Impact Factor
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusion
Supplementary Material
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2014; 4(6):642-659. doi:10.7150/thno.8527 This issue Cite
Research Paper
A Novel Tumor Targeting Drug Carrier for Optical Imaging and Therapy
Rui Li1,4, Ke Zheng3, Ping Hu1, Zhuo Chen1,4, Shanyong Zhou1, Jincan Chen1, Cai Yuan1, Song Chen1,4, Wei Zheng2, En Ma2, Fengling Zhang3, Jinping Xue3, Xueyuan Chen2,4, Mingdong Huang1,4 ![]()
1. State Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou, Fujian, 350002, China;
2. Key Laboratory of Optoelectronic Materials Chemistry and Physics, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou, Fujian, 350002, China;
3. College of Chemistry and Chemical Engineering, Fuzhou University, Fuzhou, Fujian, 350002, China;
4. University of Chinese Academy of Sciences, Beijing, 100049, China.
Received 2014-1-8; Accepted 2014-3-5; Published 2014-3-24
Abstract
Human serum albumin (HSA), a naturally abundant protein in blood plasma and tissue fluids, has an extraordinary ligand-binding capacity and is advocated as a drug carrier to facilitate drug delivery. To render it tumor targeting specificity, we generated a recombinant HSA fused with the amino-terminal fragment (ATF) of urokinase, allowing the fusion protein to bind to urokinase receptor (uPAR), which is shown to have a high expression level in many tumors, but not in normal tissues. To test the efficacy of this bifunctional protein (ATF-HSA), a hydrophobic photosensitizer (mono-substituted β-carboxy phthalocyanine zinc, CPZ) was chosen as a cytotoxic agent. A dilution-incubation-purification (DIP) strategy was developed to load the ATF-HSA with this CPZ, forming a 1:1 molecular complex (ATF-HSA:CPZ). We demonstrated that CPZ was indeed embedded inside ATF-HSA at the fatty acid binding site 1 (FA1) of HSA, giving a hydrodynamic radius of 7.5 nm, close to HSA's (6.5 nm). ATF-HSA:CPZ showed high stability and remarkable optical and photophysical properties in aqueous solution. In addition, the molecular complex ATF-HSA:CPZ can bind to recombinant uPAR in vitro and uPAR on tumor cell surfaces, and was efficient in photodynamic killing of tumor cells. The tumor-killing potency of this molecular complex was further demonstrated in a tumor-bearing mouse model at a dose of 0.080 μmol / kg, or 0.050 mg CPZ / kg of mouse body weight. Using fluorescent molecular tomography (FMT), ATF-HSA:CPZ was shown to accumulate specifically in tumors, and importantly, such tumor retention was higher than that of HSA:CPZ. Together, these results indicate that ATF-HSA:CPZ is not only an efficient tumor-specific cytotoxic agent, but also an useful tumor-specific imaging probe. This bifunctional protein ATF-HSA can also be used as a drug carrier for other types of cytotoxic or imaging agents to render them specificity for uPAR-expressing tumors.
Keywords: urokinase receptor, amino-terminal fragment of urokinase, human serum albumin, phthalocyanine zinc, tumor targeting, fluorescent molecular tomography.
Introduction
Human serum albumin (HSA) is the most abundant protein in plasma (35-50 g / L) with a molecular weight of ~67 kDa. Synthesized in the liver at a rate of ~10-15 g per day, HSA has quite a long plasma half-life of 19 days [1, 2]. HSA maintains plasma osmotic pressure and modulates fluid distribution among body compartments [3]. In addition, HSA has an extraordinary ligand-binding capacity [4, 5] and plays a critical role in the transport and metabolism of endogenous ligands, including fatty acids (FA) [6], thyroxine [7], heme [8] and bilirubin [9]. Besides, HSA binds to many exogenous drug molecules, including aspirin, carboplatin and cisplatin with affinities up to nanomolar dissociation constants [3].
The key properties of long plasma half-life and extraordinary binding capability have rendered HSA numerous successful applications as a drug carrier to extend the plasma half-life of therapeutic peptides or small proteins and to modulate plasma pharmacokinetics of drug molecules. HSA can bind to various drug molecules non-covalently through its ligand binding sites or covalently through its exposed cysteine residues (e. g., Cys34) [3, 10]. A doxorubicin derivative (INNO-206) capable of covalently binding to endogenous albumin shows a higher therapeutic index and an increased antitumor activity compared to free doxorubicin on tumor-bearing animals [11]. Levemir® is a human insulin analogue conjugated with a fatty acid (FA) and used for diabetes treatment [12, 13]. Levemir® has a much longer plasma half-life of 5-7 h compared to minutes for native human insulin [12, 13], likely due to the non-covalent binding to endogenous albumin through the conjugated FA. Non-covalent mixture of albumin with paclitaxel (an anticancer drug) renders water solubility of paclitaxel, avoids the use of the solubilizing agent cremophor which often causes hypersensitivity reactions, and increases its maximum tolerated dose [14].
Despite numerous successful applications as a drug carrier, HSA by itself does not have targeting specificity to, e.g., tumors. To render HSA capability of targeting tumors, we generated a recombinant HSA fused with a tumor targeting peptide, ATF, at its N-terminus (ATF-HSA, Figure 1). ATF is the amino-terminal fragment of urokinase-type plasminogen activator (uPA) and contains all the structural elements necessary for binding to the uPA receptor (uPAR) [15]. uPAR is expressed in a low level on the cell surfaces of quiescent normal cells, but is over-expressed on the surfaces of a wide range of invasive tumor cells, and is believed to play a critical role in tumor cell migration, adhesion and tissue remodeling [15-17]. In fact, uPAR has been widely studied as a target for tumor diagnosis [18] and tumor imaging [19], using modalities like magnetic resonance imaging, near infrared imaging and positron emission topography [20]. A number of uPAR targeting agents have also been developed, including small chemical molecules, uPA-derived peptides or phage display-derived peptides, antibodies and ATF [21-31]. ATF has been shown by us [15] and others [17] to be the primary epitope recognized by uPAR. Besides, ATF binds to uPAR very tightly with a dissociation constant of ~0.2 nM. Thus, in this study, we utilized ATF to target tumors that over-express uPAR.
A bifunctional recombinant protein ATF-HSA can target to tumors through urokinase receptor (uPAR) that is over-expressed on tumor cell surfaces. HSA has multiple ligand binding sites (the fatty acid binding sites of HSA are labeled, FA1-FA7). In this study, ATF-HSA was loaded with a large hydrophobic photosensitizer, mono-substituted β-carboxy phthalocyanine zinc (ZnPcCOOH, abbreviated as CPZ), which bound at the FA1 site (inset). The ATF-HSA:CPZ displays a cytotoxicity on uPAR-expressing tumor cells upon light illumination.
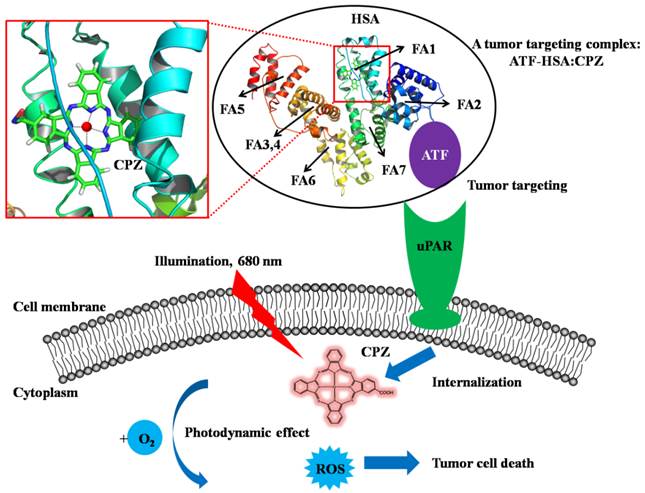
In our current study, we developed a novel strategy to load the ATF-HSA with a cytotoxic agent, monosubstituted β-carboxy phthalocyanine zinc (ZnPcCOOH, abbreviated as CPZ) [32]. They formed a 1:1 molecular complex (ATF-HSA:CPZ, Figure 1) that was completely water soluble and highly stable. We further demonstrated that CPZ bound at the FA binding site 1 (FA1) in ATF-HSA. Interestingly, the CPZ inside ATF-HSA showed a much stronger fluorescence signal (100-fold) than other water soluble phthalocyanine zinc (ZnPc) molecules. The ATF-HSA:CPZ complex was found to maintain the targeting capability to uPAR in vitro as expected. Next, we demonstrated selective cellular uptakes and enhanced phototoxicities of ATF-HSA:CPZ toward tumor cells in vitro. Furthermore, in a tumor-bearing mouse model, ATF-HSA:CPZ was found to have a potent anti-tumor effect even at a low dose of 0.080 μmol / kg, or 0.050 mg CPZ / kg of mouse body weight. Using HSA:CPZ as a control, we demonstrated unambiguously that ATF-HSA:CPZ had a tumor-specific targeting in the mouse model, which was not simply due to the enhanced permeation and retention (EPR) effect [33]. Together, these results indicate that ATF-HSA is truly an efficient tumor targeting drug carrier and can also be used for other drugs in imaging or therapeutic applications.
Materials and Methods
Materials and cell lines
Recombinant HSA was kindly provided by Zhejiang Hisun Pharmaceutical Co. Ltd. (Taizhou, Zhejiang, China). Recombinant soluble uPAR (suPAR, extracellular domain of uPAR) and ATF were prepared as previously described [34, 35]. CPZ and a CPZ derivative, pentalysine β-carbonyl phthalocyanine zinc (ZnPc-(Lys)5) were synthesized as previously described [32, 36]. All chemicals we used were purchased from Sigma-Aldrich Co. Ltd. (St. Louis, MO, USA) or Sinopharm Chemical Reagent Co. Ltd. (Shanghai, China) unless otherwise stated. The cell lines we used in the current study were human breast cancer cell line MDA-MB-231, non-small cell lung carcinoma cell line H1299 and human embryo lung fibroblasts HELF. All these cell lines were purchased from American Type Culture Collection (ATCC, Rockville, MD, USA). MDA-MB-231 and HELF cells were maintained routinely in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum (FCS) and antibiotics. H1299 cells were grown in RPMI-1640 medium supplemented with 10% FCS and antibiotics. All cells were kept at 37℃ in a humidified incubator with 5% CO2 atmosphere. The viability of cells was determined by trypan blue dye exclusion. Cells were maintained in logarithmic phase with viability > 95%.
Construction of recombinant protein expression vectors
The plasmid pPICZαA (Invitrogen, Carlsbad, CA, USA) encoding ATF (143 residues) was originally constructed as described previously [35]. The cDNA of ATF was amplified by polymerase chain reaction (PCR) from the full-length human uPA gene (kindly provided by Dr. Douglas B. Cines) using the following primers: 5'-CGGAATTCAGCAATGAACTTCATCAAGTT-3' (sense primer, the underlined nucleotides indicate the location of an EcoRI site); 5'-ACGCGTCGACTTCTTCTGGAGGAGAGGAG-3' (antisense primer, the underlined nucleotides indicate the location of a SalI site). The amplified PCR product was isolated and inserted into pPICZαA between the EcoRI and SalI sites. The construct was transformed into a competent Escherichia coli strain Top10F' (Invitrogen, Carlsbad, CA, USA), which was then screened on LLB plate (0.5% yeast extract, 1% tryptone, 0.5% NaCl, 2% agar) containing 30 μg / ml Zeocin® (Invitrogen, Carlsbad, CA, USA) to select positive colonies.
The gene encoding the mature HSA (585 residues) was generated from a human liver cDNA library by PCR using the following primers: 5'-ACGCGTCGACGGTGGTGGTGGTGATGCACACAAGAGTGAGGTTG-3' (sense primer, the underlined nucleotides indicate the location of a SalI site and the bold nucleotides indicate a four-glycine-amino acid spacer); 5'-ACGCGTCGACTAAGCCTAAGGCAGCTTGACT-3' (antisense primer, the underlined nucleotides indicate the location of a SalI site). This HSA cDNA fragment was purified and inserted into the plasmid pPICZαA containing ATF at the SalI site. This composite gene could be described from its 5' to 3' end by the succession of: (1) an EcoRI-SalI restriction fragment encoding ATF, immediately followed by a four-glycine-amino acid spacer; (2) genetically linked to a SalI-SalI restriction fragment encoding HSA. The construct was transformed into a competent Escherichia coli strain DH5α (Invitrogen, Carlsbad, CA, USA), which was screened on LLB plate containing 30 μg / ml Zeocin® to select positive colonies. The resistant strains were sequenced to confirm the presence of ATF-HSA.
The plasmid pPICZαA encoding ATF-HSA-D1 was constructed by a similar procedure and the gene encoding HSA domain I (205 residues) was generated by PCR using the following primers: 5'- ACGCGTCGACGATGCACACAAGAGTGAGGTTG-3' (sense primer, the underlined nucleotides indicate the location of a SalI site); 5'- ACGCGTCGACTTTTTGGAGACTGGCACACTT-3' (antisense primer, the underlined nucleotides indicate the location of a SalI site).
Transformation and expression of ATF-HSA in Pichia pastoris strain X-33
A competent Pichia pastoris strain X-33 (Invitrogen, Carlsbad, CA, USA) was prepared for transformation. The plasmid pPICZαA-ATF-HSA was linearized using PmeI and transformed into X-33 by electroporation (1500 V, 25 μF, 200 Ω for 6 ms). After incubation for 1 h at 30 ℃ in 1 M sorbitol without agitation, the transformed X-33 was cultured on YPD plate (1% yeast extract, 2% peptone, 2% glucose, 2% agar) containing 100 μg / ml Zeocin® at 30 ℃ for 3 days to screen for colonies that have the transformed gene integrated into the host chromosomal DNA. The colonies were cultured in 2 ml BMGY medium (1% yeast extract, 2% peptone, 100 mM potassium phosphate, pH 6.0, 1.34% YNB, 4 × 10-5% biotin, 1% v/v glycerol) to an OD600 reading of 2-6. To induce protein expression, the cells were incubated in 10 ml BMMY medium (1% yeast extract, 2% peptone, 100 mM potassium phosphate, pH 6.0, 1.34% YNB, 4 × 10-5% biotin, 1% v/v methanol). Then, the cells were induced every 24 h with 1% (v/v) methanol for identification ATF-HSA expression by SDS-PAGE. For a large scale expression, cells were first grown in 200 ml BMGY medium and were then cultured in 1 L BMMY medium. The protein expression in the cells was induced every 24 h with 1% (v/v) methanol. Recombinant ATF-HSA-D1 was expressed in the same way as ATF-HSA.
Purification and characterization of ATF-HSA
After 4 days' induction, 1 L culture medium was harvested by centrifugation at 12000 g for 10 min. The supernatant was collected with phenylmethylsulfonyl fluoride (PMSF) and ethylene diamine tetraacetic acid (EDTA) added to the final concentrations of 0.5 mM and 1 mM, respectively. After mixing, this supernatant was adjusted to pH 4.5 with a 0.5 M acetate buffer, pH 3.5 and centrifuged at 21000 g for 40 min. The supernatant was collected, filtered and diluted 4-fold with ultrapure water (Millipore, 18.2 MΩ). The supernatant was then applied to a cation exchange column, SP Sepharose Fast Flow (SPFF, GE Healthcare, Uppsala, Sweden) pre-equilibrated with 20 mM acetate buffer, pH 4.5. The column was then extensively washed with 20 mM acetate buffer, pH 4.5. The target protein was eluted by 20 mM acetate buffer, pH 4.5, with an ion gradient up to 1 M NaCl. The fraction containing ATF-HSA from SPFF was dialyzed overnight against 20 mM Tris-HCl, 50 mM NaCl, pH 8.5 at 4 ℃ and concentrated by a Millipore ultra-centrifugation tube. Then the purity and concentration of ATF-HSA were detected by SDS-PAGE, a gel filtration column superdex200 (GE Healthcare, Uppsala, Sweden) on a Bio-Rad BioLogic system (Hercules, CA, USA) and GE Nanovue® (Cambridge, UK). ATF-HSA-D1 was purified and characterized in the same way as ATF-HSA.
Preparation and purification of ATF-HSA:CPZ
Immediately prior to use, CPZ was dissolved to 10 mM in dimethylsulfoxide (DMSO) in a foil-wrapped microcentrifuge tube. ATF-HSA and CPZ were mixed in a solution containing 20 mM Tris-HCl, 50 mM NaCl, pH 8.5 and 10% DMSO, with the protein and CPZ at a molar ratio of 1 to 5 and the final concentration of CPZ at 10 μM. This solution was incubated with rotation in the dark at room temperature (RT, 25 ℃) for at least 12 h. The resulting mixing solution was then applied to a diethylaminoethyl anion exchange column (DEAE, GE Healthcare, Uppsala, Sweden) pre-equilibrated with 20 mM Tris-HCl, 50 mM NaCl, pH 8.5. The column was then extensively washed with 20 mM Tris-HCl, 50 mM NaCl, pH 8.5. ATF-HSA:CPZ was eluted with 20 mM Tris-HCl, 300 mM NaCl, pH 8.5. The fraction containing ATF-HSA:CPZ from DEAE was concentrated in a dialysis bag by sucrose powder and further dialyzed overnight against saline (pH 7.2) in the dark at 4 ℃. We also prepared and purified HSA:CPZ and ATF-HSA-D1:CPZ by a similar procedure. Unless otherwise stated, purified ATF-HSA:CPZ and HSA:CPZ were used in the following experiments.
Gel filtration chromatography and dynamic light scattering of ATF-HSA:CPZ
The molecular weights of ATF-HSA:CPZ and HSA:CPZ were analyzed by gel filtration chromatography using a superdex200 column on the Bio-Rad BioLogic system with ATF-HSA and HSA as controls, and 20 mM Tris-HCl, 300 mM NaCl, pH 7.4 as a elution buffer. The molecule sizes of these agents were further determined by dynamic light scattering (DLS) measurement (Nano ZS ZEN 3600, Malvern Instruments, Malvern, UK) equipped with a 2 ml micro-sampling cell at RT. All agents were filtered through a 0.22 μm Millipore filter membrane to remove any dust particles prior to DLS measurement.
Optical and photophysical properties of ATF-HSA:CPZ
The UV/Vis absorption spectrum of ATF-HSA:CPZ in saline was recorded from 500 to 800 nm using a quartz cuvette with 1 cm path length on a Lambda-35 UV/Vis spectrometer (PerkinElmer, Waltham, MA, USA). The fluorescence intensity of ATF-HSA:CPZ was measured on a multimodal microplate reader (Synergy 4, BioTek Instruments, Winooski, VT, USA) with λex = 610 nm and λem = 680 nm. The protein concentration of ATF-HSA:CPZ was determined with a BCA Protein Assay Kit (Solarbio Science & Technology Co. Ltd., Beijing, China). The absolute fluorescence quantum yield of ATF-HSA:CPZ was measured at RT on a FLS920 spectrometer (Edinburgh Instruments, Edinburgh, UK). ATF-HSA:CPZ's fluorescence emission and excitation spectra and decay time were recorded on a spectrometer equipped with both continuous (450 W) xenon and pulsed flash lamps (FLS920, Edinburgh Instruments, Edinburgh, UK) at RT. HSA:CPZ was characterized in the same way.
Structural modeling of HSA:CPZ
Structural model of HSA:CPZ (Figure 1) was generated with the software PyMol [37] using the crystal structure of HSA in complex with hemin (PDB ID 1O9X) [8] as a template and superimposing CPZ aromatic ring to hemin. The HSA:CPZ model was then refined by a molecular dynamic program implemented in CNS v1.2 package [38].
Tryptophan fluorescence quenching assay and probe competition assay of HSA:CPZ
In the tryptophan fluorescence quenching assay, HSA and CPZ were mixed in a solution containing 20 mM Tris-HCl, 50 mM NaCl, pH 8.5 and 10% DMSO to give HSA and CPZ at different molar ratios (1:0.1, 1:0.5, 1:1, 1:5, 1:10), with the same concentration of HSA at 1 μM. These mixing solutions were incubated with rotation in the dark at RT for at least 12 h. The tryptophan fluorescence signals of these mixing solutions were measured using a quartz cuvette with 1 cm path length on a Cary Eclipse Fluorescence spectrophotometer (Varian Inc., Walnut Creek, CA, USA) with λex = 280 nm and λem from 300 to 800 nm. In the probe competition assay, immediately prior to use, aspirin and its metabolite (salicylic acid) were dissolved at 10 mM in ultrapure water in a foil-wrapped microcentrifuge tube. 10 μM HSA:CPZ and 10 mM aspirin or salicylic acid solution were mixed in 100 mM Tris-HCl, pH 8.5 to give a HSA:CPZ and aspirin or salicylic acid at a molar ratio of 1:100 with the final pH 7.4. These solutions were incubated in the dark at RT for at least 24 h. The UV/Vis absorption spectrum of HSA:CPZ with aspirin or salicylic acid mixing solution was recorded from 500 to 800 nm using the BioTek microplate reader. 10 μM HSA:CPZ in saline, pH 7.2 was compared in parallel.
ATF-HSA:CPZ's binding to recombinant suPAR in vitro
10 μM ATF-HSA:CPZ was mixed with 10 μM suPAR at a molar ratio of 1:0.5 in 20 mM Tris-HCl, pH 7.4 without rotation in the dark at RT for 30 min. The mixing solution was then analyzed by native-PAGE. 10 μM ATF-HSA was also mixed with 10 μM suPAR at different molar ratios (0.5:1, 1:1, 2:1) and analyzed in the same way as comparison.
Cellular uptake of ATF-HSA:CPZ
Aliquots (200 μl) of exponentially growing cells (MDA-MB-231 and H1299, 1.5×105 / ml culture medium) were placed in 96-multiwell plates (Costa) and incubated overnight. Cells were then incubated with 1 µM ATF-HSA:CPZ (in saline after being filtrated by a 0.22 μm Millipore filter membrane) for various periods of time (0.5 h, 1 h, 2 h, 4 h, 12 h, 18 h, 24 h). One control column in the plate was filled with only culture medium as a blank. After incubation, the cells were washed with sterile PBS buffer, pH 7.2 before being lysed with 0.1 ml lysis buffer (0.1 M NaOH, 1% SDS) to give a homogenous solution. The fluorescence of the cell lysates was measured on the BioTek microplate reader with λex = 610 nm and λem = 680 nm. The concentration of cellular proteins was determined with the BCA Protein Assay Kit using bovine serum albumin as standard. Results were expressed as nmol CPZ / mg cellular proteins. HSA:CPZ was measured in parallel as a comparison. Four replicates at each time point were tested for each cell line and each experiment was repeated three times.
Phototoxicity and dark toxicity of ATF-HSA:CPZ in vitro
Cells (MDA-MB-231, H1299 and HELF) at a density of 5×104 / ml culture medium in a volume of 200 μl per well were placed in 96-multiwell plates and incubated overnight. The cells were incubated with ATF-HSA:CPZ or ATF-HSA at various concentrations (0.1, 0.5, 1 μM) for 24 h. One control column in the plate was filled with only culture medium as a blank. The cells were then washed with sterile culture medium before fresh medium was added. The plates were then illuminated at a light dosage of 1.5 or 3 J / cm2 using a 680 nm LED light source (100 mW, Sundynamic Inc., Qingdao, Shandong, China) for 1min or 2 min, respectively, and returned to the incubator. Dark toxicity was measured in parallel. After 12 h incubation, the cell viability was measured on the BioTek microplate reader using a MTT colorimetric assay measured at 490 nm. Four replicates in each dosage were tested for each cell line and each experiment was repeated three times. HSA:CPZ or HSA was measured in parallel for comparison.
Phototoxicity of ATF-HSA:CPZ on H1299 cells in the presence of free ATF
200 μl of H1299 cells (at a density of 5×104 / ml culture medium) were seeded in each well of 96-multiwell plates and incubated overnight. After treatment with 1 µM ATF-HSA:CPZ, and with or without free ATF at various concentrations (1 µM, 10 µM, 100 µM) for 24 h, H1299 cells were washed with sterile culture medium before fresh medium was added. One control column in the plate was filled with only culture medium as a blank. The plates were illuminated at a light dosage of 1.5 J / cm2 using the 680 nm LED light source and then returned to the incubator. After 12 h incubation, the cell viability was measured on the BioTek microplate reader using the MTT colorimetric assay measured at 490 nm. Four replicates in each ATF dosage were run and the experiment was repeated three times. HSA:CPZ was measured in parallel as a comparison
Cellular localization of ATF-HSA:CPZ in H1299 and HELF cells
Intracellular distribution of ATF-HSA:CPZ was monitored in H1299 and HELF cells on a Olympus FluoView™ FV1000 confocal laser scanning microscope (Melville, NY, USA) coupled to an inverted microscope with a 60× differential interference contrast oil immersion objective lens. 5×104 / ml H1299 and HELF cells were seeded in confocal chamber slides (NEST) for 24 h. Then the cells were incubated with 2 μM ATF-HSA:CPZ for 24 h and washed with PBS buffer, pH 7.2. The cells were subsequently incubated with a DNA fluorescent dye, DAPI (4', 6-diamidino-2-phenylindole) at RT for 5 min (the ratio and incubation time were followed under the manufacturer's instruction), washed with PBS buffer and then prepared for scanning according to the confocal laser scanning microscopy manual. Cell specimens were excited by He-Ne laser light (λ = 633 nm for CPZ) and diode laser light (λ = 405 nm for DAPI), and the emitted fluorescence was filtered with barrier filters (650/100 nm and 435/55 nm band pass, respectively). Laser line intensity, photometric gain, PMT setting and filter attenuation were kept constant throughout the experiments. HSA:CPZ was measured in parallel as a comparison. The scanning images were then analyzed to acquire the DAPI or CPZ intensity by the Image-Pro Plus software (Media Cybernetics Inc., Rockville, MD, USA).
Establishment of Hepatoma-22 (H22) tumor-bearing Kunming mouse model
Male Kunming mice (4 weeks old, 18-22 g, purchased from Shanghai SLAC Laboratory Animal Co. Ltd., Shanghai, China) were maintained and handled in accordance with the recommendations of the institutional animal care and use committee (IACUC). The animals were allowed free access to water and food throughout the course of the experiments. The mouse hepatocellular carcinoma cell line H22 was purchased from the Shanghai Institute of Cell Biology, Chinese Academy of Sciences (Shanghai, China), and passaged weekly through Kunming mice in the form of ascites. To establish the H22 tumor-bearing mouse model, ascites containing H22 cells were harvested from the peritoneal cavity of a tumor-bearing mouse 5-7 days after inoculation, diluted with sterilized saline at a ratio of 1:6 to adjust the cell concentration to 1.0×107 / ml, and 0.2 ml aliquots were subcutaneously inoculated into the right flank of Kunming mice (18-22 g). For imaging and therapy purpose, the hair of the tumor-bearing mice was removed by shaving and chemically depilating using a depilation cream (Shibi, Shanghai, China). The tumors reached a size of ~150 mm3 in typically 5-7 days after inoculation.
Fluorescent molecular tomography (FMT) imaging of ATF-HSA:CPZ in H22 tumor-bearing Kunming mice
The H22 tumor-bearing Kunming mice (established as above) were randomly divided into three groups (15 mice per group) with the equivalent average starting tumor size (~150 mm3) and body weight (~23 g). ATF-HSA:CPZ or HSA:CPZ (0.080 μmol / kg, or 0.050 mg CPZ / kg of mouse body weight) or saline was injected into the mice via tail vein. After injection, 3 mice in each group were anaesthetized with isoflurane and the tumor targeting capability of the agents within the mice was monitored at different time points (6 h, 12 h, 24 h, 48 h, 96 h) by using a fluorescent molecular tomography FMT 2500TM LX instrument (PerkinElmer, Waltham, MA, USA). A 680 nm laser diode was used to excite the CPZ molecules and the regions of interest (ROIs) were scanned with 50-60 source locations and default scan parameters (3 mm spacing between adjacent source locations). Long wavelength fluorescence emission (690-740 nm) was detected. For quantitation of the CPZ average concentrations, 1 μM ATF-HSA:CPZ in saline was used as a standard to calibrate the instrument. The recorded images were then reconstructed by the software TrueQuant v3.0 (PerkinElmer, Waltham, MA, USA) to three-dimension and to derive quantitative information in terms of CPZ average concentrations by creating ROIs around the tumor sites and non-tumor sites after the subtraction of auto-fluorescence from the tumor-bearing mice treated with saline.
Biodistribution and clearance of ATF-HSA:CPZ in H22 tumor-bearing Kunming mice
The H22 tumor-bearing Kunming mice were established and randomly divided into three groups (15 mice per group) as described above. ATF-HSA:CPZ or HSA:CPZ (0.080 μmol / kg, or 0.050 mg CPZ / kg of mouse body weight) or saline was injected through tail vein when the tumor size reached ~150 mm3. At 6 h, 12 h, 24 h, 48 h and 96 h post the injection, 3 mice in each group were sacrificed and their primary organs (liver, kidneys, heart, spleen, lung) or tissues (muscle and skin) were collected post mortem. These samples were then washed with saline, dried briefly with a paper towel, put on the sampling tray and imaged by the FMT 2500TM LX instrument using the same acquisition settings as the in vivo imaging to quantitate the CPZ average concentrations in the samples.
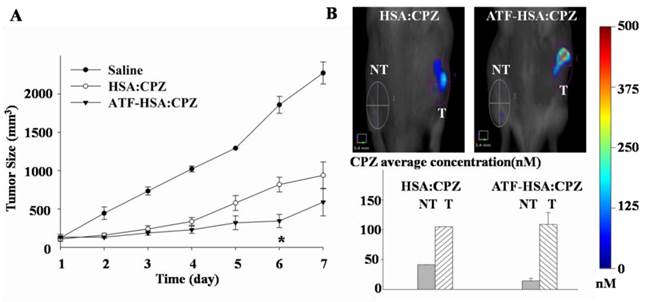
Photodynamic efficacy of ATF-HSA:CPZ in H22 tumor-bearing Kunming mice
The H22 tumor-bearing Kunming mice were established and randomly divided into three groups (8 mice per group) as described above. After injection of ATF-HSA:CPZ or HSA:CPZ at a dose of 0.080 μmol / kg, or 0.050 mg CPZ / kg of mouse body weight, or saline when the tumor size reached ~150 mm3, the tumor-bearing mice were illuminated upon tumors with a 680 nm light source (1 W, Luma Care Medical Group, Newport Beach, CA, USA) for 3 min to a light dose of 50 J / cm2. This photodynamic therapy (PDT) treatment was carried out once per day for continuous 7 days. The photodynamic efficacy of the PDT treatment was monitored by measuring the tumor sizes calculated using the ellipsoid volume formula π/6 × (length × width × height) with a caliper and body weights daily for 7 days.
Statistical analysis
All data represent group means and standard errors of the mean (SEM). The experimental data in vitro and in vivo were analyzed using the unpaired, 2-tailed Student t test. Differences at the 95% confidence level (p < 0.05) were considered to be statistically significant.
Results
Expression and purification of the uPAR-targeting recombinant fusion protein: ATF-HSA
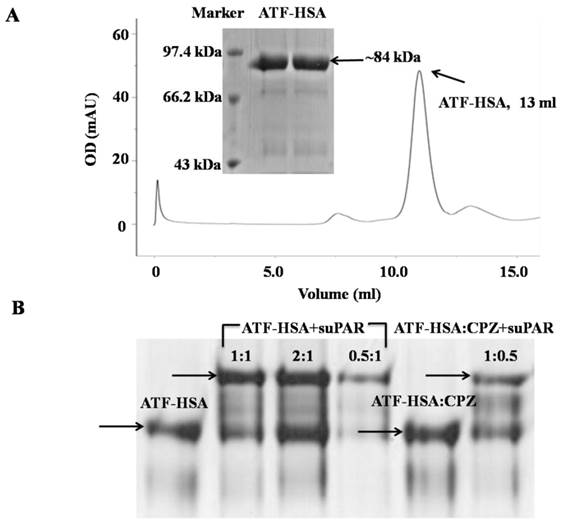
uPAR is a cell surface receptor and has been shown to play a significant role in cell migration and tumor metastasis [16, 17, 39]. Our three-dimensional crystal structural study [15], together with other studies [17], show that uPAR binds to ATF of uPA (uPA 1-143 residues). In this study, we utilized ATF as a targeting moiety for tumors that over-express uPAR. We generated a recombinant HSA in fusion with ATF. The purpose of HSA is to carry cytotoxic agents to destroy targeted tumor cells. In addition, HSA's fusion prolongs the plasma half-life of ATF, which is small (~14 kDa) and can be quickly extruded from circulation. We expressed ATF-HSA in the Pichia pastoris X-33 cells as a secreted protein. The choice of this eukaryotic expression system is to ensure the proper folding of the disulfide bonds of ATF-HSA (ATF has 4 disulfide bonds and HSA has 17 disulfide bonds) [40]. As a result, the ATF-HSA was detected as a predominant band at ~84 kDa by SDS-PAGE (Figure 2A inset) and was captured from the culture medium by a cation exchange column in one step to high purity. The ATF-HSA preparation was further analyzed on a gel filtration column and was confirmed as a single peak at 13 ml with the expected molecular weight (Figure 2A).
Recombinant protein ATF-HSA was expressed and purified to high purity as shown on SDS-PAGE (inset of Figure 2A) and a gel filtration column (Figure 2A). Figure 2B shows that either ATF-HSA or ATF-HSA:CPZ binds to suPAR (soluble and extracellular domain of uPAR) in vitro based on native-PAGE.
Loading a large hydrophobic photosensitizer (PS), CPZ, into ATF-HSA by the DIP (dilution-incubation-purification) method
HSA is an abundant serum protein that has multiple ligand binding sites inside its molecule and can accommodate a large number of endogenous metabolites and exogenous drug molecules, especially drugs with strong hydrophobicity [4]. In this work, we used PS as a class of cytotoxic agents for tumor cell destruction. PS is one of three critical components in photodynamic therapy (PDT), where a PS is injected into blood circulation and a laser light illuminates at the region of interest. Oxygen molecules in the illuminated area provide the third critical component of the PDT, which will be activated by the PS into either free radicals (type I mechanism) or singlet oxygen (type II mechanism). These free radicals or singlet oxygen are cytotoxic and can destroy the immediate cells before they are de-activated [41-45].
Loading PSs into HSA is a major challenge. PSs typically have a large aromatic ring so that they can absorb longer wavelength light to facilitate deeper penetration into tissues, and therefore are usually hydrophobic in nature and require special formulation or chemical modification to enhance their water solubility. The PS we used here (CPZ) is a potent PS and is also hydrophobic (Figure 1). Besides, CPZ tends to aggregate or even precipitate under aqueous condition due to its hydrophobic nature and is thus difficult to be loaded into HSA. Quite often, HSA and PSs form nanoparticles with a large molecule size, e.g., 88 nm with chlorin e6 [46].
We have developed a novel method to load CPZ into ATF-HSA non-covalently to form a molecular complex (ATF-HSA:CPZ) instead of nanoparticles. The method involves three steps: dilution, incubation and purification, and is named the DIP method. In order to reduce CPZ's aggregation and increase the percentage of monomeric CPZ molecules, we included 10% DMSO in solution and carried out the loading process at a very low concentration (10 μM CPZ): the dimerization of typical phthalocyanines has a dissociation constant at micromolar range [47]. This DMSO concentration was well-tolerated by ATF-HSA without causing its aggregation. We then incubated ATF-HSA with excess amounts of diluted CPZ molecules in the hope that all ATF-HSA molecules were loaded with the CPZ molecules. After incubation overnight, the mixture was purified on an ion-exchange column to enrich the ATF-HSA:CPZ preparation, separate the complexes ATF-HSA:CPZ from the free components CPZ and get rid of DMSO. The preparation from this procedure gave soluble and pure complexes ATF-HSA:CPZ in saline, showing a typical blue color of CPZ (Figure 3A left panel inset cuvette and Supplementary Material: Figure S1B). This high aqueous solubility demonstrated the successful loading of CPZ inside ATF-HSA because CPZ does not have water solubility. Furthermore, we measured a stoichiometric ratio of ~1.2:1 between CPZ and ATF-HSA in the complex based on CPZ's fluorescence and ATF-HSA's concentration, which indicated one ATF-HSA bound to only one CPZ molecule. Using this DIP method, we also prepared HSA:CPZ (without ATF) and used it as a control in our following experiments.
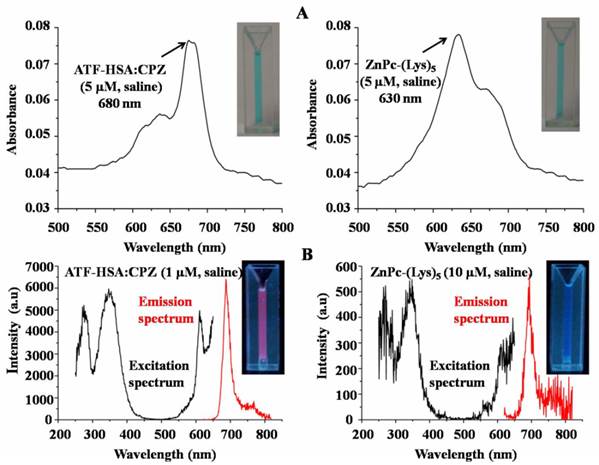
Remarkable optical and photophysical properties of ATF-HSA:CPZ. (A) The UV/Vis spectrum of ATF-HSA:CPZ in saline (left panel) shows that CPZ is mainly in monomeric state with the maximum absorption at 680 nm. The UV/Vis spectrum of ZnPc-(Lys)5 in saline (right panel) shows the maximal absorption at 630 nm, corresponding to the aggregation of ZnPc-(Lys)5 in aqueous solution. The corresponding samples in cuvettes are also shown in insets. Both samples show typical blue color of CPZ. (B) The excitation and emission fluorescence spectra of ATF-HSA:CPZ in saline (left panel) show the fluorescence signals of ATF-HSA:CPZ are ~100-fold stronger than that of ZnPc-(Lys)5 in saline (right panel). The corresponding samples in cuvettes are also shown in insets. The fluorescence signal of ATF-HSA:CPZ (red color) is stronger than that of ZnPc-(Lys)5.
To further ensure that CPZ bound inside the protein instead of the outer surface in a non-specific way or forming nanoparticles, we ran ATF-HSA:CPZ or HSA:CPZ through a gel filtration column. If CPZ bound to the protein non-specifically, it would stay on the top of the column as a blue color band and could not migrate through the column due to its insolubility in aqueous solution. The results showed no blue band on the top of the column, and both ATF-HSA:CPZ and HSA:CPZ migrated through the column as a single band with the retention time similar to ATF-HSA or HSA, suggesting that CPZ indeed bound inside ATF-HSA or HSA (Supplementary Material: Figure S1B, gel filtration chromatography has a limited resolution and cannot distinguish the molecular weights between HSA:CPZ and ATF-HSA:CPZ). We further measured the hydrodynamic molecular radii of ATF-HSA and ATF-HSA:CPZ in saline by dynamic light scattering (DLS), which gave a hydrodynamic radius of 5.6 nm and 7.5 nm, respectively. These values were compatible with the hydrodynamic radius of HSA (6.5 nm, Supplementary Material: Figure S1A). No large aggregates or nanoparticles were observed for both complexes in the DLS measurements. Thus, these experiments confirmed that CPZ was indeed buried inside HSA and they formed molecular complexes instead of large nanoparticles.
As ATF-HSA:CPZ was a non-covalent complex, next, we examined the stability of this complex to examine whether ATF-HSA would discharge buried CPZ easily. We found that this complex was remarkably stable for at least one month in saline at 4ºC without the bleaching of its blue color or protein degradation. In addition, this complex was stable during regular protein manipulations, such as protein concentration, native-PAGE and purification over chromatographic columns. We further evaluated the plasma stability of this complex and found that it was stable for at least 24 h (Supplementary Material: Figure S2). This preliminary plasma stability study of ATF-HSA:CPZ was consistent with its strong in vivo efficacy in the results described below. At extreme pH, HSA has been shown to adopt different conformations, which are less compact. Indeed, we found the discharge of the loaded CPZ at low pH (pH 4.5, Supplementary Material: Figure S3).
ATF-HSA:CPZ shows remarkable optical and photophysical properties
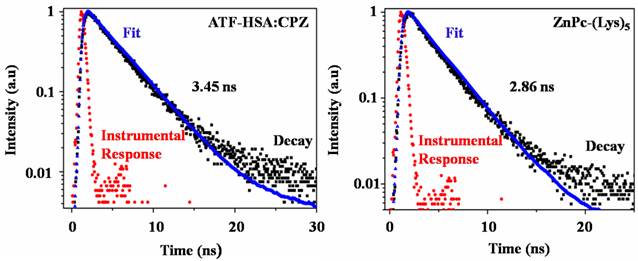
CPZ shows the maximum UV/Vis absorption at 680 nm in monomeric form, whereas its aggregated form has the maximum absorption at 630 nm. We observed the maximum absorption at 680 nm for ATF-HSA:CPZ in saline, which demonstrated the CPZ was monomeric (Figure 3A left panel). We further measured the fluorescence spectra of ATF-HSA:CPZ. Surprisingly, the fluorescence intensity of CPZ in ATF-HSA:CPZ in saline was ~100-fold higher than the control PS, ZnPc-(Lys)5 [36], which is a water soluble CPZ derivative (Figure 3B). The absolute fluorescence quantum yield of ATF-HSA:CPZ was 0.8%. The fluorescence decay time of CPZ in ATF-HSA:CPZ was 3.45 ns, while that of ZnPc-(Lys)5 was 2.86 ns, ~20% shorter than the former's (Figure 4). Such enhanced fluorescent properties of CPZ in ATF-HSA:CPZ are likely due to its monomeric state and its shielding away from the solvent by ATF-HSA, which reduce the quenching of CPZ's fluorescence signal. These results indicate that ATF-HSA:CPZ may be a promising imaging agent. HSA:CPZ showed similar optical and photophysical properties compared to ATF-HSA:CPZ (Supplementary Material: Figure S4).
The fluorescence decay time of ATF-HSA:CPZ (left panel) in saline is 3.45 ns and is ~20% longer than that of ZnPc-(Lys)5 (2.86 ns, right panel).
HSA FA1 accommodates CPZ and the integral structure of HSA is necessary to maintain the stable complex
HSA has seven major FA binding sites named from FA1 to FA7, which can bind to exogenous drug molecules (Figure 1). X-ray crystal structural studies from us [48-50] and others [8, 9] have established that only two binding sites can accommodate a large drug molecule like CPZ: the FA7 (located at HSA sub-domain IIA) and the FA1 (located at HSA sub-domain IB) [4]. The FA7 is quite large and can be further classified into three sub-sites [49]. The FA1 is the largest binding site of HSA and can bind to large molecules like heme and bilirubin [8, 9]. Our structural modeling study shows that the FA1 is large enough to accommodate CPZ (Figure 1, inset). We further designed two assays to identify the exact binding site of CPZ in HSA.
HSA has only one tryptophan residue located at its domain II and this tryptophan residue has a fluorescence signal at 350 nm when excited at 280 nm. The drug's binding to the FA7 at sub-domain IIA will perturb or even quench the tryptophan fluorescence signal [51]. In our experiment, we mixed HSA and CPZ at different molar ratios and examined their tryptophan fluorescence signals. We found that the tryptophan fluorescence of HSA was not quenched at all, even at a high molar excess of 10-fold CPZ to HSA (Supplementary Material: Figure S5). The experimental results suggested that CPZ did not bind at the FA7 site. To further cross-validate this result, we used a probe competition assay. We have previously shown that aspirin binds only at the FA7 site, but not at any other FA binding sites, whereas its metabolite, salicylic acid, binds at both the FA7 and FA1 sites [48]. If CPZ binds at the FA7 site, the addition of a large quantity of aspirin will displace CPZ from HSA. The displaced CPZ is not soluble in saline and will precipitate out, leading to the decreased UV-Vis absorption of CPZ at 680 nm. In our experiment (Supplementary Material: Figure S6), we observed that the high molar excess of aspirin (up to 100:1) did not perturb the absorption profile of HSA:CPZ. On the other hand, the addition of salicylic acid to HSA:CPZ did reduce the absorption at 680 nm. These results once again supported that CPZ bound at the FA1 instead of the FA7 site. For ATF-HSA, we also found that the binding site of CPZ was the FA1 (data were not shown).
To further verify the binding of CPZ at the FA1 site located at sub-domain IB, we generated another recombinant protein ATF-HSA-D1 that contained only HSA domain I with the FA1, but not domain II or III. We then loaded CPZ into ATF-HSA-D1 using the DIP method. Indeed, we were able to prepare the complex ATF-HSA-D1:CPZ which was completely water soluble and showed a blue color in aqueous solution like ATF-HSA:CPZ, clearly demonstrating that the CPZ was indeed incorporated into ATF-HSA-D1. However, the blue color of ATF-HSA-D1:CPZ was bleached when ATF-HSA-D1:CPZ was run through either gel filtration chromatography or ion exchange chromatography with a high salt concentration (300 mM NaCl), and at the same time the top of column became bluish (data were not shown). This showed that ATF-HSA-D1:CPZ had a limited stability, and suggested that HSA domain I was not enough to form a stable complex with CPZ and the integral structure of HSA was needed to stabilize the CPZ inside HSA. Thus, we used ATF-HSA:CPZ instead of ATF-HSA-D1:CPZ for the following experiments.
ATF-HSA:CPZ is able to bind to recombinant uPAR in vitro
To evaluate whether ATF-HSA:CPZ will bind to its targeted receptor in vitro, we carried out a gel shift experiment using native-PAGE in the absence of SDS and other reducing agents (Figure 2B). The presence of recombinant soluble uPAR (suPAR) indeed shifted the migration of either ATF-HSA:CPZ or ATF-HSA, indicating that ATF-HSA was active in its uPAR binding in vitro. In addition, the CPZ's loading did not affect the receptor binding property of the fusion protein, which was again consistent with the fact that the CPZ was buried inside the fusion protein.
Selective cellular uptake and enhanced phototoxicity of ATF-HSA:CPZ mediated by cell surface uPAR
We measured the uPAR-specific cellular uptake of ATF-HSA:CPZ using two tumor cell lines (MDA-MB-231 and H1299) that have high levels of surface uPAR in vitro [52]. We used the HSA without ATF but loaded with CPZ (HSA:CPZ) as a control. We incubated 1 µM ATF-HSA:CPZ or HSA:CPZ with the cells for various amounts of time (0.5 to 24 h, Figure 5A), washed out the unbound complexes and measured the uptake of ATF-HSA:CPZ or HSA:CPZ by the fluorescence of CPZ at 680 nm. Each experiment was repeated three times with four replicates at each time point for each cell line. Figure 5A showed ATF-HSA:CPZ was taken up by both tumor cell lines in a time dependent manner. In comparison, the cellular uptakes of HSA:CPZ by these two cell lines didn't change with time and were significantly lower than the uptakes of ATF-HSA:CPZ after 12 h. After 24 h incubation, the differences of cellular uptakes between ATF-HSA:CPZ and HSA:CPZ were ~2-fold (0.36±0.01 vs. 0.20±0.01 nmol CPZ / mg cellular proteins for H1299 cells, 0.17±0.01 vs. 0.10±0.01 nmol CPZ / mg cellular proteins for MDA-MB-231 cells, p < 0.05), clearly demonstrating the enhanced cellular uptakes of ATF-HSA:CPZ.
(A) ATF-HSA:CPZ shows higher cellular uptakes in H1299 cells (left panel) and MDA-MB-231 cells (right panel) compared to HSA:CPZ (~2 fold). The cells were incubated with 1 µM ATF-HSA:CPZ or HSA:CPZ for 0.5 to 24 h and then were washed with PBS buffer, pH 7.2. The bound ATF-HSA:CPZ or HSA:CPZ was measured by the fluorescence of CPZ at 680 nm and the results were expressed as nmol CPZ / mg cellular proteins. (B) ATF-HSA:CPZ shows enhanced phototoxicities on H1299 cells (left panel) and MDA-MB-231 cells (right panel) compared to HSA:CPZ (~2 fold at 1 µM). Both ATF-HSA and HSA have little phototoxicities on these two tumor cell lines. We incubated these agents with the cells for 24 h, then washed out the unbound agents, illuminated the cells by a 680 nm LED light source for 1 min to a dose of 1.5 J / cm2 and finally used the MTT colorimetric assay to measure the cell viability. Four replicates at each time point or in each dosage were tested for each cell line and each experiment was repeated three times. Values represent the mean of three separate experiments; bars represent standard error of the mean (SEM). The unpaired, 2-tailed Student t test was used to analyze data; *, p < 0.05.
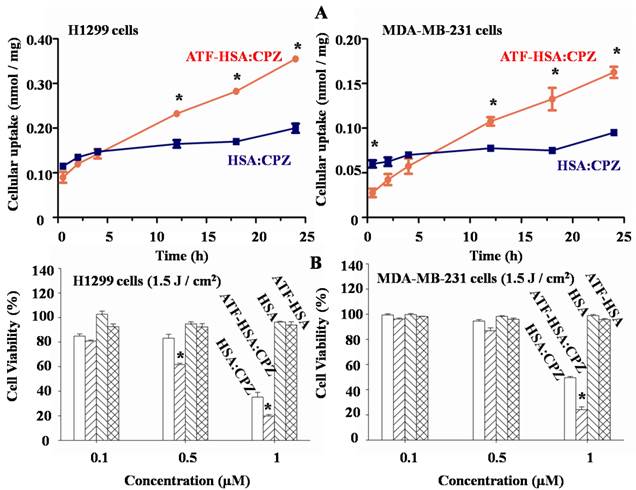
We further evaluated the phototoxicities of ATF-HSA:CPZ on the uPAR over-expressing cells (H1299 and MDA-MB-231) and normal HELF cells using ATF-HSA, HSA, and HSA:CPZ as controls. We incubated these agents (0.1-1 µM) with the cells for 24 h to allow their sufficient cellular uptakes. We then washed out the unbound agents, illuminated the cells with a 680 nm LED light source for 1 min (Figure 5B) or 2 min (Supplementary Material: Figure S7) to a dose of 1.5 or 3 J / cm2, respectively, and then used the MTT colorimetric assay to measure the cell viability. Dark toxicity was also measured in parallel. Four replicates in each dosage were measured for each cell line and each experiment was repeated three times. In the absence of CPZ, ATF-HSA or HSA showed neither phototoxicities nor dark toxicities (Figures 5B and S7). At dark, ATF-HSA:CPZ or HSA:CPZ showed no toxicities on these two tumors cell lines either (Supplementary Material: Figure S7). In the presence of light illumination to a dose of 1.5 J / cm2, ATF-HSA:CPZ showed an enhanced phototoxicity on H1299 cells at 0.5 μM compared to HSA:CPZ (61.6±1.2 vs. 83.2±3.1% cell viability, p < 0.05, Figure 5B), while there was no phototoxicity difference between ATF-HSA:CPZ and HSA:CPZ on MDA-MB-231 cells at the same concentration (86.8±2.3 vs. 94.6±1.2% cell viability). At a higher dose of 1 μM, ATF-HSA:CPZ showed ~2-fold enhanced phototoxicities on both tumor cell lines compared to HSA:CPZ (19.9±1.1 vs. 35.3±3.8% cell viability for H1299 cells, 24.2±2.2 vs. 50.0±0.9% cell viability for MDA-MB-231 cells, p < 0.05). A higher light dose (3 J / cm2) also led to higher phototoxicities for ATF-HSA:CPZ on both tumor cell lines compared to HSA:CPZ (Supplementary Material: Figure S7). These results were consistent with that ATF-HSA:CPZ had ~2-fold higher cellular uptakes in two tumor cell lines compared to HSA:CPZ. In comparison, in normal cells (HELF) that express a very low level of uPAR [52], both ATF-HSA:CPZ and HSA:CPZ showed no dark toxicities and little phototoxicities. Even at the concentration of 1 μM after illumination at the light dose of 3 J / cm2, the cell viability was more than 75% (Supplementary Material: Figure S8). Interestingly, at a lower concentration of 0.1 μM, no significant phototoxicities were observed for both complexes, even though a concentration of 0.1 μM of ATF-HSA:CPZ was enough to saturate the cell surface receptors (7100 uPAR per cell on H1299 cells [52]). This is likely due to the relatively low light doses (1.5 or 3 J / cm2) used in the study. We have previously shown that cells can recover from limited redox damage [53].
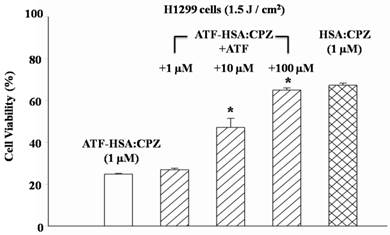
To further verify whether the enhanced phototoxicities of ATF-HSA:CPZ were uPAR-specific, we measured its phototoxicity on H1299 cells in the presence or absence of free ATF (Figure 6). At 100-fold excess of free ATF, ATF-HSA:CPZ almost lost its enhanced phototoxicity on H1299 cells (64.9±1.1 vs. 24.8±0.3% cell viability, p < 0.05). This is probably due to the blocking of the surface uPAR on tumor cells by a large quantity of free ATF, making the cell surface uPAR not available for ATF-HSA:CPZ. Consequently, we concluded that ATF-HSA:CPZ's enhanced phototoxicities were uPAR-mediated.
Enhanced phototoxicity of ATF-HSA:CPZ on H1299 cells is mediated by uPAR on tumor cell surfaces. The presence of free ATF greatly reduces the phototoxicity of ATF-HSA:CPZ on uPAR-expressing tumor cells H1299 (10 and 100 μM). Four replicates in each ATF dosage were run and the experiment was repeated three times. Values represent the mean of three separate experiments; bars represent standard error of the mean (SEM). The unpaired, 2-tailed Student t test was used to analyze data; *, p < 0.05.
It should be pointed out that there existed non-specific uptakes of HSA:CPZ by two tumor cell lines (Figure 5A). This is not too surprising. It is often observed that PSs like CPZ can be taken up by cells probably due to the hydrophobic nature of most PSs [53, 54]. In the case of HSA:CPZ, it is likely that HSA undergoes a conformational change on tumor cell surfaces and sheds out the CPZ, which is then taken up by tumor cells through non-specific pinocytosis process. Therefore, HSA:CPZ also had some phototoxicities on tumor cells when illuminated as shown in Figure 5B and Supplementary Material: Figure S7.
ATF-HSA:CPZ is endocytosed and located in cytoplasm but not in cell nucleus
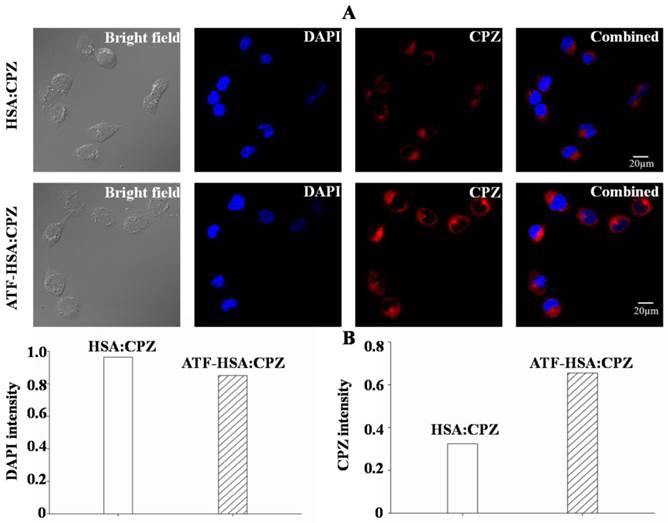
The cellular localization of ATF-HSA:CPZ after taken up by H1299 or HELF cells was studied using confocal laser microscopy. The strong fluorescence signal of CPZ was used to follow its intracellular localization. Figure 7A clearly showed that CPZ (red color) was not co-localized with cell nucleus (stained with blue color using DAPI) and existed only in the cytoplasm after 2 μM ATF-HSA:CPZ was incubated with H1299 cells for 24 h. Under the identical experimental procedure, HSA:CPZ without ATF showed a much weaker (~50%) fluorescence signal (Figure 7), supporting once again the uPAR-specific cellular uptake of ATF-HSA:CPZ in H1299 cells. In another control experiment, CPZ in both ATF-HSA:CPZ and HSA:CPZ displayed lower fluorescence signals in normal HELF cell line (Supplementary Material: Figure S9) probably due to non-specific pinocytosis process.
ATF-HSA:CPZ is localized in the cytoplasm after cellular uptake and shows a much stronger CPZ fluorescence signal than HSA:CPZ in H1299 cells (~2 fold). H1299 cells were seeded in confocal chamber slides for 24 h, then were incubated with 2 μM ATF-HSA:CPZ or HSA:CPZ for 24 h and washed with PBS buffer, pH 7.2. The cells were finally incubated with DAPI and prepared for scanning according to the confocal laser scanning microscopy manual. The scanning images were then analyzed to acquire the DAPI or CPZ intensity by the Image-Pro Plus software (Media Cybernetics Inc., Rockville, MD, USA).
ATF-HSA:CPZ displays a high tumor retention not simply related to the enhanced permeation and retention (EPR) effect in vivo
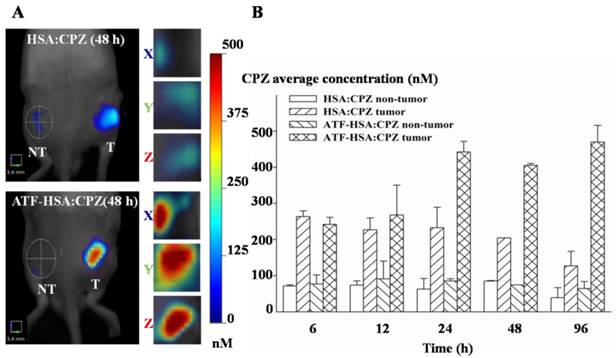
We used the well-established H22 mouse model to study ATF-HSA:CPZ in vivo. These H22 cells contain a large quantity of mouse uPAR on cell surfaces and can bind to human ATF (Supplementary Material: Figure S10). Another advantage of this mouse model is that it does not require the use of the immune compromised mice. Immune reactions are consistently observed as an important component in photodynamic effect in vivo [55, 56]. After the tumor size reached ~150 mm3, we injected ATF-HSA:CPZ, HSA:CPZ (0.080 μmol / kg, or 0.050 mg CPZ / kg of mouse body weight) or saline into the mice via tail vein. The mice were then imaged in a noninvasive manner on the FMT instrument using CPZ's fluorescence signal, which allows three-dimensional imaging (Figure 8A) and probe quantitation (Figure 8B). As seen from the data at 6 h and 12 h post the injection in Figure 8B, both HSA:CPZ and ATF-HSA:CPZ accumulated at the tumor sites. At these two time points, ATF-HSA:CPZ showed no different tumor targeting specificity from HSA:CPZ, yet with the signals of ~3 fold compared to the non-tumor sites (Figure 8B). This tumor retention is most likely due to the enhanced permeation and retention (EPR) effect [33]. HSA was previously demonstrated to be accumulated preferentially in malignant and inflamed tissues most likely due to a leaky capillary combined with an absent or defective lymphatic drainage system (named the EPR effect) [57-59]. At 24 h after the injection, ATF-HSA:CPZ showed a ~8-fold enrichment in tumors compared to the non-tumor sites. Importantly, ATF-HSA:CPZ showed a higher tumor retention than HSA:CPZ (~2-3 fold). At 96 h post the injection, this tumor retention difference was further enlarged. Thus, these results using HSA:CPZ as a control demonstrate unequivocally that ATF-HSA:CPZ has a high tumor targeting specificity in vivo not simply related to the EPR effect. Therefore, ATF-HSA:CPZ may be a promising imaging agent with tumor targeting specificity in vivo.
Tumor targeting capability of HSA:CPZ and ATF-HSA:CPZ shown by non-invasive three-dimension FMT 2500TM LX (PerkinElmer, Waltham, MA, USA) measurements of CPZ fluorescence in H22 tumor-bearing Kunming mice. (A) Specific accumulation of both HSA:CPZ (top panels) and ATF-HSA:CPZ (bottom panels) in tumors as shown in the front view of the representative mice and three-dimensional view of tumors after 48 h injection of the complexes to H22 tumor-bearing Kunming mice. ATF-HSA:CPZ showed a higher tumor retention than HSA:CPZ. The non-tumor sites (NT) are on the left and the tumor sites (T) are on the right. (B) Quantitations of CPZ average concentrations at the tumor and non-tumor sites at different time points after injection of the complexes to the mice. In our study, both HSA:CPZ and ATF-HSA:CPZ accumulated rapidly to the tumors sites, as seen from the data at 6 h and 12 h, with signals of 2-3 fold compared to the non-tumor sites. After 24 h injection, ATF-HSA:CPZ showed a higher tumor retention than HSA:CPZ (2-3 fold). In the mean time, ATF-HSA:CPZ showed a ~8-fold enrichment in tumors compared to the non-tumor sites. The H22 tumor-bearing mouse model was established and ATF-HSA:CPZ or HSA:CPZ or saline was injected through tail vein into the tumor-bearing mice. The data were averaged from 3 mice in each group at each time point; bars represent standard error of the mean (SEM).
Biodistribution and clearance of ATF-HSA:CPZ in vivo
The accumulation of PSs in skin is the main cause of skin sensitivity to sun burn, or even ambient light, for patients after the PDT. Phthalocyanine-type PSs typically have a long liver retention time due to their aggregation [60], which can lead to liver toxicity at high doses (e.g., > 4 mg / kg for Photocyanine ZnPcS2P2 [61, 62]). We thus carried out preliminary studies on the biodistribution and clearance of ATF-HSA:CPZ.
H22 tumor-bearing mice were established as stated above and injected with ATF-HSA:CPZ or HSA:CPZ (0.080 μmol / kg, or 0.050 mg CPZ / kg of mouse body weight) or saline. At different time points (6 h, 12 h, 24 h, 48 h and 96 h post the injection), the primary organs/tissues from the mice were collected post mortem and the CPZ average concentrations in these organs/tissues were quantitated by FMT. As shown in Figure 9, both ATF-HSA:CPZ and HSA:CPZ accumulated mainly in livers and kidneys but much less in other organs/tissues. The CPZ average concentrations of ATF-HSA:CPZ or HSA:CPZ in the livers and kidneys reached ~2 µM and ~0.5 µM, respectively at 6 h (Figure 9B). After 6 h, the amounts of CPZ in livers and kidneys reduced gradually with time. At 96 h post injection, the CPZ average concentrations in primary organs/tissues were reduced to low levels (~150 nM in livers, ~80 nM in kidneys and lower than 10 nM in other organs/tissues). It was remarkable that the clearance rate of ATF-HSA:CPZ or HSA:CPZ was quite fast compared to other ZnPc-type PSs [36, 63], which also showed high concentrations in livers and other organs/tissues. The fast clearance and low concentrations in organs/tissues, especially in the skin for ATF-HSA:CPZ or HSA:CPZ, are quite important and can potentially decrease the skin sensitivity to light after the PDT treatment. It is unclear why the biodistribution and clearance of ATF-HSA:CPZ or HSA:CPZ are so different from other ZnPcs, but is likely related to the incorporation of monomeric CPZ inside ATF-HSA or HSA, and further studies should be carried out in the future.
Biodistribution and clearance of ATF-HSA:CPZ and HSA:CPZ based on CPZ fluorescence in H22 tumor-bearing Kunming mice. The primary organs and tissues were dissected out and quantified by the FMT 2500TM LX instrument. The representative images are shown in Figure 9A. The quantitation of CPZ average concentrations in different organs/tissues at 6 h, 12 h, 24 h, 48 h and 96 h after injection of the complexes are shown in Figure 9B. Both ATF-HSA:CPZ and HSA:CPZ accumulated mainly in livers and kidneys but much less in other organs/tissues, and then gradually disappeared. After 96 h injection, almost all CPZ molecules were eliminated and in low concentrations. The data were averaged from 3 mice in each group at each time point; bars represent standard error of the mean (SEM).
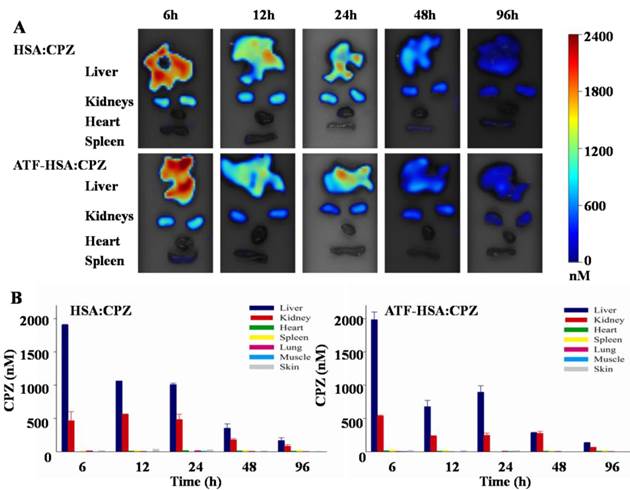
Between ATF-HSA:CPZ and HSA:CPZ, there was no significant difference on the biodistribution and clearance in most organs/tissues of the tumor-bearing mice (Figure 9), except on tumors (Figure 8). ATF-HSA:CPZ had higher CPZ average concentrations in tumors, and lower concentrations in livers and kidneys compared to HSA:CPZ at 12 h after the injection. We also observed that ATF-HSA:CPZ had a higher average concentration in livers than in tumors (~10 fold) at 6 h, but dropped to a lower concentration in livers (~0.5 fold compared to tumors) at 96 h. This is likely due to the tumor targeting effect of ATF-HSA:CPZ. ATF-HSA:CPZ accumulated initially in livers right after being injected into the bloodstream. With ATF's tumor targeting taking effect, ATF-HSA:CPZ redistributes from livers to the tumor sites and such redistribution seems faster compared to HSA:CPZ.
Potent photodynamic efficacy of ATF-HSA:CPZ in vivo
H22 tumor-bearing mice were established as stated above, and injected through tail vein with ATF-HSA:CPZ or HSA:CPZ (0.080 μmol / kg, or 0.050 mg CPZ / kg of mouse body weight) or saline. We then illuminated the tumor sites with a light source (680 nm) once a day for 3 min to a light dose of 50 J / cm2, and monitored tumor sizes (Figure 10A) and body weights (Supplementary Material: Figure S11) daily for 7 days. We observed that the ATF-HSA:CPZ-treated group displayed a significant reduced tumor growth rate compared to other groups after 6 days' PDT (Figure 10A). At the 6th day, the tumor size of the ATF-HSA:CPZ-treated group was reduced by ~4 fold and ~2 fold compared to the saline-treated group and the HSA:CPZ-treated group, respectively (p < 0.05). Interestingly, we also observed a ~2-fold tumor size reduction in the HSA:CPZ-treated group compared to the saline-treated group (Figure 10A). Again, this is probably due to the EPR effect, leading to HSA:CPZ's accumulation in tumors. It should be pointed out that, at the end of the PDT, there was actually no significant difference between the ATF-HSA:CPZ-treated group and the HSA:CPZ-treated group (Figure 10A). This result is probably due to that both ATF-HSA:CPZ and HSA:CPZ were in low concentrations at the tumor sites at the end of PDT treatment (7th day, Figure 10B). Therefore, the tumors grew quickly in H22 tumor-bearing mice when there was no therapy as shown in the saline-treated group. It is expected that a higher ATF-HSA:CPZ dose by additional injection can achieve more drastic photodynamic effects in a longer period. A higher light dose can also be used to achieve this goal because we used a relatively low light dose in this study.
The photodynamic efficacy of ATF-HSA:CPZ in H22 tumor-bearing Kunming mice (A) and the effect of the PDT on ATF-HSA:CPZ or HSA:CPZ average concentration at the tumor sites (B). In Figure 10A, the ATF-HSA:CPZ-treated group displayed a significant reduced tumor growth rate compared to the saline-treated group and the HSA:CPZ-treated group after 6 days' PDT (8 mice in each group with a daily light dose of 50 J / cm2). In Figure 10B, at the 7th day post injection, both complexes had low concentrations at the tumor sites. NT: non-tumor sites; T: tumor sites. The data were averaged from 8 mice in each group daily; bars represent standard error of the mean (SEM). The unpaired, 2-tailed Student t test was used to analyze data; *, p < 0.05.
Discussion
Species specificity of uPAR
uPAR is over-expressed on the surfaces of a wide range of invasive tumor cells and is believed to associate with tumor progression and metastasis, and plays a critical role in tumor cell migration, adhesion and tissue remodeling [15-17]. Therefore, uPAR has been widely studied as a target for tumor diagnosis [18] and tumor imaging [19]. Species specificity is well recognized between uPAR and uPA, where human uPAR binds much more strongly to human uPA than to mouse uPA [64]. However, the ATF-HSA:CPZ complex containing human ATF did react well with the mouse H22 tumors in vivo. This is not too surprising because human ATF still binds to mouse uPAR at a micromolar level dissociation constant [64]. This explains the efficacy of ATF-HSA:CPZ observed in our mouse model.
Advantages of ATF-HSA as a drug carrier to load CPZ
HSA can bind to a wide range of drug molecules. In this report, we generated a recombinant protein of HSA fused at its N-terminus with ATF, which binds to tumor cell surface uPAR.
As a proof-of-concept study of our drug carrier, we loaded CPZ as a warhead into ATF-HSA. CPZ will generate cytotoxicities under the illumination of light with a specific wavelength (680 nm), leading to tumor destruction. As an asymmetrical ZnPc, CPZ shows better photodynamic effects compared to, e.g., symmetrical ZnPc (ZnPc(SO3)4 or ZnPc). However, CPZ has a few limitations in the PDT application: CPZ does not dissolve in aqueous solution and has no tumor targeting specificity. Loading CPZ into ATF-HSA seems a reasonable way to overcome its limitations.
Through this study, we identified a number of distinguished advantages of the ATF-HSA:CPZ complex: (1) High cytotoxicity potency of ATF-HSA:CPZ. In our in vivo study, we used a low dose of 0.080 μmol / kg, or 0.050 mg CPZ / kg of mouse body weight and observed a prominent photodynamic effect (Figure 10A). Typical doses used for phthalocyanine-type PSs in in vivo studies are at the mg / kg range, e.g., 1 mg / kg for Photosense [63], 0.5 mg / kg for Photocyanine [61, 62], or 1 mg / kg for ZnPc-(Lys)5 [36]; (2) Unambiguous uPAR-specificity not simply related to the EPR effect. Albumin was shown to have retention at the tumor sites [59, 65] due to the EPR effect. In this study, we used HSA:CPZ as a control for this EPR effect and observed that ATF-HSA:CPZ had a higher retention in tumors than HSA:CPZ (Figures 8); (3) Low retention in primary organs/tissues and fast clearance rate compared to other types of PSs (Figure 9). (4) Remarkable optical and photophysical properties of ATF-HSA:CPZ compared to ZnPc-(Lys)5 (Figures 3 and 4). All these properties seem related to the fact that the CPZ is monomeric and embedded inside ATF-HSA.
A novel method to embed drug molecules inside HSA
HSA is capable of binding to FAs and many other endogenous molecules or drug molecules, and can be regarded as a chemical sponge. There are many reports to use HSA as a drug carrier [66, 67], but the approach of direct embedding drugs inside HSA is under explored. One potential reason is the difficulty to embed drug molecules inside HSA. These drug molecules usually have aromatic rings and negatively charged groups. Therefore they have a tendency to form aggregates even at a concentration around micromolar range [68, 69] and the aggregation is typically not visible to naked eyes at this concentration.
In this report, we develop a simple and straightforward DIP method to embed hydrophobic drug molecules inside HSA. We dilute the drug molecules to below the concentrations corresponding to their dissociation constants for the aggregation and thus greatly increase the percentage of monomeric drug molecules. We then incubate HSA with a large amount of diluted drug molecules to ensure all HSA molecules are loaded with the drug molecules. Next, we use ion exchange chromatography to enrich the preparations and to separate the HSA molecular complexes from the unbound drug molecules. A simple dialysis may not work because the aggregated drug molecules may be too large in size or too hydrophobic to be dialyzed out. This DIP method works quite well in this study for CPZ, generating truly molecular complexes ATF-HSA:CPZ and HSA:CPZ. The presence of ATF peptide did not affect the drug's embedding at all.
Covalent conjugation between HSA and a drug is also a common way to generate new drug candidates [66, 67]. Other PSs were previously conjugated to HSA by covalent chemical bonds to enhance their water solubility [46, 70, 71]. The unique advantage of our DIP method of embedding CPZ inside ATF-HSA is to avoid the aggregation tendency of CPZ, and thus maintain the high fluorescence intensity and photodynamic efficiency of CPZ due to its monomeric conformation.
Of course, such embedding approach will not work with very large drug molecules due to the limited sizes of HSA drug binding sites. We still anticipate that this DIP method will be feasible for many other, certainly not all, drug molecules.
Conclusion
In summary, we generated ATF-HSA as a drug carrier with targeting specificity to uPAR-expressing tumors and demonstrated that the carrier can be loaded with CPZ. ATF-HSA:CPZ showed a selective phototoxicity on uPAR-expressing tumor cells and little damage to normal cells. The laser confocal scanning images indicated CPZ was internalized into the cells and localized in cytoplasm. Moreover, ATF-HSA:CPZ clearly showed a tumor targeting specificity in mice by FMT imaging and a potent photodynamic efficacy on tumors not simply related to the EPR effect compared to HSA:CPZ. Such an uPAR-targeted drug carrier may be a useful fluorescent probe and a therapeutic agent for tumors. The ATF-HSA carrier and the DIP method developed in this work can also be used to carry other hydrophobic drugs to render their water-solubility and tumor targeting specificity.
Supplementary Material
Supplemental experiments. Fig.S1 - S11.
Abbreviations
ATF: amino-terminal fragment of urokinase; CPZ: β-carboxy phthalocyanine zinc; EPR: enhanced permeation and retention; FA: fatty acid; FMT: fluorescent molecular tomography; HSA: human serum albumin; PDT: photodynamic therapy; PS: photosensitizer; suPAR: soluble uPAR; uPA: urokinase-type plasminogen activator; uPAR: uPA receptor; ZnPc: phthalocyanine zinc.
Acknowledgements
This work was supported by grants from Natural Science Foundation of China (31161130356, 21171167), Natural Science Foundation of Fujian Province (2013J01066), Special Project of National Major Scientific Equipment Development of China (2012YQ120060), Pilot Strategic Science and Technology Project of the CAS (XDA09030307) and Scientific Equipment Development Project of the CAS (YZ201210).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Peters T Jr. Serum albumin. Adv Protein Chem. 1985;37:161-245
2. Peters T Jr. All about albumin: biochemistry, genetics, and medical applications. San Diego, USA: Academic Press, Inc. 1996
3. Fanali G, di Masi A, Trezza V. et al. Human serum albumin: from bench to bedside. Mol Aspects Med. 2012;33(3):209-90
4. Curry S. Lessons from the crystallographic analysis of small molecule binding to human serum albumin. Drug Metab Pharmacokinet. 2009;24(4):342-57
5. Fasano M, Curry S, Terreno E. et al. The extraordinary ligand binding properties of human serum albumin. IUBMB Life. 2005;57(12):787-96
6. Ashbrook JD, Spector AA, Santos EC. et al. Long chain fatty acid binding to human plasma albumin. J Biol Chem. 1975;250(6):2333-8
7. Petitpas I, Petersen CE, Ha CE. et al. Structural basis of albumin-thyroxine interactions and familial dysalbuminemic hyperthyroxinemia. Proc Natl Acad Sci U S A. 2003;100(11):6440-5
8. Zunszain PA, Ghuman J, Komatsu T. et al. Crystal structural analysis of human serum albumin complexed with hemin and fatty acid. BMC Struct Biol. 2003;3:6
9. Zunszain PA, Ghuman J, McDonagh AF. et al. Crystallographic analysis of human serum albumin complexed with 4Z,15E-bilirubin-Ⅸ alpha. J Mol Biol. 2008;381(2):394-406
10. Yamasaki K, Chuang VT, Maruyama T. et al. Albumin-drug interaction and its clinical implication. Biochim Biophys Acta. 2013;1830(12):5435-43
11. Graeser R, Esser N, Unger H. et al. INNO-206, the (6-maleimidocaproyl hydrazone derivative of doxorubicin), shows superior antitumor efficacy compared to doxorubicin in different tumor xenograft models and in an orthotopic pancreas carcinoma model. Invest New Drugs. 2010;28(1):14-9
12. Morales J. Defining the role of insulin detemir in Basal insulin therapy. Drugs. 2007;67(17):2557-84
13. Garg SK. The role of basal insulin and glucagon-like peptide-1 agonists in the therapeutic management of type 2 diabetes-a comprehensive review. Diabetes Technol Ther. 2010;12(1):11-24
14. Gradishar WJ, Tjulandin S, Davidson N. et al. Phase Ⅲ trial of nanoparticle albumin-bound paclitaxel compared with polyethylated castor oil-based paclitaxel in women with breast cancer. J Clin Oncol. 2005;23(31):7794-803
15. Huai Q, Mazar AP, Kuo A. et al. Structure of human urokinase plasminogen activator in complex with its receptor. Science. 2006;311(5761):656-9
16. Andreasen PA, Egelund R, Petersen HH. The plasminogen activation system in tumor growth, invasion, and metastasis. Cell Mol Life Sci. 2000;57(1):25-40
17. Ploug M. Structure-function relationships in the interaction between the urokinase-type plasminogen activator and its receptor. Curr Pharm Des. 2003;9(19):1499-528
18. Lund IK, Illemann M, Thurison T. et al. uPAR as anti-cancer target: evaluation of biomarker potential, histological localization, and antibody-based therapy. Curr Drug Targets. 2011;12(12):1744-60
19. Ploug M. Structure-driven design of radionuclide tracers for non-invasive imaging of uPAR and targeted radiotherapy. The tale of a synthetic peptide antagonist. Theranostics. 2013;3(7):467-76
20. Persson M, Kjaer A. Urokinase-type plasminogen activator receptor (uPAR) as a promising new imaging target: potential clinical applications. Clin Physiol Funct Imaging. 2013;33(5):329-37
21. Knor S, Sato S, Huber T. et al. Development and evaluation of peptidic ligands targeting tumour-associated urokinase plasminogen activator receptor (uPAR) for use in alpha-emitter therapy for disseminated ovarian cancer. Eur J Nucl Med Mol Imaging. 2008;35(1):53-64
22. Yang L, Mao H, Cao ZH. et al. Molecular imaging of pancreatic cancer in an animal model using targeted multifunctional nanoparticles. Gastroenterology. 2009;136(5):1514-25
23. Wang M, Lowik DW, Miller AD. et al. Targeting the urokinase plasminogen activator receptor with synthetic self-assembly nanoparticles. Bioconjug Chem. 2009;20(1):32-40
24. Jing YQ, Tong CL, Zhang J. et al. Tumor and vascular targeting of a novel oncolytic measles virus retargeted against the urokinase receptor. Cancer Res. 2009;69(4):1459-68
25. Eefting D, Seghers L, Grimbergen JM. et al. A novel urokinase receptor-targeted inhibitor for plasmin and matrix metalloproteinases suppresses vein graft disease. Cardiovasc Res. 2010;88(2):367-75
26. Hildenbrand R, Allgayer H, Marx A. et al. Modulators of the urokinase-type plasminogen activation system for cancer. Expert Opin Investig Drugs. 2010;19(5):641-52
27. Kriegbaum MC, Persson M, Haldager L. et al. Rational targeting of the urokinase receptor (uPAR): development of antagonists and non-invasive imaging probes. Curr Drug Targets. 2011;12(12):1711-28
28. Ngo JC, Jiang LG, Lin ZH. et al. Structural basis for therapeutic intervention of uPA/uPAR system. Curr Drug Targets. 2011;12(12):1729-43
29. Abdalla MO, Karna P, Sajja HK. et al. Enhanced noscapine delivery using uPAR-targeted optical-MR imaging trackable nanoparticles for prostate cancer therapy. J Control Release. 2011;149(3):314-22
30. Jakobsche CE, McEnaney PJ, Zhang AX. et al. Reprogramming urokinase into an antibody-recruiting anticancer agent. ACS Chem Biol. 2012;7(2):316-21
31. LeBeau AM, Duriseti S, Murphy ST. et al. Targeting uPAR with antagonistic recombinant human antibodies in aggressive breast cancer. Cancer Res. 2013;73(7):2070-81
32. Chen JC, Chen NS, Huang JF. et al. Derivatizable phthalocyanine with single carboxyl group: synthesis and purification. Inorg Chem Commun. 2006;9:313-5
33. Maeda H. Tumor-selective delivery of macromolecular drugs via the EPR effect: background and future prospects. Bioconjug Chem. 2010;21(5):797-802
34. Yuan C, Huai Q, Bian CB. et al. The expression, purification and crystallization of monomeric soluble human urokinase receptor. Prog Biochem Biophys. 2006;33:277-81
35. Zhao GX, Yuan C, Bian CB. et al. Protein expression and preliminary crystallographic analysis of amino-terminal fragment of urokinase-type plasminogen activator. Protein Expr Purif. 2006;49(1):71-7
36. Chen Z, Zhou SY, Chen JC. et al. Pentalysine beta-carbonylphthalocyanine zinc: an effective tumor-targeting photosensitizer for photodynamic therapy. ChemMedChem. 2010;5(6):890-8
37. Seeliger D, de Groot BL. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J Comput Aided Mol Des. 2010;24(5):417-22
38. Brunger AT. Version 1.2 of the Crystallography and NMR system. Nat Protoc. 2007;2(11):2728-33
39. Stephens RW, Pedersen AN, Nielsen HJ. et al. ELISA determination of soluble urokinase receptor in blood from healthy donors and cancer patients. Clin Chem. 1997;43(10):1868-76
40. Kobayashi K, Kuwae S, Ohya T. et al. High-level expression of recombinant human serum albumin from the methylotrophic yeast Pichia pastoris with minimal protease production and activation. J Biosci Bioeng. 2000;89(1):55-61
41. Moreira LM, dos Santos FV, Lyon JP. et al. Photodynamic therapy: porphyrins and phthalocyanines as photosensitizers. Aust J Chem. 2008;61:741-54
42. Wainwright M. Photodynamic therapy: the development of new photosensitisers. Anticancer Agents Med Chem. 2008;8:280-91
43. Garg AD, Nowis D, Golab J. et al. Photodynamic therapy: illuminating the road from cell death towards anti-tumour immunity. Apoptosis. 2010;15(9):1050-71
44. Huang Z. A review of progress in clinical photodynamic therapy. Technol Cancer Res Treat. 2005;4(3):283-93
45. Huang Z, Xu HP, Meyers AD. et al. Photodynamic therapy for treatment of solid tumors - potential and technical challenges. Technol Cancer Res Treat. 2008;7(4):309-20
46. Jeong H, Huh M, Lee SJ. et al. Photosensitizer-conjugated human serum albumin nanoparticles for effective photodynamic therapy. Theranostics. 2011;1:230-9
47. Lebedeva NS. Aggregation properties of water-soluble metal phthalocyanines: effect of ionic strength of solution. Russ Chem Bull. 2004;53:2674-83
48. Yang F, Bian CB, Zhu LL. et al. Effect of human serum albumin on drug metabolism: structural evidence of esterase activity of human serum albumin. J Struct Biol. 2007;157(2):348-55
49. Zhu LL, Yang F, Chen LQ. et al. A new drug binding subsite on human serum albumin and drug-drug interaction studied by X-ray crystallography. J Struct Biol. 2008;162(1):40-9
50. Guo SH, Shi XL, Yang F. et al. Structural basis of transport of lysophospholipids by human serum albumin. Biochem J. 2009;423(1):23-30
51. Vivian JT, Callis PR. Mechanisms of tryptophan fluorescence shifts in proteins. Biophys J. 2001;80(5):2093-109
52. Abi-Habib RJ, Singh R, Liu SH. et al. A urokinase-activated recombinant anthrax toxin is selectively cytotoxic to many human tumor cell types. Mol Cancer Ther. 2006;5(10):2556-62
53. Li LS, Luo ZP, Chen Z. et al. Enhanced photodynamic efficacy of zinc phthalocyanine by conjugating to heptalysine. Bioconjug Chem. 2012;23(11):2168-72
54. Xu P, Chen JC, Chen Z. et al. Receptor-targeting phthalocyanine photosensitizer for improving antitumor photocytotoxicity. PLoS One. 2012;7(5):e37051
55. Castano AP, Mroz P, Hamblin MR. Photodynamic therapy and anti-tumour immunity. Nat Rev Cancer. 2006;6:535-45
56. Mroz P, Hashmi JT, Huang YY. et al. Stimulation of anti-tumor immunity by photodynamic therapy. Expert Rev Clin Immunol. 2011;7(1):75-91
57. Babson AL, Winnick T. Protein transfer in tumor-bearing rats. Cancer Res. 1954;14:606-11
58. Wilkinso.P Jeremy R, Brooks FP et al. Mechanism of hypoalbuminemia in rheumatoid arthritis. Ann Intern Med. 1965;63:109-14
59. Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer-chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46:6387-92
60. Isele U, Schieweck K, Kessler R. et al. Pharmacokinetics and body distribution of liposomal zinc phthalocyanine in tumor-bearing mice: influence of aggregation state, particle size, and composition. J Pharm Sci. 1995;84(2):166-73
61. Zhang ZB, Jin HT, Bao J. et al. Intravenous repeated-dose toxicity study of ZnPcS2P2-based-photodynamic therapy in Wistar rats. Photochem Photobiol Sci. 2006;5(11):1006-17
62. Liu W, Chen NS, Jin HT. et al. Intravenous repeated-dose toxicity study of ZnPcS2P2-based-photodyamic therapy in beagle dogs. Regul Toxicol Pharmacol. 2007;47(3):221-31
63. Smirnova ZS, Oborotova NA, Makarova OA. et al. Efficiency and pharmacokinetics of photosense: a new liposomal photosensitizer formulation based on aluminum sulfophthalocyanine. Khim Farm Zh. 2005;39:3-7
64. Lin L, Gardsvoll H, Huai Q. et al. Structure-based engineering of species selectivity in the interaction between urokinase and its receptor: implication for preclinical cancer therapy. J Biol Chem. 2010;285(14):10982-92
65. Heneweer C, Holland JP, Divilov V. et al. Magnitude of enhanced permeability and retention effect in tumors with different phenotypes: 89Zr-albumin as a model system. J Nucl Med. 2011;52(4):625-33
66. Elsadek B, Kratz F. Impact of albumin on drug delivery - new applications on the horizon. J Control Release. 2012;157(1):4-28
67. Kratz F. Albumin as a drug carrier: design of prodrugs, drug conjugates and nanoparticles. J Control Release. 2008;132(3):171-83
68. Frenkel YV, Clark AD Jr, Das K. et al. Concentration and pH dependent aggregation of hydrophobic drug molecules and relevance to oral bioavailability. J Med Chem. 2005;48(6):1974-83
69. Shoichet BK. Interpreting steep dose-response curves in early inhibitor discovery. J Med Chem. 2006;49(25):7274-7
70. Brasseur N, Langlois R, La Madeleine C. et al. Receptor-mediated targeting of phthalocyanines to macrophages via covalent coupling to native or maleylated bovine serum albumin. Photochem Photobiol. 1999;69(3):345-52
71. Anatelli F, Mroz P, Liu Q. et al. Macrophage-targeted photosensitizer conjugate delivered by intratumoral injection. Mol Pharm. 2006;3(6):654-64
Author contact
![]() Corresponding author: Mingdong Huang, Email address: mhuangac.cn.
Corresponding author: Mingdong Huang, Email address: mhuangac.cn.