Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2014; 4(9):945-952. doi:10.7150/thno.9246 This issue Cite
Research Paper
Specific and Sensitive Tumor Imaging Using Biostable Oligonucleotide Aptamer Probes
Zihua Zeng1,2, Parag Parekh1,2, Zheng Li3, Zheng-Zheng Shi3, Ching-Hsuan Tung3, Youli Zu1,2 ![]()
1. Department of Pathology and Genomic Medicine, Houston Methodist Hospital;
2. Cancer Pathology Laboratory, Houston Methodist Research Institute;
3. Department of Translational Imaging, Houston Methodist Research Institute, Houston, TX 77030, USA.
Received 2014-3-28; Accepted 2014-5-29; Published 2014-7-19
Abstract

Although several imaging modalities are widely used for tumor imaging, none are tumor type-specific. Different types of cancer exhibit differential therapeutic responses, thus necessitating development of an imaging modality able to detect various tumor types with high specificity. To illustrate this point, CD30-specific oligonucleotide aptamer in vivo imaging probes were conjugated to the near-infrared IRD800CW reporter. Mice bearing xenografted CD30-positive or control CD30-negative lymphoma tumors on contralateral sides of the same mouse were developed. Following a systemic administration of aptamer probes, whole body imaging of tumor-bearing mice was performed. Imaging signal from tumor sites was analyzed and imaging specificity confirmed by tissue immunostaining. The in vivo biodistribution of aptamer probes was also evaluated. Whole body scans revealed that the RNA-based aptamer probes selectively highlighted CD30-expressing lymphoma tumors immediately after systemic administration, but did not react with control tumors in the same mouse. The resultant imaging signal lasted up to 1 hr and the aptamer probes were rapidly eliminated from the body through urinary and lower intestinal tracts. For more sensitive imaging, biostable CD30-specific ssDNA-based aptamer probes were also generated. Systemic administration of these probes also selectively highlighted the CD30-positive lymphoma tumors, with imaging signal detected 4-5 folds higher than that derived from control tumors in the same animal, and lasted for up to 24hr. This study demonstrates that oligonucleotide aptamer probes can provide tumor type-specific imaging with high sensitivity and a long-lasting signal, indicating their potential for clinical applications.
Keywords: biostable oligonucleotide aptamer, sensitive imaging, tumor-specific
Introduction
Modern imaging technologies, including computed tomography (CT), positron emission tomography (PET), and magnetic resonance imaging (MRI), are widely used to detect tumors (1,2). However, none of these imaging modalities are tumor type-specific; thus, they cannot distinguish one type of tumor from another. CT scans highlight the anatomic location of a tumor, whereas PET scans are performed by injecting patients with 18F-FDG in order to reveal hyper-metabolically active tumors. Therefore, merged CT-PET scans reveal the presence of tumors with hyper-metabolic status (3,4), indirectly indicating their malignant nature. Similar to CT, MRI scans can detect presence of tumor masses, but they are not tumor type-specific (5). Since therapeutic approaches are different for each type of tumor, there is an urgent need to develop tumor-type specific imaging for efficient therapy (6-8). Currently, molecular characterization of tumors can be conducted by histology examination of biopsy material and/or of resected tumors, followed by immunostaining of tissues for tumor type-associated biomarker(s). Although antibodies are routinely used for tissue immunostaining, their applicability for in vivo detection of tumors is largely limited due to 1) immunogenicity of protein-based antibodies, 2) poor tissue penetration due to their large molecular weight, and 3) limited capacity for chemical modification. In addition, tissue biopsy or resection may not always be possible because of tumor location in the body and other factors. To overcome these obstacles, new imaging probes that are tumor type/biomarker-specific, non-immunogenic, biocompatible, and easily conjugated with different imaging reporters are required.
Aptamers are small-molecule probes composed of short, single-stranded oligonucleotides (RNA or ssDNA ranging from 30 to 60 bases) (9-12). Oligonucleotide aptamers specifically recognize their targets, which include small molecules, biomacromolecules, viruses, and even live cells and whole organisms (13-17). Our previous studies have demonstrated that aptamers can be used as “chemical antibodies” for multi-color flow cytometric analysis of tumor cells (18,19) and for immunostaining of formalin-fixed and paraffin-embedded tumor tissues with higher efficiency than antibodies (20). Notably, synthetic oligonucleotide aptamers are easily modified and chemically conjugated with functional molecules for imaging purposes (21-28). More importantly, in contrast to protein antibodies, oligonucleotide aptamers exhibit little to none immunogenicity and off-target toxicity in vivo (29-34). These characteristics suggest that oligonucleotide aptamers might be suitable probes for in vivo specific tumor imaging. In this study, RNA- and ssDNA-based aptamer probes were generated and tested for specific in vivo imaging of tumors in an animal model.
Results
Tumor cell-specific RNA aptamer probes
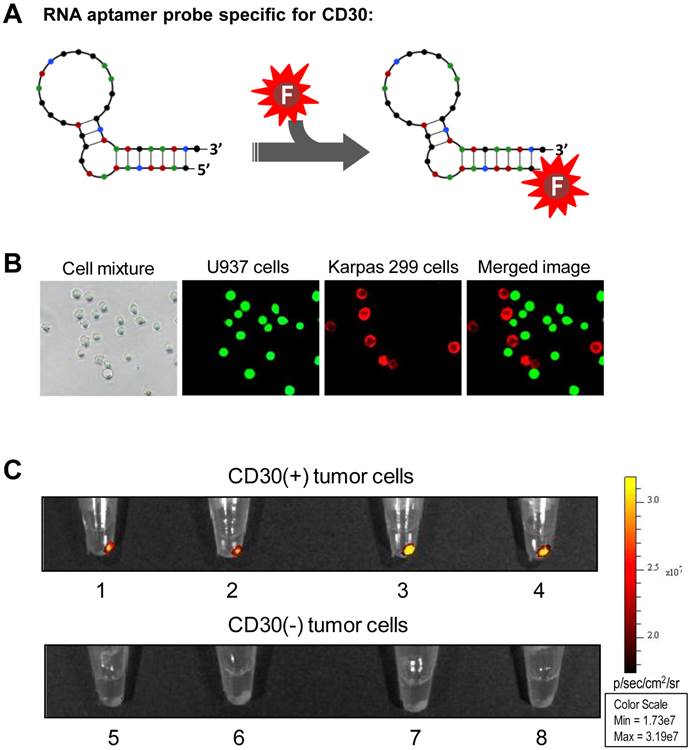
To generate an imaging probe, a 39-nucleotide RNA aptamer specific for CD30 (18,20) was conjugated with an imaging reporter (IRD800CW) or the Cy3 fluorochrome as illustrated in Figure 1A. To confirm specificity of the aptamer probe, a tumor cell mixture was prepared by diluting CD30-expressing lymphoma cells (Karpas 299) in CD30-negative leukemia cells (U937) that were pre-stained with green fluorescence for tracing purposes. The cell mixture was treated at room temperature for 30 min with aptamer probes labeled with the Cy3 fluorochrome for visualization. Fluorescent microscopy revealed that the aptamer probes selectively stained lymphoma cells (red fluorescence from Cy3 reporter), but did not react with CD30-negative control cells (green fluorescence) in the same cell mixture (Fig. 1B). For imaging validation, several cultured tumor cell lines including CD30-expressing tumor cells (Karpas 299, HDLM2, KMH2, and SU-DHL-1) and CD30-negative control cells (Jurkat, H-9, Sup-T1, and U937) were used. Cells were treated with the IRD800CW-conjugated RNA aptamer probe at room temperature for 30 min. After two washes the cell pellets were scanned using the XENOGEN IVIS 200 Imaging System with the ICG channel. As shown in Figure 1C, the RNA aptamer probes specifically highlighted pellets of CD30-expressing tumor cells (upper panel), but did not react with any of the control cells that are negative for CD30 (lower panel).
Specific and selective highlighting of lymphoma cells by aptamer probes. A, The aptamer probe was generated by conjugating a CD30-specific 39-mer RNA aptamer sequence to the Cy3 fluorochrome for cell staining and the IRD800CW reporter for cell pellet imaging. B, A cell mixture containing CD30-expressing lymphoma cells (Karpas 299) and CD30-negative control tumor cells (U937) that were pre-stained with green fluorescence was treated with the Cy3-labeled RNA aptamer probe. Fluorescent microscopy revealed that the aptamer probes selectively stained lymphoma cells (red), but did not react with CD30-negative control tumor cells (green) in the same mixture. C, Cultured CD30-expressing tumor cells (1: HDLM2; 2: Karpas 299; 3: KMH2; and 4: SU-DHL-1) and CD30-negative tumor cells (5: Jurkat; 6: H-9; 7: Sup-T1; and 8: U937) were treated with the IRD800CW-conjugated aptamer probes and cell pellets were scanned using the IVIS 200 Imaging System.
Specific and selective tumor imaging using RNA aptamer probes
For the animal model study, individual mice were subcutaneously inoculated with CD30-expressing lymphoma cells (Karpas 299) and CD30-negative control tumor cells (U937) as illustrated in Figure 2A. Development of both xenograft tumors in each mouse was confirmed by physical examination (Fig 2B, far left). The IRD800CW-conjugated RNA aptamer probes (10 µg/mouse) were systemically administrated through the tail veins of tumor-bearing mice and whole body imaging was performed using the IVIS 200 Imaging System. Imaging signals derived from aptamer probes at the regions of interest (ROIs) of tumor sites and body background were recorded at different time points. As shown in Figure 2B, the RNA aptamer probes selectively highlighted lymphoma tumor immediately after systemic administration, but did not react with the control tumor in the same mouse. The imaging signal from the ROIs of lymphoma tumors was 5-fold higher than that of control tumors in the same mouse and 8-fold higher than body background. This specific imaging signal gradually diminished and was nearly undetectable at 60 min post-aptamer administration.
Specific and selective tumor imaging using RNA aptamer probe. A, Scheme of animal imaging study: individual SCID mice were subcutaneously inoculated with CD30-expressing lymphoma cells (Karpas 299) and CD30-negative tumor cells (U937). After confirming development of both xenograft tumors, the RNA aptamer probes were systemically administered through the tail vein of tumor-bearing mice and whole body imaging was performed using the IVIS 200 Imaging System. B, Whole body imaging scans pre- and post-systemic administration of the RNA aptamer probe at different time points from 0 to 180 min. The aptamer probe selectively highlighted the lymphoma tumor, but not the control tumor in the same mouse. C, For confirmation, tumor tissues were removed 1 hr after whole body imaging and re-scanned (upper panel). Tissue immunostaining for CD30 expression was also performed (lower panel). D, To evaluate in vivo biodistribution of the aptamer probes, major organs were removed from the mice 1 hr after whole body scanning and re-imaged.
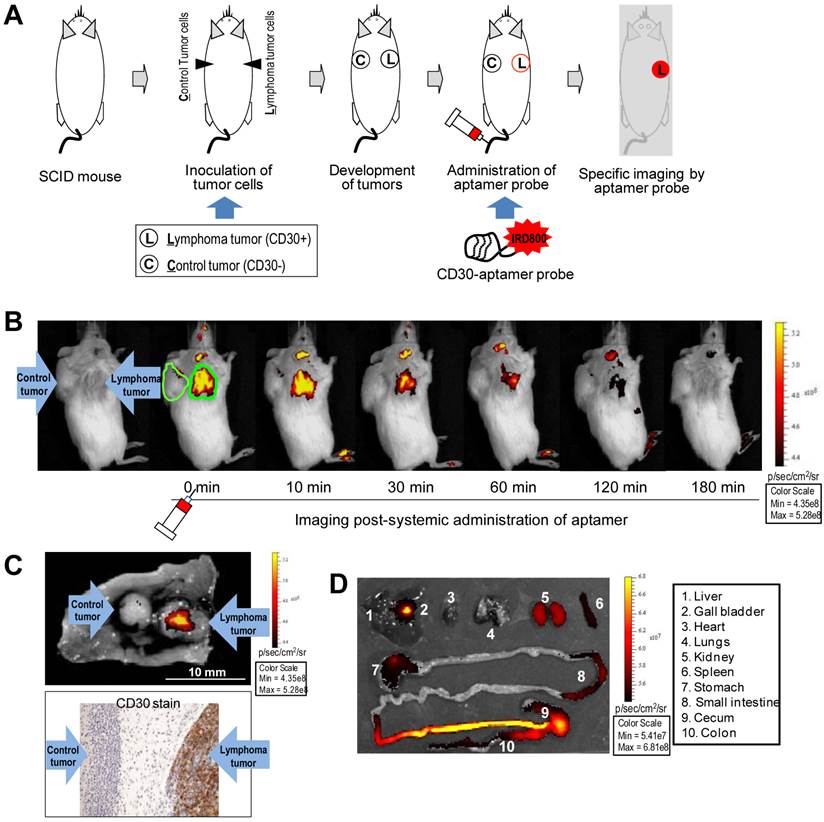
For confirmation of these imaging findings, tumors and adjacent tissues were removed from the mice and re-scanned ex vivo. In addition, tumor tissue was fixed and immunostained with anti-CD30 antibody. These studies confirmed that the RNA aptamer probe selectively highlighted the CD30-expressing lymphoma tumor and did not react with control tumor in the same mouse (Fig. 2C). To study the bio-distribution of aptamer probes, major organs of the mice were dissected and imaged. Figure 2D shows that the aptamer probes were rapidly eliminated from the mouse body mainly through the urinary tract (kidneys) and lower intestinal tract (gallbladder to intestine). There was minimal accumulation in other organs, including heart, lungs, spleen, stomach, and small intestine.
Sensitive and long-lasting tumor imaging using biostable ssDNA aptamer probes
Although RNA aptamer probes could specifically highlight tumors, they were easily degraded in vivo, leading to the rapid loss of signal intensity (Fig. 2B). To overcome this limitation, a 31-nucleotide ssDNA aptamer specific for CD30 (21) was conjugated with the IRD800CW reporter for imaging use (Fig. 3A). To compare biostability, synthetic ssDNA and RNA aptamer probes were incubated in human serum at 37°C to mimic in vivo conditions, residual aptamer probe products recovered at different time points and quantified. As shown in Figure 3B, the ssDNA aptamer probe was highly stable in human serum, with over 70% remaining after 24 hr. In contrast, the RNA aptamer probe was less stable in serum under the same conditions. These findings indicate that the ssDNA aptamers have greater biostability and are more suitable for in vivo use.
Sensitive and long-lasting tumor imaging using biostable ssDNA aptamer probes. A, A 31-nucleotide CD30-specific ssDNA aptamer sequence was conjugated with the IRD800CW reporter to generate an imaging probe. B, For the biostability assay, synthetic ssDNA and RNA aptamer probes were incubated in human serum at 37°C. The residual aptamer products were recovered at different time points and quantified. C, For more sensitive imaging, luciferase-expressing traceable lymphoma cells and breast cancer cells were used. Development of both xenograft tumors in each mouse was monitored by whole body imaging of bioluminescent signals derived from the tumor cells. The size of lymphoma tumors was limited to ≤5 mm in diameter whereas control breast cancer tumors were allowed to grow >5 mm by physical examination. D, CD30-specific ssDNA aptamer probes were systemically administered through tail veins of mice and whole body imaging was performed up to 24 hr after injection as indicated. E, Imaging signals at regions of interest (ROIs) from lymphoma tumors, breast cancer tumors, and normal body areas were recorded and statistically analyzed (n=5). Imaging signals of the ROIs of the tumors relative to body tissue background (fold-difference) are shown as mean ±SD (p<0.01). F, Additional xenograft animal models were established by using SUDHL-1 (CD30+) and LNCaP cells (CD30-) or KMH2 (CD30+) and ASPC-1 cells (CD30-) and development of both xenograft tumors in each mouse was confirmed by physical examination. Whole body imaging was performed and imaging signals of ROIs at 2 hr post-administration of ssDNA aptamer probe (a time point to allow the completion of complicated imaging procedures) are shown as mean ±SD (n=5, p<0.01).
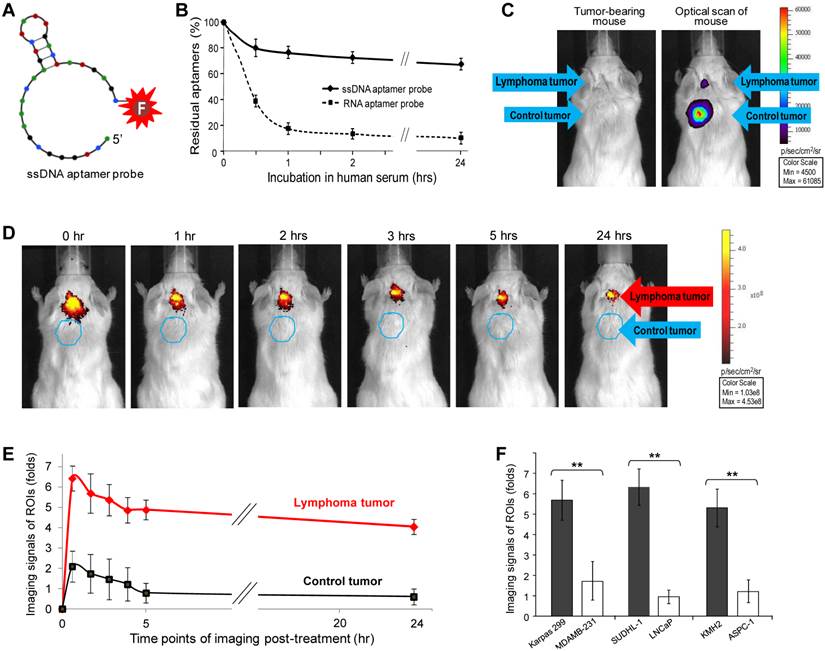
To conduct highly sensitive imaging, luciferase-expressing lymphoma cells (Karpas 299) and control breast cancer cells (231-D3H2-LN) were used to generate xenograft tumors. Tumor development was monitored by whole body bioluminescent imaging (Fig. 3C) and physical examination. The size of the CD30-expressing lymphoma tumors was limited to ≤5 mm in diameter, whereas control breast cancer tumors in the same mouse were allowed to grow larger than 5 mm. Because the ssDNA aptamer probes are more biostable than the RNA probes, a lower dose of imaging probe was administered systemically through the tail vein (1 µg/mouse vs. 10 µg/mouse of RNA imaging aptamer probes) and whole body imaging was performed as described above. Figure 3D shows that the ssDNA aptamer probes selectively highlighted CD30-positive lymphoma tumors and, more importantly, provided a long-lasting imaging signal. Immediately after administration, signals from the ROIs of lymphoma tumors were 6-fold higher than body tissue background (p<0.01) and 4-fold higher than the ROIs of control breast tumors (p<0.01) in the same mouse (Fig. 3E). Notably, the CD30-specific signal intensity detected in lymphoma tumors was stable for up to 24 hr. In contrast, signal from the CD30-negative breast tumors rapidly decreased to the level of background tissue in the same mouse.
For further validation, additional xenograft animal models were established by using CD30-expressing SUDHL-1 lymphoma cells and CD30-negateive LNCaP prostate cancer cells or CD30-exprssing KMH2 lymphoma cells and CD30-negateive ASPC-1 pancreatic cancer cells. Development of both xenograft tumors in the same mouse was confirmed by physical examination. The ssDNA aptamer probe (1 µg/mouse) was administered systemically and whole body imaging was then performed as described above. Imaging signals from the ROIs of CD30-expressing and CD30-negative tumors in the same mice were recorded and compared (n=5). Figure 3F shows that the ssDNA aptamer probes specifically highlighted CD30-expressing tumors, with their signal intensity being 4 to 5-fold higher than that derived from control tumors in the same mice (p<0.01) at 2 hr post-administration, a time point to allow the completion of complicated imaging procedures.
Discussion
Our proof-of-concept study utilized a unique animal model where animals bearing both a biomarker-expressing target tumor and a biomarker-negative control tumor. We demonstrated that oligonucleotide-based aptamers are able to selectively detect tumors by specifically targeting biomarker(s) expressed on different type of cancer cells. In addition, biostable ssDNA aptamer probes can produce a long-lasting, tumor-restricted imaging signal for up to 24 hr. Importantly, aptamer probes are synthetic oligonucleotides, which can be easily modified and chemically conjugated with a variety of reporters for different imaging approaches, including reporter molecules for PET, SPECT, CT, and MRI scans.
Notably, although radiation-based imaging approaches, such as PET and CT scans, are widely used in the clinical setting, the fluorescent-based imaging modalities may still have value in preclinical research and clinical disease due to lacking of radiation exposure. detection. In addition, compared to other fluorescent reporters the NIR imaging approach shows less background auto-fluorescence and deeper tissue penetration. Thus, it may replace radiation-based methods for imaging detection and monitoring of disease lesions under certain circumstances. For example, CT-guided fine needle aspiration and lesion biopsy are common diagnostic procedures. For example, CT-guided fine needle aspiration and tissue biopsy are common diagnostic procedures, which may take minutes to hours to complete depending on the anatomic location of the lesion. Therefore, both physicians and patients may experience an unnecessary radiation exposure with potential subsequent adverse health effects due to prolonged CT imaging. One possible solution would be to develop a specific NIR imaging aptamer probe that would exhibit an extended retention time and will selectively highlight disease lesions in order to facilitate tissue biopsy or surgical resection, thereby eliminating radiation exposure of patients and medical staff. In addition, an oligonucleotide-based aptamer can be simply conjugated with both a NIR reporter and a therapeutic compound, allowing for careful and dynamic monitoring of the drug's in vivo biodistribution.
Interestingly, SGN-35, trade name Brentuximab Vedotin, a CD30 antibody-drug conjugate, was recently approved by the FDA for the treatment of relapsed CD30-positive lymphomas (35-37). To benefit from this targeted therapy, it is critical to establish the status of CD30 biomarker expression on tumor cell and an. in vitro immunostaining of tumor tissues is currently the only method for such analysis. Therefore, a specific imaging approach for rapid, non-invasive in vivo detection of tumor CD30 biomarker expression will be valuable for determining eligibility for Brentuximab therapy, as well as for real-time monitoring of therapeutic response and early detection of disease relapse. Finally, our studies suggest that oligonucleotide aptamers might serve as a powerful vehicle for tumor-specific therapeutics delivery, opening the possibility for development of novel targeted aptamer-drug conjugates.
Materials and Methods
Reagents and cells
For imaging, a RNA-based 39-mer aptamer sequence specific for CD30 biomarker (18,20), was conjugated to the near-infrared fluorochrome IRD800CW reporter as shown in Figure 1A (IRD800CW-5′-GAUUCGUAUGGGUGGGAUCGGGAAGGGCUACGAACACCG-3'; Integrated DNA Technologies, Coralville, Iowa). The RNA aptamer was also conjugated with the Cy3 fluorochrome reporter for cell staining. Similarly, a 31-nucleotide ssDNA aptamer sequence specific for CD30-expressing tumor cells (21) was conjugated to the fluorochrome IRD800CW as shown in Figure 3A (IRD800CW-5′-ACTGGGCGAAACAAGTCTATTGACTATGAGC-3′). Control tumor cells were labeled with the carboxyfluorescein succinimidyl ester (CFSE) dye (Invitrogen, Grand Island, NY).
CD30-positive cell lines used in this study were human anaplastic large cell lymphoma cells (Karpas 299 and SUDHL-1 cell lines from Dr. Mark Raffeld at NIH) and Hodgkin lymphoma cells (HDLM2 and KMH2 cell lines from Dr. Barbara Savoldo, Baylor College of Medicine, Houston, TX). CD30-negative tumor cell lines including Jurkat (T cell lymphoma/leukemia), H-9 (T cell lymphoma), Sup-T1 (T cell lymphoblastic lymphoma), U937 (histiocytic lymphoma), LNCaP (prostate carcinoma), and ASPC-1 (pancreatic carcinoma) were purchased from ATCC. Luciferase-expressing breast cancer cells (MDA-MB-231-luc-D3H2LN), which are CD30-negative, were purchased from PerkinElmer (Santa Clara, CA). Cells were cultured in RPMI 1640 or DMEM medium with 10% FBS, 100 U/ml penicillin, and 100 µg/ml streptomycin at 37°C in an atmosphere of 5% CO2 and ≥95% humidity.
Cell staining and cell pellet scanning
For cell staining, CD30-positive Karpas 299 cells and CD30-negative U937 control cells, which were pre-stained with 10 µM CFSE at 37°C for 15 min, were mixed at a 1:1 ratio. The cell mixture was treated with the CD30-specific RNA aptamer probe labeled with Cy3 reporter (10 nM final concentration) at room temperature for 30 min. After two washes, the cells were examined under a fluorescent microscope using a fluorescein isothiocyanate (FITC) filter for CFSE of pre-stained cells and a tetramethylrhodamine isothiocyanate (TRITC) filter for cells highlighted by the Cy3-labeled aptamer probe.
To validate imaging potential of the aptamer probe, freshly cultured cells in diluted in 100 µl PBS (106 cells/test) were treated with the CD30-specific RNA aptamer labeled with IRD800CW reporter (10 nM final concentration) at room temperature. After incubation for 30 min, cell pellets were obtained by centrifugation and scanned using the IVIS 200 Imaging System (PerkinElmer) with an ICG filter to detect fluorescent signal derived from the aptamer probe as indicated in Figure 1C.
Biostability assays
To mimic an in vivo physiological condition, 1 µg of synthetic CD30-specific RNA or ssDNA aptamers was incubated in 200 µl of human serum at 37°C and residual aptamers were recovered using phenol-chloroform extraction at different time points up to 24 hr, as indicated. The residual aptamers were detected on 5% agarose gels and quantified (as % of initial amount).
Imaging studies
For imaging studies, 4-6-week old NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NOD SCID) mice were purchased from Jackson Laboratory (Bar Harbor, ME, USA). Each mouse was subcutaneously inoculated with cultured CD30-expressing lymphoma cells (Karpas 299, 6×106) and CD30-negative control tumor cells (U937, 2×106) at the same time, but at different anatomic sites, as shown in Figure 2A. Tumor development was monitored and confirmed by physical examination until the tumor size reached ≥5 mm in diameter (approximately 20 to 25 days post tumor cell implantation). The CD30-specific RNA aptamer probe labeled with IRD800CW reporter (10 µg in 100 µl PBS) was administered systemically via a tail vein injection and whole body imaging using the IVIS 200 Imaging System was performed immediately following and up to 3 hr after the injection, as indicated in Figure 2B. Imaging signal of regions of interest (ROIs) from lymphoma tumors, control tumors, and body areas without tumor was recorded in digital format using Xenogen software and the fold increase in signal intensity was calculated according to the manufacturer's instructions as follows: fold increase = Tumorcorrect_dye - Tumorcorrect_ none) / (Blankcorrect_dye - Blankcorrect_ none).
To confirm imaging findings, tumors and adjacent tissues were removed at the end-point and re-scanned. In addition, tumor tissues were fixed and immunostained for CD30 expression in our pathology laboratory following a standard protocol. To examine systemic distribution, major organs of the mice including heart, lungs, kidneys, spleen, liver and gallbladder, stomach, and small and large intestines were removed 60 min after administration of the aptamer probes and imaged.
To conduct highly sensitive imaging using the biostable CD30-specific ssDNA aptamer probe, mice were inoculated subcutaneously with luciferase-expressing lymphoma cells (Karpas 299, 6×106) and luciferase-expressing breast cancer cells (MDA-MB-231-luc-D3H2LN, 5×105) at separate sites as shown in Figure 3C. The development of xenograft tumors was monitored and confirmed by whole body bioluminescent imaging using IVIS 200 immediately after intraperitoneal injection of luciferin. For the sensitive imaging study the lymphoma tumor size was limited to ≤5 mm in diameter whereas the control breast cancer tumors in the same mouse were allowed to be >5 mm in size. The CD30-specific ssDNA aptamer probe (1 µg in 100 µl PBS) was administered through the tail vein and whole body imaging was performed immediately and up to 24 hr after injection as indicated in Figure 3D. Fold increase in signal intensity in the ROI of tumor sites was calculated as described above. Statistical analysis of results (n=5) showing mean ±SD is provided in Figure 3E.
For further confirmation, additional animal models with different xenograft tumors were employed. Similar to methods described above, CD30-exprssing SUDHL-1 lymphoma cells and CD30-negateive LNCaP prostate carcinoma cells or CD30-exprssing KMH2 lymphoma cells and CD30-negateive ASPC-1 pancreatic carcinoma cells were used in pairs and inoculated subcutaneously into mice (n=5/group). Development of both xenograft tumors in each mouse was confirmed by physical examination. The ssDNA imaging probes (1 µg/mouse) were systemically administrated and whole body imaging was performed 2 hr post-administration (a time point to allow the completion of all complicated imaging procedures). Fold increase in signal intensity in the ROI of tumor sites was calculated and statistical analysis of results showing mean ±SD is provided in Figure 3F.
Acknowledgements
The authors thank Dr. Mark Raffeld at NIH/NCI, Bethesda, MD, for kindly providing Karpas 299 and SUDHL-1 cell lines, and Dr. Barbara Savoldo at Baylor College of Medicine, Houston, TX, for kindly providing HDLM2 and KMH2 cell lines.
Grant Support
This study was supported in part by NIH grants R01CA151955 and R33CA173382 to Y. Zu.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Weissleder R, Pittet M.J. Imaging in the era of molecular oncology. Nature. 2008;452:580-589
2. Brindle K. M. New approaches for imaging tumour responses to treatment. Nat Rev Ca. 2008;8:94-107
3. Steven J Frank, KS Clifford Chao, David L Schwartz. et al. Technology Insight: PET and PET/CT in head and neck tumor staging and radiation therapy planning. Nat Clin Pract Oncol. 2005;2:526-533
4. Sandip Basu. Personalized versus evidence-based medicine with PET-based imaging. Nat Rev Clin Oncol. 2010;7:665-668
5. Blamire AM. The technology of MRI- the next 10 years? Br Inst Radiol. 2008;81:601-617
6. Tony SK. Mok. Personalized medicine in lung cancer: what we need to know. Nat Rev Clin Oncol. 2011;8:661-668
7. Wistuba I, Gelovani JG, Jacoby JJ, at al. Methodological and practical challenges for personalized cancer therapies. Nat Rev Clin Oncol. 2011;8:135-141
8. Rebecca Kirk. Genetics: Personalized medicine and tumour heterogeneity. Nat Rev Clin Oncol. 2012;9:250-250
9. Ellington AD, Szostak JW. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990;346:818-822
10. Tuerk C, Gold L. Systematic evolution of ligands byexponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990;249:505-510
11. Brody EN, Gold L. Aptamers as therapeutic and diagnostic agents. J Biotechnol. 2000;74:5-13
12. Hermann T, Patel DJ. Adaptive recognition by nucleic acid aptamers. Science. 2000;287:820-825
13. Bunka DH, Stockley PG. Aptamers come of age at last. Nat Rev. 2006;4:588-596
14. Lin Y, Nieuwlandt D, Magallanez A, Feistner B, Jayasena SD. High-affinity and specific recognition of human thyroid stimulating hormone (hTSH) by in vitro-selected 20-amino-modified RNA. Nucleic Acids Res. 1996;24:3407-3414
15. Drolet DW, Moon-McDermott L, Romig TS. An enzyme-linked oligonucleotide assay. Nat Biotechnol. 1996;14:1021-1025
16. Ulrich H, Wrenger C. Disease-specific biomarker discovery by aptamers. Cytometry A. 2009;75:727-733
17. Davis KA, Lin Y, Abrams B, Lin Y, Jayasena SD. Staining of cell surface human CD4 with 20-F-pyrimidine-containing RNA aptamers for flow cytometry. Nucleic Acids Res. 1998;26:3915-3924
18. Zhang P, Zhao N, Zeng Z, Feng Y, Chang CC, Zu Y. Using an RNA aptamer probe for flow cytometry detection of CD30-expressing lymphoma cells. Lab Invest. 2009;89:1423-1432
19. Zhang P, Zhao N, Zeng Z, Chang CC, Zu Y. Combination of an aptamer probe to CD4 and antibodies for multi-color cell phenotyping. Ame J Clin Pathol. 2010;134:586-593
20. Zeng Z, Zhang P, Zhao N, Sheehan AM, Tung CH, Chang CC, Zu Y. Using oligonucleotide aptamer probes for immunostaining of formalin-fixed and paraffin-embedded tissues. Mod Pathol. 2010;23:1553-1558
21. Parekh P, Kamble S, Zhao N, Zeng Z, Portier B, Zu Y. Immunotherapy of CD30-expressing lymphoma using a highly stable ssDNA aptamer. Biomaterials. 2013;34:8909-8917
22. Tavitian B, Ducongé F, Boisgard R, Dollé F. In vivo imaging of oligonucleotidic aptamers. Methods Mol Biol. 2009;535:241-59
23. Shi H, Tang Z, Kim Y, Nie H, Huang YF, He X. et al. In vivo fluorescence imaging of tumors using molecular aptamers generated by cell-SELEX. Chem Asian J. 2010;5:2209-13
24. Shi H, He X, Wang K, Wu X, Ye X, Guo Q. et al. Activatable aptamer probe for contrast-enhanced in vivo cancer imaging based on cell membrane protein-triggered conformation alteration. Proc Natl Acad Sci USA. 2011;108:3900-5
25. Shi H, Cui W, He X, Guo Q, Wang K, Ye X. et al. Whole cell-SELEX aptamers for highly specific fluorescence molecular imaging of carcinomas in vivo. PLoS One. 2013;8:e70476
26. Su X, Xiao X, Zhang C, Zhao M. Nucleic acid fluorescent probes for biological sensing. Appl Spectrosc. 2012;66:1249-62 Review
27. Wang T, Ray J. Aptamer-based molecular imaging. Protein Cell. 2012;3:739-54 Review
28. Wang AZ, Farokhzad OC. Current progress of aptamer-based molecular imaging. J Nucl Med. 2014;55:353-6 Review
29. Dausse E, Da Rocha Gomes S, Toulmé JJ. Aptamers: a new class of oligonucleotides in the drug discovery pipeline? Curr Opin Pharmacol. 2009;9:602-7 Review
30. Scaggiante B, Dapas B, Farra R, Grassi M, Pozzato G, Giansante C, Fiotti N, Tamai E, Tonon F, Grassi G. Aptamers as targeting delivery devices or anti-cancer drugs for fighting tumors. Curr Drug Metab. 2013;14:565-82 Review
31. Hu M, Zhang K. The application of aptamers in cancer research: an up-to-date review. Future Oncol. 2013;9:369-76 Review
32. Pei X, Zhang J1, Liu J. Clinical applications of nucleic acid aptamers in cancer. Mol Clin Oncol. 2014;2:341-348 Review
33. Esposito CL, Catuogno S, de Franciscis V, Cerchia L. New insight into clinical development of nucleic acid aptamers. Discov Med. 2011;11:487-96 Review
34. Que-Gewirth NS, Sullenger BA. Gene therapy progress and prospects: RNA aptamers. Gene Ther. 2007;14:283-91 Review
35. Younes A, Bartlett NL, Leonard JP, at al. Brentuximab Vedotin (SGN-35) for Relapsed CD30-Positive Lymphomas. N Engl J Med. 2010;363:1812-1821
36. Younes A. CD30-targeted antibody therapy. Curr Opin Oncol. 2011;23:587-93
37. Foyil KV, Bartlett NL. Brentuximab vedotin for the treatment of CD30+ lymphomas. Immunotherapy. 2011;3:475-85
Author contact
![]() Corresponding author: Youli Zu, M.D., Ph.D. Professor and Medical Director of Hematopathology, Director, Cancer Pathology Laboratory, Houston Methodist Hospital, Weill Cornell Medical College of Cornell University. Tel: 713 441 4460; Email: yzuorg
Corresponding author: Youli Zu, M.D., Ph.D. Professor and Medical Director of Hematopathology, Director, Cancer Pathology Laboratory, Houston Methodist Hospital, Weill Cornell Medical College of Cornell University. Tel: 713 441 4460; Email: yzuorg