Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2014; 4(10):1002-1013. doi:10.7150/thno.8763 This issue Cite
Research Paper
FMN-Coated Fluorescent USPIO for Cell Labeling and Non-Invasive MR Imaging in Tissue Engineering
Marianne E. Mertens*,1, Julia Frese*,2, Deniz Ali Bölükbas*,1,#, Ladislav Hrdlicka2, Susanne Golombek1, Sabine Koch2, Petra Mela2, Stefan Jockenhövel2, Fabian Kiessling1 ![]() , Twan Lammers1
, Twan Lammers1 ![]()
1. Department of Experimental Molecular Imaging, Uniklinik RWTH Aachen and Helmholtz Institute for Biomedical Engineering, RWTH - Aachen University, Pauwelsstraße 30, 52074 Aachen, Germany;
2. Department of Tissue Engineering & Textile Implants, Applied Medical Engineering, Helmholtz Institute for Biomedical Engineering, RWTH - Aachen University, Pauwelsstraße 20, 52074 Aachen, Germany.
* These authors contributed equally to this study.
# Current address: Comprehensive Pneumology Center, Institute of Lung Biology and Disease, University Hospital, Ludwig-Maximilians-University and Helmholtz Zentrum München, Member of the German Center for Lung Research (DZL), Max-Lebsche-Platz 31, 81377 Munich, Germany.
Received 2014-2-5; Accepted 2014-3-24; Published 2014-7-28
Abstract

Non-invasive magnetic resonance imaging (MRI) is gaining significant attention in the field of tissue engineering, since it can provide valuable information on in vitro production parameters and in vivo performance. It can e.g. be used to monitor the morphology, location and function of the regenerated tissue, the integrity, remodeling and resorption of the scaffold, and the fate of the implanted cells. Since cells are not visible using conventional MR techniques, ultrasmall superparamagnetic iron oxide (USPIO) nanoparticles are routinely employed to label and monitor the cells embedded in tissue-engineered implants. We here set out to optimize cell labeling procedures with regard to labeling efficiency, biocompatibility and in vitro validation during bioreactor cultivation, using flavin mononucleotide (FMN)-coated fluorescent USPIO (FLUSPIO). Efficient FLUSPIO uptake is demonstrated in three different cell lines, applying relatively short incubation times and low labeling concentrations. FLUSPIO-labeled cells were successfully employed to visualize collagen scaffolds and tissue-engineered vascular grafts. Besides promoting safe and efficient cell uptake, an exquisite property of the non-polymeric FMN-coating is that it renders the USPIO fluorescent, providing a means for in vitro, in vivo and ex vivo validation via fluorescence microscopy and fluorescence reflectance imaging (FRI). FLUSPIO cell labeling is consequently considered to be a suitable tool for theranostic tissue engineering purposes.
Keywords: MRI, FLUSPIO, tissue engineering
Introduction
Tissue engineering is an interdisciplinary field concentrating on the development of biological substitutes to restore or replace the function of damaged or diseased organs and tissues. Continued progress in the last couple of years has promoted the design and fabrication of complex tissue architectures, which have e.g. been used for the therapy of skin damage, bone and cartilage injury, vessel reconstruction and heart valve replacement (1, 2). The translation of tissue-engineered implant materials into the clinic requires new concepts for their non-invasive monitoring (3), enabling quality assessment in vitro, and a deeper understanding of the behavior and the host interactions of the engineered tissue in vivo (4). Theranostic tissue-engineered implants, which combine optimized biocompatibility and functionality with the ability of in vitro and in vivo monitoring therefore hold significant potential for facilitating clinical translation.
The incorporation of imaging agents makes tissue-engineered implants visible and traceable, not only during in vitro production, but also upon in vivo implantation. This allows for longitudinal and more informative studies in animals, and for non-invasive follow-up monitoring in patients. We previously demonstrated that the labeling of collagen-based scaffold materials with ultrasmall superparamagnetic iron oxide nanoparticles (USPIO) enables sensitive and longitudinal monitoring of their location, function and remodeling via magnetic resonance imaging (MRI) (5). In addition to the direct visualization of the scaffold material itself, however, it is also of great interest to monitor the cells colonizing them during in vitro bioreactor cultivation and upon in vivo implantation, in order to assess the location of the implanted cells and to confirm their functionality (Figure 1A). Several studies have reported cell tracking on the basis of optical imaging, upon labeling them with fluorescent imaging markers, but these approaches are limited because of poor light penetration depth in living organisms (6-8). Labeling cells with radiotracers allows unlimited tissue penetration; however, it is somewhat limited in terms of spatial resolution, and it also raises serious (genotoxic) safety concerns (9, 10). Moreover, commonly applied radiolabels (e.g. 18F) decay within a few hours, and therefore provide a very narrow time window for in vivo monitoring.
Labeling and imaging of tissue-engineered implants. A) USPIO nanoparticles can either be directly incorporated into the scaffold material (to allow a clear delineation, and enable longitudinal monitoring of its integrity and degradation), or into cells seeded onto them (to assess the location of the implanted cells, and to confirm their functionality within the tissue-engineered construct). B) Schematic structure of a FMN-coated USPIO, physicochemically stabilized via co-coating with GMP. FMN specifically binds to the riboflavin carrier protein (RCP) on metabolically active cells, enabling efficient and safe cell uptake, and it also renders the particles fluorescent.
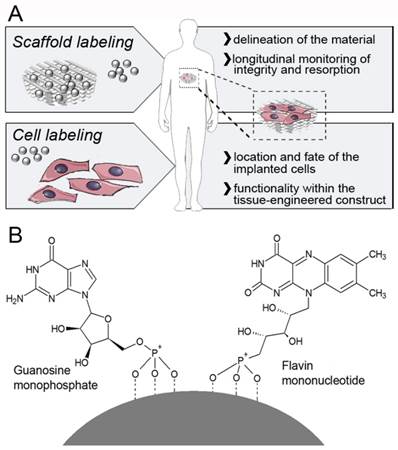
Superparamagnetic iron oxide (SPIO) nanoparticles present with negative contrast in MRI and can be sensitively detected at any depth with excellent soft tissue contrast (11). Labeling cells with SPIO has been extensively evaluated over the last decade, in particular for immune cell and stem cell tracking, and several of these approaches have been successfully tested in clinical trials (12). SPIO-labeled cells have also been employed for tissue engineering purposes, e.g. to monitor grafts upon in vivo implantation, and to determine the fate of embedded cells (13-18). The nature, the amount and the biocompatibility of the contrast agent used for cell labeling determine the sensitivity and the usefulness of MR-based imaging approaches, as well as the ability to visualize even relatively small cell populations in tissue-engineered implants. Carefully optimizing the properties of the contrast agent and the procedures for cell labeling is consequently considered to be highly important.
Ideally, contrast agents for non-invasive cell tracking in tissue engineering should provide i) high labeling efficiency, ii) low cytotoxicity, iii) good in vivo visualization, and iv) the opportunity for ex vivo validation. Here, we prepared flavin mononucleotide (FMN)-coated fluorescent USPIO (FLUSPIO), and we show that these nanoparticles are highly useful for cell labeling and subsequent cell imaging in tissue-engineered implants (Figure 1B). FMN specifically binds to the riboflavin carrier protein (RCP) on metabolically active cells (19), and it enables efficient uptake without the need for cationic (and generally relatively toxic) transfection agents. Relatively low labeling concentrations and relatively short incubation times are shown to be sufficient to sensitively visualize the cells using MRI. FMN-coating furthermore renders the particles fluorescent, allowing for a cross-validation of cell labeling and localization via optical imaging techniques. FLUSPIO-labeling showed no adverse effects on the cells tested (fibroblasts, smooth muscle cells and endothelial cells), and labeled cells were successfully used for theranostic tissue engineering purposes, i.e. for colonizing and visualizing collagen-based scaffold materials and tissue-engineered vascular grafts.
Results
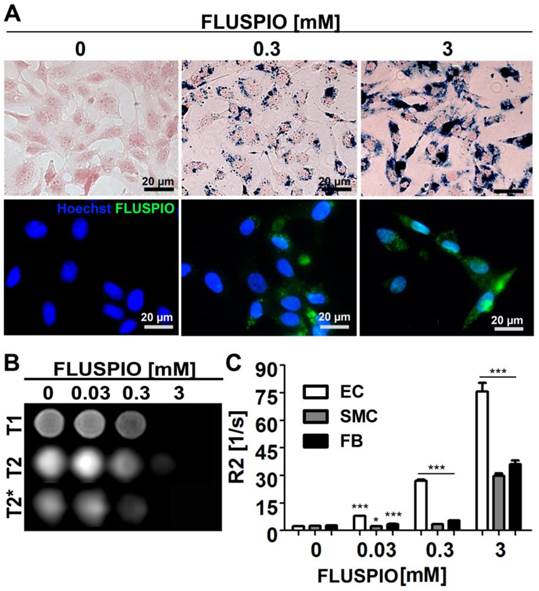
Endothelial cells (EC), smooth muscle cells (SMC) and fibroblasts (FB) were incubated with FLUSPIO at three different concentrations (0.03, 0.3 and 3 mM). As expected, Prussian Blue and immunofluorescence stainings revealed increasing intracellular iron deposits with increasing labeling concentrations. Efficient cellular uptake of the nanoparticles could be observed at concentrations from 0.3 mM onwards (Figure 2A). To determine the labeling efficacy of the cells using MRI, they were embedded in gelatin phantoms, at 2*106 cells per 300 µl phantom material. FLUSPIO-labeled cells presented with hypo-intense contrast in T2- and especially T2*-weighted imaging (Figure 2B). Transversal relaxation rates (R2) were quantified on the basis of T2-relaxometry measurements, showing a significant increase in R2 for phantoms containing FLUSPIO-labeled cells (Figure 2C). The highest increase in R2 was observed for endothelial cells, indicating the highest uptake of FLUSPIO nanoparticles.
Cell labeling using FLUSPIO. A) Prussian Blue (top panel; FLUSPIO in blue, cytoplasm in red) and immunofluorescence (bottom panel; FLUSPIO in green, nuclei in blue) staining of endothelial cells incubated with increasing FLUSPIO concentrations for 1 h. Efficient FLUSPIO internalization could be observed at concentrations of 0.3 mM onwards. B) T1-, T2- and T2*- weighted MR images of endothelial cells upon FLUSPIO incubation. FLUSPIO-labeled cells presented with a strong hypo-intense contrast. C) Corresponding R2 relaxation rates measured for FLUSPIO-labeled endothelial cells (EC), smooth muscle cells (SMC) and fibroblasts (FB). Asterisks indicate significant differences vs. non-labeled control cells (*P:0.01 - 0.05, *** P:<0.001).
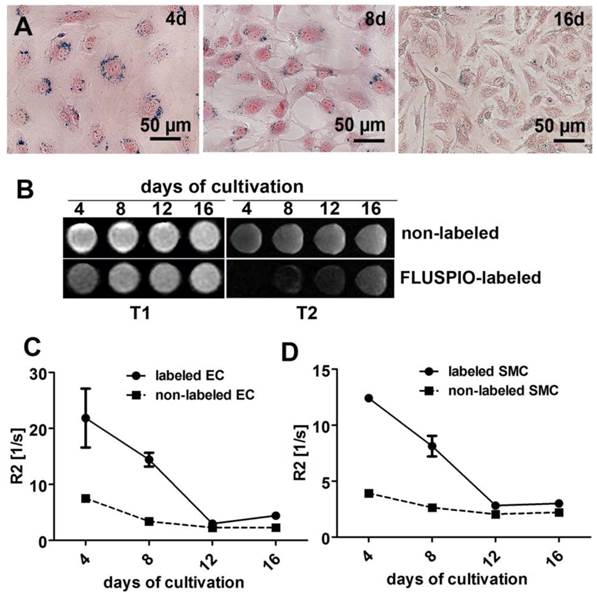
In order to determine MR contrast agent retention, cells labeled with 0.3 mM FLUSPIO were expanded in culture for 16 days and evaluated after 4, 8, 12 and 16 days. Qualitative analysis via Prussian Blue staining, T2-weighted imaging and quantitative MR analyses via R2-based relaxometry measurements were performed (Figure 3A-D). In line with previous reports (18), we observed a clear decrease in intracellular iron content during cell replication, which can be attributed to the fact that the FLUSPIO nanoparticles introduced into each cell are distributed over daughter cells during cell division. The dilution of intracellular iron correlated well with the decrease in R2 values. Nonetheless, upon using 0.3 mM of FUSPIO, cells could still be detected up to two weeks after labeling.
Stability of FLUSPIO labeling. A) Prussian Blue staining of FLUSPIO-labeled endothelial cells during the course of 16 days of cultivation, showing a decrease of intracellular iron concentrations over time, B) T1- and T2-weighted MRI measurements. Corresponding R2 values for endothelial (C) and smooth muscle (D) cells, conforming a drop in signal intensity which is proportional to cell expansion and FLUSPIO distribution over daughter cells.
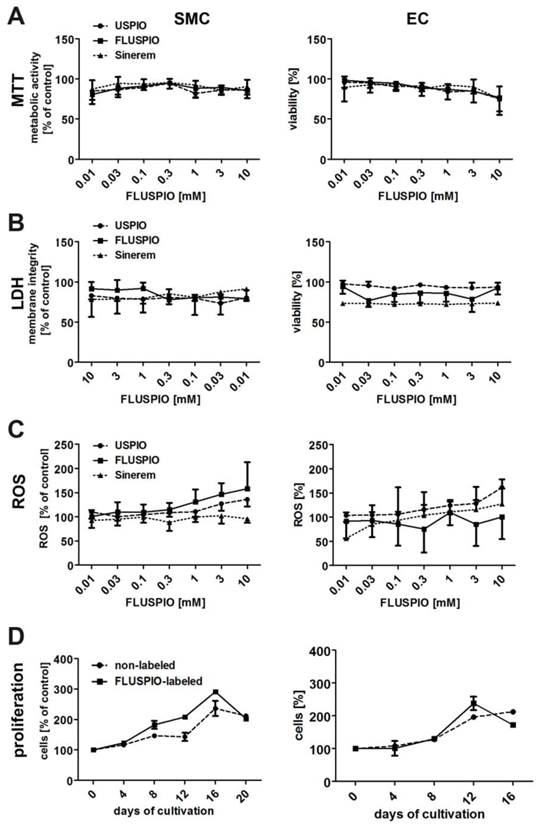
The biocompatibility of the FLUSPIO nanoparticles was compared to that of non-coated USPIO and clinically used iron oxide nanoparticles (Sinerem) (Figure 4). The metabolic activity of the labeled cells, as assessed by MTT assay, was not significantly different from that of control cells, even at very high labeling concentrations of 10 mM (Figure 4A). Additional cytotoxicity analyses, such as membrane disruption (assessed by LDH assay, Figure 4B) or the production of reactive oxygen species (assessed by ROS assay, Figure 4C), also indicated that FLUSPIO labeling is very well tolerated by cells.
Cytotoxicity evaluation of FLUSPIO labeling via A) MTT, B) LDH, C) ROS and D) proliferation assay in comparison to non-coated standard USPIO and clinically used iron oxide nanoparticles (Sinerem). FLUSPIO showed no increased cytotoxic effects and no negative influence on cell proliferation.
Next, we colonized collagen scaffolds with FLUSPIO-labeled cells. Collagen scaffolds provide a suitable pore structure for cell seeding and have been extensively used for tissue engineering purposes (10, 20-23). Colonization was carried out for 4, 8 and 12 days, and the scaffolds colonized with FLUSPIO-labeled cells were imaged using T2- and T2*-weighted MRI (Figure 5A). The T2-weighted images of the colonized scaffolds revealed a strong negative contrast upon using labeled cells, making them easily detectable in MRI, whereas scaffolds seeded with non-labeled cells could not been clearly delineated. For quantitative analysis, R2 and R2* values were determined, and they were shown to be significantly increased upon colonization with all three labeled cell types at all three time points tested (Figure 5B-D).
In vitro colonization of 3D collagen scaffolds with FLUSPIO-labeled cells. A) T2-weighted MR images of scaffolds colonized with FLUSPIO-labeled (+) and non-labeled (-) fibroblasts (FB), smooth muscle cells (SMC) and endothelial cells (EC) at day 4, 8 and 12 after cell seeding, showing strong negative contrast in case of labeled cells. B-D) Quantitative R2 and R2* analysis of collagen scaffolds seeded with labeled and non-labeled FB (B), SMC (C) and EC (D). Asterisks indicate significant differences in comparison to collagen scaffolds seeded with non-labeled cells (*P:0.01 - 0.05; ** P: 0.001 - 0.01, *** P:<0.001). E) Fluorescence reflectance imaging (FRI) of scaffolds seeded with FLUSPIO-labeled and non-labeled FB after 1, 2 and 3 d of cultivation, demonstrating increased fluorescence intensity upon seeding with FLUSPIO-labeled cells. F) Exemplary light and fluorescence microscopy analyses of collagen scaffolds seeded with FLUSPIO-labeled SMC. Cells were stained with hematoxylin and eosin (H&E), with Hoechst (blue; nuclei) and with antibodies against the proliferation marker Ki67 (red). Collagen autofluorescence is depicted in green. G) Quantitative evaluation of the fraction of proliferating cells. No qualitative and quantitative differences in colonization were observed for labeled vs. non-labeled cells.
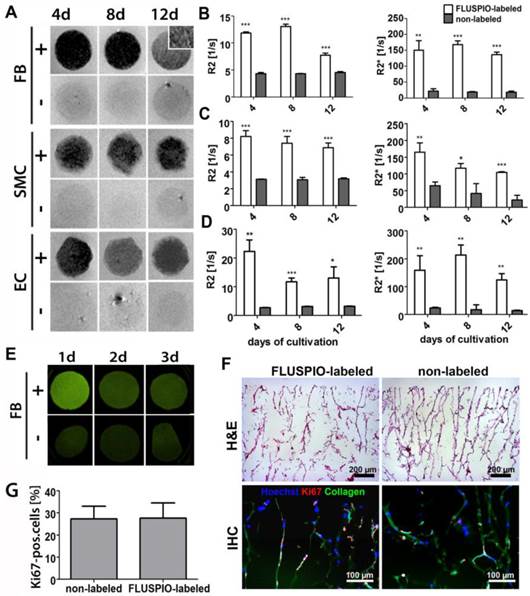
To assess whether FLUSPIO labeling also allows fluorescence imaging of tissue-engineered implants, collagen scaffolds were colonized with labeled and non-labeled cells, and they were imaged using fluorescent reflectance imaging (FRI) at 530 nm. As shown in Figure 5E, a strong fluorescence signal intensity was observed at day 1 post labeling and colonization. After 2 and 3 days, the signal became weaker, which can be attributed to the de-attachment of FMN from the particles' surface upon cellular internalization, and its subsequent metabolization in endo- and lysosomes. Nonetheless, even after 3 days of colonization with FLUSPIO-labeled cells, the fluorescence signal was still stronger than that of scaffolds colonized with non-labeled cells. It should be noted in this regard that due to collagen autofluorescence, control scaffolds also generate some fluorescence at 530 nm (Figure 5E-F). Cellular distribution and proliferation on the scaffolds were investigated by histology and immunohistochemistry. As shown in Figures 5F-G, a qualitative analysis of the H&E stainings revealed no differences in colonization behavior between labeled vs. non-labeled cells, and also the number of proliferating cells (as assessed via Ki67 staining) was found to be unaffected by FLUSPIO labeling.
Subsequently, the possibility of visualizing endothelial cells on tissue-engineered vascular grafts was evaluated. First, the labeling efficiency of primary endothelial cells harvested from an explanted ovine carotid artery was evaluated, as was the MR signal intensity over time and the detection limit of the labeled endothelial cells. Figure 6A demonstrates that FLUSPIO uptake can be observed at labeling concentrations exceeding 0.1 mM, and becomes really efficient at a concentration of 1 mM. During proliferation, as expected, R2 values decreased proportionally, but even after 16 days, values were still significantly higher as compared to control cells (R2 value for non-labeled cells 3.1 ± 0.2 s -1, versus 3.9±0.1 s -1 for FLUSPIO-labeled cells; Figure 6B). To determine the visibility of endothelial cell layers seeded at different densities, layers of FLUSPIO-labeled cells were seeded on fibrin gels with densities ranging from 5*104 to 80*104 cells/cm2. As shown in Figure 6C, the endothelial layer appeared as a black lining on the fibrin gel, and the signal intensity increased proportionally with increasing cell density. From concentrations of 40*104 cells/cm2 onwards, the cell layer was clearly depictable. Therefore, this cell seeding density was used for the bioreactor-endothelialization of the tissue-engineered vascular grafts.
FLUSPIO-labeled endothelium on a tissue-engineered vascular graft. A) R2 values determined for EC upon FLUSPIO labeling at different concentrations and incubation times. B) R2 values and proliferation of FLUSPIO-labeled EC during 16 days of cultivation. C) Fibrin gel seeded with FLUSPIO-labeled EC at different cell densities. Concentrations from 40*104 cells onwards were clearly visible as a black lining (white arrowhead). D) T2-weighted images of vascular grafts with a labeled (+) and non-labeled (-) endothelial lining, at day 1, 4 and 8 after bioreactor endothelialization. Labeled endothelium presented with hypo-intense contrast (white arrowhead) and could be delineated on the textile scaffold (black arrowhead). Hashtags indicate the graft lumen. E-F) Corresponding R2 (E) and R2*(F) values. Asterisks indicate significant differences in comparison to vascular grafts seeded with non-labeled cells (*P:0.01 - 0.05). G) 3D MRI rendering of a labeled (left) and non-labeled (right) vascular graft, H) Photograph of a tissue-engineered vascular graft after 14 days of bioreactor cultivation. I-K) Hematoxylin and eosin (I), immunofluorescence staining of the endothelial marker CD31 (J: CD31 in red, DAPI in blue) and fluorescence microscopy imaging of FLUSPIO (K, FLUSPIO in green, DAPI in blue), confirming the presence of labeled EC on the luminal side of the scaffold. Hashtags indicate the graft lumen.
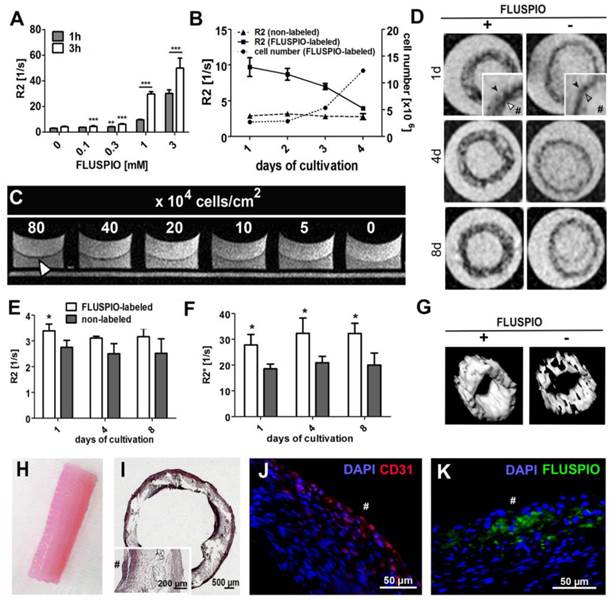
For vascular graft construction, a tubular polylactide-based textile scaffold was molded with a composite of fibrin, fibroblasts and smooth muscle cells (isolated from an ovine carotid artery), and the inner wall was colonized either with non-labeled or with FLUSPIO-labeled endothelial cells (harvested from the same ovine carotid artery). The grafts were bioreactor-cultivated under physiological flow conditions for 21 days, and monitored via MRI at day 14, 17 and 21 (i.e. day 1, 4 and 8 post endothelialization). At each time point, the endothelial cells could be successfully visualized using T2-weighted MRI (Figure 6D-G). A significant increase in R2 and in particular R2* relaxation rates could be demonstrated for grafts colonized with FLUSPIO-labeled endothelial cells (Figure 6E-F). H&E staining showed a dense cell layer on the inner surface of the graft, indicating efficient endothelialization (Figure 6I). Positive staining for the endothelial cell marker CD31 and FMN-based fluorescence microscopy analyses confirmed the localization of the labeled cells on the luminal side of the grafts (Figure 6J-K). These findings indicate that FLUSPIO-labeling is a suitable means for monitoring endothelial cells in tissue-engineered vascular grafts.
Discussion
We here show that FMN-coated fluorescent USPIO (FLUSPIO) nanoparticles are useful for labeling and monitoring cells incorporated into tissue-engineered implants. Labeling cells with iron oxide-based MR contrast agents for tissue engineering purposes has been performed on several occasions before. One of the earliest attempts describing USPIO cell labeling in tissue-engineered vascular grafts has demonstrated the possibility of monitoring these materials in vivo using labeled aortic smooth muscle cells (18). In a later study, also the use of USPIO-labeled macrophages to determine the fate of cells in a venous tissue-engineered vascular graft was evaluated (17). Most of the studies published to date, however, used dextran- or poly-L-lysine-coated USPIO for cell labeling, which are passively internalized via endocytosis. The drawback of using dextran-coated particles is that they are not effectively internalized by cells, and thus require relatively high concentrations, long incubation times and relatively toxic cationic transfection agents to achieve efficient labeling. Poly-L-lysine coatings directly render the particles cationic, which promotes cell uptake, but at the expense of increased cytotoxicity and reduced long-term viability. We here show that FLUSPIO nanoparticles are efficiently taken up by several different cell types, and enable efficient labeling at low concentrations and short incubation times. Furthermore, FLUSPIO-labeled cells could be sensitively detected using MRI, making the identification of individual cell layers in tissue-engineered implants possible. The fluorescence of FMN furthermore enables optical imaging, using both fluorescence reflectance imaging and fluorescence microscopy. This can be used to validate nanoparticle uptake in histology and also to track the labeled materials during the first few days post implantation.
Potential pitfalls associated with FLUSPIO-labeling are - as in case of other cell labeling techniques - the dilution of the label during cell division, and the risk that the label can be incorporated by other cells, e.g. by macrophages, which are responsible for removing dead cells. This can lead to a misinterpretation of the imaging findings. Here, we demonstrate that FLUSPIO-labeling can be visualized and quantified for up to two weeks. This period of time is sufficient to monitor the fate of labeled cells during in vitro cell colonization and bioreactor cultivation (in case of endothelialization typically one week), as well as during the initial stages following in vivo implantation. This is a critical time window for assuring proper production and function of tissue-engineered materials, as exemplified e.g. by non-invasive imaging information demonstrating that the cells seeded on a vascular graft can be rapidly lost upon in vivo implantation (17). Such information, in particular on the fate of vascular endothelium generated in vitro, is of great interest to better understand and improve the performance of vascular grafts. FLUSPIO nanoparticles are shown to be a suitable tool for cell labeling, and might be highly useful for providing this information. For longer follow-up monitoring, i.e. for periods of time exceeding two weeks, indirect labeling methods using reporter genes can be used, as well as direct scaffold labeling approaches. Indirect labeling e.g. refers to strategies related to the overexpression of the iron-binding protein ferritin, which can act as a reporter gene in MRI (24-26). The direct incorporation of USPIO nanoparticles in the scaffold material (e.g. in collagen matrices (5)), seems highly suitable for long-term monitoring of tissue-engineered implants, as well as assessment of their remodeling and resorption, but does not provide cellular information. Consequently, it is important to develop optimized materials and methods for non-invasive cell imaging in tissue engineering.
Conclusion
FLUSPIO nanoparticles can be used to efficiently and safely label several different cell types, enabling their visualization using MRI and optical imaging techniques. We demonstrate that FLUSPIO-labeling allows for the sensitive detection of cells on collagen scaffolds and vascular grafts, thereby facilitating their bioreactor production, and providing valuable information on their morphology, location and function.
Material & Methods
FLUSPIO synthesis
Flavin mononucleotide (FMN; for specific RCP uptake) and guanosine monophosphate (GMP; for colloidal stabilization purposes) were used as non-polymeric coating molecules (19). Coating with these materials was performed by sonication (Ultrasonic cleaner, 30 W, VWR GmbH, Germany) of 143 mM USPIO with 35 mM FMN for 1h at room temperatur. Excess FMN was F by high-gradient magnetophoresis. FMN modified particles were sonicated with 50 mM GMP for another 1 hour at room temperature. Excess of GMP was removed by high-gradient magnetophoresis to yield the final fluorescent FLUSPIO nanoparticles (TEM diameter in DLS = 5.6 ± 1.7 nm; hydrodynamic diameter in Nanosight = 97 ± 3 nm; relaxivity at 3T in water: r1 = 202.64 ± 0.48 s-1mM-1; r2 = 2.13 ± 0.04 (19).
Cell culture
Fibroblasts (NIH3T3) and a mixed population of smooth muscle cells/fibroblasts (referred to as SMC) were cultured using DMEM (Gibco, Invitrogen, Germany) cell culture medium supplemented with 10% fetal calf serum (Invitrogen, Germany), 1% non-essential amino acid (NEAA), 1% sodium pyruvate, 1% L-glutamine and 1% Pen/strep (10,000 U/ml penicillin; 10,000 µg/ml streptomycin, Invitrogen, Germany). Endothelial cells (HUVEC) were cultured using Vasculife (Lifeline Cell Technology, Germany) medium supplemented with 2% fetal bovine serum, 5% L-glutamine, 0.1% rh VEGF, 0.1% rh EGF, 0.1% rh FGF basic, 0.1% rh IGF-1, 0.1% ascorbic acid, 0.1% hydrocortisone hemisuccinate, 0.1% heparin sulfate (Lifeline Cell Technology, Germany) and 1% Pen/strep (10,000 U/ml penicillin; 10,000 µg/ml streptomycin, Invitrogen, Germany). Cells were cultured in T25 and T75 cell culture flasks (Cell star, Greiner, Germany) and incubated at 37°C, 5% CO2 and 95% relative humidity.
Cell labeling
The FLUSPIO exposure time and concentration in the medium was optimized in respect to MR visibility and an induced cytotoxicity. A series of labeling experiments with different exposure times (1 and 3 hours) and different FLUSPIO concentrations (0.1, 0.3, 1 and 3 µmol/ml) were investigated. FLUSPIO were suspended in the respective cell culture medium (10 ml) and exposed to 80% confluent cells T75 flasks. Incubation time (1 or 3 h) was carried out at 37oC, 5% CO2 and 95% relative humidity. After incubation, cells were washed twice with PBS and subsequently trypsinized using 2 ml of trypsin/EDTA (0.25%/0.5%). Trypsinization was stopped by adding cell culture medium and the cell suspension was centrifuged at 1000 rpm (Multifuge, Thermo scientific, Germany) for 5 min. The cell pellet was re-suspended in 10 ml of PBS buffer and washed once by performing the centrifugation as described above.
Prussian Blue Staining
The efficiency of particle uptake and permanence of intracellular iron was determined by Perl's Prussian blue staining. The endothelial cells were seeded on microscopic slides and incubated overnight. For staining, the cells were washed once with PBS and fixed with 10% neutral buffered formalin (NBF; Sigma-Aldrich) for 30 min. A mixture of equal parts of hydrochloric acid (20%) and 10% aqueous solution of potassium ferrocyanide (Potassium ferrocyanide, Sigma) was freshly prepared before the staining, added to the cells (200µl of the solution per well-chamber) and incubated for 30 min. After washing with PBS, a contra-staining with Nuclear Fast Red (Roth N069.1) was conducted for 5 min. Afterwards the slides were thoroughly washed with distilled water and covered with a glass cover-slip and mounting medium for further microscopic evaluation using brightfield microscope (Zeiss Imager D.1, 40x magnification, AxioCam MRc, Axio SW). Images were post-processed with license free soft-ware: IrfanView.
Immunohistochemistry and histology
Cells were seeded on coverslips and incubated overnight at 37°C, 5% CO2 and 95% relative humidity. Cells attached to the surface of the microscopic slides were washed three times with PBS and fixed in 70% ethanol for 10 min. Afterwards, the samples were stained with 1:400 Hoechst diluted in PBS for 45 min. The samples were washed with PBS, embedded in Mowiol and imaged with the Zeiss Axio Imager M2. H&E stainings were done according to standard histopathological protocols. Immunofluorescence stainings of the scaffolds were performed using a primary antibody against the proliferation marker Ki67, which was incubated for 60 min at room temperature, followed by secondary antibody staining and nuclear counterstaining using Hoechst. Stained sections were examined and photographed with the Zeiss Axio Imager M2. To evaluate the impact of FLUSPIO-labeling on proliferation, the total number of cells as well as the number of proliferating Ki67-positive cells were quantified and compared. For quantifying proliferating cells in the scaffolds, 3 micrographs (100x magnification) from 3 representative sections per scaffold were analyzed. Ki67 positive cells were counted and expressed as ratio of the total cell number.
Magnetic resonance imaging
Nuclear MR imaging was performed in a clinical 3T whole-body MR scanner (PHILIPS Achieva, The Netherlands) using a knee coil (SENSE-flex-M, PHILIPS, The Netherlands) at room temperature. For MR measurements 2 x 106cells were embedded in 0.3 ml gelatin/well (Greiner 96 well Flat Transparent Polystyrol microplate, Germany). T2-weighted images were acquired in 2D scan mode using a multi-slice, multi-shot spin-echo (SE) sequences with a 90° excitation pulse followed by a train of equally spaced 180° refocusing pulses [TR = 1500 ms, TE = 8 - 160 ms, number of echoes = 20, flip angle (FA) = 90°]. For T2* relaxometry, images at 32 echo times [TE range 4 - 94 ms] were acquired by using a multi-shot, multi-slice fast-field gradient-echo (FFE) sequence [TR = 195ms, FA = 30°]. T2 and T2* relaxation times and corresponding R2 and R2* ratios were calculated by fitting an exponential curve to the signal amplitudes as a function of the echo time (TE) for each segmented scaffold region using the Imalytics Preclinical Software (Philips Technologie GmbH, Aachen, Germany). T2- weighted turbo-spin-echo (TSE) sequences were performed using following parameters: TR = 400 ms, TE = 100 ms, FA = 90°.
Label retention study
SMCs and HUVECs were labeled with 0.3 mM FLUSPIO for 3 h. 2x106 labeled as well as non-labeled control cells were seeded on T75 flasks and incubated for 2, 4, 8 and 16 days. For each time point viable cells were counted and gelatine phantoms containing 2x106 cells/0.3 ml were prepared. MR-relaxometry of cells at each time points was performed at 3T using the MR-sequences described above.
MTT assay
Metabolic activity was investigated using the Cell Proliferation Kit 1 (MTT) Roche, Switzerland. Cells were seeded on 96 well plates and cultivated to 70% confluence. The medium was then exchanged with USPIO, FLUSPIO or Sinerem containing medium dilutions (300µl per well with concentrations ranging from 10 mM to 0.01 mM iron) and incubated for 3 h at 95% relative humidity, 5% CO2 and 37°C. As positive control 250 µl medium was supplemented with 50 µl of Triton-X 10%, non-treated cells served as negative control. After incubation, the cells were washed with 200 µl PBS until most of the particles were washed out. 100 µl cell culture medium and 50 µl of MTT solution were added to each well (1:5 dilution of MTT in PBS, according to the manufactures protocol). The solution was incubated for 4 h at 37°C. After the incubation, the MTT solution was discarded and 20 µl of DMSO were added to solve the formazan crystals. The DMSO was incubated at room temperature overnight, covered with an opaque material. 100 µl of the supernatants were transferred into new 96-well plates and the absorbance was measured at 57 nm with a reference of 690 nm using a Tecan reader.
LDH assay
Membrane integrity i.e the release of lactate dehydrogenase to the cell culture medium, was tested using the Roche Cytotoxicity Detection Kit (Roche, Switzerland). Cultivation, incubation with nanoparticles and controls were done as described for the MTT assay. After the incubation period, 200 µl of the supernatants were transferred to Eppendorf tubes and centrifuged at 14000 g for 30 min to remove cell debris and nanoparticles. 100 µl of the supernatant was transferred into a new 96 well plates. The LDH reagents were prepared according to the manufacturer protocol, added in a 1:1 ratio to the supernatant and incubated for 30 min at room temperature, covered with an opaque material. After incubation, the absorbance was measured at 490 nm with a reference of 600 nm.
ROS assay
The amount of reactive oxygen species in the cell is an indicator for cellular stress and thus can be used to test the toxicity of different biomaterials. Cultivation and incubation with nanoparticles was done as described before. 400 µl cell culture medium and 120 µl NBT (5 mg/ml) were added and incubated for 24 h in the incubator. NBT was removed thoroughly and 150 µl trypsin solution was added and incubated for 10 min in the incubator and 20 min at room temperature on the shaker (750 rpm). 170 µl of a 10 M KOH solution was then added and incubated for 30 min at room temperature on the shaker (750 rpm). 200 µl of the supernatants were transferred into fresh 96-well-plates and the absorbance was measured at 620 nm.
Seeding of 3D collagen scaffold
400 µl of cell suspensions (5000 cells/µl) were added onto a dry collagen scaffolds (diameter = 13 mm, depth = 3 mm). Scaffolds were incubated for 1 hour at 37°C, 5% CO2 and 95% relative humidity to allow efficient attachment of the cells to the surface. After 1 hour respective cell culture medium was added and incubation was continued for 4, 8 and 12 days. After the incubation period scaffolds were fixed in 4% PFA and either embedded in 10% gelatin for further MRI measurements or in Tissue Tek® for immunohistochemical analysis.
Vascular Graft Preparation: Cell isolation, cell culture and fibrin synthesis
Arterial endothelial cells, and a mixed population of smooth muscle cells (SMC)/fibroblasts, were harvested from explanted ovine carotid arteries as described previously in detail (27). SMC/fibroblasts were cultured in Dulbecco's modified Eagle's medium (DMEM; Invitrogen, Darmstadt, Germany) supplemented with 10% fetal calf serum (FCS; PAA, Colbe, Germany). Endothelial cells were treated with Endothelial cell basal medium (Endothel Basal Medium and Supplement, PAA, Cölbe, Germany plus 1% Glutamax, Invitrogen). To obtain sufficient numbers, cells were serially passaged up to 4 times using 0,25% trypsin/0.02%EDTA solution (Gibco) and cultured in 5% CO2 and 95% humidity at 37°C. Human fibrinogen (Calbiochem) was dissolved in purified water and dialysed with a cut-off membrane (Novodirect, Kehl, Germany) of 6000-8000 MW overnight against Tris-buffered saline (TBS). The fibrinogen concentration following sterile filtration was estimated by measuring absorbance at 280 nm with a spectrophotometer (Infinity M 200, Tecan, Männedorf, Switzerland). The final concentration of the fibrinogen solution was adjusted to 12 mg/ml with sterile TBS. The fibrin gel component of the vascular composite graft (4.0 ml total) consisted of 2.0 ml of fibrinogen solution (12 mg/ml), 1.4 ml TBS containing 40 x 106 carotid artery SMC/fibroblasts and 300 µl 50 nM CaCl2 (Sigma) in TBS. Fibrin polymerisation was initialized by adding 350 µl of thrombin solution (40 U/ml; Sigma).
Vascular Graft Molding: Mesh production, graft molding and endothelialization
P(L/D)LA 96/4 was used to produce a macroporous warp-knitted tubular mesh. Following manufacture, the mesh scaffold was sterilized using low-temperature hydrogen peroxide gas plasma (STERRAD® 100S Sterilisation System; Ethicon GmbH, Hamburg, Germany). Vascular composite grafts were cast in molds as described previously (27). Briefly, the inner casting cylinder of the mold was lined with the P(L/D)LA 96/4 mesh. Subsequently, the annular space between the inner and outer cylinders of the mold was filled with a SMC/fibroblast /fibrinogen suspension (10 x 106 cells/ml), which provided a uniform wall thickness throughout the graft. The solution was combined with a thrombin/CaCl2 solution to initiate polymerisation and cell encapsulation. After 45 min, the inner casting cylinder was removed and the graft, maintained in the outer cylinder was connected to a customised bioreactor system and transferred to an incubator at 37°C and 5% CO2 for mechanical conditioning. Medium flow through the graft was gradually increased from 20 to 200 ml/min over a 14-day conditioning period, with physiological pressure conditions (120 mm Hg, systolic pressure; 80 mm Hg, diastolic pressure) also maintained over the conditioning period. After 14 days of cultivation, FLUSPIO labeled ovine carotid artery endothelial cells (16x 106 per graft) were seeded on the luminal surface of the graft as described previously (28). The conditioned vascular composite graft (plus outer mold cylinder) was disconnected from the bioreactor system and filled with endothelial medium containing 16 x 106 ovine carotid artery-derived endothelial cells. The cylinder was then sealed at both ends and mounted on a programmable roller pump, which was used to rotate the vascular composite graft about a horizontal axis for 20 s, followed by a 20-min stationary phase to allow endothelial cell attachment. The cycle was repeated three times before the direction of rotation was changed. In total, endothelial cell seeding was performed for 6 h. After cell seeding, the vascular composite graft (plus outer cylinder) was reconnected to the bioreactor system under sterile conditions, allowing a further 8 days of conditioning under reduced flow conditions (50 ml/min).
MR monitoring during bioreactor cultivation
The grafts were transferred within the bioreactor circuit into a Philips clinical 3T whole-body MR scanner with small animal solenoid sense-receive mouse coil and measured after 1, 4 and 8 days after endothelialization. Following sequences were used: T2-weighted TSE [TR = 1.2 s, TE = 100 ms, FA = 90°, aquisition matrix: 1020], T2-weighted SE [TR = 1.5 s, TE = 9- 170 ms, number or echos: 20, FA = 90°], T2*-weighted FFE [TR = 395 ms, TE = 4 - 95 ms, number of echos: 32, FA = 30°].
Microscopy and immunohistochemical analysis of the vascular grafts
Carnoy's-fixed, paraffin-embedded vascular composite grafts were sectioned transversely at 4 µm thickness and were subsequently analysed by hisological staining. Graft morphology was assessed by haematoxylin & eosin (H&E) staining, while the efficiency of particle uptake and permanence of intracellular iron was determined by Perl's Prussian blue staining. Sections were viewed using routine bright field light microscopy (AxioImager D1; Carl Zeiss GmbH, Jena Germany). Images were acquired using a digital colour camera (AioCam MRc; Carl Zeiss GmbH). Non-Specific sites on Carnoy's-fixed, paraffin-embedded graft sections were blocked and the cells permeablised with 5% normal goat serum (NGS; Sigma) in 0,1% Triton-PBS. For fluorescence immunohistochemistry, sections were incubated for 1 h at 37°C with the mouse anti-human CD31 (1:100; Sigma). The sections were the incubated for 1 h at room temperature with rhodamine- conjugated secondary antibody (1:400; Molecular Probes, Leiden, The Netherlands). Cell nuclei were counterstained using DAPI nucleic acid stain (Molecular Probes). As negative controls, sampled were incubated in diluent and the secondary antibody only. Samples were viewed using a fluorescence microscope (AxioObserver Z1; Carl Zeiss GmbH), and images were acquired using a digital colour camera (AxioCam MRm; Carl Zeiss GmbH).
Statistical analysis
All values are presented as average ± standard deviation. P values were calculated using an unpaired, two-tailed t test (GraphPad Software, version 4.0). P<0.05 was considered to represent statistical significance. In the figures, * indicates P < 0.01 - 0.05; ** P < 0.001 - 0.01, and *** P < 0.001).
Acknowledgements
The authors acknowledge Dr. J. Jayapaul, M. Weiler and D. Moeckel for technical support. This work was supported by the NRW/EU-Ziel 2-Programm ((EFRE) 2007-2013: “Entwicklung und Bildgebung patienten-optimierter Implantate”), by the European Research Council (ERC Starting Grant 309495: NeoNaNo), by the ERS Boost Fund at RWTH Aachen, and by a Helmholtz-Society Portfolio grant (“Technologie und Medizin - Multimodale Bildgebung zur Aufklärung des In-vivo-Verhaltens von polymeren Biomaterialien”).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Atala A, Kasper FK, Mikos AG. Engineering complex tissues. Sci Transl Med. 2012;4(160):160rv12
2. Place ES, Evans ND, Stevens MM. Complexity in biomaterials for tissue engineering. Nat Mater. 2009;8(6):457-470
3. Udelsman BV, Maxfield MW, Breuer CK. Tissue engineering of blood vessels in cardiovascular disease: moving towards clinical translation. Heart. 2013;99(7):454-460
4. Appel AA, Anastasio MA, Larson JC, Brey EM. Imaging challenges in biomaterials and tissue engineering. Biomaterials. 2013;34(28):6615-6630
5. Mertens ME, Hermann A, Bühren A. et al. Iron Oxide-Labeled Collagen Scaffolds for Non-Invasive MR Imaging in Tissue Engineering. Adv Funct Mater. 2014;24(6):754-762
6. Sutton EJ, Henning TD, Pichler BJ, Bremer C, Daldrup-Link HE. Cell tracking with optical imaging. Eur Radiol. 2008;18(10):2021-2032
7. Michalet X, Pinaud FF, Bentolila LA. et al. Quantum Dots for Live Cells, in Vivo Imaging, and Diagnostics. Science. 2005;307(5709):538-544
8. Wu TJ, Tzeng YK, Chang WW. et al. Tracking the engraftment and regenerative capabilities of transplanted lung stem cells using fluorescent nanodiamonds. Nat Nanotechnol. 2013;8(9):682-689
9. Zhang SJ, Wu JC. Comparison of Imaging Techniques for Tracking Cardiac Stem Cell Therapy. J Nucl Med. 2007;48(12):1916-1919
10. Kircher MF, Gambhir SS, Grimm J. Noninvasive cell-tracking methods. Nat Rev Clin Oncol. 2011;8(11):677-688
11. Ahrens ET, Bulte JW. Tracking immune cells in vivo using magnetic resonance imaging. Nat Rev Immunol. 2013;13(10):755-763
12. Bulte JW. In Vivo MRI Cell Tracking: Clinical Studies. AJR Am J Roentgenol. 2009;193(2):314-325
13. Di Corato R, Gazeau F, Le Visage C. et al. High-Resolution Cellular MRI: Gadolinium and Iron Oxide Nanoparticles for in-Depth Dual-Cell Imaging of Engineered Tissue Constructs. ACS Nano. 2013;7(9):7500-7512
14. Perea H, Aigner J, Heverhagen JT, Hopfner U, Wintermantel E. Vascular tissue engineering with magnetic nanoparticles: seeing deeper. J Tissue Eng Regen Med. 2007;1(4):318-321
15. Terrovitis JV, Bulte JW, Sarvananthan S. et al. Magnetic resonance imaging of ferumoxide-labeled mesenchymal stem cells seeded on collagen scaffolds-relevance to tissue engineering. Tissue Eng. 2006;12(10):2765-2775
16. Ramaswamy S, Greco JB, Uluer MC. et al. Magnetic resonance imaging of chondrocytes labeled with superparamagnetic iron oxide nanoparticles in tissue-engineered cartilage. Tissue Eng Part A. 2009;15(12):3899-3910
17. Harrington JK, Chahboune H, Criscione JM. et al. Determining the fate of seeded cells in venous tissue-engineered vascular grafts using serial MRI. FASEB J. 2011;25(12):4150-4161
18. Nelson GN, Roh JD, Mirensky TL. et al. Initial evaluation of the use of USPIO cell labeling and noninvasive MR monitoring of human tissue-engineered vascular grafts in vivo. FASEB J. 2008;22(11):3888-3895
19. Jayapaul J, Hodenius M, Arns S. et al. FMN-coated fluorescent iron oxide nanoparticles for RCP-mediated targeting and labeling of metabolically active cancer and endothelial cells. Biomaterials. 2011;32(25):5863-5871
20. Olde Damink LH, Dijkstra PJ, van Luyn MJ, van Wachem PB, Nieuwenhuis P, Feijen J. Cross-linking of dermal sheep collagen using a water-soluble carbodiimide. Biomaterials. 1996;17(8):765-773
21. Freed LE, Vunjak-Novakovic G, Biron RJ. et al. Biodegradable Polymer Scaffolds for Tissue Engineering. Biotechnology (N Y). 1994;12(7):689-693
22. Köse GT, Korkusuz F, Özkul A. et al. Tissue engineered cartilage on collagen and PHBV matrices. Biomaterials. 2005;26(25):5187-5197
23. Chan TR, Stahl PJ, Yu SM. Matrix-Bound VEGF Mimetic Peptides: Design and Endothelial-Cell Activation in Collagen Scaffolds. Adv Funct Mater. 2011;21(22):4252-4262
24. Cohen B, Ziv K, Plaks V. et al. MRI detection of transcriptional regulation of gene expression in transgenic mice. Nat Med. 2007;13(4):498-503
25. Cohen B, Ziv K, Plaks V, Harmelin A, Neeman M. Ferritin nanoparticles as magnetic resonance reporter gene. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2009;1(2):181-188
26. Gilad AA, Ziv K, McMahon MT, van Zijl PC, Neeman M, Bulte JW. MRI reporter genes. J Nucl Med. 2008;49(12):1905-1908
27. Tschoeke B, Flanagan TC, Koch S. et al. Tissue-engineered small-caliber vascular graft based on a novel biodegradable composite fibrin-polylactide scaffold. Tissue Eng Part A. 2009;15(8):1909-1918
28. Weinandy S, Rongen L, Schreiber F. et al. The BioStent: novel concept for a viable stent structure. Tissue Eng Part A. 2012;18(17-18):1818-1826
Author contact
![]() Corresponding authors: tlammersde; fkiesslingde.
Corresponding authors: tlammersde; fkiesslingde.