Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Experimental
Results and discussion
Conclusion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2014; 4(7):701-707. doi:10.7150/thno.8803 This issue Cite
Short Research Communication
A New Method to Assay Protease Based on Amyloid Misfolding: Application to Prostate Cancer Diagnosis Using a Panel of Proteases Biomarkers
Hao Li1, Yue Huang1, Bin Zhang2, Dawei Yang1, Xiaoli Zhu2, Genxi Li1, 2 ![]()
1. Department of Biochemistry and State Key Laboratory of Pharmaceutical Biotechnology, Nanjing University, Nanjing, China.
2. Laboratory of Biosensing Technology, School of Life Sciences, Shanghai University, Shanghai, China.
Received 2014-2-11; Accepted 2014-3-26; Published 2014-4-25
Abstract

This paper reports a sensitive method with electrochemical technique to detect various proteases, which can be used for the diagnosis of prostate cancer. For the proposed assay method, the working electrode is modified with the peptide probes for the target proteases. These probes contain the substrate sequence of target proteases, as well as the seed peptide sequence that can accelerate the misfolding of amyloid-beta. If there are proteases in the test solution, after protease cleavage of the substrate peptides, the distal seed peptide will be removed from the electrode surface. So, in the absence of proteases, the seed peptides can initiate and accelerate amyloid-beta misfolding on the electrode surface. Consequently, the formed aggregates strongly block the electron transfer of the in-solution electroactive species with the electrode, resulting in suppressed signal readout. Nevertheless, in the presence of proteases, enzyme cleavage may lead to greatly mitigated protein misfolding and evident signal enhancement. Since the contrast in signal readout between the two cases can be amplified by using the protein misfolding step, high sensitivity suitable for direct detection of proteases in serum can be achieved. These results may suggest the feasibility of our new method for the detection of a panel of proteases in offering detailed diagnosis of prostate cancer and a better treatment of the cancer.
Keywords: prostate cancer, biomarker, protease, amyloid misfolding, electrochemical detection.
Introduction
Serum has long been the target of studies aiming at sensitive detection of tumor biomarkers for the diagnostics and prognostics of cancer [1-4]. Despite the enormous effort devoted to this course, early and timely diagnosis of cancer is still substantially hampered by the nature of serum biomarkers [5]. First, individual biomarker has very limited indicative value since one biomarker only represents one aspect of carcinogenesis. Second, many serum biomarkers have extremely low abundance. Nowadays, there is a growing consensus that detection of a panel of serum biomarkers, rather than one specific biomarker [3, 4, 6, 7], combined with highly sensitive protocol of detection, can improve the accuracy and efficacy of diagnostics and prognostics of cancer. In this sense, protease, among many types of biomarkers, is a better candidate as serum cancer biomarker [8, 9]. On the one hand, several different kinds of proteases are shed into serum during carcinogenesis, and the activities of these serum proteases are associated with various aspects of the cancerous process, so the overall diagnostic value of a protease marker panel is much higher than a single cancer marker. On the other, protease activity can be exploitable for designing signal alteration and amplification step [10-13], thus the sensitivity of the developed detection method can be greatly improved for the assay of the protein directly in serum samples.
Here we employ the specific interaction between a designed peptide probe and amyloidal peptide to induce amyloid misfolding, as an effective means to detect serum proteases. It is well-known that amyloidal peptides, such as amyloid-beta, can misfold into aggregates [14], while the misfolding process can be modulated by special peptide sequences [15]. So, we have designed a peptide probe by attaching the substrate sequence of the target protease to a “seed peptide” that can specifically bind with the monomer of amyloid-beta and accelerate its misfolding into aggregates [14]. Electrochemical methods are employed to develop this assay [16]. Protease cleavage of electrode surface immobilized probes protease cleavage may provide an assured change of the signal readout. To provide sufficient sensitivity for the detection of low abundant serum proteases, the signal change induced by protease cleavage needs to be amplified. The subsequent formation of aggregates on the sensing layer can greatly magnify this change. In the absence of protease, extensive formation of aggregates will block the access of in-solution electroactive species to the electrode surface, leading to very low signal readout. However, in the presence of protease, the dramatically mitigated misfolding process combined with incomplete surface layer after protease cleavage will result in large signal response of the in-solution electroactive species. The contrast in signal readout between the two cases is magnified, enabling much lower limit of detection.
This method has no special requirement for the sequence of the substrate peptides, so it is generally applicable for the assay of many kinds of proteases. To examine the feasibility of this method, it has been used along with a recently developed nonspecific absorption-resistant peptide [17] and applied in the protease assays using serum from prostate cancer victims. The protease species under inspection, namely, matriptase [18, 19], kallikrein 2 [20, 21] and prostate specific antigen (PSA) compose a marker panel and differential expression of the three enzymes can offer reliable reference to the establishment and progression of cancerous condition.
Experimental
Chemicals and Reagents
Peptide probes for matriptase, kallikrein 2 and PSA (MUA (11-mercaptoundecanoic acid)-SGKGSSGSST [17]-RQARV [22]/TFRSA [23]/SSYSS [24]-KLVFFEEEE [15] and the absorption-resistant peptide (MUA-SGKGSSGSST) were manufactured by Shanghai Science Peptide as lyophilized powder, purity>95%. Human recombinant matriptase was from Alexis, Switzerland. Recombinant kallikrein 2 and PSA were provided by R&D Systems. Plasmin was purchased from Milipore. Thrombin and amyloid-beta was from Sigma Aldrich. Analytical-grade was warranted for all of the other reagents used. Powder of the peptide probes and the absorption-resistant peptide was dissolved with 10 mM phosphate buffer solution (PBS) (pH 7.4) to prepare 5 μM stock solutions. The original buffer solutions of all the proteases were dissolved with proper solution to prepare standard samples. The buffer adopted for matriptase is 100mM Tris, pH 9.5; for kallikrein: 20 mM Tris, 0.15 M NaCl, 10 mM CaCl2, pH 7.5; for PSA: 50 mM Tris, 10 mM CaCl2, 150 mM NaCl, pH 7.5. Redistilled water for preparation of all the solutions was produced with a Milli-Q purification system, the resistance of 18 MΩ·cm was achieved to guarantee the purity. Serum samples of patients with prostate conditions were obtained from the Clinical Laboratory of the First Affiliated Hospital of Nanjing Medical University, after elected consent by the local ethical committee.
Electrode Treatment and Modification
Firstly, the gold disk electrode with a diameter of 3 mm was chemically cleaned for 5 min using Piranha rinsing solution, which was formulated by 70% sulfuric acid (concentrated) and 30% hydro-peroxide, followed with rinsing by double-distilled water. Then, the electrode was polished with fine sand paper, 1 µm and 0.3 µm alumina slurry in sequence, till achieving a mirror-smooth surface. Subsequently, water bath ultra-sonicating using both ethanol and water as the media was conducted to remove alumina powder from the electrode. Again, chemical cleaning was employed to clean the electrode: 50% nitric acid was incubated with the electrode for 30 min. Finally, electrochemical cleaning via repeated cyclic voltammetric scans in 0.5 M H2SO4 was utilized to remove any remaining impurities.
To immobilize peptide probes on the surface of the working electrode, the electrode was firstly dried under a mild stream of nitrogen with high purity, immediately following the above cleaning steps. Then, the self-assembly procedure was initiated by immerging the electrode in the peptide assembly solution (2.5 μM substrate peptide and 5mM TCEP in 10mM PBS, pH 7.4). The self-assembly step lasted for 16 h, at 4 °C and in an inert atmosphere. After rinsing with double distilled-water, the electrode was treated with the passivation solution (2.5 μM absorption-resistant peptide and 5mM TCEP in 10mM PBS, pH 7.4) at room temperature for 3 h. At last, any remaining non-specifically absorbed peptides were removed via thorough rinsing. The as-prepared electrode was thus ready for protease assay.
Protease Activity Assay
Firstly, standard or serum samples containing protease were incubated with the electrode at 37 °C. Thorough rinsing of the electrode with double distilled water and incubation with 5% Tween-20 was conducted to exclude non-specific absorption. Then, the cleaved electrode was incubated with 300 μM amyloid-beta, 100 μM peroxynitrite, in 10 mM PBS (pH 5.0) for 75 min at room temperature for amyloid-beta misfolding. After that, the electrode was again thoroughly rinsed with double-distilled water, followed by being dipped in 5% Tween-20 for 30 min. The electrode was finally ready for measurement.
Experimental Measurements
Atomic force microscopy (AFM) images are recorded using ex situ Agilent 5500 AFM system. Samples were imaged at a scan rate of 0.5-1 Hz in a tapping mode. AFM tips with resonant frequency in a range 160-260 kHz were used. Images were acquired at a resolution of 512×512 pixels. Isothermal titration calorimetry (ITC) measurements were conducted using a MicroCal ITC200 System (GE healthcare life sciences). The titration was conducted at 25 °C. The titration schedule consisted of 38 consecutive injections of 1 μL with at least a 120 s interval between injections. Heats of dilution, measured by titrating beyond saturation, were subtracted from each data set. All solutions were degassed prior to titration. The data were analyzed using Origin 7.0 software. All the electrochemical assays were conducted with a CHI660D electrochemical workstation (CH Instruments, Austin, USA) using a conventional three-electrode cell at room temperature. Square wave voltammograms (SWVs) and electrochemical impedance spectra (EIS) were recorded in 5mM [Fe(CN)6]3-/4-with 1 M KNO3 Experimental parameters are as follows. SWV: Scan range, -0.7 ~ -0.2 V, step potential, 5 mV, frequency, 15 Hz, amplitude, 25 mV. EIS: Bias potential, 0.224 V vs. SCE, amplitude, 5 mV, frequency range, 0.1 Hz ~ 10 kHz. The data were obtained from at least three times of repetition of independent experiment.
Results and discussion
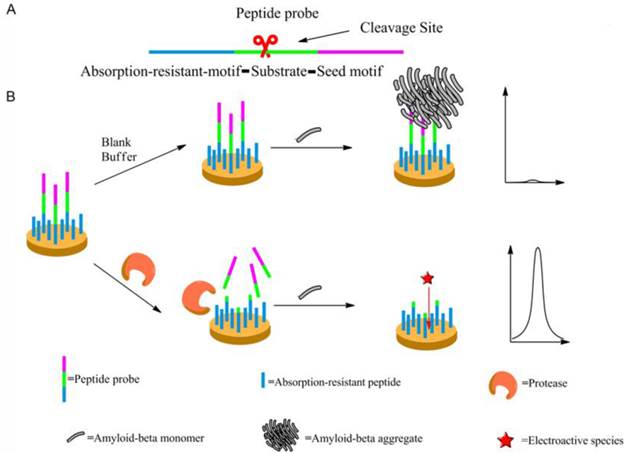
Scheme 1 may illustrate the principle of the proposed method. The peptide probe is composed of three motifs. Located at one end of the probe is the absorption-resistant sequence serving to minimize nonspecific interaction with the non-target protein and to immobilize the probe onto the surface of the working electrode. At the other end is the seed peptide sequence to initiate and accelerate amyloid-beta misfolding on the electrode surface. Between these two motifs is the substrate sequence of the target protein, i.e., protease. The absorption-resistant peptide is also employed as the blocking reagent for the passivation of the electrode surface to further reduce the interference of serum non-target proteins. The peptide probes firstly form a self-assembly monolayer on the electrode surface. After incubation with the test solution containing protease, many surface-anchored probes are cleaved and lose their seed peptide motifs. The remaining uncleaved peptide probes then use their seed motifs to induce amyloid-beta misfolding. [Fe(CN)6]3-/4- is employed to electrochemically show the resulted change of the sensing layer. Since more protease will cleave more peptide probes, resulting in more seed motifs being removed from the sensing layer, less extensive misfolding will ensue. Consequently, the resistance to electron transfer will decrease, leading to larger electrochemical response of [Fe (CN)6]3-/4-. So, the final signal response is positively related to protease abundance.
The mechanism of the designed protease assay. (A) The peptide probe showing its motifs. (B) The procedure of the protease assay. Not shown to scale.
The above-described assay procedure has been firstly verified using EIS recording the step-wise modification of the electrode during the assay procedure. Matriptase is here employed as the model target. According to Supplementary Material: Figure S1, a straight line is the case for the bare electrode. After modification by the peptide probes, an evident semicircle appears, indicating augmented resistance to interface electron transfer, due to the forming of a peptide monolayer on the electrode surface. In the absence of the protease, subsequent extensive amyloid-beta misfolding on the electrode surface seriously impedes the interface electron transfer, drastically increasing the diameter of the semicircle on EIS. By way of contrast, in the presence of the protease, the electrochemical impedance evidently decreases after catalytic cleavage, since the cleavage removes many seed motifs, each containing 4 consecutive glutamic acid residues that strongly repel [Fe(CN)6]3-/4-. Subsequently, amyloid-beta misfolding takes place in a much smaller scale, resulting in a tiny increase of electrochemical impedance, compared with the situation that protease is absent.
Changes of the sensing layer in the presence/absence of the protease have also been verified by using atomic force microscopy. As shown in Supplementary Material: Figure S2, in the absence of the protease, many a peptide probe is observed densely distributed on the sensing layer (Figure S2A), and extensive amyloid-beta misfolding can be observed (Figure S2B). In contrast, protease cleavage leaves only very few intact peptide probes on the sensing layer (Figure S2C), and subsequent misfolding is greatly mitigated (Figure S2D).
The experimental conditions have also been studied and optimized. First, since the amyloid-beta misfolding process takes very long time to complete, to improve the performance of the assay, the time for misfolding process has been greatly shortened by using chemical reagent to nitrate the protein monomer [25], so that the misfolding process is dramatically accelerated (Supplementary Material: Figure S3). Second, the concentration of amyloid-beta monomer has been optimized. To attain low electrochemical background signal in the absence of protease, more concentrated monomer protein should be used, but this will inevitably result in smaller change of the electrochemical response in the absence/presence of protease, thus making the difference among signals of different amount of protease to be much less prominent. Therefore, SWV signals obtained using different concentrations of amyloid-beta monomer in the presence/absence of protease are compared (Supplementary Material: Figure S4). From 100 μM to 600 μM, the background gradually decreases to negligible level, while the signal after protease cleavage keeps decreasing. So, 300 μM amyloid-beta monomer is selected for the assay, at which concentration the background has already been sufficiently low while the signal response has not decreased too much. Thirdly, the cleavage time of the three target proteases has been investigated as well, and the incubation time just sufficient to cleave all peptides probes on the electrode surface has been known and applied for all the following experiments (Supplementary Material: Figure S5).
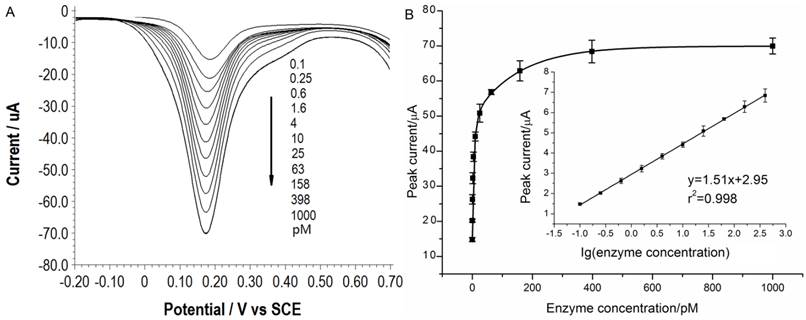
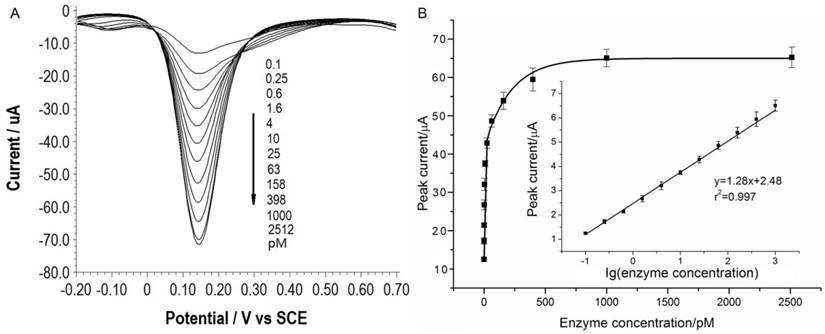
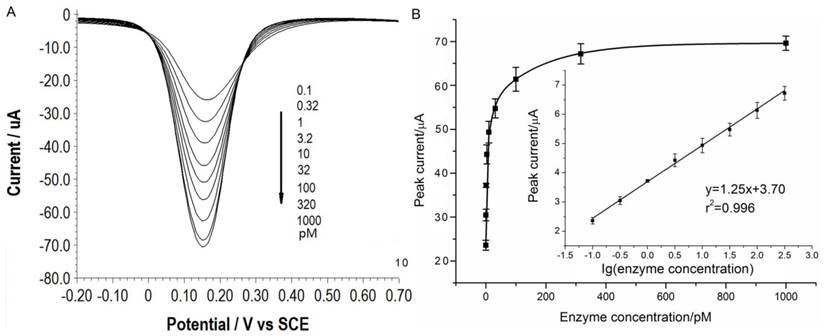
Under the optimal conditions, assays of the three target proteases are conducted using our method. Figure 1A, 2A and 3A show that the peak current of the electroactive probe [Fe(CN)6]3-/4- increases with the concentration of all the three target proteases. A linear detection range can be established for all the three proteases (Figure 1B, 2B and 3B). For matriptase, a linear range from 0.1 pM to 398 pM can be obtained, with a detection limit as low as 0.047 pM. For kallikrein 2 and PSA, the linear range are 0.1~1000 pM and 0.1~320 pM, with the limit of detection of 0.035 pM and 0.067 pM, respectively. The standard deviation of all data is within 5%. So, the sensitivity and reproducibility of our assay method for the detection of all the three proteases are satisfactory. Specificity of our method has also been examined by using some control species, all of which, including HSA, an abundant component in serum, may show no evident interference (Supplementary Material: Figure S6). Our method has been compared with the commercial ELISA as well (Supplementary Material: Table S1). In detecting all the three target proteases, our method is superior to ELISA in terms of sensitivity.
(A) SWVs of [Fe(CN)6]3-/4- to quantify matriptase. (B) Peak currents in (A) plotted as a function of protease concentration. Inset is the linear concentration range. The error bars represent standard deviation from average (n=3).
(A) SWVs of [Fe(CN)6]3-/4- to quantify kallikrein 2. All the curves and points in (B) are of the same meaning as in Figure 1.
(A) SWVs of [Fe(CN)6]3-/4- to quantify PSA. All the curves and points in (B) are of the same meaning as in Figure 1.
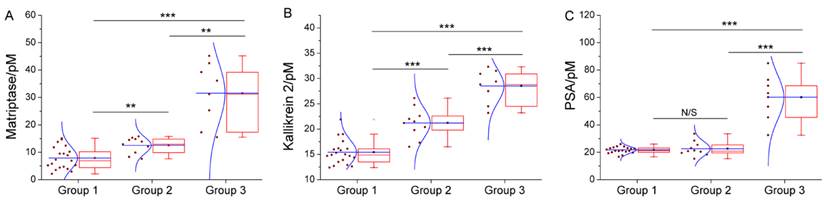
We have further employed these three proteases as a marker panel to test feasibility of our method in the diagnosis of prostate cancer. The protease assays have been conducted in the serum of several patients with prostate conditions. For each patient, concentration of the target proteases in his serum is averaged from the result of three repetitions for each protease, calculated by using the linear range obtained above. To compare our method with the traditional clinical characteristics, the detected protease level in all patients is sub-grouped according to the clinically confirmed pathological condition, namely, non-cancerous, cancerous and metastatic condition (Figure 4). For all proteases, the distribution of detected concentration agrees well with the clinical sub-grouping, and the behavior of each protease marker is coherent with the current bio-medical understanding of it. For example, as shown in Figure 4C, PSA of all groups are well-above the borderline (around 1 pM), indicating high risk of prostate cancer. But only the advanced prostate cancer (group 3) shows significantly higher PSA level, so PSA alone is insufficient for accurate diagnosis, as widely recognized. In contrast, matriptase and kallikrein 2 show evident difference between the groups (Figure 4A, 4B). Especially kallikrein 3, a promising prostate cancer marker which attracted substantial interest recently [26, 27], shows prominent inter-group difference (p < 0.01). It is also worth noting that for all groups, SWVs recorded in patient serum samples (Supplementary Material: Figure S7) are not evidently different from that of standard sample. So, the absorption-resistant peptide is functional, and the design may show reliable robustness which is indispensible for the detection of complex biological sample. So, it can be concluded that this marker panel may provide a reliable reference for the diagnosis of prostate cancer.
Box chart to show the distribution of detected concentration of (A) matriptase, (B) kallikrein 2 and (C) PSA in patient of non-cancerous (group1), cancerous (group 2) and metastatic (group 3) condition. Each box includes the maximum, minimum, mean, 1st and 99th percentile marked on the graph, in addition to the 25th, median, and 75th percentiles. The raw data is included as a column scatter plot to the left of each box. A curve corresponding to normal distribution is also displayed on top of the scatter plot. Statistical significance of the observed difference between groups is accessed by t-test: *P<0.05; **P<0.01; ***P<0.001; N/S denotes P>0.05.
Conclusion
Presented here is a simple but highly sensitive method to detect several serum proteases which can compose a marker panel to improve prostate cancer diagnosis. In the strategy of this new method, peptide is employed as the probe (Supplementary Material: Figure S8, S9), as well as the reagent to form an absorption-resistant electrode surface (Supplementary Material: Figure S10). Furthermore, through the amyloid misfolding induced signal amplification, sensitivity sufficient for direct serum detection can be attained. So, the detected expression pattern of the marker panel in patient serums may provide much more detailed prognostic information. Furthermore, the expression pattern is consistent with traditional pathological inspection, proving our method to be a promising tool for the early screening and diagnosis in cancer treatment.
Supplementary Material
Fig.S1 - Fig. S10, Table S1-Table S3.
Acknowledgements
This work is supported by the National Science Fund for Distinguished Young Scholars (Grant No. 20925520), the National Natural Science Foundation of China (Grant No. 21235003).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Hong B, Zu Y. detecting circulating tumor cells: current challenges and new trends. Theranostics. 2013;3:377-394
2. Petricoin EF, Belluco C, Araujo RP, Liotta LA. The blood peptidome: a higher dimension of information content for cancer biomarker discovery. Nat Rev Cancer. 2006;6:961-967
3. Hu Z, Lausted C, Yoo H. Quantitative liver-specific protein fingerprint in blood: a signature for hepatotoxicity. Theranostics. 2014;4:215-228
4. Novák J, Kružliak P, Bienertová-Vašků J. et al. MicroRNA-206: a promising theranostic marker. Theranostics. 2014;4:119-133
5. Diamandis EP, Blasutig IM. The long journey of cancer biomarkers from the bench to the clinic. Clin Chem. 2013;59:147-157
6. Tufman A, Tian F, Huber R. Can microRNAs improve the management of lung cancer patients? a clinician's perspective. Theranostics. 2013;3:953-963
7. Shapiro J, Nason KS, Davison JM. et al. Three-gene immunohistochemical panel adds to clinical staging algorithms to predict prognosis for patients with esophageal adenocarcinoma. Ong. J Clin Oncol. 2013;31:1576-1582
8. Storr SJ, Carragher NO, Frame MC. The calpain system and cancer. Nat Rev Cancer. 2011;1:364-374
9. Lopez-Otin C, Matrisian LM. Emerging roles of proteases in tumour suppression. Nat Rev Cancer. 2007;7:800-808
10. Kim J, Kim Y. Analysis of protease activity using quantum dots and resonance energy transfer. Theranostics. 2012;2:127-138
11. Yang K, Zhu L, Nie L. et al. Visualization of protease activity in vivo using an activatable photo-acoustic imaging probe based on CuS nanoparticles. Theranostics. 2014;4:134-141
12. Shih PM, Liu TK, Tan KT. Fluorescence amplified detection of proteases by the catalytic activation of a semisynthetic sensor. Chem Comm. 2013;49:6212-6214
13. Chen H, Mei Q, Hou Y. et al. Fabrication of a protease sensor for caspase-3 activity detection based on surface plasmon resonance. Analyst. 2013;138:5757-5761
14. Garai K, Frieden C. Quantitative analysis of the time course of A beta oligomerization and subsequent growth steps using tetramethylrhodamine-labeled A beta. Pro Nat Acad Sci USA. 2013;110:3321-3326
15. Lowe TL, Strzelec A, Kiessling LL. Structure-function relationships for inhibitors of beta-amyloid toxicity containing the recognition sequence KLVFF. Biochemistry. 2001;40:7882-7889
16. Li G, Miao P. Electrochemical analysis of proteins and cells. Berlin, Germany: Springer. 2012
17. Chelmowski R, Koster SD, Kerstan A. et al. Peptide-based SAMs that resist the absorption of proteins. J Am Chem Soc. 2008;130:14952-14953
18. Saleem M, Adhami VM, Zhong WX. et al. A novel biomarker for staging human prostate adenocarcinoma: overexpression of matriptase with concomitant loss of its inhibitor, hepatocyte growth factor activator inhibitor-1. Cancer Epidem Biomar. 2006;15:217-227
19. Tripathi M, Potdar AA, Yamashita H. et al. Laminin-332 cleavage by matriptase alters motility parameters of prostate. Cancer Cells. Prostate. 2011;71:184-196
20. Mize GJ, Wang WB, Takayama TK. Prostate-specific kallikreins-2 and-4 enhance the proliferation of DU-145 prostate cancer cells through protease-activated receptors-1 and-2. Mol Cancer Res. 2008;6:1043-1051
21. Vickers AJ, Gupta A, Savage CJ. et al. A Panel of kallikrein marker predicts prostate cancer in a large, population-based cohort followed for 15 years without screening. Cancer Epidem Biomar. 2011;20:255-261
22. Beliveau F, Desilets A, Leduc R. Probing the substrate specificities of matriptase, matriptase-2, hepsin and DESC1 with internally quenched fluorescent peptides. FEBS Journal. 2009;276:2213-2226
23. Cloutier SM, Chagas JR, Mach JP. et al. Substrate specificity of human kallikrein 2 (hK2) as determined by phage display technology. Eur J Biochem. 2002;269:2747-2754
24. Coombs GS, Bergstrom RC, Pellequer JL. et al. Substrate specificity of prostate-specific antigen (PSA). Chem Biol. 1998;5:475-488
25. Kummer MP, Hermes M, Delekarte A. et al. Nitration of tyrosine 10 critically enhances amyloid beta aggregation and plaque formation. Neuron. 2011;71:833-844
26. Emami N, Diamandis EP. Utility of kallikrein-related peptidases (KLKs) as cancer biomarkers. Clin. Chem. 2008;54:1600-1607
27. Parekh DJ, Ankerst DP, Troyer D. et al. Biomarkers for prostate cancer detection. J. urol. 2007;178:2252-2259
Author contact
![]() Corresponding author: Department of Biochemistry and State Key Laboratory of Pharmaceutical Biotechnology, Nanjing University, Nanjing 210093, China. Fax: +86 25 83592510. E-mail address: genxiliedu.cn.
Corresponding author: Department of Biochemistry and State Key Laboratory of Pharmaceutical Biotechnology, Nanjing University, Nanjing 210093, China. Fax: +86 25 83592510. E-mail address: genxiliedu.cn.