Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2014; 4(5):475-486. doi:10.7150/thno.7729 This issue Cite
Research Paper
Myocardial Protective Effect of Extracellular Superoxide Dismutase Gene Modified Bone Marrow Mesenchymal Stromal Cells on Infarcted Mice Hearts
Qiao Pan1*, Xing Qin1*, Sai Ma1*, Haichang Wang1*, Kang Cheng1, Xinxing Song1, Haokao Gao1, Qiang Wang2, Rannie Tao3, Yabin Wang1, Xiujuan Li1, Lize Xiong2 ![]() , Feng Cao1,4
, Feng Cao1,4 ![]()
1. Department of Cardiology, Xijing Hospital, Fourth Military Medical University, Xi'an, Shaanxi, China;
2. Department of Anesthesia, Xijing Hospital, Fourth Military Medical University, Xi'an, Shaanxi, China;
3. Department of Medicine & Dentistry, University of Alberta, Canada;
4. Department of Cardiology, China PLA General Hospital, Beijing 100853, China.
* Qiao Pan, Xing Qin, Sai Ma and Haichang Wang contributed equally to this work.
Received 2013-9-23; Accepted 2013-11-4; Published 2014-2-15
Abstract

Aim: Extracellular superoxide dismutase (ecSOD) is a unique scavenger of superoxide anions and a promising target of gene therapy for ischemia/reperfusion injury (I/R). However, conventional gene therapies have limitation in effectiveness and efficiency. This study aimed to investigate the protective effects of ecSOD gene modified bone marrow mesenchymal stromal cells (BMSCs) on cardiac function improvement in mice infarcted heart. METHODS & RESULTS: BMSCs were isolated from Fluc+ transgenic mice (Tg FVB[Fluc+]) and transfected by adenovirus combined with human ecSOD gene. ELISA was performed to determine ecSOD protein level. Female syngeneic FVB mice were randomized into 5 groups: (1) Sham group (sham); (2) MI group (MI); (3) MI+BMSCs group (BMSC); (4) MI+BMSCs-vector group (BMSC-vector); (5) MI+ BMSCs-ecSOD group (BMSC-ecSOD). MI was accomplished by ligation of the left anterior descending artery. BMSCs (2x106) were injected into the border zone of infarction. In vivo bioluminescence imaging (BLI) was performed to monitor transplanted BMSCs viability. Echocardiography and histological staining revealed that BMSCs-ecSOD significantly reduced myocardial infarction size and improved cardiac function. Lucigenin chemiluminescence, DHE and TUNEL staining demonstrated that BMSCs-ecSOD delivery reduced ROS level and cell apoptosis both in vivo and in vitro. Western blot assay revealed that ecSOD supplementation increased FoxO3a phosphorylation in cardiomyocytes. Moreover, quantitative real-time PCR showed that pro-apoptotic factors (bim and bax) were decreased while the anti-apoptotic factor mir-21 expression was increased after ecSOD intervention. CONCLUSION: Intra-myocardial transplantation of adenovirus-ecSOD transfected BMSCs could exert potential cardiac protection against MI, which may be partly through reduction of oxidative stress and improvement of BMSCs survival.
Keywords: human extracellular superoxide dismutase, mesenchymal stromal cells, myocardial infarction, cell-based gene therapy, reactive oxygen species.
Introduction
Considerable evidence indicated that reactive oxygen species (ROS), such as superoxide anion (O2-) and hydrogen peroxide, played an important role in mediating myocardial ischemic injury [1]. Excessive O2- formation was detected in patients with acute myocardial infarction (MI) and unstable angina pectoris [2]. Superoxide dismutase (SOD) may provide anti-oxidative protection by inactivating O2- [3]. Extracellular superoxide dismutase (ecSOD) binds to heparin sulfate proteoglycans on cellular surface as a unique scavenger of superoxide anions in the extracellular space [4]. Many studies have shown that direct gene transfer of the cDNA encoding membrane-bound human ecSOD affords transient cardio-protective effect [3, 5]. However, gene delivery via plasmid or virus containing ecSOD gene causes several problems, such as the inefficiency and instability of ecSOD expression in local tissue and resultant local immune response. Bone marrow mesenchymal stromal cells (BMSCs), characterized by their multipotential property, are promising vehicles for stem cell based gene therapy [6]. Recently, Abdel et al. demonstrated for the first time that intravenous administration of BMSCs transfected with adenovirus carrying ecSOD could improve the survival of irradiated mice [7]. We hypothesized that the transplantation of ecSOD overexpressed BMSCs may exert ehanced protective effects on myocardium against infarction injury.
Materials and methods
Animals
ß-Actin-luc transgenic mice (8-10 weeks old, 22-28g, SPF)[FVB/N-Tg (ß-Actin-fluc)-Xen, FVB/N background, Caliper Life Sciences, USA], which constitutively expressed firefly luciferase (Fluc) in all tissues and organs, were used for BMSC isolation. Syngeneic female FVB mice with the same genetic background as ß-Actin-luc transgenic mice (8 weeks old, 20 to 25g) underwent LAD ligation for the MI model and served as hosts for cellular therapy. All procedures were performed in accordance with the National Institutes of Health Guidelines on the Use of Laboratory Animal. The experimental protocol was approved by the Fourth Military Medical University Committee on Animal Care.
MI model and cell transplantation
FVB mice (n = 120) were divided into 5 groups: (1) Sham group (n = 20); (2) MI group (MI, n = 20); (3) MI+BMSCs group (BMSC, n = 20); (4) MI+BMSCs-vector group (BMSC-vector, n = 20); 5) MI+ BMSCs-ecSOD group (BMSC-ecSOD, n = 40). MI was accomplished by ligation of the left anterior descending (LAD) artery with 6-0 silk sutures after left thoracotomy as described before [8]. The infarction was confirmed when the anterior wall of left ventricle (LV) turned pale. After 30 minutes, injections were made at 2 sites near the border zone of infarction (medial and lateral zones) with a total volume of 50 μL containing 2x106 cells or same volume PBS using a Hamilton syringe with a 29-gauge needle. All surgical procedures were performed blindly by an expert with several years of experience on myocardial infarction model.
Cell culture, transfection and determination of ecSOD, VEGF, bFGF, IGF-1 and HGF
Bone marrow tissues from Fluc+ transgenic mice (Tg[Fluc+]) were washed thoroughly with phosphate buffered saline (PBS) containing 1% antibiotic/antimycin to prevent bacterial contamination. Then the suspension was centrifuged at 200×g/min for 10 min. The cellular pellet was incubated in Dulbecco's modified Eagle's medium/F-12 (DMEM/F-12, Hyclone, USA) with 10% (vol/vol) fetal bovine serum (FBS, Hyclone) for 72 h at 37°C/5%CO2. Adherent cells were digested, plated (2×104/cm2) in 75cm2 flask (Corning, USA) and cultured in DMEM/F-12/10%FBS supplemented with 2 ng/ml basic fibroblast growth factor (bFGF, Peprotech, USA) and 1% antibiotic/ antimycotic. The medium was refreshed every two days. When cells reached 80% confluence, they were detached by 0.05% trypsin + 0.02% EDTA (Invitrogen, USA), and then re-plated at a density of 5×104/cm2 in cell culture flasks. Replication-deficient adenoviruses with/without human ecSOD expression, driven by the human CMV promoter/enhancer, were constructed using standard procedures [9]. BMSCs were then transfected with adenovirus at various multiplicity of infections (MOI) (defined as plaque-forming units/cell) of 0, 1, 10, 20, 50, 100, 300, 500 or 2000 for 48 hours [7]. The virus-containing medium was removed 24 hours later, and the cells were washed 3 times with PBS and further incubated in fresh medium for 24, 48, 72 h respectively. The medium was collected and analyzed for ecSOD activity using a SOD activity assay kit (R&D Systems, USA). The concentrations of VEGF, bFGF, IGF-1 and HGF secreted by BMSCs were determined by enzyme-linked immunosorbent assay (ELISA, TSZ, USA) according to the manufacturer's instructions.
In vivo optical bioluminescence imaging (BLI)
BLI of Fluc was a reliable noninvasive imaging tool to quantitatively monitor transplanted BMSCs viability with the sensitivity of 500 viable cells [10, 11]. BLI of engrafted cells was detected using the Xenogen In Vivo Imaging System (IVIS, Caliper Life Sciences, USA). Recipient mice were anesthetized with 3% isoflurane and placed in the imaging chamber. After acquisition of a baseline image, mice were injected with D-Luciferin (200 mg/kg) intraperitoneally. Mice were imaged on day 1 and weekly until sacrifice at week 4 after cell transplantation. Average signals (photons/s/cm2/sr) from a fixed region of interest (ROI) were evaluated as described [10]. In order to further confirm the relationship between BMSCs cell number and Fluc activity in vitro, BLI was performed as described [12]. Briefly, BMSCs of different quantities were suspended in 500 μL phosphate-buffered saline (PBS), incubated with reporter probe D-luciferin (150 ng/L, Invitrogen, USA), and then imaged for 1 minute using Xenogen Kinetic In Vivo Imaging System (IVIS, Caliper Life Sciences, USA).
Echocardiography determination for cardiac function
Echocardiography was conducted at 24 h after infarction as followed [13]. FVB mice (3% isoflurane) were studied on an M-mode ultrasound system (Sequoia Acuson, Siemens; 15-MHz linear transducer). Left ventricular end-diastolic diameter (LVEDd) and left ventricular end-systolic diameter (LVESd) were measured on the parasternal left ventricular long axis view. All measurements were based on 5 consecutive cardiac cycles. Left ventricular ejection fraction (LVEF) and left ventricular fractional shortening (FS) were calculated by computer algorithms. All of these measurements were performed by a blinded investigator.
Histological analysis for apoptosis and infarction area
To determine the underlying mechanism of BMSCs-ecSOD transplantation protection against myocardial infarction, 24 h after first operation, apoptosis ratio was examined using the terminal deoxy-nucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) assay (ApopTag; Millipore, Billerica, MA) according to manufacturer's instructions. Apoptotic rate within in the peri-infarction regions was calculated by laser confocal microscope (FV1000, Olympus, Tokyo, Japan). Caspase-3 activity was determined by caspase-activity assay kit (Chemicon, Billerica, USA) and was expressed as nmol pNA/h/mg protein. 28 days after MI operation, myocardial fibrosis was examined to indicate scar formation within the LV. Heart sections (5 μm) were stained with Masson's trichrome (Sigma-Aldrich; St. Louis, MO). MI area was measured using computer morphometry (Bioquant 98) and the results were expressed as MI/LV ratio.
Lucigenin chemiluminescence and DHE staining for ROS production
ROS are well known to be involved in cardiovascular disease. To examine the potential mechanism of ecSOD's protective effects against MI, ROS production of the 5 group were tested by lucigenin-enhanced chemiluminescence as described previously [14]. Results were expressed as relative light units (RLU) per second per milligram heart weight (RLU/s/mg protein). In addition, the superoxide sensitive fluorescent dye dihydroethidium (DHE) was used to evaluate in situ formation of O2- as followed [15]. Harvested heart tissue was made into sections and incubated with DHE (DHE, 1:1000 dilution, Beyotime Institute of Biotechnology, Nanjing, China) at 37°C for 30 minutes. Tissue sections were visualized with confocal microscope and fluorescence was detected with a wavelength of 590-nm pass filter. Images were collected and stored digitally.
Western blot analysis and ELISA assay
Myocardium tissues were harvested for Western blot as described previously [12]. Cells of each group were harvested at appropriate time. Cells were washed three times with PBS and collected after ice-cold lysis buffer digestion. Protein lysates were separated on 12% SDS-PAGE gels and transferred onto nitrocellulose (NC) membrane. Membranes were blocked with 5% milk in 1×TBS-Tween-20 buffer and incubated overnight at 4°C with primary antibodies (dilution 1:2000 for anti-Akt, 1:1000 for anti-ERK1/2 (ERK, #4695), phospho-ERK1/2 (Ser473, #4370), phospho-FoxO3a (Thr32, #2599), Akt (#4685), phospho-Akt (#4060), ecSOD (#2920) and Tubulin (#4967), all from Cell Signaling Technology, Danvers, MA, USA), followed by incubation with the corresponding secondary antibodies at room temperature for 1 h. The signal was detected with ECL-Plus reagent. Nuclear and cytoplasmic fractions of the cells were isolated using Nuclear and Cytoplasmic Protein Extraction Kit (Beyotime Biotechnology, Jiangsu, China). Immunoreactivity was detected by sequential incubation with HRP-conjugated antibodies and enzymatic chemiluminescence (#7003). Integrated optical density (IOD) of immunoblot was quantified using QuantiOne imaging software (Bio-Rad, USA).
Cardiomyocytes hypoxia/serum deprivation injury
Primary cardiomyocytes (CMs) were isolated and cultured as described previously [16]. CMs were divided into seven groups as followed: control group, H/SD group (H/SD), H/SD+BMSCs group (BMSC), H/SD+BMSCs-vector group (BMSC-vector), H/SD+BMSCs-esSOD group (BMSC-esSOD), H/SD+BMSCs-ecSOD+PI3K inhibitor LY294002 group (ecSOD+LY) and H/SD+BMSCs-ecSOD+MEK1/2 inhibitor U0126 group (ecSOD+U). CMs of the following group (BMSC, BMSC-vector, BMSC-ecSOD, ecSOD+LY and ecSOD+U) were co-cultured with BMSCs [17] for 24h and subjected to H/SD injury according to previous study description [18]. Briefly, cardiomyocytes cultured in Hanks buffer were exposed to hypoxia (approximately 94% N2, 2% O2 and 5% CO2) in an anaerobic system (Thermo Forma) at 37°C for 6h. Cardiomyocytes maintained in normoxia conditions (95% air-5% CO2) were considered as control group.
Cell proliferation assays
MTT and WST-8 assays were performed to investigate whether co-culture with BMSCs-ecSOD could promote CMs proliferation after H/SD injury. CMs were plated overnight in 96-well culture plates at the concentration of 1×104 cells/mL. After H/SD treatment, medium were replaced by 100 μl MTT (Sigma-Aldrich, USA) and plates were placed at 37°C for 4 h. Then, MTT solution was replaced by formazan crystals dissolved in DMSO. Formazan was quantified spectroscopically at the wavelength of 490nm by spectrophotometer. For WST-8 [2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfo-phenyl)-2H-tetrazolium, mono-sodium salt] (also named as CCK- 8) assays, CMs were plated on 96-well culture plates and subjected to starvation with DMEM (0.5% NCS) for 24 h. After starvation, CMs were incubated with WST-8 (Dojindo, Labforce, Nunningen, Switzerland) at 37°C for 2 h. Cells released orange formazan into the medium when incubated with WST-8. Formazan was quantified spectroscopically at the wavelength of 490nm by spectrophotometer to indicate cell proliferation capacity.
Quantitative real-time PCR
Total RNA was isolated from CMs. The cDNA was synthesized using the QuantiTect reverse transcription kit (Qiagen). Real-time PCR was performed using the KAPA SYBR fast qPCR kit (KAPA Biosystems, Woburn, MA, USA). Data were normalized to house-keeping gene GAPDH. Primers used in this experiment were purchased from Qiagen (Qiagen, Hilden, Germany). The primer sequences are: bim forward 5'- ATCTCAGTGCAATGGCTTCCA- 3' and reverse 5'- GCTCCTGTGCGATCCGTATC- 3'; bax forward 5'- GCCCTTTTGCTTCAGGGTTT- 3' and reverse 5'- TCCAATGTCCAGCCCATGAT- 3'; mir-21 and RNU5 miScript Primer Assays (Qiagen, Hilden, Germany) were used to study the amplification and to normalize the miR-21 expression. REST MCS software was utilized for the calculation of the relative differences between the test groups.
Statistics
Results are expressed as mean ± standard deviation (SD). SPSS15.0 (SPSS Inc., USA) and Prism5.0 (GraphPad Software, USA) were used to perform the one-way analysis of variance (ANOVA) for evaluating the differences among different experimental groups and different time points within each group. Pairwise multiple comparisons were to identify the parameters differences between the two groups using ANOVA-conjuncted Tukey test. Data expressed as proportion was assessed with Chi-square testing. A two-tailed P-value <0.05 was considered significant. Polynomial regression analysis was performed to evaluate the correlation between cell number and optical radiance in vitro.
Results
Dose-dependent secretion of ecSOD in Ad-ecSOD transfected BMSCs
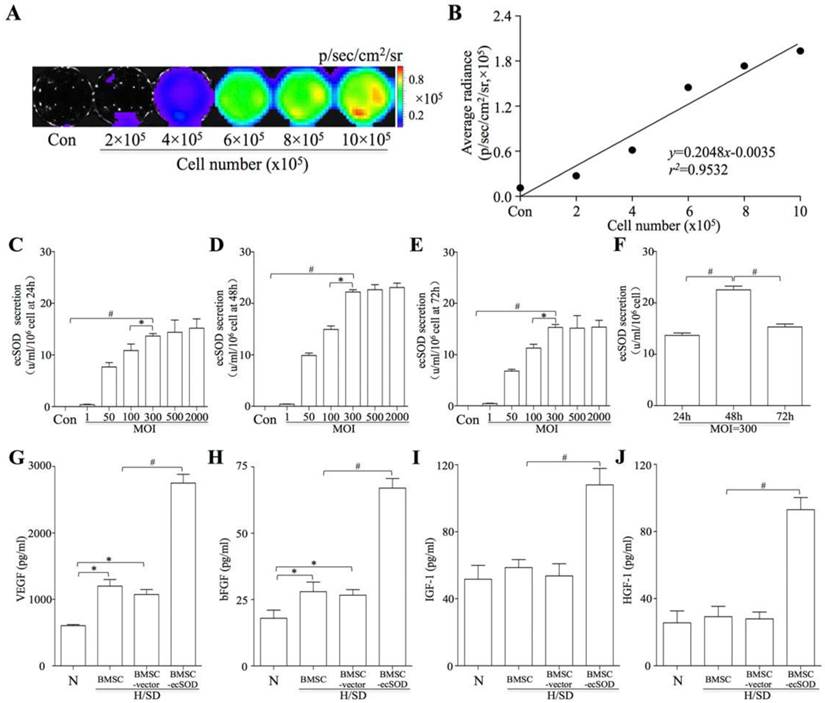
BMSCs, isolated from ß-Actin-Fluc transgenic FVB mice, could constitutively express firefly luciferase (Fluc) within the cytosol. In vitro BLI results displayed a robust linear correlation between the number of BMSCsFlu+ and Flu average radiance (r2 = 0.9532; SDC, Fig. 1A-B). BMSCs were transfected with Ad5CMVecSOD. When the MOI was less than 300, the secretion of ecSOD was dose-dependent at 24, 48 and 72 h (Fig. 1C-E). However, the secretion of ecSOD no longer increased in a dose-dependent fashion when MOI was up to 300. Secretion of ecSOD from BMSCs-ecSOD at MOI=300 was higher at the 48h time point than 24h or 72h (Fig. 1F). There was no significant difference of ecSOD secretion between MOI = 500 group and MOI = 2000 group. Therefore, BMSCs transfected with Ad5CMV-ecSOD at the MOI of 300 for 48h were used in the following experiments.
In vitro bioluminescence imaging (BLI) and ecSOD expression. A: BLI of BMSCs from different cell number group in vitro. Color scale bar represents optical radiance intensity in photons/second/cm2/steridian (p/sec/cm-2/sr); B: Linear correlation of cell number with Fluc signal intensity; C-E: Secretion of ecSOD in BMSCs-ecSOD group of different MOI. When MOI was less than 300, the secretion of ecSOD displayed a dose-dependent secretion at 24, 48, 72 h; F: At MOI=300, the secretion of ecSOD from BMSCs-ecSOD reached its peak in 48h. G-J: Ad-ecSOD transfection promoted VEGF (G), bFGF (H), IGF-1 (I) and HGF (J) secretion in BMSCs after H/SD injury. *p < 0.05 vs. control group, #p < 0.05 vs. H/SD group.
Elevation of growth factors secreted by BMSCs-ecSOD
ELISA assays were performed to evaluate the effect of ecSOD on cytokine secretion in BMSCs. As shown in Fig. 1G-J, the levels of VEGF (1.20 ± 2.17×103 vs. 0.60 ± 0.28×103 pg/ml, p < 0.05) and bFGF (28.16 ± 3.25 vs. 18.53 ± 2.19 pg/ml, p < 0.05) were increased after H/SD injury in MI group compared with control group. Moreover, the secretion of VEGF, bFGF, IGF-1 and HGF were further elevated after H/SD in BMSC- ecSOD group as compared with H/SD, BMSC and BMSC-vector group (p < 0.05). There was no significant difference of cytokine secretion between BMSC group and BMSC-vector group (p > 0.05).
Higher survival of BMSCs after ecSOD gene modifying
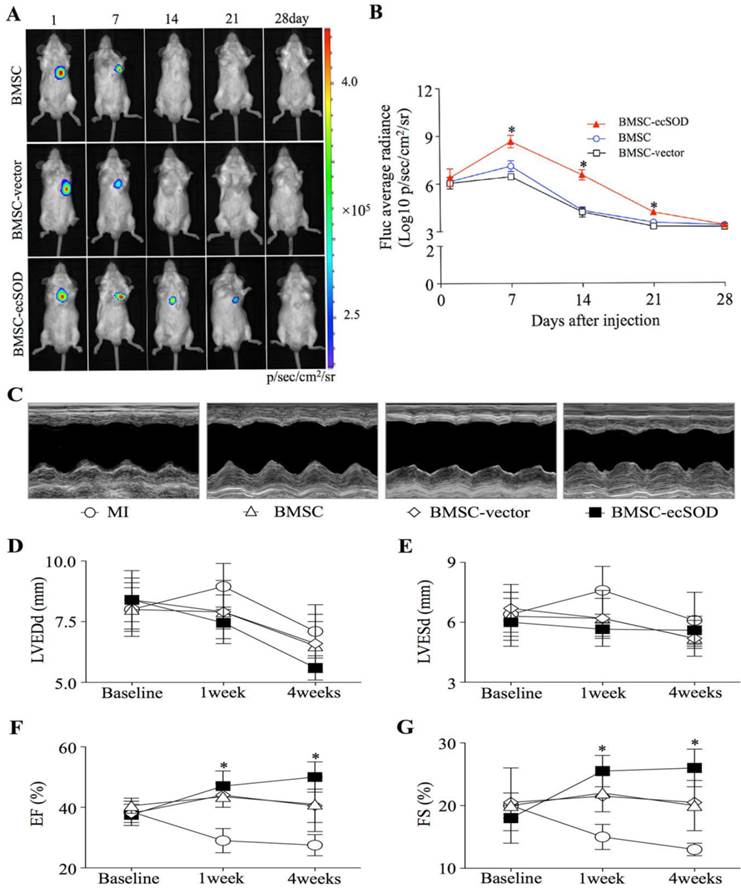
BLI results showed that there was no significant difference among each group on day 1. BLI signaling was significantly decreased in BMSC and BMSC-vector groups on day 7, 14, 21 and 28 as compared with day 1, indicating reduced BMSCs viability post transplantation (p < 0.05). In ecSOD group, BLI signaling did not display significant difference on day 1, 7, 14, 21, 27 (p > 0.05).
BMSCs-ecSOD significantly improved cardiac function after MI
Echocardiography was performed to evaluate cardiac function at baseline and post MI operation (Fig. 2C-G). Our results showed that there was no significant difference among all groups at baseline (p > 0.05). On week 1 and 4 post operation, a marked reduction of LV diameters, including LVEDd and LVESd were noted in the BMSC-ecSOD group compared with MI group (p < 0.05). After 4 weeks, we observed significant reduction in EF and FS parameters in BMSC and BMSC-vector group compared with those of control animals (p < 0.05). BMSC-ecSOD group showed tremendous improvement of cardiac functional parameters (EF and FS) compared with MI, BMSC and BMSC-vector group at 1 week after MI (p<0.05) and this tendency sustained until day 28 post operation.
Survival of transplanted BMSCs after MI: A: In vivo BLI of BMSCs survival after transplantation; B: Quantitative analysis of BLI. *p < 0.05 vs. BMSC group. C: Representative images of M-mode echocardiography. D-G: Quantitative analysis of cardiac function of LVEDd (D), LVESd (E), EF (F)and FS (G) at baseline, 1week and 4 weeks after MI. n=20/group, *p < 0.05 vs. MI group.
Transplantation of BMSCs-ecSOD significantly reduced fibrosis and apoptosis after MI
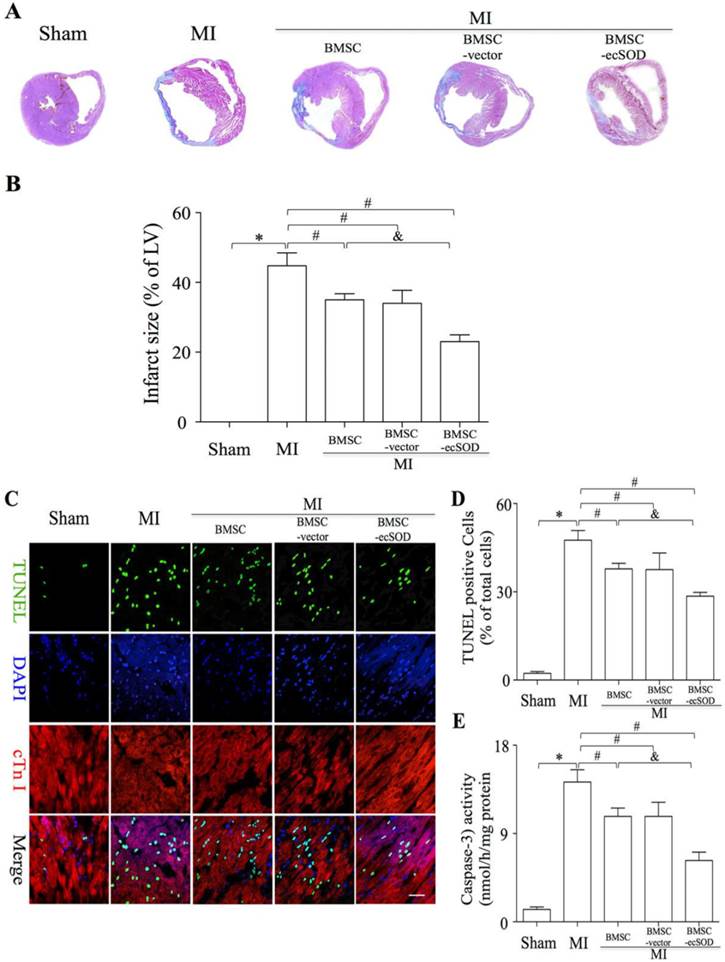
To analyze if BMSC-ecSOD modified fibrosis in infarcted myocardium, we performed Masson trichrome staining on day 28 post LAD ligation. Masson trichrome staining results showed that fibrosis was markedly alleviated in BMSC-ecSOD group (23±2%) compared with MI, BMSC and BMSC-vector group (p<0.05). As is shown in representative TUNEL images (Fig. 3C-D), the apoptosis index (AI) was significantly increased in MI group compared with control group (p<0.001). A sharp decrease of AI was noted in BMSC, BMSC-vector and BMSC-ecSOD group compared with MI group (p<0.05), indicating that BMSCs transplantation could suppress MI induced apoptosis. Furthermore, this anti-apoptotic effect was more pronounced in BMSC-ecSOD group compared with BMSC and BMSC-vector groups (p<0.05). Caspase-3 activity assays revealed that activation of caspase-3 was attenuated in BMSC-ecSOD group compared with MI, BMSC and BMSC-vector group (p<0.05) (Fig. 3E).
Significant reduction of fibrosis and CMs apoptosis after BMSC-ecSOD transplantation. A: Representative images of Masson's trichrome staining of each group 4 weeks after MI; B: Quantitative analysis of Masson's trichrome staining; C: Representative images of immunostaining for apoptotic cells (green, TUNEL; blue, DAPI; scale bar, 100μm). D: quantification of apoptotic cells; E: Analysis of caspase-3 activity. *p < 0.05 vs. control group, #p< 0.05 vs. MI group, &p< 0.05 vs. BMSC group.
Transplantation of BMSCs-ecSOD induced a sustained increase in ecSOD expression accompanied with reduction of MI-induced oxidative stress in the injured myocardium
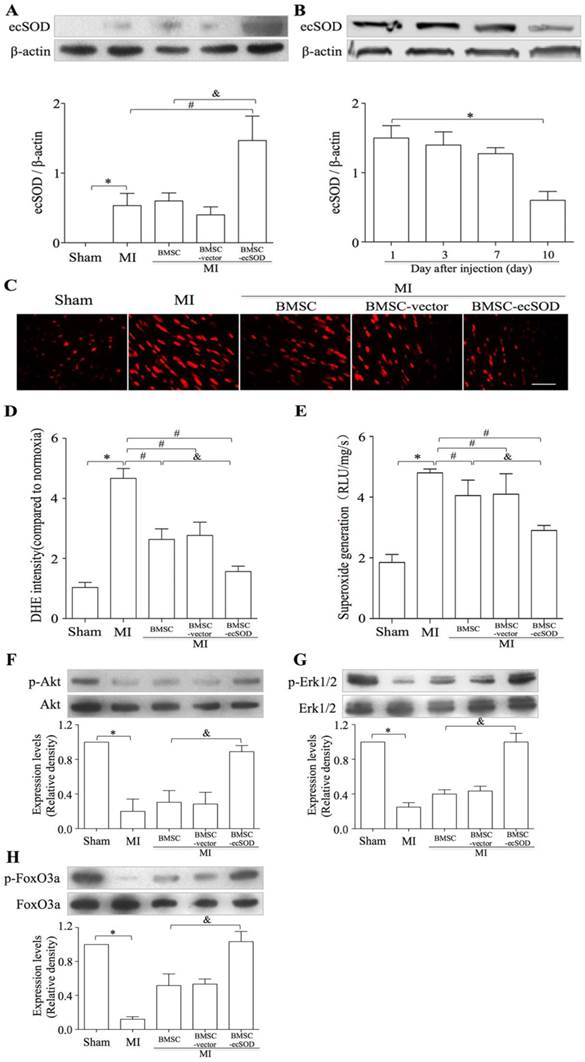
Western blot results revealed that the expression of ecSOD in BMSC-ecSOD group was significantly increased compared with control, MI, BMSC and BMSC-vector group (p < 0.05) and this increase sustained until day 10. (Fig.4A-B). DHE staining showed that in situ ROS production was elevated in MI group, while decreased in BMSC-ecSOD group (p < 0.05) (Fig. 4C-D). According to lucigenin chemiluminescence results (Fig. 4E), total ROS amount in BMSC-ecSOD group was significantly decreased compared to MI groups (p < 0.05).
A: The transplantation of BMSCs-ecSOD promoted the expression of ecSOD in myocardial tissues. B: The expression of ecSOD in BMSC-ecSOD group on 1, 3, 7 and 10 days after transplantation. C: Representative images of DHE staining. D: Quantification of DHE staining. E: Analysis of O2- production by lucigenin-enhanced chemiluminescence. F-G: Western blot and quantification of phosphorylation of Akt, Erk1/2 and FoxO3a. *p < 0.05 vs. control group; #p< 0.05 vs. MI group; &p< 0.05 vs. BMSC group.
The transplantation of BMSCs-ecSOD activated the PI3K/Akt and MEK/Erk1/2 signaling pathways in vivo
The activation of PI3K and MEK signaling pathways were evaluated by measuring the phosphorylation of Akt and Erk1/2. As shown in representative radiograms of Western blot assay and quantification analysis (Fig. 4F-G), there was a significant increase in phospho-Akt and phospho-Erk1/2 (normalized to total Akt and Erk1/2 respectively) in BMSC-ecSOD group as compared to MI, BMSC and BMSC-vector group (p < 0.05). Furthermore, the amount of phospho-FoxO3a (normalized to total FoxO3a) was also markedly elevated in BMSC-ecSOD group (p < 0.05) (Fig. 4H).
Co-culture with BMSCs-ecSOD significantly improved CMs proliferation after H/SD injury in vitro
MTT and WST-8 assays were performed to evaluate CMs proliferation. As is shown in Fig. 5A-B, CMs viability displayed a tremendous reduction in H/SD group compared with control group (p < 0.05). Significant increase of CMs viability was observed in BMSC, BMSC-vector and BMSC-ecSOD group compared with H/SD group and BMSC+ecSOD group showed the maximum increase (p < 0.05). Furthermore, the beneficial effect of BMSCs on CMs viability was abolished when PI3K inhibitor LY294002 and MEK inhibitor U0126 were used (p < 0.05).
Co-culture with BMSCs-ecSOD significantly promoted CMs survival and reduced ROS overproduction after H/SD injury. A: Cell viability measurement by MTT assay. B: Cell viability measurement by WST-8 assay. C: Representative images of immunostaining for apoptotic cells (green, TUNEL; blue, DAPI; scale bar, 100μm). D: Quantification of TUNEL assay. E: Caspase-3 activity assay. F: Representative images of DHE staining. G: Quantification of DHE staining. H: Analysis of O2- production by lucigenin-enhanced chemiluminescence. *p < 0.05 vs. control group; #p< 0.05 vs. BMSC group; &p< 0.05 vs. BMSC-ecSOD group.
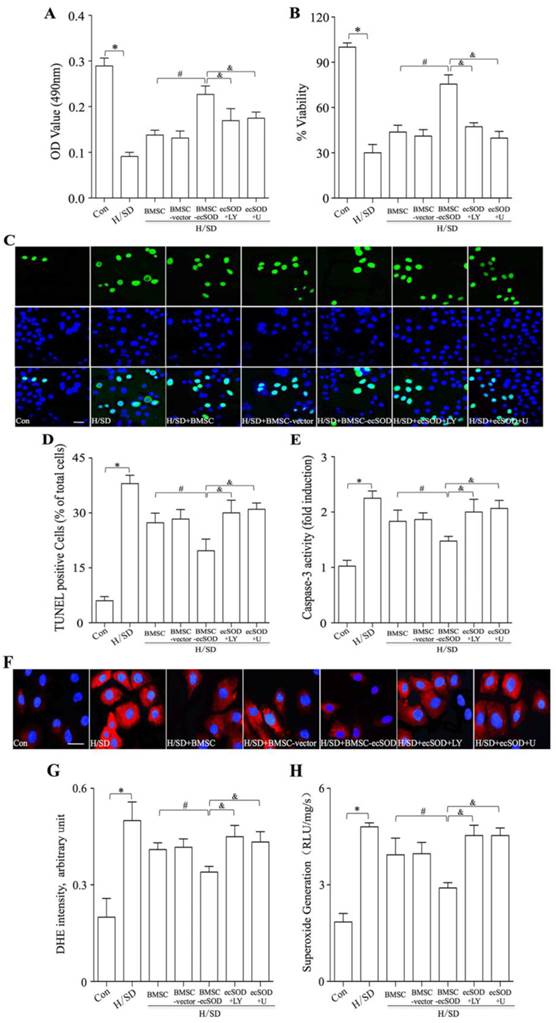
ecSOD inhibited apoptosis of CMs induced by H/SD injury
TUNEL staining and caspase 3 activity assay were performed to evaluate apoptosis of CMs in vitro. As is shown in Fig. 5C-E, H/SD injury markedly increased apoptosis in CMs, while co-culture with BMSCs inhibited H/SD induced apoptosis (p < 0.05). Moreover, LY294002 and U0126 abolished the apoptosis decrease caused by BMSCs co-incubation (p < 0.05).
ecSOD decreased H/SD-induced ROS overproduction in CMs
ROS production was measured by DHE staining and lucigenin-enhanced chemiluminescence assay (Fig. 5F-H) in vitro. Fig.5F-H revealed that intracellular ROS amount of CMs was increased after H/SD injury, while the induction of ROS production could be attenuated by co-culture with BMSCs-ecSOD (p < 0.05). Furthermore, LY294002 and U0126 abolished the ROS generation decline caused by BMSCs co-incubation (p < 0.05).
ecSOD supplement increases Akt and Erk1/2 phosphorylation in CMs after H/SD injury
The phosphorylation of Akt, Erk1/2 and FoxO3a in CMs of all groups were measured by Western blot assay (Fig. 6A-C). Our results showed that co-culture with BMSCs-ecSOD increased PI3K/Akt, MER/Erk1/2 and FoxO3a phosphorylation in CMs after H/SD injury (p < 0.05) (Fig.6A-C). The effect of promoting Akt, Erk1/2 and FoxO3a phosphorylation by ecSOD on CMs could be attenuated by LY294002 and U0126.
Co-culture with BMSCs-ecSOD increased the phosphorylation of Akt, Erk and FoxO3a in CMs after H/SD injury. A-C: Western blot and quantification of phosphorylation of Akt, Erk1/2 and FoxO3a (normalized to total Akt Erk1/2 and FoxO3a respectively). D-F: The mRNA expression of pro-apoptotic factors bim and bax and anti-apoptotic factor mir-21 by real-time PCR. *p < 0.05 vs. control group; #p< 0.05 vs. BMSC group; &p< 0.05 vs. BMSC-ecSOD group. G: Proposed scheme for the mechanisms of ecSOD mediated CMs survival.
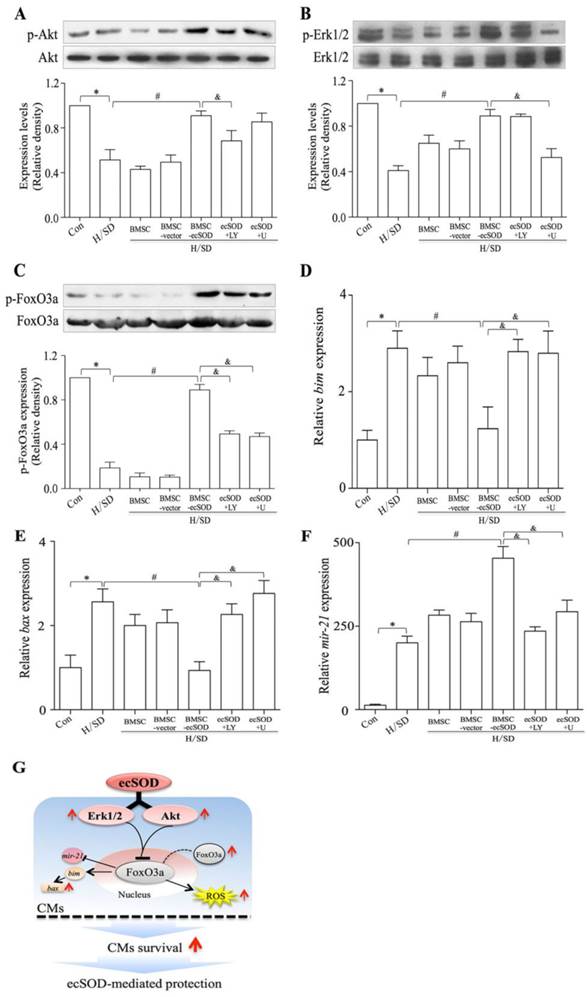
ecSOD regulated apoptotic signaling pathways
bim, bax and mir21 are apoptosis associated factors, so we analyzed mRNA expression of these genes to figure out whether ecSOD regulated apoptotic signaling pathways. Based on our data (Fig. 6D-E), the mRNA expression of pro-apoptotic factors bim and bax were increased after H/SD injury (p < 0.01), while BMSC-ecSOD inhibited this change (p < 0.05). We then checked the mRNA expression of anti-apoptotic factor mir-21. As is shown in figure 6F, there was a significant decline in mir-21 expression after H/SD injury, while BMSC-ecSOD elevated mir-21 mRNA amount (p < 0.05). However, the anti-apoptotic effect of BMSC-ecSOD was eliminated when LY294002 and U0126 were used (p < 0.05), indicating that the anti-apoptotic effect of ecSOD was via PI3K and MARKERK pathways.
Discussion
In the present study, we demonstrated for the first time that the intra-myocardial transplantation of ecSOD gene modified BMSCs could exert potential cardiac protection against MI. Echocardiography and histological staining results revealed that BMSC-ecSOD significantly reduced cardiac fibrosis, decreased cardiomyocyte apoptosis and improved cardiac function after MI injury. Concurrently, ecSOD increased the survival of transplanted BMSCs in the regional myocardial tissue. Furthermore, both in vivo and in vitro results verified that BMSCs-ecSOD reduced oxidative stress and cardiomyocyte apoptosis, partly through the activation of Akt, Erk1/2 and FoxO3a signaling pathways. .
Adenovirus is widely used as vector in gene therapy for its high transfection efficiencies. However, it was also reported that the use of adenovirus raised the undesirable possibilities of inflammatory response or random transgenic alteration in many cell types [3, 19]. Therefore, cell-based gene therapy is considered as an ideal biological approach for disease treatment. According to some recent studies, the application of BMSCs as vehicle could stably increase therapeutic protein expression [6, 19]. Furthermore, BMSCs were able to home to the viable myocardium and attenuate cardiac dysfunction and ventricular remodeling associated with MI [20]. In our study, we used adenovirus-ecSOD modified BMSCs as a new approach for MI treatment, which not only eliminated adenovirus induced inflammation response, but also ensured accurate distribution of ecSOD in infarcted myocardium [21]. Previous study recommended MOI = 300 for BMSCs transfection [7]. However, we found that there was no significant difference of ecSOD secretion between MOI = 500 and MOI = 2000. Consequently, we chose MOI = 500 for ad-ecSOD transfection. As was indicated in our results, there was sustained ecSOD secretion in ad-ecSOD transfected BMSCs. Growth factors, such as VEGF, bFGF, IGF-1 and HGF, were significantly increased in ecSOD modified BMSCs, indicating the effectiveness of adenovirus mediated ecSOD modification.
Previous studies reported low survival of BMSCs after transplantation, which limited the protective effects of BMSCs on infracted myocardium [22, 23]. In our present study, we observed prolonged BMSCs survival after ad-ecSOD modification. BLI results revealed that BMSCs viability was significantly elevated in BMSC-ecSOD group: BLI signal of BMSC-ecSOD group completely returned to baseline by the 28th day post-MI, whereas that of BMSCs group returned to baseline by day 21 post-MI. We hypothesized that ecSOD exerted an anti-oxidative role in improving microenvironment for BMSCs survival. The improved BMSCs viability further contributed to cardiac functional recovery after MI. As was revealed by echocardiography results, BMSC-ecSOD markedly improved cardiac function post-MI and this beneficial effect sustained until day 28 after transplantation. This sustained cardiac functional recovery may be attributed to prolonged BMSCs survival and increased ecSOD expression. Horiuchi M reported that ecSOD amount was positively related with myocardial recovery after MI injury and our results were in accordance with theirs [24, 25].
Our Masson trichrome staining showed that ecSOD modified BMSCs markedly reduced cardiac fibrosis induced by MI injury, verifying long-term protective effects of BMSC-ecSOD on infarcted myocardium. To figure out the underlying mechanism of BMSC-ecSOD intervention, we analysed oxidative stress and apoptosis level both in vivo and in vitro. DHE staining revealed that BMSC-ecSOD transplantation significantly reduced ROS production in peri-infarction myocardium, which contributed to the cardiac protective role of BMSD-ecSOD. Besides, both in vivo and in vitro TUNEL and caspase-3 activity assay revealed that BMSC-ecSOD reduced cardiomyocyte apoptosis, which may account for another mechanism for BMSC-ecSOD protection. Oxidative and apoptosis related proteins, such as p-Akt, p-Erk and FoxO3, were significantly increased in BMSC-ecSOD group as compared with MI or H/SD group. Moreover, this increase was diminished when PI3K inhibitor LY294002 and MEK inhibitor U0126 were used, indicating that BMSC-ecSOD reduced ROS generation and apoptosis in infarcted myocardium via the activation of PI3K-Akt, Erk1/2 and FoxO3a signaling pathways. Previous study reported that Akt and Erk1/2 signaling cascades were able to mediate anti-apoptotic and pro-proliferative effects via the phosphorylation and translocation of FoxO3a [26]. Our study further confirmed that BMSC-ecSOD decreased ROS production and cardiomyocyte apoptosis induced by MI through Akt, Erk1/2 and FoxO3a signaling pathways.
Our study demonstrated the potential clinical perspective of combination of ecSOD gene therapy with BMSCs for cardiac protection. However, it is still unclear about the relationship between prolonged BMSCs survival and increased ecSOD expression after transplantation. Further in-depth and expansive studies are still needed for more conclusive evidence.
In summary, our study firstly demonstrated that the intra-myocardial transplantation of ad-ecSOD transduced BMSCs could reduce oxidative stress and apoptosis, improved cardiac function and ventricular remolding after MI injury, partly through the activation of Akt, Erk and FoxO3a signaling pathways. This may provide a new approach for clinical MI gene therapy.
Acknowledgements
This work was supported by National Funds for Distinguished Young Scientists of China (81325009), National Nature Science Foundation of China (No. 81270168, 81090274, 81090270), (FCAO BWS12J037); Innovation Team Development Grant by China Department of Education (2010CXTD01, IRT1053); National Basic Research Program of China (2012CB518101).
Abbreviations
BMSCs: bone marrow mesenchymal stromal cells; BLI: bioluminescence imaging;
DHE: Dihydroethidium; ecSOD: extracellular superoxide dismutase;
EF: ejection fraction; FoxO3a: forkhead box protein O3a;
FS: fractional shortening; H/SD: hypoxia and serum deprivation;
I/R: ischemia/reperfusion injury; LVEDd: left ventricular end-diastolic diameter;
LVESd: left ventricular end-systolic diameter; MOI: multiplicity of infections;
MI: myocardial infarction; O2-: superoxide anion;
RLU: relative light units; ROS: reactive oxygen species.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bolli R. Oxygen-derived free radicals and myocardial reperfusion injury: an overview. Cardiovasc Drugs Ther. 1991;5(Suppl 2):249-68
2. Dubois-Rande JL, Artigou JY, Darmon JY, Habbal R, Manuel C, Tayarani I. et al. Oxidative stress in patients with unstable angina. Eur Heart J. 1994;15:179-83
3. Li Q, Bolli R, Qiu Y, Tang XL, Murphree SS, French BA. Gene therapy with extracellular superoxide dismutase attenuates myocardial stunning in conscious rabbits. Circulation. 1998;98:1438-48
4. Fattman CL, Schaefer LM, Oury TD. Extracellular superoxide dismutase in biology and medicine. Free Radic Biol Med. 2003;35:236-56
5. Li Q, Bolli R, Qiu Y, Tang XL, Guo Y, French BA. Gene therapy with extracellular superoxide dismutase protects conscious rabbits against myocardial infarction. Circulation. 2001;103:1893-8
6. Bivalacqua TJ, Deng W, Kendirci M, Usta MF, Robinson C, Taylor BK. et al. Mesenchymal stem cells alone or ex vivo gene modified with endothelial nitric oxide synthase reverse age-associated erectile dysfunction. Am J Physiol Heart Circ Physiol. 2007;292:H1278-90 doi:10.1152/ajpheart.00685.2006
7. Abdel-Mageed AS, Senagore AJ, Pietryga DW, Connors RH, Giambernardi TA, Hay RV. et al. Intravenous administration of mesenchymal stem cells genetically modified with extracellular superoxide dismutase improves survival in irradiated mice. Blood. 2009;113:1201-3 doi:10.1182/blood-2008-07-170936
8. Gao E, Lei YH, Shang X, Huang ZM, Zuo L, Boucher M. et al. A novel and efficient model of coronary artery ligation and myocardial infarction in the mouse. Circ Res. 2010;107:1445-53 doi:10.1161/CIRCRESAHA.110.223925
9. Chu Y, Iida S, Lund DD, Weiss RM, DiBona GF, Watanabe Y. et al. Gene transfer of extracellular superoxide dismutase reduces arterial pressure in spontaneously hypertensive rats: role of heparin-binding domain. Circ Res. 2003;92:461-8 doi:10.1161/01.RES.0000057755.02845.F9
10. Cao F, Lin S, Xie X, Ray P, Patel M, Zhang X. et al. In vivo visualization of embryonic stem cell survival, proliferation, and migration after cardiac delivery. Circulation. 2006;113:1005-14 doi:10.1161/CIRCULATIONAHA.105.588954
11. Cao F, Li Z, Lee A, Liu Z, Chen K, Wang H. et al. Noninvasive de novo imaging of human embryonic stem cell-derived teratoma formation. Cancer Res. 2009;69:2709-13 doi:10.1158/0008-5472.CAN-08-4122
12. Fan W, Cheng K, Qin X, Narsinh KH, Wang S, Hu S. et al. mTORC1 and mTORC2 Play Different Roles in the Functional Survival of Transplanted Adipose-derived Stromal Cells in Hindlimb Ischemic Mice via Regulating Inflammation in vivo. Stem Cells. 2012 doi:10.1002/stem.1265
13. Sun D, Shen M, Li J, Li W, Zhang Y, Zhao L. et al. Cardioprotective effects of tanshinone IIA pretreatment via kinin B2 receptor-Akt-GSK-3beta dependent pathway in experimental diabetic cardiomyopathy. Cardiovasc Diabetol. 2011;10:4. doi:10.1186/1475-2840-10-4
14. Liu Y, Ma Y, Wang R, Xia C, Zhang R, Lian K. et al. Advanced glycation end products accelerate ischemia/reperfusion injury through receptor of advanced end product/nitrative thioredoxin inactivation in cardiac microvascular endothelial cells. Antioxid Redox Signal. 2011;15:1769-78 doi:10.1089/ars.2010.3764
15. Luo JD, Wang YY, Fu WL, Wu J, Chen AF. Gene therapy of endothelial nitric oxide synthase and manganese superoxide dismutase restores delayed wound healing in type 1 diabetic mice. Circulation. 2004;110:2484-93 doi:10.1161/01.CIR.0000137969.87365.05
16. Tao L, Gao E, Hu A, Coletti C, Wang Y, Christopher TA. et al. Thioredoxin reduces post-ischemic myocardial apoptosis by reducing oxidative/nitrative stress. Br J Pharmacol. 2006;149:311-8 doi:10.1038/sj.bjp.0706853
17. Rasmusson I, Le Blanc K, Sundberg B, Ringden O. Mesenchymal stem cells stimulate antibody secretion in human B cells. Scand J Immunol. 2007;65:336-43 doi:10.1111/j.1365-3083.2007.01905.x
18. Zhang Z, Li W, Sun D, Zhao L, Zhang R, Wang Y. et al. Toll-like receptor 4 signaling in dysfunction of cardiac microvascular endothelial cells under hypoxia/reoxygenation. Inflamm Res. 2011;60:37-45 doi:10.1007/s00011-010-0232-2
19. Deng W, Bivalacqua TJ, Chattergoon NN, Jeter JR Jr, Kadowitz PJ. Engineering ex vivo-expanded marrow stromal cells to secrete calcitonin gene-related peptide using adenoviral vector. Stem Cells. 2004;22:1279-91 doi:10.1634/stemcells.2004-0032
20. Boomsma RA, Swaminathan PD, Geenen DL. Intravenously injected mesenchymal stem cells home to viable myocardium after coronary occlusion and preserve systolic function without altering infarct size. Int J Cardiol. 2007;122:17-28 doi:10.1016/j.ijcard.2006.11.022
21. Tyukavin AI, Galagudza MM, Mikhailov VM, Venkov AA, McHedlidze G, Burkova NV. Mechanism of targeted migration of mesenchymal stem cells. Bull Exp Biol Med. 2012;153:577-80
22. Mangi AA, Noiseux N, Kong D, He H, Rezvani M, Ingwall JS. et al. Mesenchymal stem cells modified with Akt prevent remodeling and restore performance of infarcted hearts. Nat Med. 2003;9:1195-201 doi:10.1038/nm912
23. van der Bogt KE, Sheikh AY, Schrepfer S, Hoyt G, Cao F, Ransohoff KJ. et al. Comparison of different adult stem cell types for treatment of myocardial ischemia. Circulation. 2008;118:S121-9 doi:10.1161/CIRCULATIONAHA.107.759480
24. Horiuchi M, Tsutsui M, Tasaki H, Morishita T, Suda O, Nakata S. et al. Upregulation of vascular extracellular superoxide dismutase in patients with acute coronary syndromes. Arterioscler Thromb Vasc Biol. 2004;24:106-11 doi:10.1161/01.ATV.0000104240.56460.AB
25. Sheng H, Brady TC, Pearlstein RD, Crapo JD, Warner DS. Extracellular superoxide dismutase deficiency worsens outcome from focal cerebral ischemia in the mouse. Neurosci Lett. 1999;267:13-6
26. Yang JY, Zong CS, Xia W, Yamaguchi H, Ding Q, Xie X. et al. ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nat Cell Biol. 2008;10:138-48 doi:10.1038/ncb1676
Author contact
![]() Corresponding author: Feng Cao, Department of Cardiology, Xijing Hospital, Fourth Military Medical University, Xi'an, Shaanxi, 710032, China. Department of Cardiology, China PLA General Hospital, Beijing, 100853, China. Phone: 86-29-84771024; Fax: 86-29-84775183; e-mail: wind8828com. Co-correspondence: Lize Xiong, M.D., Ph.D. Email: mzkxlzcom.
Corresponding author: Feng Cao, Department of Cardiology, Xijing Hospital, Fourth Military Medical University, Xi'an, Shaanxi, 710032, China. Department of Cardiology, China PLA General Hospital, Beijing, 100853, China. Phone: 86-29-84771024; Fax: 86-29-84775183; e-mail: wind8828com. Co-correspondence: Lize Xiong, M.D., Ph.D. Email: mzkxlzcom.