Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2013; 3(9):618-632. doi:10.7150/thno.6810 This issue Cite
Research Paper
Improved PET Imaging of uPAR Expression Using new 64Cu-labeled Cross-Bridged Peptide Ligands: Comparative in vitro and in vivo Studies
Morten Persson1,2,3,4, Masood Hosseini1,5, Jacob Madsen4, Thomas J. D. Jørgensen6, Knud J Jensen1,5, Andreas Kjaer1,3,4, Michael Ploug1,2, ![]()
1. The Danish-Chinese Center for Proteases and Cancer;
2. Finsen Laboratory, Rigshospitalet & BRIC, Copenhagen Biocenter, Denmark;
3. Cluster for Molecular Imaging, Faculty of Health Sciences, University of Copenhagen, Copenhagen, Denmark;
4. Department of Clinical Physiology, Nuclear Medicine and PET, Center for Diagnostic Investigations, Rigshospitalet, Copenhagen, Denmark;
5. Department of Chemistry, University of Copenhagen, Denmark;
6. Department of Biochemistry and Molecular Biology, University of Southern Denmark, Denmark.
Received 2013-5-31; Accepted 2013-7-21; Published 2013-8-3
Abstract

The correlation between uPAR expression, cancer cell invasion and metastases is now well-established and has prompted the development of a number of uPAR PET imaging agents, which could potentially identify cancer patients with invasive and metastatic lesions. In the present study, we synthesized and characterized two new cross-bridged 64Cu-labeled peptide conjugates for PET imaging of uPAR and performed a head-to-head comparison with the corresponding and more conventionally used DOTA conjugate. Based on in-source laser-induced reduction of chelated Cu(II) to Cu(I), we now demonstrate the following ranking with respect to the chemical inertness of their complexed Cu ions: DOTA-AE105 << CB-TE2A-AE105 < CB-TE2A-PA-AE105, which is correlated to their corresponding demetallation rate. No penalty in the uPAR receptor binding affinity of the targeting peptide was encountered by conjugation to either of the macrobicyclic chelators (IC50 ~ 5-10 nM) and high yields and radiochemical purities (>95%) were achieved in all cases by incubation at 95ºC. In vivo, they display identical tumor uptake after 1h, but differ significantly after 22 hrs, where the DOTA-AE105 uptake remains surprisingly high. Importantly, the more stable of the new uPAR PET tracers, 64Cu-CB-TE2A-PA-AE105, exhibits a significantly reduced liver uptake compared to 64Cu-DOTA-AE105 as well as 64Cu-CB-TE2A-AE105, (p<0.0001), emphasizing that our new in vitro stability measurements by mass spectrometry predicts in vivo stability in mice. Specificity of the best performing ligand, 64Cu-CB-TE2A-PA-AE105 was finally confirmed in vivo using a non-binding 64Cu-labeled peptide as control (64Cu-CB-TE2A-PA-AE105mut). This control PET-tracer revealed significantly reduced tumor uptake (p<0.0001), but identical hepatic uptake compared to its active counterpart (64Cu-CB-TE2A-PA-AE105) after 1h. In conclusion, our new approach using in-source laser-induced reduction of Cu(II)-chelated PET-ligands provides useful information, which are predictive for the tracer stability in vivo in mice. Furthermore, the increased stability of our new macrobicyclic 64Cu-CB-TE2A-PA-AE105 PET ligand is paralleled by an excellent imaging contrast during non-invasive PET scanning of uPAR expression in preclinical mouse cancer models. The translational promises displayed by this PET-tracer for future clinical cancer patient management remains, however, to be investigated.
Keywords: urokinase-type plasminogen activator receptor, CD87, positron emission tomography, biomarker, CB-TE2A, Ly-6, SPR, tumor targeting.
Introduction
Development of novel PET ligands for cancer diagnosis and therapy monitoring has accelerated considerably during the last decade, reflecting the steadily growing knowledge on molecular drivers and biomarkers of pathogenesis in several human cancer diseases [1]. One receptor found to correlate to cancer pathogenesis due to its ability to modulate extracellular matrix degradation, which is considered to impact tumor invasion and metastasis, is the urokinase-type plasminogen activator receptor (uPAR) [2, 3]. This high-affinity receptor for uPA is tethered to the cell surface via a glycosyl-phosphatidylinositol (GPI) membrane anchor [4]. Besides facilitating focal plasminogen activation by receptor-bound urokinase-type plasminogen (uPA) [5], increasing evidence also implicates uPAR in cell adhesion and migration by modulating various cell signaling pathways [6]. Levels of shed forms of uPAR in blood furthermore predict poor prognosis in various cancer types such as breast, lung, prostate and colorectal cancer [7]. Since uPAR is implicated in the remodeling of the extracellular matrix during cancer invasion, it is considered to be a relevant anti-cancer target [5, 8], and various cytotoxic intervention strategies targeting uPAR have accordingly been developed, including “intelligent” pro-drugs requiring proteolytic activation by receptor-bound uPA [9], radiotherapeutics [10, 11], and uPAR-mediated internalization of functionalized nanoparticles [12, 13, 14]. Another virtue from a therapeutic perspective is the assumption that uPAR is readily druggable due to its protein architecture with a large hydrophobic uPA-binding cavity [15], its accessibility on the cell surface, and the mild overt phenotypes associated with its genetic ablation [16, 17]. Importantly, its expression levels in solid tumors are also considered an important biomarker for poor prognosis in various human cancers [7] rendering it an obvious candidate for non-invasive biomarker targeted PET imaging in the assessment of cancer patients with aggressive disease [5].
We have previously developed and characterized 64Cu- [18,19], 68Ga- [20], and 18F-labelled [21] peptide PET probes for non-invasive imaging of uPAR expressing in tumor bearing animals based on the macrocyclic chelators DOTA, NODAGA, and NOTA conjugated to a high-affinity 9-mer linear peptide (denoted AE105), which specifically targets human uPAR [22] i.e. 64Cu-DOTA-AE105 [23], 68Ga-DOTA/NODAGA-AE105 [24], and 18F-AlF-NOTA-AE105. Initially, we obtained very promising results using 64Cu-DOTA-AE105, where a high tumor-to-background was found and a significant correlation between tumor uptake values and levels of uPAR expression were noted, thus validating the in vivo specificity of this particular PET probe [19]. However, the use of DOTA as a general chelator for 64Cu has been challenged in a number of studies, where its relatively low kinetic stability allegedly enables a significant transchelation and/or transmetallation of 64Cu in vivo to superoxide dismustase and metallothionien in liver as well as albumin in the blood circulation. This suboptimal chelation-chemistry consequently causes an elevated non-specific uptake in these compartments [25, 26]. In line with these limitations, we also noted a relatively high uptake in both liver and blood in tumor bearing mice using 64Cu-DOTA in our studies [18, 19].
This inherent in vivo instability of the 64Cu-DOTA complex has prompted an extensive search for improved radionuclide metal chelators with higher in vitro and in vivo stability [27]. First, 1,4,8,11-tetraazacyclotetradecane-1,4,8,11-tetraacetic acid (TETA) demonstrated higher in vivo stability compared to DOTA, but its baseline 64Cu dissociation still resulted in binding to superoxide dismutase in vivo [28]. Second, introduction of several tetra-amine based macrobicyclic chelators, in which an ethylene bridge connects two nonadjacent nitrogens, significantly improve their in vivo stability compared to the monocyclic DOTA and TETA [29-32]. In particular, the use of CB-TE2A as chelator for 64Cu has been used with success in a number of in vivo studies for PET imaging of integrins [33, 34], somatostatin receptor subtype 2 (SSTR2) [35, 36], epidermal growth factor receptor (EGFR) [37] and gastrin-releasing peptide receptor (GRPr) [38, 39], where superior tumor-to-background ratios were reported in all cases compared to the DOTA counterparts. Recently, a CB-TE2A analogue with a propionamide functionalized linker (CB-TE2A-PA) was developed by Boswell et al. [40] (Figure 1). The virtue of this macrobicyclic chelator is that it engages all six coordination sites of Cu2+ thus forming an uncharged Cu complex in the final peptide conjugated PET probe, in contrast to the positively charged and five coordinated complex using the original CB-TE2A.
Chemical structures of the uPAR targeting ligands: CB-TE2A-AE105, CB-TE2A-PA-AE105 and inactive control peptide CB-TE2A-PA-AE105mut, where Phe→Glu and Trp→Glu replacements grossly impairs the AE105-uPAR binding interface defined by the corresponding X-ray crystal structure [15].
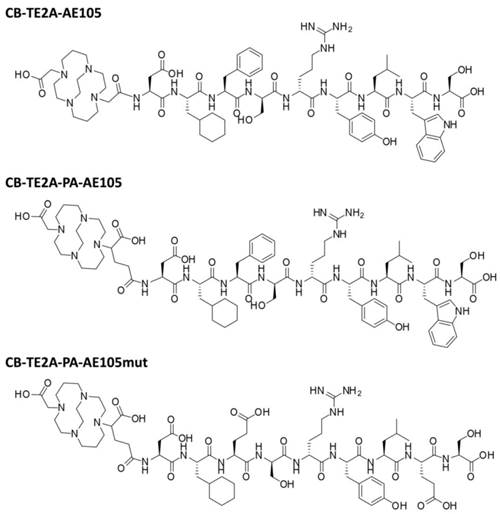
The aims of the present study are to perform the first head-to-head in vitro and in vivo evaluation of the two cross-bridge cyclam chelators CB-TE2A and CB-TE2A-PA, conjugated to AE105 for uPAR PET imaging, and to compare the tumor-to-background ratios with the DOTA-conjugate analogue in vivo in mice carrying human U87MG glioblastoma xenotransplants.
Materials and methods
Chemical reagents
All commercial chemicals were of analytic grade and were purchased from Sigma-Aldrich, CheMatech, Fluka, Iris Biotech GmbH, or Rapp Polymere GmbH, and used without further purification unless otherwise stated. NMR was performed using a Bruker Advance 300 spectrometer and matrix assisted laser desorption mass spectrometry (MALDI-MS) was performed on a Bruker autoflex II TOF/TOF (Bruker Daltonics, Germany). Analytical HPLC was performed on a Dionex UltiMate 3000, using a Phenomenex Gemini 110 Å C18 column (3 µm, 4.6 × 50 mm) with a flow rate of 1.0 mL/min and a 10 minutes linear gradient going from 95% H2O/5% acetonitrile with 0.1% HCOOH to 100% acetonitrile 0.1% HCOOH. Preparative HPLC was performed on a Dionex UltiMate 3000, equipped with a Phenomenex Gemini-NX C18 110 Å column operated at a flow rate of 10.0 mL/min and a 30 minutes linear gradient from 95% H2O/5% acetonitrile with 0.1% TFA to 100% acetonitrile with 0.1% TFA. 2-(4,7,10-tris(2-tert-butoxy-2-oxoethyl)-1,4,7,10-tetraazacyclo-dodecan-1-yl)-acetic acid (DOTA-tris(tBu) ester) was purchased from CheMatech (Dijon, France). 64CuCl2 in 0.1 M HCl was obtained from Risø (DTU, Denmark). Recombinant human uPAR and pro-uPA were expressed in Drosophila S2-cells and purified as described [41, 42].
General methods for solid-phase peptide synthesis
Peptide synthesis was performed using Nα-Fmoc protected amino acids and HBTU/HOBt activation using a polystyrene resin with 2-Cl-Trityl linker (0.61 mmol/g) or Tentagel resin with a trityl linker (0.20 mmol/g). HBTU (3.7 equiv.) HOBt/HOAt (4:1, 4 equiv.), Fmoc-AA-OH (4 equiv.), DIEA (7.2 equiv.) in N-methyl-2-pyrrolidone (NMP) were used. Preactivation (5 min) and couplings (90 min). Fmoc deprotection was performed by two incubations (15 min each) in piperidine/NMP (1:4). Following peptide assembly and coupling to the respective macrocyclic chelator, the resin was washed extensively with NMP and CH2Cl2, before the conjugates were released by 2 hours incubation with TFA/H2O/triethylsilane (95:2.5:2.5). The released peptide was subsequently collected by filtration and pooled with the two washes with TFA used to clean the resin. This TFA-peptide mixture was stirred at room temperature for 24 hours before TFA was removed under a stream of nitrogen and the peptide was precipitated with diethyl ether. The synthesized peptides were dissolved in a minimum amount of H2O/acetonitrile (2:1) before being purified by preparative HPLC and lyophilized.
Synthesis of cross-bridged macrobicyclic chelators
Compound 2: 1,4,8,11-tetraazabicyclo[6.6.2]hexadecane (CB-Cyclam) [29, 43].
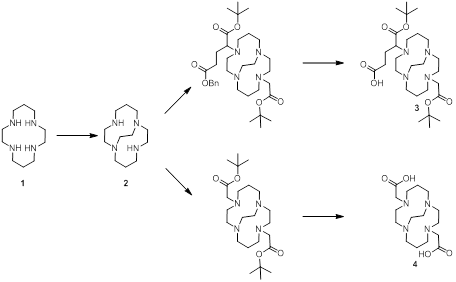
In brief; a solution of 5.5 g 1,4,8,11-tetraazacyclotetradecane (27.5 mmol) [cyclam 1] in 100 mL EtOH was added to 5.5 mL glyoxal (40 % in H2O) and the mixture was left stirring for 16 h. The mixture was evaporated and re-dissolved in 150 mL acetonitrile. To this solution was added 25 mL benzyl bromide and 300 mg tetrabutylammonium iodide (1.8 mmol). The mixture was stirred for 40 h before the N,N'-dibenzylated product was precipitated from the reaction mixture and collected by filtration (10 g, 90%).
A fraction of this precipitate (2.1 g, 5.1 mmol) was dissolved in 50 mL EtOH, added 400 mg NaBH4 (10.5 mmol), stirred at room temperature until the reaction was complete according to LCMS (1 h), and finally quenched by adding 5 mL NH4Cl (aq.), and left stirring overnight. After addition of 100 mL saturated NaHCO3 (aq.), the aqueous phase was extracted with 3 x 100 mL CH2Cl2. The product was isolated by evaporation (2.1 g), dissolved in 20 mL AcOH and 5 % (w/w) Pd/C, degassed (Ar), connected to hydrogen balloon, and stirred overnight (16 h). The spent catalyst was removed by filtration, the volatile solvent was evaporated, and the remaining solid redissolved in 20 mL H2O. The pH of the solution was adjusted to 14 by solid NaOH, extracted with 6 x 25 mL benzene, and the organic phase was finally dried (NaSO4) and evaporated under reduced pressure to produce a clear yellow oil (550 mg, 47%). NMR was in accordance with previous published data [31].
Compound 3: 5-(tert-butoxy)-4-(11-(2-(tert-butoxy)-2-oxoethyl)-1,4,8,11-tetraazabicyclo[6.6.2]hexadecan-4-yl)-5-oxopentanoic acid (protected CB-TE2A-PA). A solution of 800 mg CB-Cyclam (3.53 mmol) [compound 2] in 200 mL acetonitrile received 2 g K2CO3 and 1325 mg (3.71 mmol) α-bromoglutaric acid-1-tert-butylester-5-benzyl ester (in 4 portions over 24 h). 790 mg (4.05 mmol) tert-butyl bromoacetate was subsequently added to this suspension, left stirring at room temperature (4 h) before being acidified with AcOH and concentrated by evaporation. The benzyl group was removed by hydrogenation (THF, Pd/C, 4 bar, 18 h), the catalyst removed by filtration, and the mixture purified by HPLC yielding 1022 mg [compound 3] (55%).
Compound 4: 4,11-bis(carboxymethyl)-1,4,8,11-tetraazabicyclo[6.6.2]hexadecane (CB-TE2A). To a solution of 500 mg CB-Cyclam (2.21 mmol) [2] in 100 mL acetonitrile were added 1.5 g K2CO3 and 900 mg (4.66 mmol) tert-butyl bromoacetate. The mixture was stirred 24 h at room temperature, acidified with AcOH, concentrated by evaporation, and finally purified by HPLC. Yield 382 mg, 38%. The tert-butyl groups were removed prior to use to generate [compound 4] by treatment with TFA at room temperature for 6 hours (important for complete removal).
Synthesis of uPAR-targeting peptides with N-terminal macrocyclic chelators
The uPAR-targeting core peptide AE105 (Asp-Cha-Phe-(D)Ser-(D)Arg-Tyr-Leu-Trp-Ser) and a corresponding inactive variant AE105mut (Asp-Cha-Glu-(D)Ser-(D)Arg-Tyr-Leu-Glu-Ser) were synthesized using Nα-Fmoc protected amino acids and a polystyrene resin with 2-Cl-Trityl linker (0.61 mmol/g). These peptides (0.1 mmol) were conjugated in situ on the resin by adding either 137 mg tert-butyl protected CB-TE2A (0.40 mmol) or 211 mg CB-TE2A-PA (0.40 mmol), which were pre-activated with 137 mg N[(1H-benzotriazol-1-yl)(dimethylamino)methylene]-N-methylmethanamonium hexafluorophosphate-N-oxide (0.36 mmol) and 0.12 mL N,N-diisopropylethylamine (0.72 mmol) in 2 mL N-methyl-2-pyrrolidinone. The mixture was shaken for 2 hours, the peptide conjugates released with TFA/H2O/triethylsilane (95:2.5:2.5), and purified by preparative HPLC using a Phenomenex Gemini-NX C18 110 Å column running at a flow rate of 10 mL/min and a 30 minutes linear gradient from 95% H2O/5% acetonitrile with 0.1% TFA to 100% acetonitrile with 0.1% TFA. Yields of the purified peptides were: CB-TE2A-AE105 (10 mg; MH+: 1550.74 Da/Δm 0.09 Da), CB-TE2A-AE105mut (24 mg; MH+: 1547.81 Da/Δm 0.08 Da ), CB-TE2A-PA-AE105 (16 mg; MH+: 1622.91 Da/Δm 0.06 Da), CB-TE2A-PA-AE105mut (4 mg; MH+: 1547.81 Da/Δm 0.04 Da). DOTA-AE105 was synthesized as previously described [19].
Radiochemistry
Radiolabeling of all uPAR-targeting peptides conjugated to various macrocylic chelators (DOTA, CB-TE2A and CB-TE2A-PA) (Figure 1) with 64Cu were done by adding 2 nmol conjugated peptide to a vial containing 500 µL sodium acetate buffer (0.5 M, pH 8) and 50 µL (approx. 100 MBq) 64CuCl2 in 0.1 M HCl. The solution was heated to 95°C for 30 min. After completion of the synthesis a small aliquot was retrieved for analysis using RP-HPLC as previously described [19]. The reaction mixture was then passed through a C18 SepPak cartridge and free Cu2+ was eluded with 5 mL water. The radiolabeled peptide was finally eluted with 0.5 mL ethanol and diluted in water to reduce the ethanol concentration below 5% before tail vein injection. The radiochemical purity of the final product was determined by RP-HPLC. This protocol typically yielded 95 MBq of radiolabeled peptide with a radiochemical purity of 95-97%. The amount of unlabeled Cu2+ was less than 1% in the final product.
Potency and specificity of the targeting constructs
To assess the penalty on the uPAR-binding affinity of AE105 by conjugation of CB-TE2A or CB-TE2A-PA to its α-amino-group, the inhibitory effect (IC50) of 3-fold dilution series the corresponding conjugates on the interaction between immobilized pro-uPA and 0.5 nM uPAR were assessed by surface plasmon resonance and compared to that of the unmodified AE105. These studies were enabled by choosing running conditions that confer strict mass transport limitations on the binding reaction between pro-uPA and uPAR, as outlined previously in detail [44]. The direct binding between immobilized uPARwt (28 fmol/mm2) and 2-fold dilution series of the various targeting constructs were also measured by surface plasmon resonance using a Biacore T200 operated at 20°C and a flow rate of 50 µL/min running buffer (i.e. 10 mM HEPES, 150 mM NaCl, 3 mM EDTA, and 0.1 % (v/v) surfactant P-20 at pH 7.4). Regeneration of the CM5 sensor chip was accomplished by two consecutive 20 µL injections of 1 M formic acid. Kinetic rate constants (kon and koff) were derived by fitting the double referenced data to a 1:1 binding model using the evaluation software supplied with the instrument.
Cell line and animal model
U87MG glioblastoma cancer cells expressing high levels of uPAR expression [18, 19] were obtained from the American Type Culture Collection (Manassas, VA, USA) and culture media was obtained from Invitrogen Co. (Carlsbad, CA, USA). The cell line was cultured in DMEM supplemented with 10% (v/v) fetal bovine serum and 1% (v/v) penicillin/streptomycin at 37°C and 5% CO2. Xenografts of human U87MG glioblastoma cancer cells was established by injection of 200 µl cells (1 x 108 cells/mL) suspended in 100 µL Matrigel (BD Biosciences, San Jose, CA, USA), subcutaneously into the left and right flank of female NMRI nude mice obtained from Taconic, under anesthesia by Fentanyl (Hypnorm®) / Midazolam (Doricum®). All animal experiments were performed under a protocol approved by the Animal Research Committee of the Danish Ministry of Justice.
MicroPET/CT Imaging
Ten minutes static PET scans were acquired on a small animal PET Focus 120 scanner (Siemens Medical Solutions, Malvern, PA) 1 and 22 hrs post i.v. injection of approximately 10 MBq radiolabeled peptide. PET/CT scanning conditions were all set as described previously [19]. Results were analyzed using Inveon software (Siemens Medical Solutions) and PET data was expressed as percent of injected dose per gram tissue (%ID/g) based on manual region-of-interest drawing on fused PET-CT images for better anatomical localization of organs/tissues of interest.
Biodistribution studies
Biodistribution studies were performed as described [19]. In brief, Nude mice bearing U87MG xenografts received 2-3 MBq of radiolabeled peptide by tail-vein injections. All mice were euthanized after 1 or 22 hrs post tracer injection. Blood, tumor and major organs were collected (wet-weight) and the radioactivity was measured using a γ-counter from Perkin Elmer, MA, USA (n=4 mice/group).
uPAR ELISA
Levels of uPAR in resected U87MG tumors lysates were determined by a validated ELISA as described [19] with one minor improvement. In the present study, we used a mechanical tissue homogenization (Precellys, Bertin Technologies) to pulverize the resected and frozen tumor xenograft as compared to the manual homogenization used previously. All data measurements were performed in duplicates.
Statistical analysis
All quantitative data are expressed as means ± SEM (standard error of the mean) and means are compared using one-way ANOVA. P values ≤ 0.05 were considered statistically significant.
Results
Impact on uPAR-targeting affinity by chemical conjugation to various macrocyclic chelators
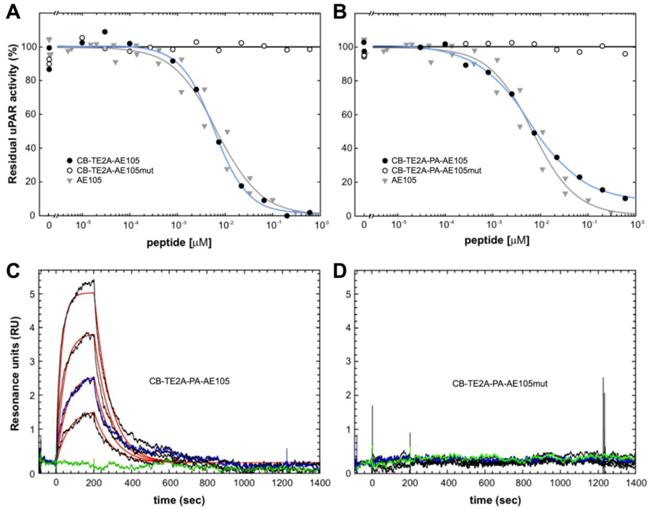
In the present study, we synthesized and purified two new PET derivatives of a 9-mer linear peptide AE105 [22], which targets human uPAR with high affinity and specificity in vivo, by conjugating it to different macrobicyclic chelators using its NH2-terminus (Figure 1). Our synthesis protocol yielded mg quantities of pure products. The impact on uPAR binding by conjugating AE105 to these macrocyclic chelators was subsequently tested by employing two different assay platforms based on surface plasmon resonance. In the first array of analyses, we measured the inhibitory effect (IC50-values) of these peptides in solution on the binding of 0.5 nM uPAR to immobilized pro-uPA. Under such equilibrium binding conditions, we observed no significant differences in the inhibitory potencies of the various AE105 conjugates yielding IC50-values between 5.9 and 6.7 nM (Figures 2A, 2B, and Table 1). No inhibition was observed for the corresponding control peptides at the highest concentrations tested (600 nM). In the second array of analyses, we measured the direct binding of the conjugated peptides to immobilized uPAR on the sensor chip to obtain the kinetic rate constants for these interactions. In general, neither kon nor koff differs profoundly between the different AE105 derivatives giving rise to KD-values in the lower nanomolar range, which is in excellent agreement with the recorded IC50-values (Table 1). Nevertheless, CB-TE2A-PA-AE105 did present a slightly weaker binding to immobilized uPAR (KD 27 nM) as compared to the other derivatives (KD 8-12 nM) indicating that the more bulky propionamid linker in this conjugate may introduce a moderate imperfection of the uPAR•AE105 interface. Importantly, none of the control peptides revealed any specific interaction with immobilized uPAR in this direct binding assay. Representative real-time binding profiles for these kinetic analyses are shown in Figures 2C and 2D for CB-TE2A-PA-AE105 and its corresponding non-binding CB-TE2A-PA-AE105mut.
Binding properties of selected uPAR targeting peptide-based PET probesa
| Ligand | kon (106 M-1s-1) | koff (10-3 s-1) | KD (10-9M) | IC50 (10-9M) |
|---|---|---|---|---|
| AE105 | 0.62 ± 0.34 | 7.6 ± 0.7 | 12 | 6.7 ± 1.6 |
| DOTA-AE105 | 0.46 ± 0.19 | 4.3 ± 0.6 | 9.4 | 6.7 ± 1.0 |
| DOTA-AE105mut | nbb | nbb | nbb | >> 103 |
| CB-TE2A-AE105 | 0.60 ± 0.30 | 4.6 ± 0.1 | 7.7 | 5.9 ± 1.0 |
| CB-TE2A-AE105mut | nbb | nbb | nbb | >> 103 |
| CB-TE2A-PA-AE105 | 0.40 ± 0.13 | 10.8 ± 0.1 | 27 | 6.1 ± 1.0 |
| CB-TE2A-PA-AE105mut | nbb | nbb | nbb | >> 103 |
aThe IC50-values for competing the binding between 0.5 nM uPARwt and immobilized pro-uPA were determined as outlined in Figures 2A and 2B. Kinetic rate constants (kon and koff) for the direct interaction with immobilized uPARwt were determined by surface plasmon resonance using a Biacore T200 as illustrated for CB-TE2A-PA-AE105 and its inactive version in Figure 2C and 2D. The equilibrium dissociation constant (KD) was calculated as koff/kon. bnb: no measurable binding up to 200 nM peptide.
Binding properties of uPAR-targeting PET probes using surface plasmon resonance. The inhibitory profiles of CB-TE2A-AE105 and CB-TE2A-PA-AE105 and their corresponding inactive control peptides are shown in panels A and B, respectively. In these experiments the binding of 0.5 nM recombinant uPAR to immobilized pro-uPA (4300 RU ~ 93 fmol/mm2) are recorded under conditions conferring severe mass transport limitations. The amount of residual, unoccupied uPAR was calculated from the linear association rates in the initial phases of the binding reaction. The IC50-values were calculated by fitting the data to a four-parameter logistic model. The kinetics of the interaction between immobilized uPAR and a 2-fold dilution series of CB-TE2A-AE105 and its corresponding inactive control is shown in panels C and D, respectively. Highest concentration of the peptide tested is 100 nM and 25 nM is measured twice to validate reproducibility. Repeat measurements are shown in blue and buffer runs in green.
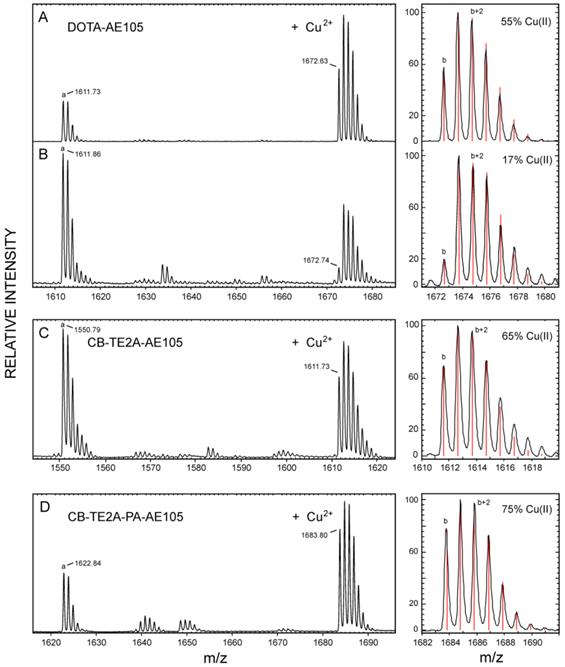
In vitro stability of Cu-complexes of the various uPAR-targeting peptides
Our three purified, active uPAR-targeting peptides DOTA-AE105, CB-TE2A-AE105 and CB-TE2A-PA-AE105 were subsequently loaded with the natural occurring isotopes of Cu(II) (69.2 % 63Cu and 30.8 % 65Cu) by incubating them with 10 mM CuCl2 for 60 minutes at 70°C and the complexes were subsequently purified by RP-HPLC (data not shown). The pure complex preparations were analyzed by MALDI-MS after co-crystallization with α-cyano-4-hydroxycinnamic acid (ACHA) or sinnapinic acid (SA) using identical laser pulse energies and delayed extraction times (130 ns) for recording the MS spectra (shown in Figure 3). Despite our pure peptide-complex preparations are stable as judged after repeat RP-HPLC chromatography, the MALDI-MS spectra recorded in ACHA, nevertheless, clearly reveals the occurrence of a variable amount of demetallated peptide with the following ranking according to stability: CB-TE2A-PA-AE105 (19 %) > CB-TE2A-AE105 (36 %) >> DOTA-AE105 (57 %), where the observed loss of Cu induced by laser desorption from ACHA are shown in brackets (Figure 3B to 3D and Table 2). The difference between the monoisotopic masses of the demetallated species and the corresponding Cu-complexes is 60.9 Da in both matrices for all three constructs corresponding to the mass of 63Cu2+ minus two protons. The isotope envelopes recorded for the three Cu-complexes are, nonetheless, very different and simulations of the isotope distributions (Figure 3, left) estimate the abundances of complexed Cu(II) to be markedly diminished for 63Cu-DOTA-AE105 (17 %) compared to both 63Cu-CB-TE2A-AE105 (65 %) and 63Cu-CB-TE2A-PA-AE105 (75 %). The molecular basis for this instability of the complexes during the matrix-assisted desorption process is presumably an in-source decay, where gas phase collisions in the electron dense MALDI plume during delayed extraction cause a reduction of Cu(II) to Cu(I) by electron transfer [45-47]. The reduction of complexed Cu(II) to Cu(I) by gas phase electron transfer during the MALDI process depends on the recombination energy of Cu(II), which is correlated to the thermodynamic probability of electron uptake. Particularly relevant to our present study is the observation, that high affinity binding to Cu(II) lowers its recombination energy, which accordingly renders Cu(II) tethered to strong chelating groups less prone to electron uptake [47]. Our experimental data in Fig. 3 clearly show, that reduction of Cu(II) to Cu(I) occurs much more readily when bound to DOTA as compared to the two macrobicyclic chelators and we ascribe this higher reactivity to a higher recombination energy, which in turn reflects an attenuated gas-phase binding affinity. In this respect, it should be emphasized that the binding energy of Cu(II) generally is much higher compared to Cu(I), which is illustrated by the differences in the total binding energies between Cu(I) and Cu (II) to three imidazoles in the gas phase (625 kJ/mol and 2000 kJ/mol, respectively [47]). In summary, we envision that reduction of Cu(II) to Cu(I) in the MALDI process substantially weakens the interaction between the copper ion and the macrocyclic chelator and this drives the loss of copper (demetallation) via the Cu(I)-peptide complexes during the internal energy build-up in the MALDI plume. Sinapinic acid is a “softer” matrix yielding ions with lower internal energy and the extent of demetallation of Cu(I)-peptide complexes is thus lower for this matrix. From these experiments it is evident that both our cross-bridged macrobicyclic chelators are by far more resilient towards reduction mediated demetallation than DOTA, with CB-TE2A-PA-AE105 showing the best performance (Figure 3 and Table 2).
Laser-induced in-source reduction of Cu(II) complexed to various macrocyclic chelators.
| Ligand - matrix1 | Demetallation (%)2 | 63Cu(II)/ 63Cu (%)3 | Isotope control (%)4 |
|---|---|---|---|
| DOTA-AE105 | |||
| CHCA | 57.1 ± 8.0 | 15.3 ± 5.2 | 50.9 ± 0.4 |
| SA | 18.1 ± 6.9 | 41.1 ± 4.9 | 51.1 ± 1.2 |
| CB-TE2A-AE105 | |||
| CHCA | 36.2 ± 7.8 | 44.8 ± 3.1 | 52.2 ± 1.6 |
| SA | 23.2 ± 3.3 | 46.0 ± 2.1 | 51.3 ± 0.9 |
| CB-TE2A-PA-AE105 | |||
| CHCA | 19.2 ± 2.9 | 45.6 ± 1.4 | 51.0 ± 0.9 |
| SA | 4.4 ± 1.2 | 46.6 ± 1.5 | 50.0 ± 3.5 |
1The following MALDI matrices were used in this study α-cyano-4-hydroxycinnamic acid (CHCA) and sinapinic acid (SA). 2Estimation of the degree of in-source prompt dissociation of Cu from the macrocyclic chelators. The relative areas of the isotope envelopes representing the demetallated peptide (M) and the Cu-complexed peptide (M+Cu) were calculated for each matrix: [M/M+(M+Cu)]x100. The data are shown for 8 independent sample depositions in CHCA and SA. 3Surrogate marker for the degree of laser-induced Cu(II) to Cu(I) reduction that still persists in the Cu-complexes leaving the source of the mass spectrometer. The relative peak heights corresponding to 63Cu(II)/63Cu were calculated for both matrices (n=8) as [b/(b+(b+1)]x100 in Figure 3. 4Quality control for the above surrogate marker assessment of Cu(II) reduction. The relative peak heights corresponding to the demetallated peptides were measure for both matrices in the same MALDI spectra as [a/(a+(a+1)]x100 in Figure 3 (n=8).
Chemical stability of different macrocyclic Cu-complexes probed by MALDI-MS. MALDI-MS spectra are recorded under identical conditions for Cu-DOTA-AE105 in SA (panel A) and ACHA (panel B), Cu-CB-TE2A-AE105 in ACHA (panel C), and for Cu-CB-TE2A-PA-AE105 in ACHA (panel D). To the left of each panel is shown the best fit obtained for the isotope envelopes using the oxidation state of the bound Cu as the only floating parameter (Cu(II) or Cu(I)). The lower case letters in the MALDI-MS spectra indicates the monoisotopic peaks of the following singly charged molecular species: a) corresponds to the demetallated peptide, [M + H]+; b) corresponds to the Cu-chelated peptide with 63Cu(II), [M - H + 63Cu(II)]+.
64Cu radiolabeling of ligands
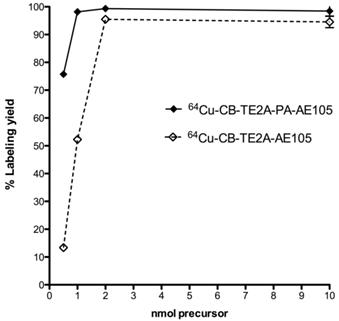
The radiochemical yield obtained for all three uPAR-targeting ligands were >95% using 2 nmol peptid-conjugate and 30 min total reaction time (95oC, pH 8). Using lower amounts of peptide-conjugate significantly reduced the overall yields, with less than 10% yield using 0.5 nmol CB-TE2A-AE105 (Figure 4). The radiolabeled peptides were purified on a Sep-Pak C18 cartridge to remove unbound 64Cu, which resulted in > 95% pure products. The specific activities of the final radiolabeled conjugates were all approximately 50 GBq/µmol.
Radiolabeling uPAR targeting peptides. Radiochemical yields for 64Cu labeling using different amounts of CB-TE2A-AE105 and CB-TE2A-PA-AE105 incubated 30 min at 95°C and pH 8.0. Above 2 nmol conjugated peptide, no significantly difference in labeling yield were observed, whereas CB-TE2A-PA-AE105 gave a higher yield below 2 nmols.
Head-to-head in vivo PET comparison
In the first experiment, a head-to-head in vivo PET imaging study of 64Cu-DOTA-AE105, 64Cu-CB-TE2A-AE105 and 64Cu-CB-TE2A-PA-AE105 for uPAR PET imaging was performed in parallel on a cohort of nude mice inoculated at the same time with human U87MG tumor cells. Such xenografts generally express high levels of uPAR [19]. All three ligands showed a high tumor uptake, with 64Cu-CB-TE2A-AE105, 64Cu-CB-TE2A-PA-AE105, and 64Cu-DOTA-AE105 having uptake values of 3.5±0.8 %ID/g, 4.2±0.6 %ID/g and 4.8±0.7 %ID/g, respectively (Figure 5A) providing a good contrast for imaging tumor localization by PET scanning already after 1h p.i. (Figure 5E). After 22 hrs the relative tumor uptake values declined as expected for the two macrobicyclic derivatives to 2.0±0.3 %ID/g and 1.6±0.3 %ID/g, whereas the DOTA-based probe remained unaltered at 4.5±2.2 %ID/g.
Quantitative ROI analysis from PET imaging of all three uPAR ligands. Tumor uptake values (%ID/g) after 1 hr and 22 hrs are shown in panel A, tumor-to-blood ratio in panel B, liver uptake values in panel C, and tumor-to-liver ratio in panel D. Representative transverse images of 64Cu-DOTA-AE105 and 64Cu-CB-TE2A-AE105, 64Cu-CB-TE2A-AE105 are shown in the lower panel for 1 h p.i.. White arrow indicates tumor. ** p<0.01, *** p<0.001. (n=8 tumors).
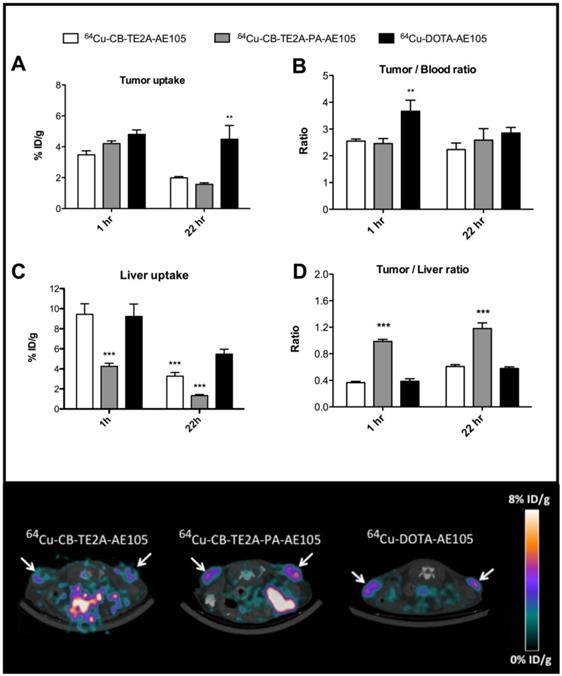
As reported previously by others [26, 34, 38], we also observed a significant difference in the hepatic uptake value for 64Cu-CB-TE2A-AE105 as compared to 64Cu-DOTA-AE105 at 22hrs (p<0.001). This reduced hepatic uptake was, however, even more pronounced for 64Cu-CB-TE2A-PA-AE105 (Figure 5C) indicating that this particular PET tracer exhibits the better in vivo stability of the three tested uPAR ligands. Unexpectedly, no significant differences in tumor-to-liver ratios were found in this study between 64Cu-DOTA-AE105 and 64Cu-CB-TE2A-AE105 after either 1h (0.39±0.10 and 0.37±0.06) or 22hrs (0.58±0.06 and 0.61±0.09), respectively (Figure 5D). In contrast, a significantly improved tumor-to-liver ratio was importantly found for 64Cu-CB-TE2A-PA-AE105 (1h: 0.99±010, p<0.001 and 22hrs: 1.18±0.27, p<0.001) compared to both 64Cu-DOTA-AE105 and 64Cu-CB-TE2A-AE105. 64Cu-DOTA-AE105 presents a significantly higher tumor-to-blood ratio (3.66±1.09, p<0.01) compared to 64Cu-CB-TE2A-AE105 (2.55±0.25) and 64Cu-CB-TE2A-PA-AE105 (2.46±0.59) 1h p.i. (Figure 5B), but this difference is not retained in images recorded after 22 hrs.
Biodistribution
Biodistribution studies were performed in nude mice bearing uPAR-positive U87MG xenografts. 64Cu-CB-TE2A-AE105 and 64Cu-CB-TE2A-PA-AE105 were administered by a single i.v. injection and organs and tissue were collected after 1h and 22 hrs p.i. Figures 6A and 6B show the radionuclide uptake after 1 h and 22 hrs, respectively. Both ligands cleared rapidly from the blood (<2.0 %ID/g after 1 h). Kidney uptake decreased more slowly, with approximate 14 %ID/g after 1 h and 5% ID/g at 22 hrs p.i. Overall, a very similar general uptake was observed for each of the ligands, although 64Cu-CB-TE2A-PA-AE105 has a tendency to clear faster from non-target tissues.
Biodistribution profiles of uPAR PET tracers. Results for 64Cu-CB-TE2A-AE105 and 64Cu-CB-TE2A-PA-AE105 are shown after 1 h (panel A) and 22 hrs (panel B). A significantly higher tumor-to-liver ratio was found for 64Cu-CB-TE2A-PA-AE105 after both 1h (p>0.01) and 22 h (p>0.001) (panel C). (n=4 mice/group).
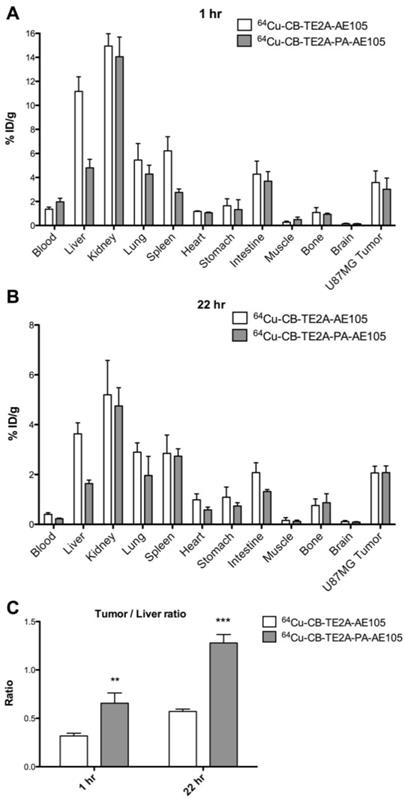
A 2-fold increase in hepatic uptake was consistently observed after 1 h and 22 hrs for 64Cu-CB-TE2A-AE105 (1 h: 11.2±1.2 %ID/g, 22 hrs: 3.6±0.4 %ID/g) compared to 64Cu-CB-TE2A-PA-AE105 (1 h: 4.8±0.2 %ID/g, 22 hrs: 1.6±0.1 %ID/g). Importantly, this property improves the tumor-to-liver ratio significantly for 64Cu-CB-TE2A-PA-AE105 (1 h: p<0.01 and 22 hrs: p<0.001, Figure 6C), which is excellently aligned with the previous PET scanning profiles recorded for a similar experiment (Fig. 5E) thus validating the improved in vivo partitioning of this particular uPAR-targeting PET tracer. All data from the biodistribution study are shown in Table 3.
Biodistributions of new cross-bridged uPAR PET tracersa .
| 64Cu-CB-TE2A-AE105 | 64Cu-CB-TE2A-PA-AE105 | |||
|---|---|---|---|---|
| Organ | 1 h (%ID/g) | 22 hrs (%ID/g) | 1 h (%ID/g) | 22 hrs (%ID/g) |
| Blood | 1.4 ± 0.2 | 0.4 ± 0.1 | 2.0 ± 0.3 | 0.2 ± 0.1 |
| Liver | 11.2 ± 1.2 | 3.6 ± 0.4 | 4.8 ± 0.7 | 1.6 ± 0.1 |
| Kidney | 15.0 ± 1.0 | 5.2 ± 1.4 | 14.0 ± 1.6 | 4.8 ± 0.7 |
| Lung | 5.5 ± 1.4 | 2.9 ± 0.4 | 4.3 ± 0.7 | 2.0 ± 0.8 |
| Spleen | 6.2 ± 1.2 | 2.9 ± 0.7 | 2.8 ± 0.3 | 2.7 ± 0.3 |
| Heart | 1.2 ± 0.1 | 1.0 ± 0.2 | 1.1 ± 0.1 | 0.6 ± 0.1 |
| Stomach | 1.7 ± 0.6 | 1.1 ± 0.4 | 1.3 ± 0.8 | 0.7 ± 0.1 |
| Intestine | 4.3 ± 1.1 | 2.1 ± 0.4 | 3.7 ± 0.8 | 1.3 ± 0.1 |
| Muscle | 0.3 ± 0.1 | 0.2 ± 0.1 | 0.5 ± 0.2 | 0.1 ± 0.1 |
| Bone | 1.1 ± 0.4 | 0.8 ± 0.3 | 0.9 ± 0.1 | 0.9 ± 0.3 |
| Brain | 0.2 ± 0.1 | 0.1 ± 0.1 | 0.1 ± 0.1 | 0.1 ± 0.1 |
| Tumor | 3.6 ± 1.0 | 2.1 ± 0.3 | 3.0 ± 0.9 | 2.0 ± 0.3 |
| Tumor-to-nontumor ratios | ||||
| Tumor-to-blood | 2.6 ± 0.5 | 5.2 ± 0.5 | 1.5 ± 0.4 | 9.3 ± 1.7 |
| Tumor-to-liver | 0.3 ± 0.1 | 0.6 ± 0.1 | 0.7 ± 0.3 | 1.3 ± 0.2 |
| Tumor-to-kidney | 0.2 ± 0.1 | 0.4 ± 0.1 | 0.2 ± 0.1 | 0.5 ± 0.1 |
| Tumor-to-intestine | 0.8 ± 0.1 | 1.0 ± 0.2 | 0.9 ± 0.3 | 1.6 ± 0.2 |
| Tumor-to-muscle | 1.4 ± 0.2 | 17.5 ± 8.0 | 7.5 ± 5.0 | 22.2 ± 15.3 |
aThe biodistributions profiles and tumor to non-tumor ratios for 64Cu-CB-TE2A-AE105 and 64Cu-CB-TE2A-PA-AE105 were established in nude mice bearing U87MG xenografts by gamma counting of resected organs.
In vivo specificity study
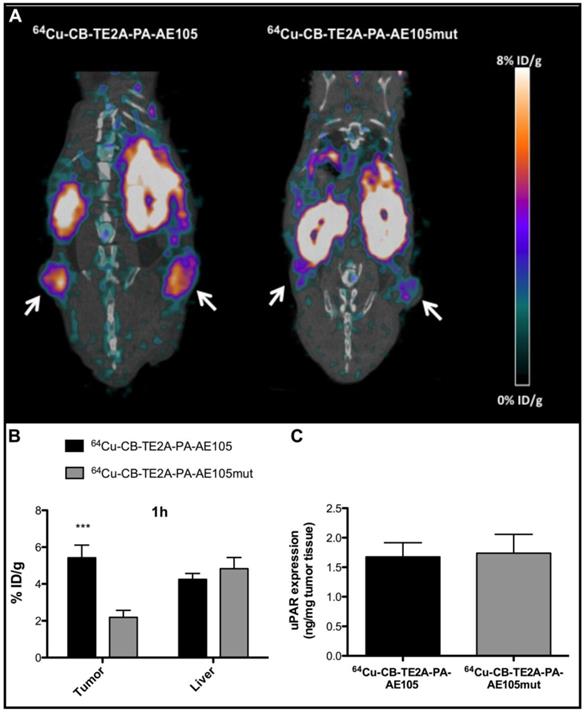
To demonstrate that the tumor uptake of the best performing PET-tracer (i.e. 64Cu-CB-TE2A-PA-AE105) is indeed caused by a specific targeting of uPAR rather than passive accumulation of free 64Cu2+, we measured the in vivo uptake by PET scanning of a non-binding version of our targeting probe labeled with 64Cu to the same specific activity. The successful abrogation of the targeting affinity of the designed control peptide towards uPAR was confirmed by surface plasmon resonance studies revealing the IC50 to be reduced from 5-10 nM to >> 1µM for the dysfunctional peptide and no direct binding can accordingly be measured (Figure 2 and Table 1). When parallel PET/CT images of this control PET probe were reconstructed for 1 h and compared to the corresponding active uPAR-targeting probe, a significantly reduced tumor uptake was found (p<0.001, Figure 7B). This effect is clearly visualized by the reconstructed PET images (Figure 7A). The tumor uptake value was 5.4±0.7 %ID/g for 64Cu-CB-TE2A-PA-AE105 after 1 h. In contrast, the tumor uptake value in mice injected in parallel with the non-binding version 64Cu-CB-TE2A-PA-AE105mut having the same specific radioactivity was only 2.2±0.4 %ID/g after 1 h. Importantly, the hepatic uptake values of these active and inactive PET-tracers were comparable after 1 h (Figure 7B). Noteworthy, the hepatic uptake values (representing a non-target organ) were significantly lower for the 64Cu-CB-TE2A-PA-AE105 PET-tracer compared to both 64Cu-DOTA-AE105 and 64Cu-CB-TE2A-AE105 (Figure 5C). Moreover, the difference in tumor uptake was clearly not related to differences in uPAR expression between the two groups as ELISA measurements on resected tumors lysates revealed comparable uPAR levels (Figure 7C). Combined these data unambiguously confirms the uPAR-targeting specificity of 64Cu-CB-TE2A-PA-AE105.
Specificity of uPAR imaging by 64Cu-CB-TE2A-PA-AE105 targeting. Representative sagittal PET/CT images for 64Cu-CB-TE2A-PA-AE105 and its corresponding inactive control peptide 64Cu-CB-TE2A-PA-AE105mut are shown after 1h (panel A). White arrows indicate location of the tumors. The corresponding quantitative ROI analysis of tumor and liver are shown (panel B). Levels of uPAR expression determined by ELISA in resected tumor lysates from mice sacrificed after termination of each imaging experiment are shown in (panel C). (n = 6 tumors (3 mice in each group). *** p<0.001.
Discussion
Our present study reports on the synthesis and characterization of two new 64Cu labeled cross-bridged peptide antagonists that specifically targets human uPAR. Importantly, our data reveal that the new 64Cu-CB-TE2A-PA-AE105 PET-tracer in our experiments exhibits a much improved tumor-to-liver ratio compared to both 64Cu-CB-TE2A-AE105 and 64Cu-DOTA-AE105, which is in accordance with the in vitro stabilities we measured by MALDI-MS.
The rational design of these uPAR-targeting PET-tracers was originally guided by our detailed knowledge on the structure-functional relationships that drive the biochemistry of this receptor - as reviewed recently [48]. Accordingly, we observed no significant impact on the affinity and specificity of the uPAR-targeting core-peptide by conjugation to any of the macrocyclic chelators employed in this study. Nonetheless, in the direct binding assay CB-TE2A-PA-AE105 did present a slightly weaker affinity to immobilized uPAR (KD 27 nM) as compared to the other derivatives (KD 8-12 nM) indicating that the propionamid linker in this particular conjugate may introduce a moderate imperfection of the uPAR•AE105 binding interface. Despite this notion, we did not observe a similar difference in the IC50-values for the solution phase competition experiments (Table 1) emphasizing the similar potencies of the various peptide conjugates. The differences observed in the direct binding to the immobilized receptor could therefore pertain to changes in the conformation of the inherently flexible uPAR [49] introduced by the immobilization chemistry, which is sensed only by the more bulky peptide conjugate i.e. CB-TE2A-PA-AE105. Importantly, none of the control peptides revealed any specific interaction with immobilized uPAR in this direct binding assay further substantiating their usefulness as reporters of non-specific probe accumulation in vivo.
Radionuclide labeling of these uPAR targeting ligands with 64Cu2+ was completed within 30 min at 95°C, pH 8 (yields > 95 %) using as little as 2 nmol conjugated peptide (Figure 4). This reaction kinetic is somewhat faster than those generally reported, where 1 h to 2 hrs of incubation is typically required for completion [40, 50]. However, the relatively harsh radiolabeling conditions still limits radiolabeling to relatively stable peptide conjugates excluding radiolabeling of proteins and antibodies. To overcome these limitations, a second-generation cross-bridge chelators incorporating methanephosphoric acid pendent arms (CB-TE2P and CB-TE1A1P) was recently published [51, 52]. These chelators enable copper radiolabeling at room temperature within 1 h and data shows improved biodistribution compared with CB-TE2A [53]. However, no conjugation to any proteins using these chelators has yet been reported.
The introduction of a propionamide arm in CB-TE2A-PA neutralizes the charge of the resultant copper complex in contrast to the original CB-TE2A chelator [31, 40], where one of the pendant carboxylates is used for conjugation to the peptide. In our present study, we find that this modification does not affect the tumor uptake value, which is comparable to that of the CB-TE2A derivative, but importantly we observe a significantly reduced uptake of the CB-TE2A-PA derivative in non-target organs such as the liver (p<0.001). Since hepatic accumulation of copper is a valid surrogate marker for compromised in vivo stability [26, 53] we propose that CB-TE2A-PA-AE105 possess a superior in vivo stability compared to CB-TE2A-AE105. Both our new macrobicyclic PET-tracers targeting uPAR do, nevertheless, exhibit a higher stability than 64Cu-DOTA-AE105 (Figure 5C). In line with this proposition is our new in vitro stability data based on MALDI in-plume reduction (Figure 3, Table 2) and a previous report, which states that positively charged 64Cu-labeled cyclam and Et-cyclam chelates generally exhibit a higher liver accumulation compared to the corresponding neutral complex, 64Cu-labeled 3,9-DOC in rats [25].
Somewhat surprisingly, we found no significant differences in the tumor-to-liver ratios between 64Cu-CB-TE2A-AE105 and 64Cu-DOTA-AE105 (figure 5D), despite a significant reduction in the hepatic uptake of 64Cu-CB-TE2A-AE105 at 22 hrs (Figure 5C). The well-established lower kinetic stability of the copper-DOTA complex in vivo [25, 26] may, however, represent a major confounding factor in such experiments. The possible contribution from un-bound 64Cu during PET imaging is illustrated by high and persistent tracer accumulations in e.g. U87MG xenotransplants after tail vein injection of free 64Cu2+; 3.5±0.4 %ID/g and 3.8±0.4 %ID/g at 1 h and 22 hrs, respectively [54]. Accordingly, we speculate that the commonly observed high tumor uptake of 64Cu-DOTA-conjugated ligands in particular, that we and others [51, 52] observe in mice 22 hrs after tracer administration, represents a composite signal from specific receptor targeting as well as non-specific accumulation of free 64Cu in the tumor tissue. A direct comparison of tumor uptake values in mice between 64Cu-labeled DOTA conjugates and new metal chelating systems such as the cross-bridged conjugates used in this study should therefore be interpreted with some precaution due to the different magnitude of confounding effects inherent to tracer leakage in vivo from the employed macrocyclic chelators.
At present only very few publications address the in vivo use of CB-TE2A-PA. In one study Liu et al [43] synthesize CB-TE2A-PA conjugated to cRGDyK (i.e. 64Cu-CB-TE2A-PA-c(RGDyK)) and used this PET-probe for imaging of αvβ3 integrin expression in PC3 prostate cancer xenografts in vivo, but a parallel comparison to the corresponding CB-TE2A conjugate was regrettably not attempted [43]. Nonetheless, by comparing the reported liver uptake data in mice from Liu et al. [43], (1 h: 1.4±0.1 %ID/g) with the liver uptake data of 64Cu-CB-TE2A-c(RGDyK) published by Anderson et al [55], (1 h: 3.9±2.0 %ID/g) a significantly reduced liver uptake (2.8 fold, p<0.05) was indeed seen between the two chelators. Although this may be considered circumstantial evidence, it is intriguing that we find a similar difference in the hepatic uptake in our controlled pairwise comparison of these chelators using a different receptor targeting ligand (2.2 fold, p<0.001).
In conclusion, the present study selects 64Cu-CB-TE2A-PA-AE105 as the most promising peptide tracer for the specific detection of human uPAR expression in xenotransplanted mouse model systems by non-invasive PET imaging among those 64Cu- and 68Ga-labeled derivatives of AE105 that we have tested [18-20]. It should, nevertheless, be emphasized that the confounding effects from the reduced stability of Cu-DOTA complexes that generally affects preclinical imaging in mouse model systems, do not necessarily translate directly to the clinical situation. Accordingly, the use of 64Cu-DOTA-TATE for PET imaging of somatostatin receptor expression in neuroendocrine cancer patients provides very high quality images with a high tumor-to-background contrast and low non-specific hepatic uptake [56]. Selection of the optimal PET tracer for future clinical imaging of uPAR expression in patients is therefore a composite process involving multiple factors and considerations.
Acknowledgements
We thank Gitte Juhl Funch for expert technical assistance and John Post for graphical artwork. This work was supported by The Danish National Research Foundation (Centre for Proteases and Cancer), Danish Medical Research Council, the Danish National Advanced Technology Foundation, the Novo Nordisk Foundation, the Lundbeck Foundation, and the A.P. Moeller Foundation and the Svend Andersen Foundation.
Abbreviations
CB-TE2A: 4,11-bis(carboxymethyl)-1,4,8,11-tetraazabicyclo[6.6.2]hexadecane;
CB-TE2A-PA: 4-carboxymethyl-11-(1,3-dicarboxypropyl)-1,4,8,11-tetraazabicyclo[6.6.2]hexadecane;
DIEA: N,N-diisopropylethylamine;
DOTA: 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid;
Fmoc: 9-fluorenylmethyloxycarbonyl;
HBTU: N[(1H-benzotriazol-1-yl)(dimethylamino)-methylene]-N-methylmethanamonium hexafluorophosphate N-oxide;
HOAt: 3-hydroxy-3H-1,2,3-triazolo[4,5-b]pyridine or 1-hydroxy-7-azabenzotriazole;
HOBt: 1-hydroxybenzotriazole;
PET: positron emission tomography;
uPAR: urokinase-type plasminogen activator receptor.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Kramer-Marek G, Capala J. The role of nuclear medicine in modern therapy of cancer. Tumour Biol. 2012;33:629-40
2. Rømer J, Nielsen B S, Ploug M. The urokinase receptor as a potential target in cancer therapy. Curr. Pharm. Des. 2004;10:2359-76
3. Mekkawy AH, Morris DL, Pourgholami MH. Urokinase plasminogen activator system as a potential target for cancer therapy. Future Oncol. 2009;5:1487-99
4. Ploug M, Rønne E, Behrendt N, Jensen A L, Blasi F, Danø K. Cellular receptor for urokinase plasminogen activator. Carboxyl-terminal processing and membrane anchoring by glycosyl-phosphatidylinositol. J Biol Chem. 1991;266:1926-33
5. Kriegbaum M C, Persson M, Haldager L, Alpízar-Alpízar W, Jacobsen B, Gårdsvoll H, Kjaer A, Ploug M. Rational targeting of the urokinase receptor (uPAR): development of antagonists and non-invasive imaging probes. Curr. Drug Targets. 2011;12:1711-28
6. Smith H W, Marshall C J. Regulation of cell signalling by uPAR. Nat. Rev. Mol. Cell. Biol. 2010;11:23-36
7. Rasch MG, Lund IK, Almasi CE, Høyer-Hansen G. Intact and cleaved uPAR forms: diagnostic and prognostic value in cancer. Front. Biosci. 2008;13:6752-62
8. Mazar AP.Urokinase plasminogen activator receptor choreographs multiple ligand interactions. implications for tumor progression and therapy. Clin Cancer Res. 2008;14:5649-55
9. Su Y, Ortiz J, Liu S, Bugge TH, Singh R, Leppla SH, Frankel AE. Systematic urokinase-activated anthrax toxin therapy produces regressions of subcutaneous human non-small cell lung tumor in athymic nude mice. Cancer Res. 2007;67:3329-36
10. Knör S, Sato S, Huber T, Morgenstern A, Bruchertseifer F, Schmitt M, Kessler H, Senekowitsch-Schmidtke R, Magdolen V, Seidl C. Development and evaluation of peptidic ligands targeting tumour-associated urokinase plasminogen activator receptor (uPAR) for use in alpha-emitter therapy for disseminated ovarian cancer. Eur J Nucl Med Mol Imaging. 2008;35:53-64
11. Persson M, Rasmussen P, Madsen J, Ploug M, Kjaer A. New peptide receptor radionuclide therapy of invasive cancer cells: in vivo studies using 177Lu-DOTA-AE105 targeting uPAR in human colorectal cancer xenografts. Nucl Med Biol. 2012;39:962-9
12. Yang L, Peng XH, Wang YA, Wang X, Cao Z, Ni C, Karna P, Zhang X, Wood WC, Gao X, Nie S, Mao H. Receptor-targeted nanoparticles for in vivo imaging of breast cancer. Clin Cancer Res. 2009;15:4722-32
13. Lee GY, Qian WP, Wang L, Wang YA, Staley CA, Satpathy M, Nie S, Mao H, Yang L. Theranostic nanoparticles with controlled release of gemcitabine for targeted therapy and MRI of pancreatic cancer. ACS Nano. 2013;7:2078-89
14. Hansen L, Unmack Larsen EK, Nielsen EH, Iversen F, Liu Z, Thomsen K, Pedersen M, Skrydstrup T, Nielsen NC, Ploug M, Kjems J. Targeting of peptide conjugated magnetic nanoparticles to urokinase plasminogen activator receptor (uPAR) expressing cells. Nanoscale. 2013
15. Llinas P, Le Du M H, Gårdsvoll H, Danø K, Ploug M, Gilquin B, Stura E A, Ménez A. Crystal structure of the human urokinase plasminogen activator receptor bound to an antagonist peptide. EMBO J. 2005;24:1655-63
16. Connolly B M, Choi E Y, Gårdsvoll H, Bey AL, Currie BM, Chavakis T, Liu S, Molinolo A, Ploug M, Leppla SH, Bugge TH. Selective abrogation of the uPA-uPAR interaction in vivo reveals a novel role in suppression of fibrin-associated inflammation. Blood. 2010;116:1593-603
17. Ploug M, Plesner T, Rønne E, Ellis V, Høyer-Hansen G, Hansen NE, Danø K. The receptor for urokinase-type plasminogen activator is deficient on peripheral blood leukocytes in patients with paroxysmal nocturnal hemoglobinuria. Blood. 1992;79:1447-55
18. Li ZB, Niu G, Wang H, He L, Yang L, Ploug M, Chen X. Imaging of urokinase-type plasminogen activator receptor expression using a 64Cu-labeled linear peptide antagonist by microPET. Clin. Cancer Res. 2008;14:4758-66
19. Persson M, Madsen J, Østergaard S, Jensen MM, Jørgensen JT, Juhl K, Lehmann C, Ploug M, Kjaer A. Quantitative PET of human urokinase-type plasminogen activator receptor with 64Cu-DOTA-AE105: implications for visualizing cancer invasion. J. Nucl. Med. 2012;53:138-45
20. Persson M, Madsen J, Østergaard S, Ploug M, Kjaer A. 68Ga-labeling and in vivo evaluation of a uPAR binding DOTA- and NODAGA-conjugated peptide for PET imaging of invasive cancers. Nucl. Med. Biol. 2012;39:560-9
21. Persson M, Liu H, Madsen J, Cheng Z, Kjaer A. First 18F-labeled ligand for PET imaging of uPAR: In vivo studies in human prostate cancer xenografts. Nucl Med Biol. 2013;40:618-24
22. Ploug M, Østergaard S, Gårdsvoll H, Kovalski K, Holst-Hansen C, Holm A, Ossowski L, Danø K Peptide-derived antagonists of the urokinase receptor. Affinity maturation by combinatorial chemistry, identification of functional epitopes, and inhibitory effect on cancer cell intravasation. Biochemistry. 2001;40:12157-68
23. 64Cu-1,4,7,10-Tetraazacyclododecane-N,N',N'',N'''-tetraaceticaid-Asp-cyclohexylalanine-Phe-D-Ser-D-Arg-Tyr-Leu-Trp-Ser-NH2 (AE105-NH2)64Cu-DOTA-AE105-NH2; Molecular Imaging and Contrast Agent Database (MICAD). Leung K. http://www.ncbi.nlm.nih.gov/books/NBK100136/
24. 68Ga-1,4,7-Triazacyclononane,1-glutaric acid-4,7-acetic acid-Asp-cyclohexylalanine-Phe-D-Ser-D-Arg-Tyr-Leu-Trp-Ser-NH2 (AE105-NH2) 68Ga-NODAGA-AE105-NH2; Molecular Imaging and Contrast Agent Database (MICAD). Leung K. http://www.ncbi.nlm.nih.gov/books/NBK100135/
25. Jones-Wilson TM, Deal KA, Anderson CJ, McCarthy DW, Kovacs Z, Motekaitis RJ, Sherry AD, Martell AE, Welch MJ. The in vivo behavior of copper-64-labeled azamacrocyclic complexes. Nucl Med Biol. 1998;25:523-30
26. Boswell CA, Sun X, Niu W, Weisman GR, Wong EH, Rheingold AL, Anderson CJ. Comparative in vivo stability of copper-64-labeled cross-bridged and conventional tetraazamacrocyclic complexes. J. Med. Chem. 2004;47:1465-74
27. Anderson CJ, Wadas TJ, Wong EH, Weisman GR. Cross-bridged macrocyclic chelators for stable complexation of copper radionuclides for PET imaging. Q. J. Nucl. Med. Mol. Imaging. 2008;52:185-92
28. Bass LA, Wang M, Welch MJ, Anderson CJ. In vivo transchelation of copper-64 from TETA-octreotide to superoxide dismutase in rat liver. Bioconjug. Chem. 2000;11:527-32
29. Weisman GR, Rogers ME, Wong EH, Jasinski JP, Paight ES. Cross-Bridged Cyclam - Protonation and Li+ Complexation in a Diamond-Lattice Cleft. J. Am. Chem. Soc. 1990;112:8604-8605
30. Weisman GR, Hill DC, Wong EH. Synthesis and transition metal complexes of new cross-bridged tetraamine ligands. Abstr. Pap. Am. Chem. 1996;211:570
31. Wong EH, Weisman GR, Hill DC, Reed DP, Rogers ME, Condon JS, Fagan MA, Calabrese JC, Lam KC, Guzei IA, Rheingold AL. Synthesis and characterization of cross-bridged cyclams and pendant-armed derivatives and structural studies of their copper(II) complexes. J. Am. Chem. Soc. 2000;122:10561-10572
32. Sprague JE, Peng Y, Fiamengo AL, Woodin KS, Southwick EA, Weisman GR, Wong EH, Golen JA, Rheingold AL, Anderson CJ. Synthesis, characterization and in vivo studies of Cu(II)-64-labeled cross-bridged tetraazamacrocycle-amide complexes as models of peptide conjugate imaging agents. J. Med. Chem. 2007;50:2527-2535
33. Zheleznyak A, Wadas TJ, Sherman CD, Wilson JM, Kostenuik PJ, Weilbaecher KN, Anderson CJ. Integrin αvβ3 as a PET Imaging Biomarker for Osteoclast Number in Mouse Models of Negative and Positive Osteoclast Regulation. Mol. Imaging Biol. 2012;14:500-508
34. Dumont RA, Deininger F, Haubner R, Maecke HR, Weber WA, Fani M. Novel 64Cu- and 68Ga-labeled RGD conjugates show improved PET imaging of αvβ3 integrin expression and facile radiosynthesis. J. Nucl. Med. 2011;52:1276-1284
35. Eiblmaier M, Andrews R, Laforest R, Rogers BE, Anderson CJ. Nuclear uptake and dosimetry of 64Cu-labeled chelator somatostatin conjugates in an SSTr2-transfected human tumor cell line. J. Nucl. Med. 2007;48:1390-1396
36. Wadas TJ, Eiblmaier M, Zheleznyak A, Sherman CD, Ferdani R, Liang K, Achilefu S, Anderson CJ. Preparation and biological evaluation of 64Cu-CB-TE2A-sst2-ANT, a somatostatin antagonist for PET imaging of somatostatin receptor-positive tumors. J. Nucl. Med. 2008;49:1819-27
37. Kumar SR, Gallazzi FA, Ferdani R, Anderson CJ, Quinn TP, Deutscher SL. In vitro and in vivo evaluation of 64Cu-radiolabeled KCCYSL peptides for targeting epidermal growth factor receptor-2 in breast carcinomas. Cancer Biother. Radiopharm. 2010;25:693-703
38. Garrison JC, Rold TL, Sieckman GL, Figueroa SD, Volkert WA, Jurisson SS, Hoffman TJ. In vivo evaluation and small-animal PET/CT of a prostate cancer mouse model using 64Cu bombesin analogs: Side-by-side comparison of the CB-TE2A and DOTA chelation systems. J. Nucl. Med. 2007;48:1327-1237
39. Abiraj K, Mansi R, Tamma ML, Fani M, Forrer F, Nicolas G, Cescato R, Reubi JC, Maecke HR. Bombesin antagonist-based radioligands for translational nuclear imaging of gastrin-releasing peptide receptor-positive tumors. J. Nucl. Med. 2011;52:1970-1978
40. Boswell CA, Regino CA, Baidoo KE, Wong KJ, Bumb A, Xu H, Milenic DE, Kelley JA, Lai CC, Brechbiel MW. Synthesis of a cross-bridged cyclam derivative for peptide conjugation and 64Cu radiolabeling. Bioconjug. Chem. 2008;19:1476-1484
41. Jacobsen B, Gårdsvoll H, Juhl Funch G, Østergaard S, Barkholt V, Ploug M. One-step affinity purification of recombinant urokinase-type plasminogen activator receptor using a synthetic peptide developed by combinatorial chemistry. Protein Expr. Purif. 2007;52:286-296
42. Gårdsvoll H, Gilquin B, Le Du MH, Ménez A, Jørgensen TJ, Ploug M. Characterization of the functional epitope on the urokinase receptor. Complete alanine scanning mutagenesis supplemented by chemical cross-linking. J Biol Chem. 2006;281:19260-19272
43. Liu W, Hao G, Long MA, Anthony T, Hsieh JT, Sun X. Imparting multivalency to a bifunctional chelator: a scaffold design for targeted PET imaging probes. Angew. Chem. Int. Ed. Engl. 2009;48:7346-7349
44. Gårdsvoll H, Jacobsen B, Kriegbaum MC, Behrendt N, Engelholm L, Østergaard S, Ploug M. Conformational regulation of urokinase receptor function: Impact of receptor occupancy and epitope-mapped monoclonal antibodies on lamellipodia induction. J. Biol. Chem. 2011;286:33544-56
45. Zhang J, Frankevich V, Knochenmuss R, Friess SD, Zenobi R. Reduction of Cu(II) in matrix-assisted laser desorption/ionization mass spectrometry. Am. Soc. Mass Spectrom. 2003;14:42-50
46. Knochenmuss R, Zenobi R. MALDI ionization: The role of in-plume processes. Chem. Rev. 2003;103:441-452
47. Dong J, Vachet RW. Coordination sphere tuning of the electron transfer dissociation behavior of Cu(II)-peptide complexes. J. Am. Soc. Mass. Spectrom. 2012;23:321-9
48. Ploug M. Structure-driven design of radionuclide tracers for non-invasive imaging of uPAR and targeted radiotherapy: The tale of a synthetic peptide antagonist. Theranostics. 2013;3:467-76
49. Mertens HD, Kjaergaard M, Mysling S, Gårdsvoll H, Jørgensen TJ, Svergun DI, Ploug M. A flexible multidomain structure drives the function of the urokinase-type plasminogen activator receptor (uPAR). J. Biol. Chem. 2012;287:34304-34015
50. Wadas TJ, Anderson CJ. Radiolabeling of TETA- and CB-TE2A-conjugated peptides with copper-64. Nat. Protoc. 2006;1:3062-3068
51. Ferdani R, Stigers DJ, Fiamengo AL, Wei L, Li BT, Golen JA, Rheingold AL, Weisman GR, Wong EH, Anderson CJ. Synthesis, Cu(II) complexation, 64Cu-labeling and biological evaluation of cross-bridged cyclam chelators with phosphonate pendant arms. Dalton Trans. 2012;41:1938-1950
52. Guo Y, Ferdani R, Anderson CJ. Preparation and biological evaluation of 64Cu labeled tyr(3)-octreotate using a phosphonic Acid-based cross-bridged macrocyclic chelator. Bioconjug. Chem. 2012;23:1470-1477
53. Sun X, Wuest M, Weisman GR, Wong EH, Reed DP, Boswell CA, Motekaitis R, Martell AE, Welch MJ, Anderson CJ. Radiolabeling and in vivo behavior of copper-64-labeled cross-bridged cyclam ligands. J. Med. Chem. 2002;45:469-77
54. Jørgensen JT, Persson M, Madsen J, Kjaer A. High tumor uptake of 64Cu: Implications for molecular imaging of tumor characteristics with copper-based PET tracers. Nucl. Med. Biol. 2013;40:345-350
55. Wei L, Ye Y, Wadas TJ, Lewis JS, Welch MJ, Achilefu S, Anderson CJ. 64Cu-labeled CB-TE2A and diamsar-conjugated RGD peptide analogs for targeting angiogenesis: comparison of their biological activity. Nucl. Med. Biol. 2009;36:277-285
56. Pfeifer A, Knigge U, Mortensen J, Oturai P, Berthelsen AK, Loft A, Binderup T, Rasmussen P, Elema D, Klausen TL, Holm S, von Benzon E, Højgaard L, Kjaer A. Clinical PET of neuroendocrine tumors using 64Cu-DOTATATE: first-in-humans study. J. Nucl. Med. 2012;53:1207-12015
Author contact
![]() Corresponding author: Michael Ploug, PhD, Dr.Sci. Finsen Laboratory & BRIC, Copenhagen Biocenter, Ole Maaløes Vej 5, 2200 - Copenhagen N, Denmark. E-mail: m-plougdk Tel: +45-35456037.
Corresponding author: Michael Ploug, PhD, Dr.Sci. Finsen Laboratory & BRIC, Copenhagen Biocenter, Ole Maaløes Vej 5, 2200 - Copenhagen N, Denmark. E-mail: m-plougdk Tel: +45-35456037.