Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Acknowledgements
Supplementary Material
Abbreviations
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2013; 3(7):477-486. doi:10.7150/thno.6617 This issue Cite
Research Paper
Scavenger Receptor B1 is a Potential Biomarker of Human Nasopharyngeal Carcinoma and Its Growth is Inhibited by HDL-mimetic Nanoparticles
Ying Zheng1,2,*, Yanyan Liu3,4,*, Honglin Jin1,2,*, Shaotao Pan1,2, Yuan Qian1,2, Chuan Huang1,2, Yixin Zeng3,4, Qingming Luo1,2, Musheng Zeng3,4, ![]() , Zhihong Zhang1,2,
, Zhihong Zhang1,2, ![]()
1. Britton Chance Center for Biomedical Photonics, Wuhan National Laboratory for Optoelectronics-Huazhong University of Science and Technology, Wuhan 430074, China;
2. MoE Key Laboratory for Biomedical Photonics, Department of Biomedical Engineering, Huazhong University of Science and Technology, Wuhan 430074, China
3. State Key Laboratory of Oncology in South China, Sun Yat-sen University Cancer Center, Guangzhou, China;
4. Department of Experimental Research, Sun Yat-sen University Cancer Center, Guangzhou, China.
* These authors contributed equally to this work.
Received 2013-5-5; Accepted 2013-6-4; Published 2013-6-25
Abstract

Nasopharyngeal carcinoma (NPC) is a very regional malignant head and neck cancer that has attracted widespread attention for its unique etiology, epidemiology and therapeutic options. To achieve high cure rates in NPC patients, theranostic approaches are actively being pursued and improved efforts remain desirable in identifying novel biomarkers and establishing effective therapeutic approaches with low long-term toxicities. Here, we discovered that the scavenger receptor class B type I (SR-B1) was overexpressed in all investigated NPC cell lines and 75% of NPC biopsies, demonstrating that SR-B1 is a potential biomarker of NPC. Additional functional analysis showed that SR-B1 has great effect on cell motility while showing no significant impact on cell proliferation. As high-density lipoproteins (HDL) exhibit strong binding affinities to SR-B1 and HDL mimetic peptides are reportedly capable of inhibiting tumor growth, we further examined the SR-B1 targeting ability of a highly biocompatible HDL-mimicking peptide-phospholipid scaffold (HPPS) nanocarrier and investigated its therapeutic effect on NPC. Results show that NPC cells with higher SR-B1 expression have superior ability in taking up the core constituents of HPPS. Moreover, HPPS inhibited the motility and colony formation of 5-8F cells, and significantly suppressed the NPC cell growth in nude mice without inducing tumor cell necrosis or apoptosis. These results indicate that HPPS is not only a NPC-targeting nanocarrier but also an effective anti-NPC drug. Together, the identification of SR-B1 as a potential biomarker and the use of HPPS as an effective anti-NPC agent may shed new light on the diagnosis and therapeutics of NPC.
Keywords: Cancer theranostics, nasopharyngeal carcinoma, biomarker, SR-B1, lipoprotein nanocarrier.
Introduction
Nasopharyngeal carcinoma (NPC) is a very regional malignant head and neck cancer that has a much higher incidence in Southern Asia (25 cases per 100,000) than that in Europe and North America (0.5-2 cases per 100,000) [1]. The etiology of NPC is complex and generally includes genetic susceptibility, environmental factors (consumption of salted fish, preserved foods, Chinese herbs, heavy metal and alcohol), fungal infections and Epstein-Barr virus (EBV) infection [2]. This increased understanding has been accompanied with the discovery of a series of prognostic/predictive biomarkers for NPC, such as EBV-associated biomarkers, plasma RNA integrity, certain tumor suppressor genes and vascular endothelial growth factor (VEGF) [2-6]. Although these biomarkers play a key role in the diagnostics of NPC, the number of existing biomarkers is generally limited. Moreover, along with the improved cure rates in patients using current therapeutic methods, such as radiotherapy, chemotherapy, surgery and immunotherapy [7-11], the issue of long-term side effects remains problematic [12]. Therefore, the next challenge of NPC is to seek novel biomarkers for early diagnosis and highly biocompatible drugs for therapy [12-14].
Scavenger receptor class B type I (SR-B1), with a molecular weight of ~82 kD, is a membrane glycoprotein composed of 509 amino acids [15, 16]. It has been reported that SR-B1 is highly expressed in a variety of tumor cells of prostate, breast, colorectal and ovarian cancers [17-20]. Moreover, the overexpression of SR-B1 potentiates the binding to high density lipoprotein (HDL) and leads to a significant increase in the uptake of HDL-cholesteryl ester (CE) [21], an essential nutrient for malignant cell proliferation and metastasis [22-24]. Recent studies pointed out that the HDL level was negatively correlated with the risk of cancer development and metastasis [25-27]. For these reasons, we hypothesize that SR-B1 might be overexpressed in NPC.
Previously, we have reported a HDL-mimicking peptide-phospholipid scaffold (HPPS) nanocarrier that can selectively bind to SR-B1 expressing cancer cells with high affinities [28, 29]. As a key component of HPPS, apoA-1 biomimetic peptides not only enable HPPS to mimic the behavior of HDL but also are capable of inhibiting tumor growth and development in vivo [20, 30, 31]. Although HPPS has been loaded with various therapeutic drugs and imaging agents for theranostic applications [29, 32, 33], use of itself as an anti-cancer drug has never been explored. Besides, treatment with a dosage of 2000 mg/kg HPPS has not induced adverse effects in mice [32]. Thus, it might be interesting to explore HPPS itself as a biocompatible drug for SR-B1 expressing tumors.
This study aims to identify SR-B1 as a potential biomarker for NPC and testify the therapeutic efficacy of HPPS in treating NPC. This finding and establishment of a novel therapeutic approach would be expected to open up a new avenue for the diagnosis and therapy of NPC in clinical settings.
Materials and Methods
Cell Lines
Immortalized nasopharyngeal epithelial cell line N2-Bmi1 was established by inducing oncogene Bmi1 overexpression in a normal nasopharyngeal epithelial cell line N2 as described preciously [5], and cultured in Keratinocyte/serum-free medium (Invitrogen). A highly differentiated NPC cell line (CNE1), five poorly differentiated NPC cell lines (CNE2, HONE1, HNE1 SUNE1and SUNE2), two cell subclones of SUNE1 (6-10B, low metastasis potential NPC cell line; 5-8F, high metastasis potential NPC cell line) and a human fibrosarcoma cell line HT1080 (SR-B1-) were cultured in RPMI 1640 (Invitrogen) containing 10% fetal bovine serum (FBS, Hyclone). The stable SR-B1 gene silenced NPC cell line 5-8F-shSR-B1 was generated by infection of 5-8F cells with the retroviral vector pRS (retro-super) (Oligoengine) containing SR-B1 short-hairpin RNAs (shRNAs). 5-8F-shSR-B1 cells were cultured in RPMI 1640 containing 10% FBS and 0.5 µg/L puromycin (Sigma). Sequences for shRNAs were as follows: CCATGACCCTGAAGCTCAT and GCTGAGCCTCTACATGAAA.
Western Blot Analysis
Proteins in the immortalized nasopharyngeal epithelial cell line and various NPC cell lines were collected by using PIPA lysate (P0013C, beyotime). The concentrations of these samples were quantified by a Lowry Protein Assay Kit and 10 µg of protein was used for gel loading. GAPDH primary antibody and SR-B1 primary antibody (Novus Biologicals) were used at a dilution of 1:2000. The secondary antibody was used at a dilution of 1:4000. The next steps were performed as previously mentioned [5].
Real-Time RT-PCR Analysis
Total RNA from different cell lines was extracted using Trizol reagents (Gibco). cDNA was synthesized from 2.5 µg of total RNA using random hexamers. Real-time PCR was carried out using a sequence detection system (ABI PRISM 7500, Applied Biosystems). Reactions were run in triplicate in three independent experiments. The geometric mean of the housekeeping gene GAPDH was used as an internal control to normalize the variability in expression levels. A 10 µL reaction system was prepared according to the following components: 5 µL PCR mix (2×), 1 µL SR-B1 primer set (2.5 nM), 1 µL cDNA template, 3 µL sterile ddH2O, and total reaction volume 10 µL. The procedure of Real-Time PCR included a pre-denaturation step at 50°C for 2 min and 95°C for 5 min, followed by 40 cycles of denaturation at 95°C for 15 s, annealing and extension at 60°C for 30 s. Custom primers were purchased with sequence as follows: GAPDH, forward primer 5'-GACTCATGACCACAGTCCATGC-3' and reverse primer 5'-AGAGGCAGGGATGATGTTCTG-3'; SR-B1, forward primer 5'-TCCTCACTTCCTCAACGCTG-3' and reverse primer 5'-TCCCAGTTTGTCCAATGCC-3'.
Immunohistochemistry
Twenty cases of fresh NPC biopsies were obtained from the Sun Yat-sen University Cancer Center (SYSUCC), and were prepared for frozen sections. Pathological diagnosis was verified by two different pathologists. Normal nasopharynx tissue samples were collected from 5 cases of healthy volunteers. For the use of these clinical samples, written Ethics Approval and Patient Consent from SYSUCC Research Ethics Committee were obtained. Briefly, the frozen sections were defrosted at room temperature for 10 min, fixed by acetone for 10 min and dried. Then the samples were washed with phosphate buffered saline (PBS) three times (3 min each), immersed in H2O2 for 20 min, and subjected to a second round of washing with PBS (three times at 3 min for each). The slides were blocked with 10% BSA in Tris-buffered saline (TBS) for 20 min at room temperature. Subsequently, frozen sections were exposed to the primary antibody diluted 50 times at 4°C for 16 h and washed with PBS. After that they were incubated with horseradish peroxidase (HRP)-conjugated secondary antibody that was dropwisely added at room temperature for 1 h. After three times washing with PBS, they were incubated in Diaminobenzidine (DAB) about 1 min. The stain intensity was monitored under the microscope. The specimens were then stained by hematoxylin, washed with water, cleared in xylene, sealed by neutral resin and observed using a microscope with an objective of 40 times.
Cell Growth Ability Test
5-8F and 5-8F-shSR-B1 cells were seeded onto 96-well plates (2×103 per well) with RPMI 1640 culture media containing 10% FBS. The culture media for 5-8F-shSR-B1 cells was additionally added with 0.5 µg/L puromycin. The average growth abilities of these cell lines were measured at a fixed time each day using a MTS kit (Promega), and the measurement lasted for 6 days.
Nanoparticle Synthesis
The Fluo-BOA dye was synthesized as previously reported [29]. HPPS-CO and HPPS-(Fluo-BOA) were synthesized via a previously reported method. In brief, 2 mg DMPC and 0.3 µmol Fluo-BOA (MW: 565) or cholesterol oleate (CO) in 200 µL chloroform were mixed in a glass tube, and the mixture was dried under nitrogen gas streams for 1 h to form a uniform thin phospholipid film, which was further dried under vacuum for 1 h. Subsequently, 1 mL PBS (pH 7.5) was added to the dry film, which was vortexed for 10 s and sonicated at 48°C for 1 h. The resulting solution was then mixed with 2 ml R4F (Ac-FAEKFKEAVKDYFAKFWD; 2 mg/mL in PBS) to produce a transparent peptide-phospholipids solution, and stored at 4°C overnight. The mixture was then filtered and purified with a FPLC system (AKTA purifier 10, GE Healthcare) at a flow rate of 1 mL·min-1. Fractions collected from 55-65 min were concentrated with centrifugal filter tubes (30 kDa, Sartorius), sterilized and kept in the 4°C for further use.
Confocal Imaging
5-8F, 5-8F-shSR-B1 and HT1080 cells (1×104/300 µL per well) were seeded onto an 8-well chamber slide. Cells were cultured at 37°C in a 5% CO2 incubator for 48 h, incubated with 10 µM HPPS-(Fluo-BOA) in 100 µL complete medium for 2 h, and washed with PBS for three times. After replacing the PBS with 100 µL medium, cells were imaged using a laser confocal microscope (FV1000, Olympus).
Flow Cytometry Analysis
5-8F, 5-8F-shSR-B1 and HT1080 (1×105, 2 mL per well) were seeded onto a 6-well dish. Cells were maintained at 37°C in a 5% CO2 incubator for 48 h, incubated with 10 µM HPPS-(Fluo-BOA) in 2 mL complete medium for 2 h at the same condition, and washed three times with PBS. After the treatment with trypsin, cells were detached from the substrate and resuspended in 300 µL PBS, which was filtered through a 200 mesh screen (Nylon).
Wound Healing Assay
5-8F and 5-8F-shSR-B1 were seeded onto a 6-well plate (1.5×105 per well) to cover 90% area of the substrate on the next day, and hungered for 24 h. The cell monolayer was scratched by a tip (20 µL), washed three times with PBS, and cultured in the complete medium. Wounds were imaged on a microscope (IX71, Olympus) (4× objective) at different time points. The drug treatment groups followed the same procedure and the experiment was repeated three times.
Colony Formation Assay
The 6-well plate was coated with 2 mL 0.6% soft agar in RPMI 1640 culture media supplemented with 10% FBS. Next, 5-8F cells that treated with PBS, HPPS-(Fluo-BOA), HPPS-CO and complex for 48 h were seeded onto 6-well plates, and cultured in 2 mL 0.3% soft agar in RPMI 1640 culture media supplemented with 10% FBS and each drug (concentration unchanged) at 37°C in a 5% CO2 incubator. 1 mL culture medium containing various drugs was added to the well on the next day. Cells were cultured for 14 days and the culture medium was changed every two days. Images were taken at 4× objective using a microscope (IX71, Olympus).
Mouse Xenograft Models and Anti-Tumor Therapy
All animal studies were conducted in compliance with protocols approved by the Hubei Provincial Animal Care and Use Committee and the animal experiment guidelines of the Animal Experimentation Ethics Committee of Huazhong University of Science and Technology. Female 4-6 weeks Balb/C nu/nu mice were purchased from the Department of Experimental Animals, Hunan Slack King Laboratory Animal Co., Ltd. 2 ×106 5-8F cells in 100 µL PBS were implanted in back leg roots of nude mice. The tumor bearing mice were randomly treated as following groups: HPPS-(Fluo-BOA) (125 mg/kg dissolved in 300 µL PBS, concentration calculation was based on the content of phospholipids), HPPS-CO (125 µg/kg in 300 µL PBS), complex (125 µg/kg dissolved in 300 µL PBS) and PBS (300 µL). The treatment was conducted by tail vein injection every three days since the fourth day after implanting tumor cells. The body weight was recorded, and tumor size was measured with a micrometer on the day when tumor was implanted (day 0) and day 4, 7, 10 and 12. The tumor volume was calculated by the formula below: Tumor volume = 0.5 × Length × Width × Height.
In Vivo Whole Body Fluorescence Imaging
About 60 h after the last injection of 125 mg/kg HPPS-(Fluo-BOA) in 300 µL PBS, the mice treated with HPPS-(Fluo-BOA) were killed and imaged by using a home-made whole body imaging system. Filters for green fluorescence included the excitation filter (wavelength 469/35 nm) and emission filter (wavelength 538/40 nm) with an exposure time of 10 s. Filters for the elimination of background fluorescence included excitation filter (wavelength 434/17 nm), and emission filter (wavelength 538/40 nm) [34].
Statistical Analysis
The two-tailed student's t test was used to analyze significant difference in experiment. A P value less than 0.05 was considered statistically significant.
Results
SR-B1 is highly expressed in NPC cell Lines and pathological tissues
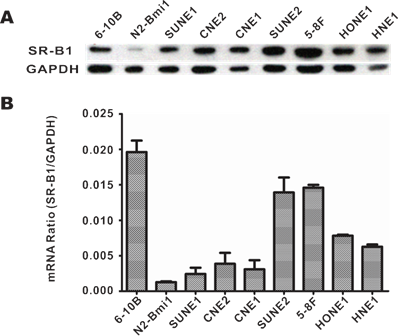
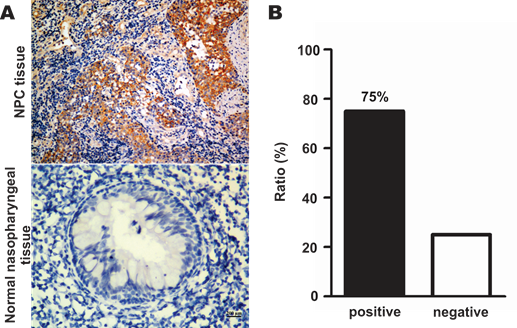
SR-B1 expression at both the protein and mRNA levels in NPC cell lines, as well as SR-B1 protein expression in clinical tissue sections was investigated using western blot analysis, RT-PCR and an immunohistochemistry assay. As shown in Fig. 1A, western blot analysis revealed that SR-B1 was highly expressed in all the investigated NPC cell lines and had a weak expression in the epithelial cell line N2-Bmi1, and these results were consistent with the mRNA expression level (Fig. 1B). Among these cell lines, 5-8F cells had the most prominent SR-B1 expression level, which was slightly higher than HepG2 cells (data not shown), and thus were selected as a model for the following studies. Moreover, SR-B1 was found over expressed in 15 of 20 NPC tissue sections (75%) from patients, but lower in the stromal or non-cancerous epithelial cells (Fig. 2A 2B and Supplementary Material: Fig. S1). These results suggested that SR-B1 is a potential biomarker of NPC.
SR-B1 expression level in NPC cell lines. a) SR-B1 protein and b) mRNA expression level in NPC cell lines and a nasopharyngeal epithelial cell line (N2-Bmi1).
SR-B1 expression level in NPC patients. a) Representative images of SR-B1 positive NPC tissue sections and normal nasopharynx tissue sections. The nuclei stained with hematoxylin are shown in blue staining and the SR-B1 appears grown. b) Percentages of SR-B1 positive NPC tissue sections in investigated NPC patients. Scale bar, 200 µm.
SR-B1 silencing affects cell motility in 5-8F Cells
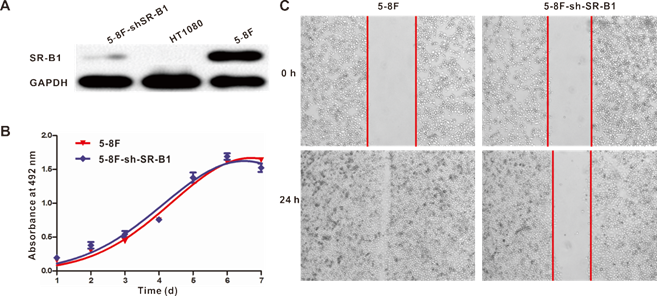
In order to probe the effect of SR-B1 expression on the behaviors of NPC cells, several parameters (including SR-B1 expression, cell growth ability and cell motility) were compared between SR-B1 silenced NPC stable cell line (5-8F-shSR-B1) and 5-8F cells. Results showed that SR-B1 expression in 5-8F-shSR-B1 cells was slightly higher than that in HT1080 cells (SR-B1-), while showing a much lower level than 5-8F cells (Fig. 3A). The cell proliferation assay result indicated that 5-8F-shSR-B1 and 5-8F cells had no significant difference in growth ability (P>0.05) (Fig. 3B). However, 5-8F-shSR-B1 cells showed reduced cell motility compared to 5-8F cells according to the scratch wound healing assay result (Fig. 3C), suggesting that SR-B1 expression level affects cell motility of NPC cells.
Effects of SR-B1 expression on NPC cell growth ability and cell motility. a) SR-B1 expression level in 5-8F, 5-8F-shSR-B1 and HT1080 cells. b) Comparison of the growth ability of 5-8F and 5-8F-shSR-B1 cell lines in vitro. No significant difference is found (P>0.05). c) Cell mobility between 5-8F and 5-8F-shSR-B1. Data are representative of at least three independent experiments.
Selective uptake of HPPS by NPC cells
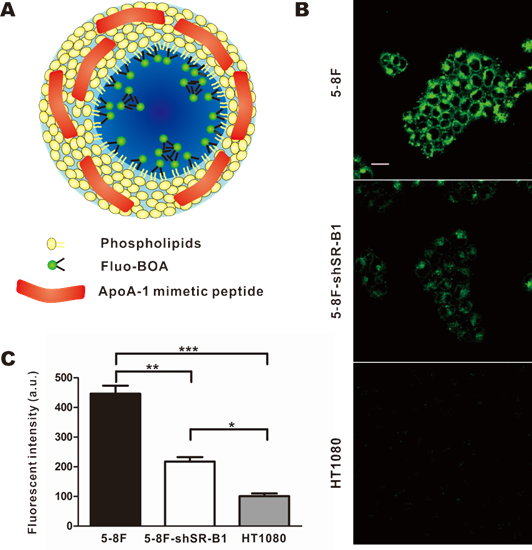
Next, HPPS nanoparticles were prepared, and their uptake abilities by NPC cells were investigated using confocal imaging and flow cytometry. HPPS nanoparticles could be formulated with a core cargo cholesterol oleate (CO) or a fluorescent dye Fluo-BOA, denoted as HPPS-CO and HPPS-(Fluo-BOA) (Fig. 4A), respectively. The purified HPPS-(Fluo-BOA) (18.53±0.43 nm) nanoparticles possessed similar size to HPPS-CO (18.32±0.63 nm) and could be directly applied for imaging and quantitative studies. Confocal images showed that 5-8F, 5-8F-shSR-B1 and HT1080 cells had differentiated uptake abilities to HPPS-(Fluo-BOA) (5-8F>5-8F-shSR-B1>HT1080), and this trend correlated well with their SR-B1 expression level, in which high-level expression of SR-B1 followed strong uptake abilities (Fig. 4B). The quantitative result showed that uptake of HPPS-(Fluo-BOA) by 5-8F cells was 4.4 and 2.1 times of that by HT1080 and 5-8F-shSR-B1 cells, respectively (Fig. 4C). In addition, cellular uptake of HPPS was 2-6 times as much as the lipid emulsion complex containing all components of HPPS only without R4F peptides with various concentrations, demonstrating the targeting role of R4F in the formulation (Supplementary Material: Fig. S2). Thus, selectively targeting to NPC cells could be achieved by using HPPS nanoparticles via SR-B1.
Selective uptake of HPPS by NPC Cells. a) A schematic diagram of HPPS-(Fluo-BOA). b) Confocal images and c) flow cytometry analysis of HPPS-(Fluo-BOA) uptake by 5-8F (SR-B1++), 5-8F-shSR-B1 (SR-B1+) and HT1080 (SR-B1-) cells (*P<0.05, **P<0.01 and ***P<0.001). Data represent mean of three independent experiments. (Scale bar, 20 µm).
Inhibitory effects of HPPS on NPC cells
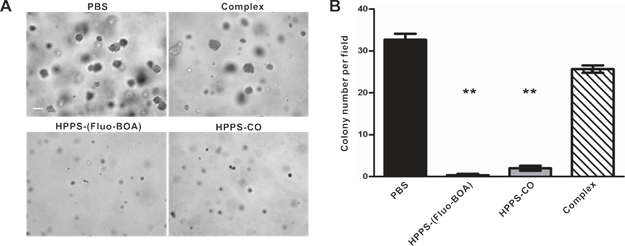
The effect of HPPS on NPC cell growth, cell motility and colony formation abilities were examined using a cell counting kit-8 (CCK-8), a scratch wound healing assay and a colony formation assay, respectively, taking PBS treated cells and the emulsion complex treated cells as control groups. Statistical analysis revealed that HPPS-(Fluo-BOA) and HPPS-CO had no significant impact on the viability of 5-8F cells with a concentration of 100 nM (Supplementary Material: Fig. S3). Next, we performed a scratch wound healing study under similar conditions. After 6 h of wounding, a large amount of cells that were exposed to PBS and complex had already migrated to the wound space, and these migrating cells became confluent at 24 h (Fig. 5). In comparison, HPPS-(Fluo-BOA) and HPPS-CO treated cells had slight migration at 6 h, and distinct gaps remained observable at 24 h post wounding, revealing that HPPS is capable of inhibiting NPC cell motility. Furthermore, the colony formation results showed that the effective colony number (diameters greater than 50 pixels) in HPPS-(Fluo-BOA) and HPPS-CO treated cells was 1.0% and 6.1% of that in PBS control, respectively, whereas this number was comparable to the PBS control for the complex group (78.6%) (Fig. 6A and 6B), demonstrating the effectiveness of HPPS in suppressing the colony formation of NPC cells.
Evaluation of mobilities of 5-8F cells treated with HPPS-(Fluo-BOA), HPPS-CO, complex or PBS. Data are representative of at least three independent experiments.

Colony formation of 5-8F cells treated with HPPS-(Fluo-BOA), HPPS-CO, complex or PBS. a) Bright field imaging and b) statistic counts of colony formation (**P<0.01, compared with control). Scale bar, 200 µm.
In vivo anti-tumor efficacy of HPPS
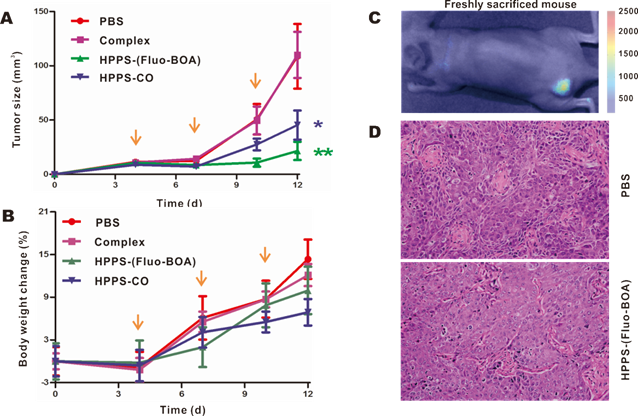
To verify the inhibitory effect of HPPS on tumor growth, HPPS-(Fluo-BOA) and HPPS-CO were intravenously administered to nude mice using mice bearing 5-8F subcutaneous tumors, and PBS and complex treated groups were set as control groups. As shown in Fig. 7A, both treatment with HPPS-(Fluo-BOA) and HPPS-CO resulted in significant decreases in tumor size compared to the PBS control. In contrast, no significant difference was observed in tumor size changes between the complex and PBS groups, demonstrating the unique role of HPPS in suppressing tumor growth. Moreover, all these treatments did not lead to significant body weight changes compared to PBS group (Fig. 7B). Additionally, the tumor shape in HPPS-(Fluo-BOA) treated group could be directly imaged and monitored by fluorescence imaging (Fig. 7C). Finally, we did not observe substantial pathologic differences between HPPS and PBS treated groups according to the H&E stain, suggesting that the mechanism of inhibition of tumor growth by HPPS does not rely on the induction of tumor cell necrosis or apoptosis.
Evaluation of anti-tumor efficacy of HPPS on nude mice bearing 5-8F tumors in vivo. Real-time monitoring a) the tumor size and b) body weight changes on the mice treated with HPPS-(Fluo-BOA), HPPS-CO, complex, or PBS (n=6; **P<0.01, *P<0.05). c) A representative whole body fluorescent image of 5-8F-bearing nude mice treated with HPPS-(Fluo-BOA). d) Representative HE staining images of PBS and HPPS-(Fluo-BOA) treated tumor tissue sections.
Discussion
SR-B1 is a transmembrane protein that can specifically bind to HDL to facilitate cellular transport of cholesterol [35], an essential nutrient for malignant cell proliferation and metastasis [24], and its overexpression can effectively increase HDL binding and HDL-CE uptake. Moreover, SR-B1 is highly expressed in a variety of tumor cell lines [17-20, 36, 37]. As high expression of SR-B1 was observed in all investigated NPC cell lines in both mRNA and protein levels, and even in majority of clinically pathological tissue sections, we concluded that SR-B1 is a novel potential biomarker of NPC. Knockdown of SR-B1 expression was found not affecting cell growth ability, suggesting that it might not be a bona fide oncogene. As cell motility often has close relation with cellular cytoskeleton [38], it could be inferred that SR-B1 might be participated in cell cytoskeleton correlated physiological activities due to the observation that 5-8F-shSR-B1 cell line had reduced motility compared with 5-8F cells. On the other hand, current NPC biomarkers are generally used as diagnostic purpose and very few have been applied for therapeutic applications except some oncogenes, such as epidermal growth factor receptor (EGFR), a direct therapeutic target of NPC using anti-EGFR techniques [2, 10, 12, 19]. However, these biomarkers are seldom utilized for receptor mediated drug delivery for NPC treatment. In this regard, use of SR-B1 as a NPC biomarker has an attractive aspect that it has rich history in receptor mediated drug delivery in view of the strong binding affinity between SR-B1 and HDL nanocarriers [27, 39].
As HDL mimetic peptide, which is a key component of HPPS [29], is reportedly capable of inhibiting the development of colorectal cancer and ovarian cancer [20, 30], it is predictable that HPPS may exhibit similar function. Here, the inhibitory effect of HPPS on NPC cell growth was illustrated at the following two levels. At the cellular level, the cell motility (Fig. 5) and colony formation (Fig. 6C and 6D) were largely suppressed by HPPS. At the in vivo level, HPPS restrained the growth of NPC compared to the complex and PBS control groups. In addition, there was no significant difference in body weight change, which is in good agreement with a previous report that HPPS has excellent biocompatibility under the treatment of HPPS containing 2000 mg·kg-1 DMPC without affecting liver functions and blood biochemical parameters [32], making HPPS a relatively safe drug against NPC. Interestingly, the mechanism involved was not associated with tumor cell necrosis or apoptosis according to the H&E stain (Fig. 7D). We infer that the inhibition of NPC cell motility and colony formation abilities may contribute to the in vivo antitumor effect of HPPS. It is suggested that the anti-tumor role of HDL mimicking peptides may be contributed by reducing the plasma level of lysophosphatidic acid (LPA) [20], a proinflammatory lysophospholipid that facilitates cell migration, invasion and colony formation [40]. Recently, it is reported that HDL mimicking peptides are able to induce the expression and increase the activity of an antioxidant enzyme MnSOD to reduce the cellular oxidant state [41], thereby decreasing in vivo oxidative damage to exert anticancer effect [42]. More recently, another group highlighted the promotion of cellular cholesterol flux by synthetic HDL in the process of treating lymphoma [43], and this mechanism seems like a more plausible explanation for our case since cholesteryl oleate-loaded synthetic HDL nanoparticles (HPPS-CO) had slightly higher antitumor ability than fluorescent dye-loaded HPPS (HPPS-(Fluo-BOA)) (Fig. 7A). Despite these findings, most of these studies share the same view that severe toxicity and apoptosis are not involved [41], and the exact mechanism is still an unresolved issue to be illustrated.
The utilization of HPPS in NPC treatment may have more advantageous properties than HDL mimicking peptides in several aspects. Previously, we have reported that HPPS can be loaded with various cargos [28], such as NPC chemotherapeutic drug paclitaxel [32], small interfering RNA and photosensitizers [33, 44], and these approaches can be easily combined to achieve more potent therapeutic efficacy for NPC therapy[45], taking advantages of the high expression of SR-B1 in NPC cells and drug loading abilities of HPPS vehicles. Furthermore, HPPS has a long circulation time (~15 h) in blood due to its nano-ranged size (~20 nm) [29], and this may enable HPPS a much higher tumor accumulation than HDL mimicking peptides, which often have short biologic half-lives (several minutes) [46, 47]. Finally, as SR-B1 is highly expressing in some normal organs, such as liver and adrenal glands [16, 35], incorporation of additional targeting ligands (such as EGF) to HPPS is likely to further improve the therapeutic window for treating NPC [48, 49].
In summary, we identified SR-B1 as a potential biomarker of human NPC for the first time, and developed an effective and safe therapeutic approach against NPC by using HPPS nanovehicles. Our findings may shed new light onto the diagnosis and therapeutics of NPC.
Acknowledgements
This work was supported by the National Basic Research Program of China (Grant No. 2011CB910401), Science Fund for Creative Research Group of China (Grant No.61121004), National Natural Science Foundation of China (Grant No. 81172153, 30911120489, 81102053), and Natural Science Foundation of Guangdong Province (Grant No.S2011040004520). The authors thank Jianwei Fu and Xiaoquan Yang for whole-body imaging assistance. The authors also thank Kenneth K. Ng for English proof and Prof. Gang Zheng for paper discussion. The authors also thank the Analytical and Testing Center (Huazhong University of Science and Technology) for spectral measurements.
Supplementary Material
Methods, Fig.S1 - S3.
Abbreviations
SR-B1: scavenger receptor class B type I; CO: cholesteryl oleate; FBS: fetal bovine serum; HDL: high-density lipoprotein; HPPS: HDL-mimicking peptide-phospholipid scaffold; NPC: nasopharyngeal carcinoma.
Competing interests
The authors have declared that no competing interest exists.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Chan AT. Nasopharyngeal carcinoma. Ann Oncol. 2010;21:vii308-12
2. Tao Q, Chan AT. Nasopharyngeal carcinoma: molecular pathogenesis and therapeutic developments. Expert Rev Mol Med. 2007;9:1-24
3. Li F, Guan Y, Chen Z. Proteomics in nasopharyngeal carcinoma. Cell Mol Life Sci. 2008;65:1007-12
4. Song LB, Li J, Liao WT, Feng Y, Yu CP, Hu LJ. et al. The polycomb group protein Bmi-1 represses the tumor suppressor PTEN and induces epithelial-mesenchymal transition in human nasopharyngeal epithelial cells. J Clin Invest. 2009;119:3626-36
5. Song LB, Zeng MS, Liao WT, Zhang L, Mo HY, Liu WL. et al. Bmi-1 is a novel molecular marker of nasopharyngeal carcinoma progression and immortalizes primary human nasopharyngeal epithelial cells. Cancer Res. 2006;66:6225-32
6. Tao Q, Young LS, Woodman CB, Murray PG. Epstein-Barr virus (EBV) and its associated human cancers--genetics, epigenetics, pathobiology and novel therapeutics. Front Biosci. 2006;11:2672-713
7. Cho WC. Nasopharyngeal carcinoma: molecular biomarker discovery and progress. Mol Cancer. 2007;6:1
8. Guigay J. Advances in nasopharyngeal carcinoma. Curr Opin Oncol. 2008;20:264-9
9. Henry NL, Hayes DF. Cancer biomarkers. Mol Oncol. 2012;6:140-6
10. Ma BB, Hui EP, Chan AT. Systemic approach to improving treatment outcome in nasopharyngeal carcinoma: current and future directions. Cancer Sci. 2008;99:1311-8
11. Suarez C, Rodrigo JP, Rinaldo A, Langendijk JA, Shaha AR, Ferlito A. Current treatment options for recurrent nasopharyngeal cancer. Eur Arch Otorhinolaryngol. 2010;267:1811-24
12. Razak AR, Siu LL, Liu FF, Ito E, O'Sullivan B, Chan K. Nasopharyngeal carcinoma: the next challenges. Eur J Cancer. 2010;46:1967-78
13. Hainaut P, Plymoth A. Targeting the hallmarks of cancer: towards a rational approach to next-generation cancer therapy. Curr Opin Oncol. 2013;25:50-1
14. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57-70
15. Krieger M. Charting the fate of the "good cholesterol": identification and characterization of the high-density lipoprotein receptor SR-BI. Annu Rev Biochem. 1999;68:523-58
16. Murao K, Terpstra V, Green SR, Kondratenko N, Steinberg D, Quehenberger O. Characterization of CLA-1, a human homologue of rodent scavenger receptor BI, as a receptor for high density lipoprotein and apoptotic thymocytes. J Biol Chem. 1997;272:17551-7
17. Cao WM, Murao K, Imachi H, Yu X, Abe H, Yamauchi A. et al. A mutant high-density lipoprotein receptor inhibits proliferation of human breast cancer cells. Cancer Res. 2004;64:1515-21
18. Mooberry LK, Nair M, Paranjape S, McConathy WJ, Lacko AG. Receptor mediated uptake of paclitaxel from a synthetic high density lipoprotein nanocarrier. J Drug Target. 2010;18:53-8
19. Shahzad MM, Mangala LS, Han HD, Lu C, Bottsford-Miller J, Nishimura M. et al. Targeted delivery of small interfering RNA using reconstituted high-density lipoprotein nanoparticles. Neoplasia. 2011;13:309-19
20. Su F, Kozak KR, Imaizumi S, Gao F, Amneus MW, Grijalva V. et al. Apolipoprotein A-I (apoA-I) and apoA-I mimetic peptides inhibit tumor development in a mouse model of ovarian cancer. Proc Natl Acad Sci U S A. 2010;107:19997-20002
21. Valacchi G, Sticozzi C, Lim Y, Pecorelli A. Scavenger receptor class B type I: a multifunctional receptor. Ann N Y Acad Sci. 2011;1229:E1-7
22. Connelly MA, Williams DL. SR-BI and cholesterol uptake into steroidogenic cells. Trends Endocrinol Metab. 2003;14:467-72
23. Gospodarowicz D, Lui GM, Gonzalez R. High-density lipoproteins and the proliferation of human tumor cells maintained on extracellular matrix-coated dishes and exposed to defined medium. Cancer Res. 1982;42:3704-13
24. Yeagle PL. Modulation of Membrane-Function by Cholesterol. Biochimie. 1991;73:1303-10
25. Cust AE, Kaaks R, Friedenreich C, Bonnet F, Laville M, Tjonneland A. et al. Metabolic syndrome, plasma lipid, lipoprotein and glucose levels, and endometrial cancer risk in the European Prospective Investigation into Cancer and Nutrition (EPIC). Endocr Relat Cancer. 2007;14:755-67
26. Fiorenza AM, Branchi A, Sommariva D. Serum lipoprotein profile in patients with cancer. A comparison with non-cancer subjects. Int J Clin Lab Res. 2000;30:141-5
27. Lacko AG, Nair M, Prokai L, McConathy WJ. Prospects and challenges of the development of lipoprotein-based formulations for anti-cancer drugs. Expert Opin Drug Deliv. 2007;4:665-75
28. Ng KK, Lovell JF, Zheng G. Lipoprotein-Inspired Nanoparticles for Cancer Theranostics. Acc Chem Res. 2011;44:1105-13
29. Zhang ZH, Cao WG, Jin HL, Lovell JF, Yang M, Ding LL. et al. Biomimetic Nanocarrier for Direct Cytosolic Drug Delivery. Angew Chem Int Ed Engl. 2009;48:9171-5
30. Su F, Grijalva V, Navab K, Ganapathy E, Meriwether D, Imaizumi S. et al. HDL mimetics inhibit tumor development in both induced and spontaneous mouse models of colon cancer. Mol Cancer Ther. 2012;11:1311-9
31. Zhang XX, Eden HS, Chen X. Peptides in cancer nanomedicine: Drug carriers, targeting ligands and protease substrates. J Control Release. 2012;159:2-13
32. Yang M, Chen J, Cao WG, Ding LL, Ng KK, Jin HL. et al. Attenuation of nontargeted cell-kill using a high-density lipoprotein-mimicking peptide phospholipid nanoscaffold. Nanomedicine. 2011;6:631-41
33. Yang M, Jin HL, Chen JA, Ding LL, Ng KK, Lin QY. et al. Efficient Cytosolic Delivery of siRNA Using HDL-Mimicking Nanoparticles. Small. 2011;7:568-73
34. Zheng Y, Huang C, Cheng ZY, Chen M. Establishment of Visible Animal Metastasis Models for Human Nasopharyngeal Carcinoma Based on a Far-Red Fluorescent Protein. J Innov Opt Health Sci. 2012;05:1250019
35. Cao G, Garcia CK, Wyne KL, Schultz RA, Parker KL, Hobbs HH. Structure and localization of the human gene encoding SR-BI/CLA-1. Evidence for transcriptional control by steroidogenic factor 1. J Biol Chem. 1997;272:33068-76
36. Bocharov AV, Baranova IN, Vishnyakova TG, Remaley AT, Csako G, Thomas F. et al. Targeting of scavenger receptor class B type I by synthetic amphipathic alpha-helical-containing peptides blocks lipopolysaccharide (LPS) uptake and LPS-induced pro-inflammatory cytokine responses in THP-1 monocyte cells. J Biol Chem. 2004;279:36072-82
37. Witt W, Kolleck I, Fechner H, Sinha P, Rustow B. Regulation by vitamin E of the scavenger receptor BI in rat liver and HepG2 cells. J Lipid Res. 2000;41:2009-16
38. Sahai E. Illuminating the metastatic process. Nat Rev Cancer. 2007;7:737-49
39. Sabnis N, Lacko AG. Drug delivery via lipoprotein-based carriers: answering the challenges in systemic therapeutics. Ther Deliv. 2012;3:599-608
40. Li H, Wang D, Zhang H, Kirmani K, Zhao Z, Steinmetz R. et al. Lysophosphatidic acid stimulates cell migration, invasion, and colony formation as well as tumorigenesis/metastasis of mouse ovarian cancer in immunocompetent mice. Mol Cancer Ther. 2009;8:1692-701
41. Ganapathy E, Su F, Meriwether D, Devarajan A, Grijalva V, Gao F. et al. D-4F, an apoA-I mimetic peptide, inhibits proliferation and tumorigenicity of epithelial ovarian cancer cells by upregulating the antioxidant enzyme MnSOD. Int J Cancer. 2012;130:1071-81
42. Behrend L, Henderson G, Zwacka RM. Reactive oxygen species in oncogenic transformation. Biochem Soc Trans. 2003;31:1441-4
43. Yang S, Damiano MG, Zhang H, Tripathy S, Luthi AJ, Rink JS. et al. Biomimetic, synthetic HDL nanostructures for lymphoma. Proc Natl Acad Sci U S A. 2013;110:2511-6
44. Jin HL, Chen J, Lovell JF, Zhang ZH, Zheng G. Synthesis and Development of Lipoprotein-Based Nanocarriers for Light-Activated Theranostics. Isr J Chem. 2012;52:715-27
45. Melancon MP, Stafford RJ, Li C. Challenges to effective cancer nanotheranostics. J Control Release. 2012;164:177-82
46. Luo H, Shi J, Jin H, Fan D, Lu L, Wang F. et al. An (125)I-labeled octavalent peptide fluorescent nanoprobe for tumor-homing imaging in vivo. Biomaterials. 2012;33:4843-50
47. Luo H, Yang J, Jin H, Huang C, Fu J, Yang F. et al. Tetrameric far-red fluorescent protein as a scaffold to assemble an octavalent peptide nanoprobe for enhanced tumor targeting and intracellular uptake in vivo. FASEB J. 2011;25:1865-73
48. Yu B, Tai HC, Xue W, Lee LJ, Lee RJ. Receptor-targeted nanocarriers for therapeutic delivery to cancer. Mol Membr Biol. 2010;27:286-98
49. Zhang Z, Chen J, Ding L, Jin H, Lovell JF, Corbin IR. et al. HDL-mimicking peptide-lipid nanoparticles with improved tumor targeting. Small. 2010;6:430-7
Author contact
![]() Corresponding author: czyzzhhust.edu.cn; zengmshorg.cn.
Corresponding author: czyzzhhust.edu.cn; zengmshorg.cn.