.01.011 S0169409X04000201 [pii]
17. Perez AT, Domenech GH, Frankel C, Vogel CL. Pegylated liposomal doxorubicin (Doxil) for metastatic breast cancer: the Cancer Research Network, Inc, experience. Cancer Invest. 2002;20(Suppl 2):22-9
18. Theodoulou M, Hudis C. Cardiac profiles of liposomal anthracyclines: greater cardiac safety versus conventional doxorubicin? Cancer. 2004;100:2052-63 doi:10.1002/cncr.20207
19. Safra T. Cardiac safety of liposomal anthracyclines. Oncologist. 2003;8(Suppl 2):17-24
20. Batist G. Cardiac safety of liposomal anthracyclines. Cardiovasc Toxicol. 2007;7:72-4 doi:CT:7:2:72 [pii] 10.1007/s12012-007-0014-4
21. Xiang G, Wu J, Lu Y, Liu Z, Lee RJ. Synthesis and evaluation of a novel ligand for folate-mediated targeting liposomes. Int J Pharm. 2008;356:29-36 doi:S0378-5173(07)01073-3 [pii] 10.1016/j.ijpharm.2007.12.030
22. Wu J, Lu Y, Lee A, Pan X, Yang X, Zhao X. et al. Reversal of multidrug resistance by transferrin-conjugated liposomes co-encapsulating doxorubicin and verapamil. J Pharm Pharm Sci. 2007;10:350-7
23. Xiong XB, Huang Y, Lu WL, Zhang X, Zhang H, Nagai T. et al. Enhanced intracellular delivery and improved antitumor efficacy of doxorubicin by sterically stabilized liposomes modified with a synthetic RGD mimetic. J Control Release. 2005;107:262-75 doi:S0168-3659(05)00121-5 [pii] 10.1016/j.jconrel.2005.03.030
24. Elwood PC. Molecular cloning and characterization of the human folate-binding protein cDNA from placenta and malignant tissue culture (KB) cells. J Biol Chem. 1989;264:14893-901
25. Lacey SW, Sanders JM, Rothberg KG, Anderson RG, Kamen BA. Complementary DNA for the folate binding protein correctly predicts anchoring to the membrane by glycosyl-phosphatidylinositol. J Clin Invest. 1989;84:715-20 doi:10.1172/JCI114220
26. Wu M, Gunning W, Ratnam M. Expression of folate receptor type alpha in relation to cell type, malignancy, and differentiation in ovary, uterus, and cervix. Cancer Epidemiol Biomarkers Prev. 1999;8:775-82
27. Ross JF, Chaudhuri PK, Ratnam M. Differential regulation of folate receptor isoforms in normal and malignant tissues in vivo and in established cell lines. Physiologic and clinical implications. Cancer. 1994;73:2432-43
28. Parker N, Turk MJ, Westrick E, Lewis JD, Low PS, Leamon CP. Folate receptor expression in carcinomas and normal tissues determined by a quantitative radioligand binding assay. Anal Biochem. 2005;338:284-93 doi:S0003-2697(05)00006-0 [pii] 10.1016/j.ab.2004.12.026
29. Leamon CP. Folate-targeted drug strategies for the treatment of cancer. Curr Opin Investig Drugs. 2008;9:1277-86
30. Zhao XB, Lee RJ. Tumor-selective targeted delivery of genes and antisense oligodeoxyribonucleotides via the folate receptor. Adv Drug Deliv Rev. 2004;56:1193-204 doi:10.1016/j.addr.2004.01.005 S0169409X04000213 [pii]
31. Pan XQ, Zheng X, Shi G, Wang H, Ratnam M, Lee RJ. Strategy for the treatment of acute myelogenous leukemia based on folate receptor beta-targeted liposomal doxorubicin combined with receptor induction using all-trans retinoic acid. Blood. 2002;100:594-602
32. Wang S, Low PS. Folate-mediated targeting of antineoplastic drugs, imaging agents, and nucleic acids to cancer cells. J Control Release. 1998;53:39-48 doi:S0168-3659(97)00236-8 [pii]
33. Henne WA, Doorneweerd DD, Hilgenbrink AR, Kularatne SA, Low PS. Synthesis and activity of a folate peptide camptothecin prodrug. Bioorg Med Chem Lett. 2006;16:5350-5 doi:S0960-894X(06)00864-X [pii] 10.1016/j.bmcl.2006.07.076
34. Bae Y, Nishiyama N, Kataoka K. In vivo antitumor activity of the folate-conjugated pH-sensitive polymeric micelle selectively releasing adriamycin in the intracellular acidic compartments. Bioconjug Chem. 2007;18:1131-9 doi:10.1021/bc060401p
35. Leamon CP, Cooper SR, Hardee GE. Folate-liposome-mediated antisense oligodeoxynucleotide targeting to cancer cells: evaluation in vitro and in vivo. Bioconjug Chem. 2003;14:738-47 doi:10.1021/bc020089t
36. Gabizon A, Horowitz AT, Goren D, Tzemach D, Shmeeda H, Zalipsky S. In vivo fate of folate-targeted polyethylene-glycol liposomes in tumor-bearing mice. Clin Cancer Res. 2003;9:6551-9
37. Lee RJ, Low PS. Folate-mediated tumor cell targeting of liposome-entrapped doxorubicin in vitro. Biochim Biophys Acta. 1995;1233:134-44
38. Gabizon A, Horowitz AT, Goren D, Tzemach D, Mandelbaum-Shavit F, Qazen MM. et al. Targeting folate receptor with folate linked to extremities of poly(ethylene glycol)-grafted liposomes: in vitro studies. Bioconjug Chem. 1999;10:289-98 doi:10.1021/bc9801124 bc9801124 [pii]
39. Liu Y, Xu S, Teng L, Yung B, Zhu J, Ding H. et al. Synthesis and evaluation of a novel lipophilic folate receptor targeting ligand. Anticancer Res. 2011;31:1521-5 doi:31/5/1521 [pii]
40. Wu J, Liu Q, Lee RJ. A folate receptor-targeted liposomal formulation for paclitaxel. Int J Pharm. 2006;316:148-53 doi:S0378-5173(06)00150-5 [pii] 10.1016/j.ijpharm.2006.02.027
41. Masuda T, Akita H, Nishio T, Niikura K, Kogure K, Ijiro K. et al. Development of lipid particles targeted via sugar-lipid conjugates as novel nuclear gene delivery system. Biomaterials. 2008;29:709-23 doi:S0142-9612(07)00785-5 [pii] 10.1016/j.biomaterials.2007.09.039
42. Haran G, Cohen R, Bar LK, Barenholz Y. Transmembrane ammonium sulfate gradients in liposomes produce efficient and stable entrapment of amphipathic weak bases. Biochim Biophys Acta. 1993;1151:201-15
43. Sorbi D, Boynton J, Lindor KD. The ratio of aspartate aminotransferase to alanine aminotransferase: potential value in differentiating nonalcoholic steatohepatitis from alcoholic liver disease. Am J Gastroenterol. 1999;94:1018-22 doi:S0002927099000611 [pii] 10.1111/j.1572-0241.1999.01006.x
44. Sheth SG, Flamm SL, Gordon FD, Chopra S. AST/ALT ratio predicts cirrhosis in patients with chronic hepatitis C virus infection. Am J Gastroenterol. 1998;93:44-8 doi:S0002-9270(97)00009-9 [pii] 10.1111/j.1572-0241.1998.044_c.x
45. Dhawan S, Tschen SI, Bhushan A. Bmi-1 regulates the Ink4a/Arf locus to control pancreatic beta-cell proliferation. Genes Dev. 2009;23:906-11 doi:23/8/906 [pii] 10.1101/gad.1742609
46. Leung C, Lingbeek M, Shakhova O, Liu J, Tanger E, Saremaslani P. et al. Bmi1 is essential for cerebellar development and is overexpressed in human medulloblastomas. Nature. 2004;428:337-41
47. Sangiorgi E, Capecchi MR. Bmi1 is expressed in vivo in intestinal stem cells. Nat Genet. 2008;40:915-20
48. Sangiorgi E, Capecchi MR. Bmi1 lineage tracing identifies a self-renewing pancreatic acinar cell subpopulation capable of maintaining pancreatic organ homeostasis. Proc Natl Acad Sci U S A. 2009;106:7101-6 doi:0902508106 [pii] 10.1073/pnas.0902508106
49. Daemen T, Hofstede G, Ten Kate MT, Bakker-Woudenberg IA, Scherphof GL. Liposomal doxorubicin-induced toxicity: depletion and impairment of phagocytic activity of liver macrophages. Int J Cancer. 1995;61:716-21
50. Park IK, Morrison SJ, Clarke MF. Bmi1, stem cells, and senescence regulation. J Clin Invest. 2004;113:175-9
51. Chen Y, Wu JJ, Huang L. Nanoparticles targeted with NGR motif deliver c-myc siRNA and doxorubicin for anticancer therapy. Mol Ther. 2010;18:828-34 doi:mt2009291 [pii] 10.1038/mt.2009.291
52. Wang Y, Gao S, Ye WH, Yoon HS, Yang YY. Co-delivery of drugs and DNA from cationic core-shell nanoparticles self-assembled from a biodegradable copolymer. Nat Mater. 2006;5:791-6 doi:nmat1737 [pii]10.1038/nmat1737
53. Kim C, Shah BP, Subramaniam P, Lee KB. Synergistic induction of apoptosis in brain cancer cells by targeted codelivery of siRNA and anticancer drugs. Mol Pharm. 2011;8:1955-61 doi:10.1021/mp100460h
54. Xiong XB, Lavasanifar A. Traceable multifunctional micellar nanocarriers for cancer-targeted co-delivery of MDR-1 siRNA and doxorubicin. ACS Nano. 2011;5:5202-13 doi:10.1021/nn2013707
55. Meng H, Liong M, Xia T, Li Z, Ji Z, Zink JI. et al. Engineered design of mesoporous silica nanoparticles to deliver doxorubicin and P-glycoprotein siRNA to overcome drug resistance in a cancer cell line. ACS Nano. 2010;4:4539-50 doi:10.1021/nn100690m
56. Taratula O, Kuzmov A, Shah M, Garbuzenko OB, Minko T. Nanostructured lipid carriers as multifunctional nanomedicine platform for pulmonary co-delivery of anticancer drugs and siRNA. J Control Release. 2013;171:349-57 doi:S0168-3659(13)00237-X [pii] 10.1016/j.jconrel.2013.04.018
Author contact
![]() Corresponding author: xcredu.cn or gyxiang1968com.
Corresponding author: xcredu.cn or gyxiang1968com.