Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2013; 3(11):903-913. doi:10.7150/thno.5938 This issue Cite
Research Paper
Cardioprotective C-kit+ Bone Marrow Cells Attenuate Apoptosis after Acute Myocardial Infarction in Mice - In-vivo Assessment with Fluorescence Molecular Imaging
Angelique Ale1*, Frank Siebenhaar2*, Katja Kosanke3, Michaela Aichler4, Karin Radrich1, Sina Heydrich2, Matthias Schiemann5,6, Isabella Bielicki3, Peter B. Noel3, Rickmer Braren3, Marcus Maurer2, Axel K. Walch4, Ernst J. Rummeny3, Vasilis Ntziachristos1, Moritz Wildgruber3,7 ![]()
1. Institute for Biological and Medical Imaging, Helmholtz Zentrum München and Technische Universität München, Germany
2. Department of Dermatology and Allergy, Charité - Universitätsmedizin Berlin, Germany
3. Department of Radiology, Klinikum Rechts der Isar, Technische Universität München, Germany
4. Research Unit Analytical Pathology, Institute of Pathology, Helmholtz Zentrum München, Germany
5. Institute for Medical Microbiology, Immunology and Hygiene, Technische Universität München, Germany
6. Clinical Cooperation Groups ''Antigen-Specific Immunotherapy'' and “Immune Monitoring” Helmholtz Zentrum München, Germany
7. International Graduate School of Science and Engineering, Technische Universität München, Garching, Germany
* These authors contributed equally to this work.
Received 2013-1-23; Accepted 2013-4-28; Published 2013-11-2
Abstract
Cardiomyocyte loss via apoptosis plays a crucial role in ventricular remodeling following myocardial infarction (MI). Cell-based therapy approaches using bone marrow derived c-kit+ pluripotent cells may attenuate apoptosis following ischemic injury. We therefore thought to examine the early course of apoptosis following myocardial infarction - in-vivo - and non-invasively determine the effect of c-kit+ bone marrow cells on post-MI remodeling. We studied apoptosis in wild-type Kit+/+, c-kit mutant KitW/KitW-v and KitW/KitW-v mice after cell therapy with bone-marrow derived c-kit+ cells after ischemia-reperfusion injury. Mice were followed by hybrid Fluorescence Molecular Tomography/X-ray Computed Tomography (FMT-XCT) at 6h, 24h and 7 days after ischemia-reperfusion injury using an Annexin V-based fluorescent nanosensor targeting phosphatidylserine. KitW/KitW-v mice showed increased and prolonged apoptosis compared to control Kit+/+ mice while c-kit cell therapy was able to attenuate the altered apoptosis rates. Increased apoptosis was accompanied by severe decline in heart function, determined by cardiac Magnetic Resonance Imaging, and cell therapy was able to rescue the animals from deleterious heart failure. Post-mortem cryoslicing and immunohistochemistry localized the fluorescence signal of the Annexin V sensor within the infarcted myocardium. Flow cytometry of digested infarct specimens identified apoptotic cardiomyocytes as the major source for the in-vivo Annexin V signal.
In-vivo molecular imaging using hybrid FMT-XCT reveals increased cardiomyocyte apoptosis in KitW/KitW-v mice and shows that c-kit+ cardioprotective cells are able to attenuate post-MI apoptosis and rescue mice from progressive heart failure.
Keywords: apoptosis, heart failure, infarction, imaging, reperfusion.
Introduction
Acute myocardial infarction (MI) is associated with a rapid loss of cardiomyocytes. Cell death after ischemic injury can occur by apoptosis, necrosis and to some degree by autophagy [1]. Apoptosis of cardiomyocytes after acute MI is mediated both via the extrinsic pathway, initiated by the binding of death ligands to cell surface receptors, and the intrinsic pathway which is modulated predominantly by mitochondria and the endoplasmatic reticulum. Both pathways lead to an activation of procaspase-3 and other downstream procaspases resulting in apoptosis of cardiomyocytes [2]. The subsequent loss of the contractile unit assembly can result in progressive adverse myocardial remodeling and heart failure. It has been demonstrated that transient inhibition of apoptosis after acute MI improves myocardial remodeling and survival by increasing the number of myofibroblasts in the infarcted area [3].
With the limited capacity of the heart for regeneration, cardiac cell therapy has emerged as a promising tool for the treatment of ischemic heart disease [4, 5]. Hematopoietic stem/progenitor cells (HSPCs) expressing c-kit have the capacity to drive efficient cardiac repair through neovascularisation/angiogenesis and myogenesis[6-8]. 'Cardioprotective' c-kit+ cells establish a proangiogenic milieu in the infarct border zone and promote myofibroblast repair after ischemic damage [7]. C-kit defective mice can be used as a model to study the effects of cell therapy on the healing heart: after reconstitution of c-kit defective KitW/KitW-v mice with c-kit+ bone marrow cells from wild-type littermates, the transplanted cells migrate to the injured myocardium and promote its endogenous repair thereby preventing progressive heart failure [6, 8, 9].
In the current study we investigate apoptosis in the early course after ischemia-reperfusion injury in KitW/KitW-v mice in-vivo using hybrid Fluorescence Molecular Tomography - X-ray Computed Tomography (FMT-XCT) and a phosphatidylserine sensing fluourescent probe based on Annexin V [10]. The in-vivo imaging approach renders quantitative molecular information and correctly localizes the apoptosis to the left ventricular (LV) wall using the similarly acquired XCT data. The novel hybrid FMT-XCT technology significantly improves imaging performance by using a dual prior inversion method. This approach enables to track cardiomyocyte apoptosis in the injured myocardium as well as therapeutic effects of bone marrow derived c-kit+ cells on the healing myocardium at the molecular level with high sensitivity.
Methods
An expanded methods section can be found within the supplementary material.
Mice
Kit+/+ mice, KitW/KitW-v mice and reconstituted Kit+/+ BM → KitW/KitW-v mice, all on a WBB6F1 background were studied by in-vivo FMT-XCT imaging at 6h, 24h and 7 days after ischemia-reperfusion injury. After imaging the mice were sacrificed at each time point for ex-vivo assessment by cryoslicing and histology. Additionally mice were studied by flow cytometry for determination of the cellular source of the molecular apoptosis signal. Cardiac MRI was used to track a subgroup of mice for 21 days following myocardial infarction to track cardiac function, respectively cardiac failure. Myocardial infarction was induced by transient ligation of the left anterior descending artery (LAD) in KitW/KitW-v mice (n=37), corresponding wild-type Kit+/+ mice (n=36) and bone marrow reconstituted Kit+/+ BM → KitW/KitW-v mice (n=34), as previously described [11] (Supplementary Material: Figure S1).
For bone marrow reconstitution KitW/KitW-v mice were injected under aseptic conditions with 107 fresh bone marrow cells obtained from Kit+/+ donors. Successful population of the heart was confirmed by flow cytometry (Supplementary Material: Figure S2). While KitW/KitW-v mice almost lack c-kit+ cells in the heart, 6-8 weeks after bone marrow reconstitution a significant repopulation of c-kit+ cells can be detected. The amount of c-kit+ cells within the heart was comparable between wild-type Kit+/+ mice and bone marrow reconstituted Kit+/+ BM → KitW/KitW-v mice. After myocardial infarction a slightly higher percentage of c-kit+ cells are found within the heart compared to naïve mice without MI (p=0.014, 2-way ANOVA).
Fluorescence Molecular Tomography/X-ray Computed Tomography
In-vivo imaging was performed under isoflurane anesthesia with hybrid FMT-XCT. The hybrid FMT-XCT system is a combination of a Fluorescence Molecular Tomography system consisting mainly of a 750nm diode laser (B&W tek) and a Charge Coupled Device (CCD) camera in trans-illumination set-up, and a micro-CT system (General Electric), both mounted on the same 360° rotating gantry. XCT imaging was performed with a tube voltage of 60kV and a tube current of 450μA. 400 views were acquired over a 360° rotation angle, the exposure time was 400ms. Spatial resolution resulted in an effective isotropic voxel size of 90μm.
The seamless co-registration of FMT and XCT provides accurate anatomical localization of the fluorescence signal. The hybrid reconstruction method developed specifically for this modality obtains accurate reconstructions of the fluorescence distribution using XCT based optical attenuation maps to correct for the influence of optical properties per anatomical region in the forward model calculation and XCT based structural prior information in the inversion.
During FMT acquisition we scanned a grid of ~21 laser positions across the mouse chest for 18 projection angles of the gantry over the full 360° range. For reconstruction of the fluorescence signal we segmented the X-ray CT volume in anatomical regions: heart, lung, bone, liver and remaining tissue. Optical properties and reconstruction parameters are shown in Supplementary Material: Table S1 and were determined as previously described. Signal quantification was calculated based on the FMT-XCT reconstructions on a voxel-based analysis by calculating a voxel as positive for the injected molecular apoptosis probe if the signal intensity (SI) in the voxel was higher than the mean SI in the heart + two standard deviations. Additionally we calculated the fluorescence ratio (FR) of the maximum fluorescence SI observed in the infarcted heart over the average background fluorescence SI in the lung. For the detection of apoptosis we used a fluorescent Annexin V based molecular sensor targeting phosphatidylserine (Annexin-Vivo750, Perkin Elmer, and Excitation: 755nm, Emission 772nm). 4h prior to FMT-XCT imaging mice were injected with 2nmol fluorescent probe/25g mouse. For contrast enhanced XCT a long circulating CT contrast agent (Exitron Nano 12000, Viscover) was injected immediately prior to imaging (100μl /25g mouse).
Cardiac Function Assessment
Cardiac function was assessed in infarcted as well as healthy mice by cardiac Magnetic Resonance Imaging using a clinical 1.5T MRI equipment adapted for small animal imaging (Philips Medical Systems, Best, Netherlands). Short-axis Cine Imaging was performed ECG-triggered under free-breathing conditions. Endocardial and epicardial contours both in the end-systolic and end-diastolic phase were tracked for calculation end-systolic volume (ESV), end-diastolic volume (EDV), stroke volume (SV), ejection fraction (EF) and left ventricular mass. After completion of MRI at day 21 mice were sacrificed and prepared for ex-vivo TTC staining.
Cryoslicing
A subgroup of sacrificed mice were frozen to -80°C and embedded in a mixture of O.C.T. (Optimal Cutting Temperature) medium and India Ink. Cryoslice imaging of the mice was performed using a multispectral imaging system. For ~20 transversal slices per mouse, 250μm apart, we acquired planar RGB images and planar fluorescence images using a filtered white light source and a sensitive CCD camera.
Flow Cytometry
To determine the cellular source of the AnnexinVivo750 signal mice injected with the probe were sacrificed and the hearts processed for further analysis by flow cytometry, as previously described. In brief, infarct tissue and healthy hearts were harvested, minced with fine scissors, and incubated in a cocktail of collagenase I, collagenase XI, DNase I, and hyaluronidase (Sigma- Aldrich) and digested at 37°C for 1 h. Cells were then triturated through nylon mesh and centrifuged (15 min, 500 g, 4°C). Total cardiac cell numbers were determined with Trypan blue (Mediatech, Inc.). For calculation of total cell numbers in the heart, normalization to tissue weight was performed. Single-cell suspensions were subsequently stained with propidium iodine and the following antibodies: FITC - cTroponin-T (Clone 7E7, HyTest Ltd.), PE - c-kit (Clone ACK45, BD Bioscience), Pacific Blue CD11b (Clone M1/70, BioLegend) and PE - Caspase 3 (Clone C92-605, BD Bioscience). We further excluded lineage positive cells by using antibodies against CD90, CD49b, B220, NK1.1. and Ly6G (all from BD Bioscience). For the detection of intracellular caspase-3 and cTroponin-T, fixation and permeabilization of cells was performed prior to antibody staining.
Data were acquired on FACS Aria (BD Biosciences) with a 635nm red laser and 755/LP and 780/60 BP filter configuration to detect AnnexinVivo750.
Histology
For histology hearts were frozen in liquid nitrogen immediately after sacrifice and cut into serial coronal sections of 12µm thickness. Slices were stained with both hematoxylin and eosin (H&E), or DAPI and dUTP nick end labeling (TUNEL) for the detection of apoptosis or used for detection of AnnexinVivo750. Fluorescence images were captured using an Axio Imager Z2 upright microscope system (Carl Zeiss, Oberkochen, Germany). AnnexinVivo750 was detected with a modified Cy 7 filter set (modification: emission filter: 785 LP; AHF Analysetechnik AG, Tübingen, Germany), TUNEL staining with filter set 09 (488009-9901-000, Carl Zeiss), while nuclei were identified with Hoechst 33342 (filter set 01, 488001-9901-000, Carl Zeiss).
Heart Morphometry
For ex-vivo assessment of infarct extension and heart geometry, excised hearts were prepared for staining with TTC. 1mm thick slices, cut perpendicular to the septum were then incubated with 1% Triphenyltetrazolium-Chloride solution (37°C, pH=7,4) for 20min followed by formalin fixation (4% paraformaldehyde, 10min, room temperature). Slices were photographed with a high-resolution digital camera and planimetered using Image J software (http://rsbweb.nih.gov/ij/). Sizes of non-ischemic and ischemic area were calculated for each slice to further calculate the volume of infarcted and remote myocardium.
Data Analysis/Statistics
Statistical analysis was performed using GraphPad Prism 5.0a (GraphPad Software Inc., La Jolla, CA). Results are expressed as mean ± SEM or in case of cardiac function assessment as median and range. For multiple group comparisons analysis of variance (ANOVA), either 1-way or 2-way as appropriate, followed by Bonferroni post-hoc test was performed. All statistical tests were performed two-sided and p-values <0.05 were considered indicating statistical significance.
Results
Apoptosis is increased and prolonged in c-kit deficient KitW/KitW-v mice after MI
To assess and compare Kit+/+ and KitW/KitW-v mice for apoptosis after MI we performed imaging by FMT/XCT with the fluorescent nanosensor Annexin-Vivo750. Mice were studied at 6h, 24h and 7 days after ischemia-reperfusion injury. Apoptosis could be detected in all infarcted mice and was localized to the anterior-lateral wall of the left ventricle on contrast-enhanced XCT (Figure 1A). Quantification of the apoptosis signal was performed using voxel-based analysis and fluorescence ratio (FR). Apoptosis decreased from 6h (17.7 ± 1.5% positive voxels) to 24h (16.1 ± 1.7%) and further at day 7 (11.7 ± 1.5%) post MI in Kit+/+ mice. In contrast KitW/KitW-v mice showed a significantly prolonged and elevated apoptosis at 24h (21.8 ± 2.2%) and 7 days (20.5 ± 0.8%) after MI (p<0.001, Figure 1B).
FMT-XCT enables in-vivo detection and quantification of Annexin-Vivo750 targeting apoptosis in the early course after myocardial infarction. (A) FMT-XCT images of Kit+/+ and KitW/KitW-v mice show the fluorescent apoptosis signal within the left ventricular wall at 6h, 24h and 7 days after myocardial infarction. The molecular apoptosis signal is accurately mapped onto the corresponding anatomic XCT images. L=lung, LV=left ventricle, RV=right ventricle, I/→=Infarct. Color bar at the right side of the images represents signal intensities in arbitrary units (AU). (B) Quantification of the fluorescence molecular signal on a voxel-based analysis reveals significantly higher apoptosis in Kit+/+ mice compared to c-kit deficient KitW/KitW-v mice (p<0.001, 2-way ANOVA), data are given as mean ± SEM. While sustained apoptosis levels are detected in KitW/KitW-v mice over the first 7 days after MI, the apoptosis signal decreases in Kit+/+ mice over time with significantly lower levels at day 7 post MI (p<0.01, Bonferroni post-hoc test). (C) Post-mortem cryoslicing is performed for validation of FMT reconstruction. Transverse Cryoslicing (large panel) confirms the molecular apoptosis signal within the left ventricular myocardium comparable to the corresponding in-vivo FMT-XCT imaging at 6h post MI (upper left panel, arrow points towards corresponding apoptosis signal in FMT-XCT and Cryoslicing image). Fluorescence signal can be further observed in the area of the thoracotomy. (D) Comparison of the fluorescence ratio, calculated as maximum signal observed in the myocardium divided by average signal in the lung, between in-vivo FMT-XCT and post-mortem cryoslicing. Ratios from FMT-XCT data sets were obtained from n=3-6 mice per group (from 4 independent experiments). For calculation of the ratios based on the cryoslicing 6-8 slices per mouse were analyzed, data points show averaged ratios over all analyzed slices obtained from n=2-3 mice. Linear fits of the data show decrease of the molecular apoptosis signal in Kit+/+ mice as compared to sustained apoptosis levels in KitW/KitW-v mice. *P < 0.05
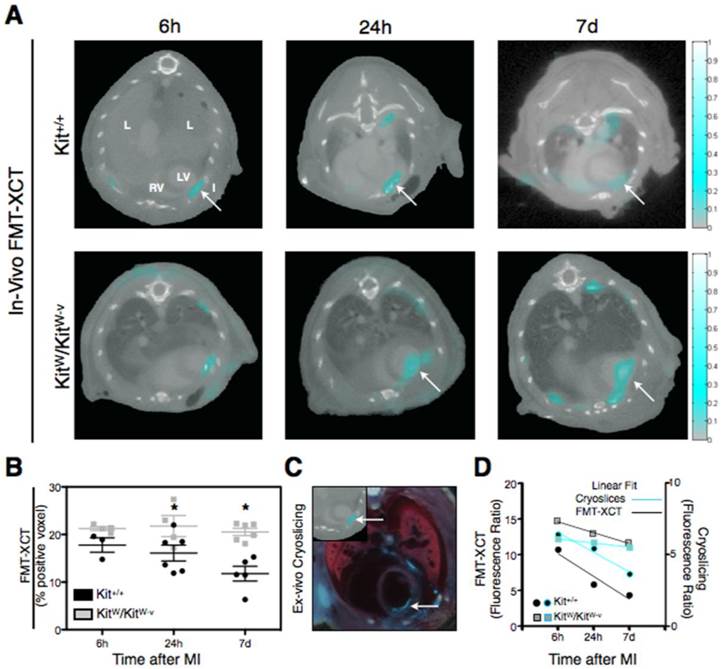
Ex-vivo cryoslicing confirmed the localization of apoptosis to the anterior-lateral wall of the left ventricle (Figure 1C). To compare the in-vivo molecular apoptosis signal obtained by FMT-XCT with ex-vivo cryoslicing we determined the fluorescence ratio of both methods over time. Linear regression of the data shows a decrease of the fluorescence ratio over time, both for in-vivo FMT-XCT measurements as well as ex-vivo cryoslicing (Figure 1D). However the decrease of the fluorescence ratio was more profound in Kit+/+ mice (Slope of linear fit: -3.2 ± 1.0 for FMT-XCT and -1.4 ± 0.2 for Cryoslicing) while the KitW/KitW-v mice retained a high fluorescence ratio within 7 days post MI (Slope of linear fit: -1.5 ± 0.1 for FMT-XCT and -0.3 ± 0.02 for Cryoslicing).
Cell therapy with c-kit+ bone marrow cells attenuates apoptosis in KitW/KitW-v mice
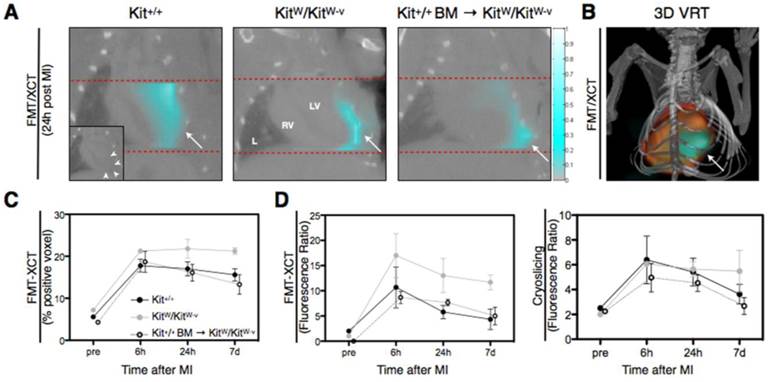
We next assessed the effect of single treatment with 107 fresh bone marrow cells from Kit+/+ donors on post-MI apoptosis in mutant KitW/KitW-v mice. Kit+/+ BM → KitW/KitW-v mice were similarly imaged with FMT/XCT at 6h, 24h and 7 days after ischemia-reperfusion injury and sacrificed at each time point immediately after imaging for further ex-vivo analysis. While a high fluorescence signal for Annexin-Vivo750 was detected in the mutant KitW/KitW-v mice, signal was markedly attenuated in the treated mice (Figure 2A/B), exemplary images from each group at 24h post MI). Signal quantification using a voxel-based analysis revealed a significantly lower molecular Annexin-Vivo750 signal of the treated mice (13.3 ± 2.4% positive voxel) compared to the mutant KitW/KitW-v mice at day 7 post MI (p<0.01, Figure 2C). Determination of the fluorescence ratio both for in-vivo apoptosis assessment (Figure 2D, left panel) and ex-vivo cryoslicing (right panel) show a significant decrease in treated mice as compared to the c-kit deficient KitW/KitW-v mice.
Cell therapy of c-kit deficient mice attenuates apoptosis in KitW/KitW-v mice, as shown by in-vivo FMT-XCT and ex-vivo cryoslicing. (A) Representative coronal views of control Kit+/+, c-kit deficient KitW/KitW-v mice and treated KitW/KitW-v → Kit+/+ mice obtained at 24h post MI show weaker FMT signal in c-kit treated animals compared to KitW/KitW-v mice. L=lung, LV=left ventricle, RV=right ventricle, → = Infarct. Color bar at the right side of the images represents signal intensities in arbitrary units (AU). (B) Volume Rendering Technique (VRT) reconstruction of the hybrid FMT-XCT data sets shows 3D distribution of the molecular apoptosis signal in the area of myocardial infarction. (C) Voxel-based quantification of the Annexin-Vivo750 signal demonstrates that while c-kit deficient KitW/KitW-v mice suffer from increased and prolonged apoptosis after experimental MI, cell therapy with c-kit+ cells obtained from the bone marrow of control Kit+/+ mice attenuates the molecular apoptosis signal in treated KitW/KitW-v → Kit+/+ mice (p<0.01, 2-way ANOVA). (D) Plotted fluorescence ratios for both FMT-XCT (left panel) and post-mortem cryoslicing (right panel) show decrease of detected apoptosis in treated KitW/KitW-v → Kit+/+ mice compared to c-kit deficient KitW/KitW-v mice (FMT-XCT: p<0.01, 2-way ANOVA, Cryoslicing: p<0.05, 2-way ANOVA). Graphs show mean ± SEM, pre=naïve mouse before myocardial infarction. n=17 KitW/KitW-v → Kit+/+ mice studied in 8 independent experiments.
Cellular ex-vivo studies confirm in-vivo apoptosis sensing and identify cardiomyocytes as predominant cells responsible for the fluorescence apoptosis signal
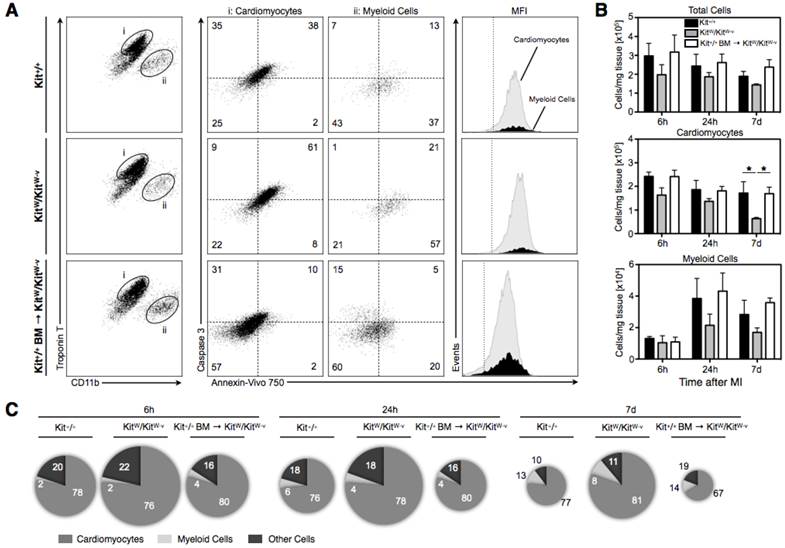
To verify the cellular origin of the molecular apoptosis signal, infarcted animals were injected with Annexin-Vivo750. After in-vivo distribution of the molecular apoptosis probe for 4h animals were sacrificed, and the cell suspensions obtained from digested infarct specimens additionally stained with cardiac Troponin T, Caspase-3, CD11b as well as lineage antibodies and analyzed by flow cytometry. We first excluded propidium iodine positive cells as well as lineage positive cells (data not shown), then separated and visualized the cell populations of cardiomyocytes, CD11b positive myeloid cells as well as remaining other cells. Third, after gating on specific cell populations, the staining intensities of the in vivo injected apoptosis sensor were determined for each gated population (Figure 3A). Percentage of AnnexinVivo750/Caspase-3 double-positive cells was highest in hearts from KitW/KitW-v mice, while the cells suspensions from hearts of Kit+/+ and treated mutant mice showed lower percentages of apoptotic cells (Figure 3A). After normalization for the weight of digested tissue KitW/KitW-v mice revealed a significant loss of viable cardiomyocytes and a markedly reduced infiltration of myeloid cells. Cell therapy with c-kit+ bone marrow cells instead was able to rescue KitW/KitW-v mice from a progressive loss of cardiomyocytes and promoted an increased influx of myeloid cells (Figure 3B). Additionally, the representation of the relative contribution of each cell population to the overall fluorescence signal illustrated that cardiomyocytes were the predominant source of the fluorescent apoptosis signal while myeloid and other cells contributed only negligibly to the overall Annexin-Vivo750 signal at all-time points (Figure 3C). The area of the pie chart indicates the total fluorescence signal in the given group with the highest apoptosis signal in KitW/KitW-v mice and a markedly reduced apoptosis signal in treated mice, comparable to the wild-type Kit+/+ mice (Figure 3C).
Flow cytometry shows that cardiomyocytes contribute predominantly to the Annexin V imaging signal. (A) Dot plots show living cells from Kit+/+ (first row), KitW/KitW-v (second row) and treated KitW/KitW-v → Kit+/+ mice (third row) after staining for CD11b and intracellular cardiac Troponin T and after excluding lin+ and propidium iodine positive dead cells (representative plots from all groups at 24h post MI, n=3-4 mice per group and time point). Cells were identified as cardiomyocytes (TropT+/CD11b-, gate i) and myeloid cells (TropT-/CD11b+, gate ii). Subsequent dot plots show intracellular Caspase-3 staining, and the Annexin-Vivo750 signal after injection in-vivo. Numbers in each quadrant show mean percentages of cells that are positive/negative. For each cell population, representative histograms depict the fluorescence intensities of Annexin-Vivo750. For Annexin-Vivo750 a negative control (animal not injected with sensor), shown as a dashed vertical line, demarcates the border between autofluorescence and positive signal. For Caspase-3 the dashed horizontal lines indicate negative staining using isotype control. Mean fluorescent intensities are shown. (B) Bar graphs display the cell number per 1 mg infarcted heart tissue, calculated as total cell numbers per 1 mg tissue multiplied by percent of gated population. KitW/KitW-v mice suffer from a progressive loss of cardiomyocytes compared to Kit+/+ mice (p<0.01, 2-way ANOVA) while cell therapy was able to prevent progressive myocyte loss in treated mice with significantly higher number of viable cardiomyocytes on day 7 in treated KitW/KitW-v → Kit+/+ mice compared to c-kit deficient KitW/KitW-v mice (*p<0.05, Bonferroni post-hoc test). Graphs show mean ± SEM. (C) Relative contribution to the overall fluorescence for each cell population (cardiomyocytes, myeloid cells and other cells). Pie chart area corresponds to the mean total Annexin-Vivo750 signal (calculated as cell number per 1 mg that is positive for the sensor). The relative contribution of each cell population is shown as percentage of overall signal and pictorially represented by the magnitude of each slice. Cardiomyocytes dominated as cellular contributor to the fluorescence signal.
In-vivo Annexin V signal corresponds to regions of increased cardiomyocyte death
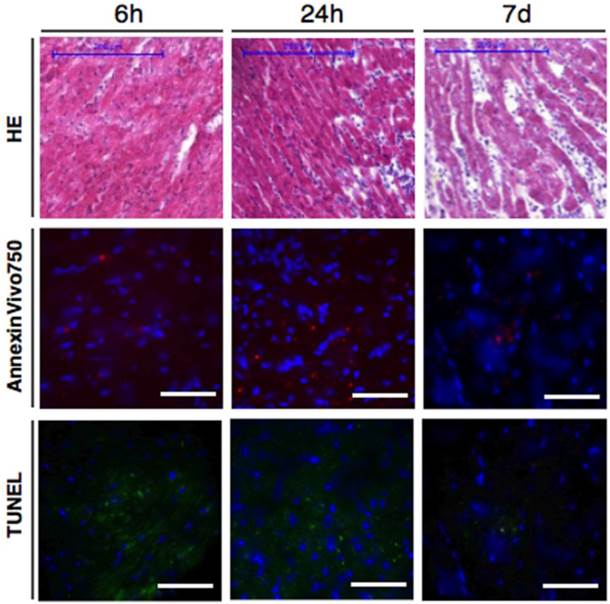
To confirm that the in-vivo injected apoptosis sensor is localized in areas of increased apoptosis within the injured myocardium we performed histology of mice after in-vivo imaging with Annexin-Vivo750 (Figure 4). Annexin-Vivo750 could be localized in areas with increased TUNEL staining and myocardial infarction was confirmed by hematoxilin eosin staining.
Ex-vivo validation of myocardial infarction and apoptosis by histology (representative Hematoxilin Eosin stainings in first row) and fluorescence microscopy. Panels in second row show NIRF signal of the in-vivo injected Annexin-Vivo750. Presence of apoptosis was confirmed by TUNEL staining (green signal, third row). Representative images are shown from 6h, 24h and 7 days following myocardial infarction. Blue channel in fluorescence microscopy images = DAPI. Scale bar in H&E stainings indicates 200μm, scale bar in fluorescence images is 50μm.
Decreased apoptosis in Kit+/+ BM → KitW/KitW-v mice is associated with reduced scar expansion and improved cardiac function at 21 days after ischemia-reperfusion injury
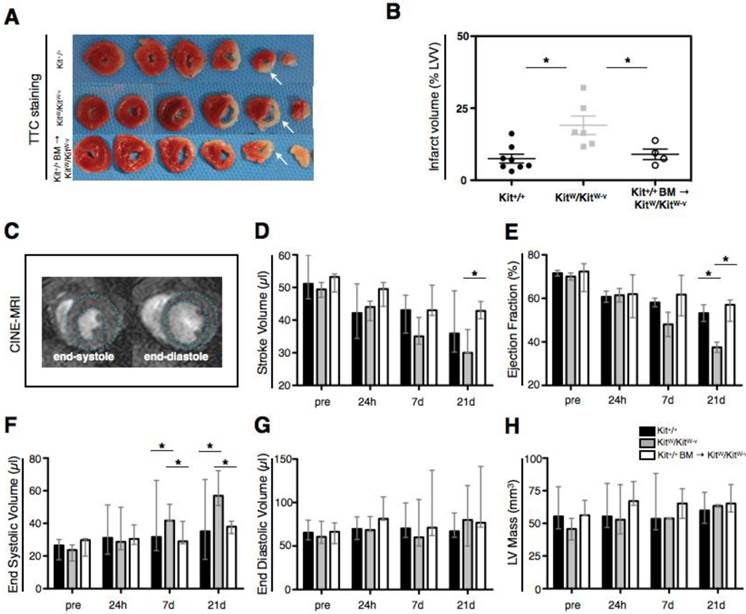
A separate set of Kit+/+, KitW/KitW-v and treated Kit+/+ BM → KitW/KitW-v mice (n=5-8 per group) was studied after ischemia-reperfusion injury for assessment of heart function by cardiac MRI before, 1, 7 and 21 days after MI. Subsequent heart morphometry was performed after sacrifice. While all Kit+/+ mice (8/8) and treated mice (4/4) survived experimental MI until day 21, 3/9 of the KitW/KitW-v died of cardiac failure between day 7 and day 21 post-MI. Ex-vivo morphometry revealed increased LV dilatation and increased infarct expansion in KitW/KitW-v mice as compared to Kit+/+ and cell treated mice (Figure 5A). Infarct volume was markedly increased in KitW/KitW-v (19.1 ± 3.2% of total LVV) compared to Kit+/+ mice (7.5 ± 1.5%, p<0.01) while therapy with c-kit+ bone marrow cells was able to attenuate infarct expansion (9.0 ± 1.8%, p<0.05, Figure 5B). In the period of 21 days after ischemic injury cardiac MRI (Figure 5C) revealed a rapid decline of left ventricular function in KitW/KitW-v mice with decreasing stroke volume (Figure 5D) and ejection fraction (Figure 5E) and increasing end-systolic volume (Figure 5F). Treatment with c-kit+ cells however was able to rescue KitW/KitW-v mice from their cardiomyopathic phenotype (Figure 5D-F). On cardiac MRI there was no significant difference between the entire LV mass and end-diastolic volume between the three groups (Figure 5G, H). Significant differences for comparison of cardiac function parameters between the different time points are provided in Supplementary Material: Table S2.
Increased apoptosis of cardiomyocytes post MI in c-kit deficient KitW/KitW-v mice is accompanied by progressive infarct expansion and progressive heart failure, while c-kit cell therapy rescues KitW/KitW-v mice from their deleterious phenotype. (A) Representative post-mortem Triphenyltetrazolium-Chloride (TTC) stainings show increased infarct expansion and progressive thinning of the left-ventricular wall at 21 days post MI (white arrows). (B) Infarct volume on POD 21 (as % of the entire LV volume) obtained from consecutive 1mm thick TTC sections reveals significantly reduced infarct volume in cell treated mice as compared to c-kit deficient KitW/KitW-v mice (p<0.01, 1-way ANOVA followed by Bonferroni post-hoc test, graph shows mean ± SEM, n=4-8 per group, data are derived from 4 independent experiments). (C) Heart function was determined by cardiac MRI. CINE images show representative short axis views at the end-systole and end-diastole at the midventricular level. KitW/KitW-v mice develop progressive heart failure up to 21 days following ischemia-reperfusion injury, determined by calculating functional parameters such as stroke volume (D), ejection fraction (E), end-systolic volume (F) and end diastolic volume (G) from cardiac MRI datasets. Cell therapy significantly improves the heart function of KitW/KitW-v mice and rescues the mice from progressive heart failure. (H) The entire left-ventricular mass (infarcted and remote myocardium) was comparable between the three groups. Significant differences of functional parameters were obtained by 2-way ANOVA followed by Bonferroni post-hoc test. *P <0.05, p-values indicates statistical significant differences obtained by the post-hoc test. Bar graphs in D-H show median and range of values. Data were obtained from 4 independent experiments.
Discussion
Both clinical and experimental trials using bone marrow derived stem/progenitor cells have received a lot of attention within the last decade as they have been shown to be the only possibility up to now to regenerate ischemically injured myocardium. The most frequently used cell type in these experiments are c-kit+/lin- cells, either derived from the bone marrow or resident cardiac cells. In various murine models using wild-type Kit+/+ compared to c-kit deficient KitW/KitW-v mice it has been shown that after myocardial infarction c-kit+ cells from the bone marrow migrate to the injured myocardium, establish a pro-angiogenic milieu in the infarct border zone, promote neovascularization and the accumulation of myofibroblasts[6, 7]. Additionally it has been shown that cell death assessed by TUNEL staining, is increased in the hearts of KitW/KitW-v mice on POD 3 and 7 after myocardial infarction [7, 9] and that bone marrow reconstitution of KitW/KitW-v mice with cells form Kit+/+ donors induces an up-regulation of various anti-apoptotic genes such as AKT1-3, DAD1, BAX and TIE 1[9]. Several studies suggest that bone marrow derived stem cells inhibit cardiomyocyte apoptosis through paracrine effects, respectively secreting cell survival factors such as AKT and BCL-2, thereby preventing LV remodeling[12, 13].
In this study we investigate the early course of apoptosis following myocardial infarction in Kit+/+, KitW/KitW-v and KitW/KitW-v mice treated with c-kit+ bone marrow cells (Kit+/+ BM → KitW/KitW-v mice) in-vivo. We found that hybrid Fluorescence Molecular Tomography/X-ray Computed Tomography resolved altered apoptosis in KitW/KitW-v mice after ischemia-reperfusion injury using a phosphatidylserine targeted sensor based on Annexin V. Shortly after reperfusion of ischemically injured myocardium apoptotic cardiomyoctes start to bind Annexin V, which is preceded by caspase activation [14]. We show that after ischemia-reperfusion injury KitW/KitW-v mice suffer from increased and prolonged cardiomyocyte apoptosis, which is accompanied by a rapid decline in heart function and in a significant number of animals by fatal cardiac failure. A single-shot cell therapy with bone marrow cells from Kit+/+ donors significantly attenuated apoptosis in the hearts of Kit+/+ BM → KitW/KitW-v mice, led to a decreased loss of cardiomyocytes and rescued the mice from progressive heart failure. Previous studies investigating the effects of c-kit+ cells on the injured myocardium relied predominantly on ex-vivo cellular and molecular analysis [5-9, 15]. We present a new technique that is able to visualize and quantify the externalization of phosphatidylserine into the outer leaflet of the cell membrane, a hallmark of apoptosis, non-invasively and in-vivo. Optical tomographic imaging using fluorescent sensors emitting light in the near-infrared spectrum has been demonstrated to have a high capacity for mapping various molecular and cellular mechanisms in myocardial healing and remodeling [16]. The novel hybrid FMT-XCT technique employed in this study attains important advantages: 1) the co-registered XCT data allow correct mapping of the molecular information onto anatomic maps. 2) The performance of the FMT system is significantly improved by the measured XCT data, the latter employed in a dual prior inversion method previously described [17], which significantly improves the reconstruction accuracy over stand-alone FMT. 3) Attenuation correction of the NIR fluorescence light allows more robust quantification of the molecular signal. Co-registration of the molecular optical signal together with anatomical XCT data permits the setup of a proper model of attenuation correction [18, 19]. The XCT data allow to segment the different organs in the chest to create a 3D model of tissue structure in which each organ is attributed with its' correct optical properties [17, 20]. This model enables to accurately calculate light propagation, thereby correct for tissue attenuation rendering full quantitative data of the molecular signal.
Additional ex-vivo studies were performed to validate the molecular in-vivo imaging signature. Ex-vivo cryoslicing confirms the molecular fluorescence signal in the LV myocardium. Signal localization in the ex-vivo cryoslices does not always match exactly the in-vivo FMT-XCT images as it is not possible to keep the mouse chest geometry identical between in-vivo imaging and the freezing of the animal. Flow cytometry has proven as an elegant tool to determine the cellular source of the fluorescent in-vivo signal as the fluorescence is assessed after in-vivo distribution within the intact animal [11, 21]. Cytometric analysis of digested infarct specimens identified cardiomyocytes as the main source of the Annexin-Vivo750 signal, while CD11b+ myeloid and other remaining cells contributed only negligibly. The fractional contribution of myeloid cells to the Annexin-Vivo750 imaging signal is not surprising, as it is known that phosphatidylserine externalization occurs also in activated macrophages and stressed cells [22]. Interestingly, the course of apoptosis showed similar patterns compared with the apoptosis of cardiomyocytes with increased apoptosis of myeloid cells in KitW/KitW-v mice and significantly attenuated apoptosis in BM → KitW/KitW-v mice. This is supported by the findings reported by Sun et al who found increased numbers of macrophages in the infarct border zone in infarcted mice treated with pluripotent BMCs [23]. Flow cytometry confirmed the course of apoptosis in the infarcted myocardium with significantly higher apoptosis in KitW/KitW-v mice and reduced cardiomyocyte apoptosis and subsequently reduced cell loss in c-kit treated mice. A general weakness of the flow cytometry approach is, that a clear distinction between the infarct core and the infarct border zone is not possible. Therefore, Annexin V positive cells from the border zone contribute similarly to the overall fluorescence signal.
We additionally performed fluorescence microscopy to confirm the localization of the Annexin V sensor within TUNEL positive infarct tissue.
The increased apoptosis levels in KitW/KitW-v mice resulted in larger infarct expansion at 21 days post MI. This result is attributed to the subsequent loss of cardiomyocytes in KitW/KitW-v mice over time. Similarly, Fazel and Cimini noted an increasing infarct size in c-kit mutant mice at 14 days post ischemic injury, with no significant differences in infarct size initially at 24h and at 7 days post MI [6, 7]. The infarct size progression was accompanied by a significant decline in heart function in these mice, while c-kit cell therapy rescued the defective cardiac repair in the treated Kit+/+ BM → KitW/KitW-v mice and preserved the animals from deleterious heart failure.
The mechanism by which c-kit+ bone marrow derived pluripotent cells modulate apoptosis is not yet fully elucidated. Up regulation of anti-apoptotic genes as revealed by Ayach et al is certainly a key factor [9]. Further it has been shown that c-kit+/AT2+ double positive cells increase in the infarct border zone in response to ischemic injury and that these cells inhibit apoptosis of co-cultured cardiomyocytes in-vitro[24]. While transplantation of c-kit+ HSPCs has been shown to effectively decrease apoptosis following myocardial infarction, both endogeneous overexpression of the c-kit ligand SCF as well as systemic application of SCF have proven effective in dampening post-MI apoptosis and thereby preventing progressive heart failure[13, 25]. Intramyocardial administration of SCF in mice improves myocardial homing and enhances the recruitment of c-kit+/lin- cells to the heart[26]. This approach is potentially translatable to clinical cardiology where endovascular administration of SCF could promote the accumulation c-kit+ cardioprotective cells, thereby decreasing the apoptosis of cardiomyocytes following myocardial infarction.
Beside exogenously applied c-kit+ cells, resident cardiac stem cells additionally contribute to myocardial healing. Cardiomyogenesis within the developing heart as well as regeneration after myocardial infarction is mediated via resident c-kit+ cardiac stem cells [27, 28]. Additionally it has been shown that the upregulation of the antiapoptotic gene Akt leads to a significant expansion of a resident cardiac progenitor cell [29]. An increased Akt transcription has been observed after the reconstitution of c-kit mutant mice with BMCs from c-kit+ donors [9]. This suggests that beside direct effects of the exogenously applied c-kit positive BMCs, these cells exert paracrine effects by stimulating the expansion of cardiac resident c-kit+ cells. However, in our setup, exogenous application of c-kit+ BMC was necessary to compensate the altered apoptosis in the c-kit mutant KitW/KitW-v mice, and to rescue the mice from progressive heart failure.
Conclusion
In summary we demonstrate successful in-vivo assessment of the apoptosis response to myocardial infarction and the beneficial effect of cell therapy with c-kit+ bone marrow cells on the healing infarct. The findings may be relevant in the care of patients with myocardial infarction. It has been shown that these patients have a decreased regenerative capacity of bone marrow derived mononuclear cells [30], comparable to that of the mutant KitW/KitW-v mice. Novel therapies using cell-based approaches warrant new diagnostic tools to assess their specific effects as well as their efficacy [31]. The novel molecular imaging approach presented in this report is able to detect apoptosis in the healing myocardium with high sensitivity non-invasively and in-vivo. This technique can be used to rapidly translate major findings in cell and molecular biology to more complex in vivo models. In the dawn of personalized medicine, molecular imaging tools will help physicians to individually control the healing process of myocardial infarction and evaluate novel therapy approaches.
Supplementary Material
Supplementary Methods, Supplementary References, Supplementary Figures and Tables.
Acknowledgements
The authors thank Uli Buchholz (Helmholtz Zentrum München) for help with Fluorescence Microscopy and Drs.Filip Swirski and Matthias Nahrendorf (Center for Systems Biology, Massachusetts General Hospital, Harvard Medical School) for careful revision of the manuscript.
Funding Sources
The work was funded in part by the 'Kommission für Klinische Forschung' (to M.W.), the 'Ernst und Berta Grimmke Stiftung' (to M.W.) and by the Deutsche Forschungsgemeinschaft SFB 824-Z2 (to M.A. and A.K.W.).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Whelan RS, Kaplinskiy V, Kitsis RN. Cell death in the pathogenesis of heart disease: mechanisms and significance. Annu Rev Physiol. 2010;72:19-44 doi:10.1146/annurev.physiol.010908.163111
2. Foo RS, Mani K, Kitsis RN. Death begets failure in the heart. J Clin Invest. 2005;115:565-71 doi:10.1172/JCI24569
3. Hayakawa K, Takemura G, Kanoh M, Li Y, Koda M, Kawase Y. et al. Inhibition of granulation tissue cell apoptosis during the subacute stage of myocardial infarction improves cardiac remodeling and dysfunction at the chronic stage. Circulation. 2003;108:104-9 doi:10.1161/01.CIR.0000074225.62168.68 01.CIR.0000074225.62168.68 [pii]
4. Laflamme MA, Murry CE. Heart regeneration. Nature. 2011;473:326-35 doi:nature10147 [pii] 10.1038/nature10147
5. Fazel S, Tang GH, Angoulvant D, Cimini M, Weisel RD, Li RK. et al. Current status of cellular therapy for ischemic heart disease. Ann Thorac Surg. 2005;79:S2238-47 doi:S0003-4975(05)00367-X [pii] 10.1016/j.athoracsur.2005.02.085
6. Cimini M, Fazel S, Zhuo S, Xaymardan M, Fujii H, Weisel RD. et al. c-kit dysfunction impairs myocardial healing after infarction. Circulation. 2007;116:I77-82 doi:116/11_suppl/I-77 [pii] 10.1161/CIRCULATIONAHA.107.708107
7. Fazel S, Cimini M, Chen L, Li S, Angoulvant D, Fedak P. et al. Cardioprotective c-kit+ cells are from the bone marrow and regulate the myocardial balance of angiogenic cytokines. J Clin Invest. 2006;116:1865-77 doi:10.1172/JCI27019
8. Fazel SS, Chen L, Angoulvant D, Li SH, Weisel RD, Keating A. et al. Activation of c-kit is necessary for mobilization of reparative bone marrow progenitor cells in response to cardiac injury. FASEB J. 2008;22:930-40 doi:fj.07-8636com [pii] 10.1096/fj.07-8636com
9. Ayach BB, Yoshimitsu M, Dawood F, Sun M, Arab S, Chen M. et al. Stem cell factor receptor induces progenitor and natural killer cell-mediated cardiac survival and repair after myocardial infarction. Proc Natl Acad Sci U S A. 2006;103:2304-9 doi:0510997103 [pii] 10.1073/pnas.0510997103
10. Schutters K, Reutelingsperger C. Phosphatidylserine targeting for diagnosis and treatment of human diseases. Apoptosis. 2010;15:1072-82 doi:10.1007/s10495-010-0503-y
11. Nahrendorf M, Sosnovik DE, Waterman P, Swirski FK, Pande AN, Aikawa E. et al. Dual channel optical tomographic imaging of leukocyte recruitment and protease activity in the healing myocardial infarct. Circ Res. 2007;100:1218-25 doi:01.RES.0000265064.46075.31 [pii] 10.1161/01.RES.0000265064.46075.31
12. Uemura R, Xu M, Ahmad N, Ashraf M. Bone marrow stem cells prevent left ventricular remodeling of ischemic heart through paracrine signaling. Circ Res. 2006;98:1414-21 doi:01.RES.0000225952.61196.39 [pii] 10.1161/01.RES.0000225952.61196.39
13. Xu M, Uemura R, Dai Y, Wang Y, Pasha Z, Ashraf M. In vitro and in vivo effects of bone marrow stem cells on cardiac structure and function. J Mol Cell Cardiol. 2007;42:441-8 doi:S0022-2828(06)00964-3 [pii] 10.1016/j.yjmcc.2006.10.009
14. Dumont EA, Reutelingsperger CP, Smits JF, Daemen MJ, Doevendans PA, Wellens HJ. et al. Real-time imaging of apoptotic cell-membrane changes at the single-cell level in the beating murine heart. Nat Med. 2001;7:1352-5 doi:10.1038/nm1201-1352 nm1201-1352 [pii]
15. Xaymardan M, Cimini M, Fazel S, Weisel RD, Lu WY, Martin U. et al. c-Kit function is necessary for in vitro myogenic differentiation of bone marrow hematopoietic cells. Stem Cells. 2009;27:1911-20 doi:10.1002/stem.106
16. Weissleder R, Ntziachristos V. Shedding light onto live molecular targets. Nat Med. 2003;9:123-8 doi:10.1038/nm0103-123 nm0103-123 [pii]
17. Ale A, Ermolayev V, Herzog E, Cohrs C, de Angelis MH, Ntziachristos V. FMT-XCT: in vivo animal studies with hybrid fluorescence molecular tomography-X-ray computed tomography. Nat Methods. 2012;9(6):615-20 doi:nmeth.2014 [pii] 10.1038/nmeth.2014
18. Ale A, Schulz RB, Sarantopoulos A, Ntziachristos V. Imaging performance of a hybrid x-ray computed tomography-fluorescence molecular tomography system using priors. Med Phys. 2010;37:1976-86
19. Schulz RB, Ale A, Sarantopoulos A, Freyer M, Soehngen E, Zientkowska M. et al. Hybrid system for simultaneous fluorescence and x-ray computed tomography. IEEE Trans Med Imaging. 2010;29:465-73 doi:10.1109/TMI.2009.2035310
20. Freyer M, Ale A, Schulz RB, Zientkowska M, Ntziachristos V, Englmeier KH. Fast automatic segmentation of anatomical structures in x-ray computed tomography images to improve fluorescence molecular tomography reconstruction. J Biomed Opt. 2010;15:036006. doi:10.1117/1.3431101
21. Christen T, Nahrendorf M, Wildgruber M, Swirski FK, Aikawa E, Waterman P. et al. Molecular imaging of innate immune cell function in transplant rejection. Circulation. 2009;119:1925-32 doi:CIRCULATIONAHA.108.796888 [pii] 10.1161/CIRCULATIONAHA.108.796888
22. Laufer EM, Reutelingsperger CP, Narula J, Hofstra L. Annexin A5: an imaging biomarker of cardiovascular risk. Basic Res Cardiol. 2008;103:95-104 doi:10.1007/s00395-008-0701-8
23. Sun J, Li SH, Liu SM, Wu J, Weisel RD, Zhuo YF. et al. Improvement in cardiac function after bone marrow cell thearpy is associated with an increase in myocardial inflammation. American journal of physiology Heart and circulatory physiology. 2009;296:H43-50 doi:10.1152/ajpheart.00613.2008
24. Altarche-Xifro W, Curato C, Kaschina E, Grzesiak A, Slavic S, Dong J. et al. Cardiac c-kit+AT2+ cell population is increased in response to ischemic injury and supports cardiomyocyte performance. Stem Cells. 2009;27:2488-97 doi:10.1002/stem.171
25. Xiang FL, Lu X, Hammoud L, Zhu P, Chidiac P, Robbins J. et al. Cardiomyocyte-specific overexpression of human stem cell factor improves cardiac function and survival after myocardial infarction in mice. Circulation. 2009;120:1065-74 doi:CIRCULATIONAHA.108.839068 [pii] 10.1161/CIRCULATIONAHA.108.839068
26. Lutz M, Rosenberg M, Kiessling F, Eckstein V, Heger T, Krebs J. et al. Local injection of stem cell factor (SCF) improves myocardial homing of systemically delivered c-kit + bone marrow-derived stem cells. Cardiovasc Res. 2008;77:143-50 doi:cvm027 [pii] 10.1093/cvr/cvm027
27. Ferreira-Martins J, Ogorek B, Cappetta D, Matsuda A, Signore S, D'Amario D. et al. Cardiomyogenesis in the developing heart is regulated by c-kit-positive cardiac stem cells. Circ Res. 2012;110:701-15 doi:10.1161/CIRCRESAHA.111.259507
28. Limana F, Germani A, Zacheo A, Kajstura J, Di Carlo A, Borsellino G. et al. Exogenous high-mobility group box 1 protein induces myocardial regeneration after infarction via enhanced cardiac C-kit+ cell proliferation and differentiation. Circ Res. 2005;97:e73-83 doi:10.1161/01.RES.0000186276.06104.04
29. Gude N, Muraski J, Rubio M, Kajstura J, Schaefer E, Anversa P. et al. Akt promotes increased cardiomyocyte cycling and expansion of the cardiac progenitor cell population. Circ Res. 2006;99:381-8 doi:10.1161/01.RES.0000236754.21499.1c
30. Heeschen C, Lehmann R, Honold J, Assmus B, Aicher A, Walter DH. et al. Profoundly reduced neovascularization capacity of bone marrow mononuclear cells derived from patients with chronic ischemic heart disease. Circulation. 2004;109:1615-22 doi:10.1161/01.CIR.0000124476.32871.E3 01.CIR.0000124476.32871.E3 [pii]
31. Leuschner F, Nahrendorf M. Molecular imaging of coronary atherosclerosis and myocardial infarction: considerations for the bench and perspectives for the clinic. Circ Res. 2011;108:593-606 doi:108/5/593 [pii] 10.1161/CIRCRESAHA.110.232678
Author contact
![]() Corresponding author: Moritz Wildgruber MD, PhD, Department of Radiology, Klinikum Rechts der Isar, Technische Universität München, Ismaninger Strasse 22, D-81675 München, Germany. Phone: +49-89-4140-2621 Fax: +49-89-4140-4834 email: moritz.wildgruberde
Corresponding author: Moritz Wildgruber MD, PhD, Department of Radiology, Klinikum Rechts der Isar, Technische Universität München, Ismaninger Strasse 22, D-81675 München, Germany. Phone: +49-89-4140-2621 Fax: +49-89-4140-4834 email: moritz.wildgruberde