Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results and Discussion
Conclusion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2013; 3(6):409-419. doi:10.7150/thno.5616 This issue Cite
Research Paper
Lung Surfactant Microbubbles Increase Lipophilic Drug Payload for Ultrasound-Targeted Delivery
Shashank R. Sirsi1, Chinpong Fung2, Sumit Garg1, Mary Y. Tianning2, Paul A. Mountford1, Mark A. Borden1, ![]()
1. Department of Mechanical Engineering, University of Colorado, Boulder, CO 80309, USA;
2. Department of Chemical Engineering, Columbia University, New York, NY, USA.
Received 2012-11-27; Accepted 2013-3-27; Published 2013-5-20
Abstract
The cavitation response of circulating microbubbles to targeted ultrasound can be used for noninvasive, site-specific delivery of shell-loaded materials. One challenge for microbubble-mediated delivery of lipophilic compounds is the limitation of drug loading into the microbubble shell, which is commonly a single phospholipid monolayer. In this study, we investigated the use of natural lung surfactant extract (Survanta®, Abbott Nutrition) as a microbubble shell material in order to improve drug payload and delivery. Pulmonary surfactant extracts such as Survanta contain hydrophobic surfactant proteins (SP-B and SP-C) that facilitate lipid folding and retention on lipid monolayers. Here, we show that Survanta-based microbubbles exhibit wrinkles in bright-field microscopy and increased lipid retention on the microbubble surface in the form of surface-associated aggregates observed with fluorescence microscopy. The payload of a model lipophilic drug (DiO), measured by flow cytometry, increased by over 2-fold compared to lipid-coated microbubbles lacking SP-B and SP-C. Lung surfactant microbubbles were highly echogenic to contrast enhanced ultrasound imaging at low acoustic intensities. At higher ultrasound intensity, excess lipid was observed to be acoustically cleaved for localized release. To demonstrate targeting, a biotinylated lipopolymer was incorporated into the shell, and the microbubbles were subjected to a sequence of radiation force and fragmentation pulses as they passed through an avidinated hollow fiber. Lung surfactant microbubbles showed a 3-fold increase in targeted deposition of the model fluorescent drug compared to lipid-only microbubbles. Our results demonstrate that lung surfactant microbubbles maintain the acoustic responsiveness of lipid-coated microbubbles with the added benefit of increased lipophilic drug payload.
Keywords: contrast agent, echogenicity, acoustic radiation force, Survanta®, SP-B and SP-C.
Introduction
Microbubbles are used commonly as intravascular ultrasound (US) imaging probes and are becoming increasingly popular tools for targeted drug delivery [1-5]. Owing to their small size and biocompatible encapsulation, microbubbles circulate systemically for several minutes and perfuse vasculature throughout the body [6]. Addition of ligands to the microbubble surface provides receptor-mediated targeting [7, 8]. Imaging of microbubbles is performed typically at low US intensity, where they volumetrically oscillate in a mode of stable cavitation [9-12]. US radiation forces can be applied at low acoustic intensities and long duty cycles near the microbubble resonance frequency to facilitate targeting [13-17]. Furthermore, drug delivery can be achieved at higher acoustic intensity, where they violently collapse and fragment in a mode of inertial cavitation [18]. Inertial cavitation may also promote site-specific extravasation of therapeutic molecules to tissue [19]. The ability to focus ultrasound to image, target and fragment microbubbles and increase drug delivery makes this a promising theranostic system.
Several studies have shown that delivery is improved by loading the drug onto the microbubble shell, instead of simply mixing or co-injecting them in their free form prior to injection [20-22]. If the drug is injected in an unattached state, it rapidly becomes diluted throughout the volume of the blood pool. If loaded onto the microbubbles, however, the drug remains concentrated on the microbubble surface during intravascular transit and in the local volume immediately following US-mediated microbubble fragmentation. Furthermore, drug loading into the microbubble shell can shield the molecules from the immune system and enzymatic degradation [6, 22-25]. It is also possible that the drug molecules are more effectively convected into tissue during inertial cavitation [23].
The major limitation of this system, however, is the low drug loading capacity of the microbubbles owing to the limited surface area (~50 µm2 for a 4-µm diameter sphere). DNA loading, for example, is limited to approximately 0.005 pg/µm2 onto cationic microbubbles [26, 27], resulting in only 0.25 pg of DNA release per microbubble fragmentation event. Several novel microbubble formulations have been developed over the last decade in order to improve drug loading capacity [6, 23, 25, 28]. However, these loading strategies typically focus on hydrophilic drugs such as DNA and may involve numerous steps to manufacture. While multiple steps may be necessary to encapsulate hydrophilic drugs, it may not be necessary for hydrophobic and lipophilic drugs. For hydrophobic/lipophilic drugs, one common approach has been to form polymer-shelled microbubbles, in which the drug is mixed with a hydrophobic polymer in an organic phase prior to microbubble generation [29-31]. Polymer-coated microbubbles exhibit higher loading capacities for lipophilic drugs than lipid-coated microbubbles [6, 28]. However, the polymer shells are typically much thicker and more rigid than lipid shells, resulting in lower acoustic responsiveness for ultrasound imaging, targeting and fragmentation [6, 32, 33]. Therefore, methods are desired for generating acoustically active microbubbles with higher payload for lipophilic drugs.
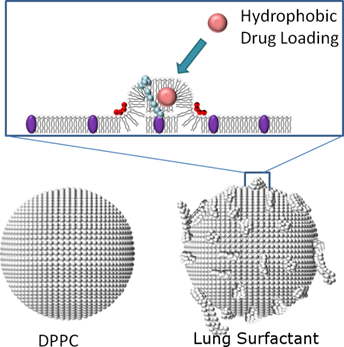
We previously described the generation of microbubbles from the natural lung surfactant extract Survanta® (Abbott Nutrition). Survanta is primarily composed of dipalmitoylphosphatidylcholine (DPPC), in addition to other lipids and the hydrophobic surfactant proteins SP-B and SP-C. Both SP-B and SP-C are known to facilitate lipid transfer between the lipid monolayer plane and attached lipid bilayer folds during repetitive compression and expansion of the alveolar lining while breathing [34-36]. SP-B and SP-C are thought to stabilize nanostructural defects within the monolayer plane [36, 37], as well as hinge and pin bilayer folds attached to the monolayer to form multilayer folds that remain strongly coupled through interpenetration of the protein hydrophobic regions between apposed monolayer and bilayer lamella [34, 35, 38]. Thus, these hydrophobic proteins serve as “anchors” that facilitate lipid retention onto the monolayer surface. Our inspection of lung surfactant microbubbles also showed the presence of lipid folds [39]. We therefore hypothesized that lung surfactant could be used to increase the payload of lipophilic drugs on the surface of lipid-coated microbubbles (Fig. 1). Furthermore, we hypothesized that the lipid folds could be shed during microbubble cavitation, thus acting as ultrasound-cleavable linkages for targeted drug delivery applications. Below, we describe a comparative study of Survanta microbubbles with DPPC microbubbles to determine the effects of SP-B and SP-C on size distribution, ultrasound imaging contrast, the payload of a model lipophilic drug (DiI and DiO), ultrasound-mediated release and targeted deposition in physiologically relevant flow.
Schematic comparing normal lipid (DPPC) and Survanta microbubble shell architectures. Hydrophobic surfactant proteins SP-B and SP-C are shown in red and light blue, respectively. We hypothesize SP-B and SP-C can form lipid folds that increase drug loading capacity and cleave upon high-intensity ultrasound exposure.
Materials and Methods
Materials
Lung surfactant extract (Survanta®, Abbot Nutrition) was purchased from McKesson (North Delran, NJ). Survanta is an organic extract of minced bovine lungs that has been fortified with ~70 wt % dipalmitoyl phosphatidylcholine (DPPC), ~10 wt % palmitic acid and tripalmitin [40-42]. Survanta has lower-than-native amounts of SP-B (0.04-0.13 wt %) and native amounts of SP-C (0.9-1.65 wt %) [41, 43, 44]. Survanta can change somewhat from batch to batch due to variations in extraction and purification, as well as variations in the source materials [40]. Synthetic phospholipids and lipopolymers were purchased from Avanti Polar Lipids (Alabaster, AL). All other reagents were purchased from Sigma-Aldrich (St. Louis, MO) and were used without further purification, unless otherwise stated.
Microbubble Preparation
The stock Survanta suspension was warmed to room temperature, taken directly from the vial with a sterile syringe and diluted to 2 mg/mL in 0.2-µm filtered PBS (10 mM NaCl, pH 7.4). Control lipid suspensions were made using 2 mg/mL of 90 mol% synthetic DPPC and 10 mol% 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (DSPE-PEG2000). Previous work by our group showed that PEG-lipid is not required for generating high yields of Survanta microbubbles [39], and therefore it was not added unless biotinylation was required. Lipid fluorescent dyes, 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI) or 3,3'-dioctadecyloxacarbocyanine perchlorate (DiO) (Invitrogen; Carlsbad, CA) were added to the lipid suspensions (6 µM dye concentration, ~ 0.25 wt % of lipid) as fluorescent markers and model lipophilic drugs. DiO was used for flow cytometric analysis and drug deposition studies. DiI was used to show drug position (instead of DiO) on the microbubbles using fluorescence microscopy owing to better photostability. Dye was added by micropipetting just after dilution of Survanta, and the solution was heated to 60 °C for 20 min to allow the dye to fully integrate into the lipid-protein mixture. The solution was then bath sonicated (Model 250A, Branson Ultrasonics; Danbury, CT) until the initially opaque lipid solution turned translucent. 2 mL of Survanta or DPPC:DSPE-PEG2000 lipid suspensions were placed in 3-mL glass serum vials (Wheaton, Millville, NJ), and the vials were immediately capped and sealed. The air headspace was replaced with perfluorobutane (PFB) gas (Fluoromed; Rockford, TX) using a custom gas-exchange apparatus. Microbubbles were produced by a shaking method using a VialMix shaker (Bristol-Myers Squibb, New York, NY) [45]. The vial was shaken at 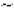 4300 Hz for 45 s and then collected into a 3-mL syringe (BD; Franklin Lane, NJ). Microbubbles were washed 3 times to remove free lipid and unincorporated dye from buoyant microbubbles using centrifugation (300g for 3 min; Model 5804, Eppendorf, Westbury, NY) [46].
4300 Hz for 45 s and then collected into a 3-mL syringe (BD; Franklin Lane, NJ). Microbubbles were washed 3 times to remove free lipid and unincorporated dye from buoyant microbubbles using centrifugation (300g for 3 min; Model 5804, Eppendorf, Westbury, NY) [46].
Particle Characterization
Particle size analysis was performed using a Multisizer III (Beckmann Coulter; Opa Locka, FL), which operates on the principle of changing electrical impedance as individual particles flow through a small aperture. Model dye uptake into the microbubbles was measured using an Accuri C6 flow cytometer (Accuri Cytometers Inc.; Ann Arbor, MI). The forward-scatter height (FSC-H) threshold was adjusted to delineate the microbubble populations from instrument and sample noise. The size class (1-2, 4-5 and 6-8 µm diameter) of the microbubbles from the density scatter plots was determined as previously described [47]. Microbubble suspensions were diluted to equal concentrations (107 mL-1), and 100,000 events were counted per trial. All measurements were repeated in triplicate.
Phosphorus Determination
The lipid content per unit surface area of Survanta and DPPC microbubbles was determined from a quantitative phosphorus assay and size distribution of a microbubble sample. The phosphorus assay was adapted from a previously published protocol [48]. Briefly, microbubbles were destroyed after size analysis by heating to 80 °C until the suspension became translucent. Digestion of the lipid to inorganic phosphate was performed by adding 100 μL of lipid solution to 0.5 mL of 8.9 N H2SO4 and heated to 200 °C in an oil bath for 25 minutes. The solution was then cooled to room temperature, mixed with 150 μL H2O2 (Thermo Fisher) and heated to 200 °C for 30 minutes. The solution was then cooled to room temperature and mixed with 3.9 mL of DI water and 0.5 mL of 2.5% ammonium molybdate (VI) tetrahydrate solution. The solution was then vortexed and mixed with 0.5 mL of 10% L-ascorbic acid. The solution was then heated to 100 °C for 7 minutes and cooled to room temperature. The absorbance of the sample was measured at 820 nm (J-815 CD Spectrometer; Jasco Inc.; Easton, M). Phosphorus standards with known concentrations were assayed along with the lipid samples to determine the total amount of phosphorus. The phosphorus amount was normalized to microbubble surface area (modeled as a perfect sphere from size distribution analysis) in each sample in order to calculate the surface density of lipid. All measurements were repeated in triplicate.
Fluorescence Microscopy
Epi-illumination fluorescence optical microscopy was performed using an Olympus BX-60 upright microscope with an oil-immersion 100X objective [39]. Images were acquired with a high-resolution camera (Orca HR; Hamamatsu, Japan). Z-stacks were obtained using an automated stage at 0.5-µm increments and then converted to maximum intensity projection (MIP) images using ImageJ software (NIH).
Microbubble Pressurization
Microbubbles were pressurized using a technique similar to the one described by Rychak et al. [49] to reduce the gas core volume and induce lipid monolayer compression. Pressurization of individual microbubbles was observed with bright-field microscopy. Microbubbles were perfused through a capillary tube (200 µm diameter; Spectrum Labs; Rancho Dominguez, CA) mounted on an inverted 1X-71 microscope, as described by Kwan et al [50]. After the microbubbles were perfused into the chamber and individual microbubbles were in focus, the exit was clamped to allow a small pressure build-up within the tube (~20 kPa). Pressure was applied slowly and was monitored using a 0-15 psi digital pressure gauge (DPG1000DAR-30G-1N, Omegadyne, Sansbury, OH). Images of the microbubbles before and after pressurization were recorded using a high-resolution camera (Orca HR, Hamamatsu).
The effect of pressurization on model drug uptake for microbubble suspensions was measured with flow cytometry. Microbubbles were placed in open serum vials at a concentration of 109 mL-1. The serum vial then was placed into a 60-mL syringe (BD Biosciences) with the syringe body connected to a digital pressure gauge (HHP241-100G, Omega, Stamford, CN). Pressure was increased by slowly pressing the syringe plunger to achieve 100 kPa of gauge pressure (~30 seconds). The pressure was held constant for 1 minute and then slowly released to atmospheric pressure (~30 seconds). Microbubbles were removed and immediately analyzed by flow cytometry following a single pressure cycle. Measurements were repeated in triplicate.
Ultrasound Imaging
The echogenicity of the Survanta bubbles was determined using an Acuson Sequoia 512 clinical ultrasound scanner (Siemens Healthcare USA, Malvern, PA). A 15-MHz 15L8 linear transducer was placed over a custom agarose gel phantom made with a hollow center to allow flow from a syringe pump to a collection beaker. A bolus of microbubbles was introduced at 5x106 mL-1 concentration. Imaging was performed using a non-linear contrast pulse sequencing (CPS) mode imaging at 7 MHz, -5 dB CPS gain and mechanical index of 0.49. The mean contrast was measured at the peak intensity following the bolus injection using ImageJ software.
Ultrasound-Triggered Release
Ultrasound-triggered release of a model drug (DiO) was demonstrated in vitro using a flow-through system to monitor fluorescent microbubbles exposed to ultrasound. A custom apparatus was developed to monitor microbubbles in a capillary tube, similar to the experimental setup described by Lum et al [51]. An acrylic water tank was placed onto an Olympus 1X-71 inverted microscope, and microbubbles were perfused through a 200-μm diameter cellulose capillary tube (Spectrum Labs) positioned within the tank directly above a 100X water-immersion objective (Olympus). Microbubbles were diluted to 106 mL-1 and perfused through the capillary tube. An ultrasound transducer (V314; Olympus NDT; Waltham, MA) with a 1.0-MHz center frequency was used to apply ultrasound pulses to microbubbles in the microscope field of view. The ultrasound transducer was driven by an arbitrary waveform generator (AWG2021; Tektronix; Beaverton, OR) with the RF signal amplified by a linear amplifier (ENI 525LA; Rochester, NY). The peak negative pressure (PNP) of the ultrasound field was measured using a needle hydrophone (HNC-0100; Ondacorp; Sunnyvale, CA) attached to an oscilloscope (HP54502A; Santa Clara, CA). Single ultrasound pulses were applied at 200 kPa PNP at a 0.5-Hz repetition frequency. Images were taken using an analog Sony CCD camera (DXC-9000; Parkridge, NJ) mounted on the inverted scope (640x480 resolution; 32 frames per second). The application of US and image acquisition was controlled using a custom Labview program and data acquisition board (National Instruments, Austin, TX). Analog video from the camera was captured and recorded using an analog-to-digital USB video adapter (Imaging Source; Charlotte, NC) and Labview drivers supplied by the manufacturer.
Targeted Drug Deposition
Ultrasound-mediated deposition of the shell-loaded material (DiO) from the microbubbles was demonstrated using the same US pulsing and imaging experimental setup as above. In this experiment, Survanta or DPPC microbubbles were biotinylated by incorporating DSPE-PEG2000-Biotin into the microbubble shell. For Survanta, DSPE-PEG2000-Biotin and DSPE-PEG2000 were added into lipid solution prior to generating bubbles (final concentrations estimated to be ~5 mol%). Adsorption of avidin onto the tube wall was done by flowing a 10-µL (5 mg/mL) plug into the dry hollow fiber and incubating for 20 minutes. The avidin-coated tube was then washed with 10 mL of DI water to remove free avidin. Microbubbles were introduced into the capillary tube (107 mL-1) at 1 mL/hr (3.5 dyn/cm2 wall shear stress, 13.2 mm/sec centerline velocity) using a 3-mL disposable syringe (BD Sciences; Franklin Lakes, NJ) attached to a syringe pump (Genie Plus; Kent Scientific, Torrington, CT). Ultrasound radiation forces (USRF) were applied (1.5 MHz, 50 kPa PNP, 3 seconds at 100% duty cycle) in order to push the flowing biotinylated microbubbles against the avidin-coated capillary wall [15, 51]. Following the USRF pulse, 5 fragmentation pulses (5 cycles per pulse, 1.5 MHz, 250 kPa PNP) were applied to destroy microbubbles bound to the tube wall. The USRF-fragmentation sequence was repeated every 10 seconds for 10 minutes. Microbubbles under flow were observed to move through the center of the tube. For 6-8 µm diameter microbubbles, the centerline velocity was approximately 1000 times larger than the calculated Stokes rise velocity. Also, ultrasound radiation force and microscopic analysis was directed at the side of the tube, orthogonal to the gravitational vector. Therefore, flotation was ruled out as a significant cause of microbubble binding to the capillary tube. The dye accumulated from the shell material remaining on the capillary wall was imaged using fluorescence microscopy before and after application of the USRF-fragmentation pulses. Lipid fragments generated by fragmentation of the microbubbles contained DiO and biotinlayted lipid. These vesicles were “shed” from the microbubble surface, but remained adherent to the avidinated cellulose tube wall following the US fragmentation pulses. However, fluorescence imaging was not performed during USRF-fragmentation pulsing in order to avoid bleaching of the fluorescent signal. Z-stacked images of the capillary wall were taken at 10-µm spacing, and the total pixel intensity was summed for all the images within the stack using ImageJ software. Experiments were repeated in triplicate.
Statistical Analysis
All statistical analyses were performed using Graphpad™ Prism software (La Jolla, CA). Students' t-tests were performed to evaluate significant differences between groups. A p-value of less than 0.05 was considered statistically significant.
Results and Discussion
Excess Lipid and Model Drug Payload
Microbubbles formed using Survanta or DPPC showed similar size distributions and yield (Fig. 2A). However, fluorescence microscopy of DiI-labeled microbubbles demonstrated an observable difference in surface structure between DPPC and Survanta-based microbubbles (Fig. 2B). Previous freeze-fracture TEM and higher magnification fluorescence microscopy images showed dark domains on the microbubble shell that excluded the lipoid dye [39], but at the current magnification the microbubbles appeared uniformly coated with dye. Lipid folds extending into the aqueous medium were clearly seen here for Survanta microbubbles, but not for microbubbles coated with DPPD:DSPE-PEG2000. The surface structure of the Survanta microbubbles was attributed to lipid folds stabilized by the surfactant proteins SP-B and SP-C. In order to confirm this hypothesis, we measured the lipid density per surface area of the microbubbles using a phosphorus assay. The lipid surface density was ~2.3-fold higher for Survanta microbubbles in comparison to DPPC microbubbles (P<0.05) (Fig. 2C).
A) Representative size distribution (number%) of DPPC and Survanta based microbubbles using an impedance-based Multisizer particle sizing system. B) Fluorescent microscopy images of normal DPPC bubbles and bulky Survanta bubbles loaded with model fluorescent drug DiI. Images are maximum-intensity projections from a 20 fluorescent image z-series taken at 0.5-µm spacing. Scale bars are 10 µm. C) Results of phosphorus assay used to determine lipid density per unit surface area of DPPC and Survanta microbubbles (3 trials per group). *Indicates P<0.05 between DPPC and Survanta groups using student's t-test
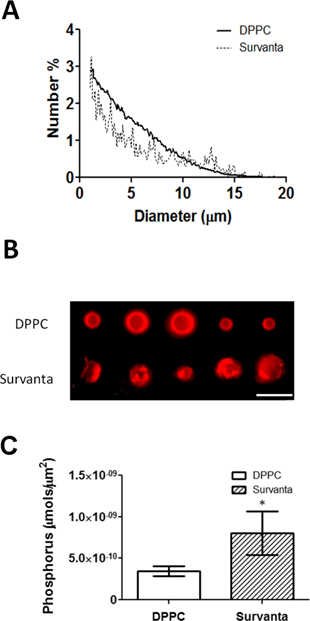
We hypothesized that an increase in the lipid surface density would improve the loading capacity of lipophilic drugs. To test this, we used the lipoid dye DiO as a model drug. DPPC and Survanta microbubbles were analyzed using flow cytometry to measure dye uptake. DPPC bubbles showed a familiar serpentine pattern in the forward-scatter vs. side-scatter plots (Fig. 3A), as previously observed [46, 47]. Survanta microbubbles displayed the same pattern. However, an increased number of events was observed outside of the typical microbubble serpentine gate. This may be another indication of excess lipid bulk on the Survanta microbubble shell, as side-scatter is often associated with cell-surface granularity in normal flow cytometry applications [47]. However, because the optical side-scatter in nonlinearly dependent on microbubble size, an unambiguous quantitative assessment of lipid fold distribution was difficult. Fluorescent dye uptake in the microbubble shell was analyzed for varying microbubble size classes (Fig. 3B&C). The relatively small error bars in Fig. 3B indicate that dye incorporation was consistent between samples. Survanta microbubbles showed significantly higher levels of dye loading in the shell compared to DPPC microbubbles for every size class (2.1-fold higher for 1-2 µm, 2.7-fold higher for 4-5 µm and 2.3-fold higher for 6-8 µm), indicating that microbubble size did not affect the degree of lipid retention on the surface. The dye uptake for the Survanta microbubbles showed a wider distribution compared to the DPPC microbubbles (Fig. 3B). This result is consistent with fluorescence microscopy images (Fig. 2B and [39]), which showed bubble-to-bubble variation in the number and size of the lipid folds. Interestingly, the increase in level of dye accumulation on the microbubble surface was similar to the values observed from the phosphorous assay (~2.3-fold increase in lipid surface density), thus the results seem to indicate that fluorescence intensity correlates to lipid surface density.
Flow cytometric characterization of DPPC and Survanta microbubbles. A) Representative density scatter plots. Gates are shown for varying size classes of microbubbles. Survanta microbubbles demonstrated an increased variability in scattering compared to DPPC microbubbles. B) Fluorescent intensity histograms of model drug DiO-loaded microbubbles corresponding to the gates in panel A. D) Cumulative data comparing DiO lipophilic dye uptake in the microbubble shell (relative dye loading vs. microbubble surface area) (N=3 trials per group). *Indicates P<0.05 between DPPC and Survanta groups using a student's t-test.
One of the primary functions of SP-B and SP-C proteins is to facilitate in lipid retention on the alveolar lining during compression [34]. Such retention may be used to increase the surface-area-to-gas-volume ratio of echogenic microbubbles for improved drug loading capacity. To explore this possibility, we employed a technique developed by Rychak et al. [49] to generate “wrinkled” microbubble structures (defined by the presence of observable lipid folds or protrusions from the microbubble surface) by applying pressure and causing partial gas loss from the microbubble core (Fig. 4A). We observed that Survanta microbubbles under slight pressure exhibit a “wrinkled” topology under bright-filed microscopy (Fig. 4B), which was not observed for DPPC bubbles. In order to visualize lipid retention, dye-labeled (DiI) microbubbles were observed using fluorescence microscopy before and after pressurization. Fluorescent microscopy showed no observable lipid buildup on DPPC microbubbles before or after pressurization (Fig. 4C). This is consistent with previous freeze-fracture TEM work, which showed that phospholipid forms a monomolecular layer around the microbubbles, rather than multilayers [52]. Survanta microbubbles showed obvious excess lipid buildup (bright areas of localized dye accumulation), both before and after pressurization. Qualitatively, pressurized Survanta microbubbles appeared to have more fluorescent dye buildup than their unpressurized counterparts.
Microbubble pressurization. A) The microbubble gas core volume was reduced by applying 100 kPa of external pressure for 1 minute, causing partial perfluorocarbon gas dissolution into the surrounding media. B) Bright-field microscopy demonstration of Survanta microbubble “wrinkling” under slight application of pressure (~20 kPa). C) Fluorescence microscopy images of selected DiI-laoded microbubbles before and after pressurization. DPPC bubbles showed no observable change in lipid shell structure before and after pressurization. Conversely, Survanta microbubbles exhibited an excess surface lipid both before and after pressurization. Images are maximum-intensity projections from a 20 image z-series taken at 0.5-µm spacing. Scale bars are 10 µm.
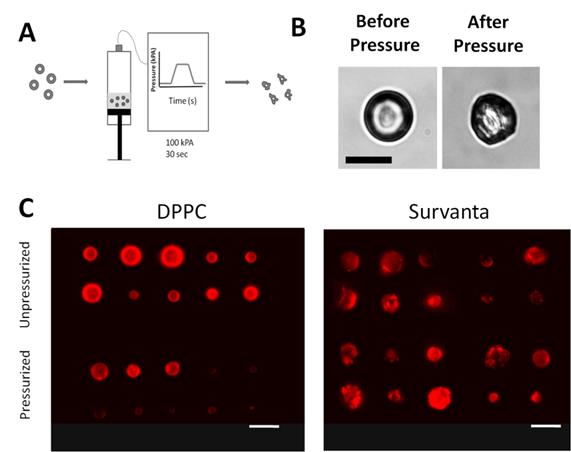
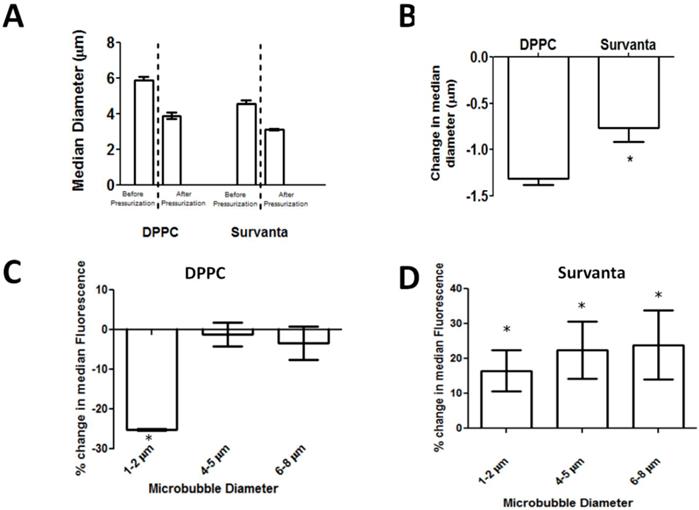
Partial dissolution of the gas core following pressurization was confirmed by measuring particle size using impedance-based sensing (Multisizer) (Fig. 5A). To further investigate if wrinkling in Survanta microbubbles can enhance the degree of hydrophobic drug uptake, we quantified the dye content of microbubbles after pressurization using a flow cytometer. We expected that pressurized microbubbles would shift to smaller size owing to partial gas loss (for example, events detected in the 6-8 µm diameter gate would subsequently be detected in the 4-5 µm diameter gate following pressurization, see Supplemental Data). Microbubbles that undergo lipid shedding during pressurization would amount to little change in the median fluorescence intensity values for each microbubble size class because excess dye is expected to be released with the lipid. On the other hand, retention of lipid on the microbubble surface would amount to an increase in the median intensity value. As expected, DPPC microbubbles showed no significant change in the median fluorescence intensity before and after pressurization for microbubbles greater than 4 µm (P=0.70 and 0.32 for 4-5 and 6-8 µm, respectively). Interestingly, the median fluorescence values for the 1-2 µm bubbles decreased by 23.0 ± 3.8% (P<0.01) (Fig. 5C). We speculate that the decrease in fluorescence for the smaller microbubbles, which are known to be more stable [46, 53, 54], may be due to molecular rearrangements within the lipid monolayer during compression rather than lipid shedding from the monolayer. Conversely, Survanta microbubbles showed an increase in median fluorescence intensity following pressurization for all size classes (+16.2 ± 4.2% for 1-2 µm, +22.4 ± 8.2% for 4-5 µm, and +23.8 ± 9.1% for 6-8 µm; P<0.05 for all groups) (Fig. 5D). This increase in median fluorescence following pressurization is an indication of dye retention on the microbubble surface and demonstrates that Survanta microbubbles can be treated to increase drug payload.
Quantification of lipid retention in pressurized microbubbles. A) Median diameters of bubbles as reported by Multisizer particle sizing systems. Median diameters are reported before and after single pressurization cycle for DPPC and Survanta microbubbles. B) Change in median diameter before and after pressurization. (C,D) Percent change in median fluorescence intensity measured by flow cytometry before and after pressurization. N= 3 trials per group for A-D. * Indicates significantly different from baseline value (P<0.05) using a student's t-test.
Acoustic Responsiveness
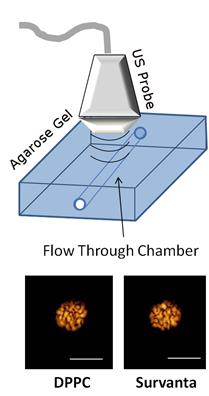
We first demonstrated that Survanta microbubbles are echogenic and retain similar contrast agent characteristics as DPPC microbubbles with matched size and concentration. Ultrasound CPS imaging showed similar echogenic properties of Survanta and DPPC microbubbles, as measured by the mean grayscale pixel intensity in an agarose phantom: 210 ± 15 relative video intensity units (ranges from 0-255 for 8-bit pixel intensity values) for Survanta microbubbles and 192 ± 32 for DPPC microbubbles (Fig. 6).
Ultrasound detection of DPPC and Survanta microbubbles in a flow-through agarose gel phantom using CPS mode ultrasound imaging.
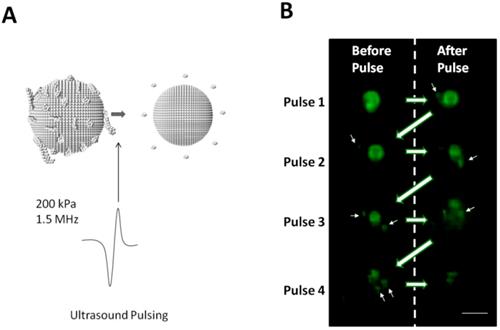
The ability of individual Survanta microbubbles to shed their excess lipid bulk via ultrasound-mediated release was examined with microscopy while applying ultrasound pulses every two seconds (Fig. 7A). Following each pulse, large fluorescent structures were observed to “pinch off” from the microbubble body (Fig. 7B, white arrows), presumably from dye-containing lipid fragments released by rapid volumetric contraction of the microbubble [18, 55]. Therefore, we conclude that the excess lipid retained on the Survanta microbubbles can act as an ultrasound-cleavable linker for on-demand drug release.
Acoustic cleavage of lipid bilayer folds on Survanta microbubbles. A) Ultrasound was applied to Survanta microbubbles on the microscopy stage using single ultrasound pulses (1.5 MHz, 200 kPa PNP) at 0.5 Hz pulse repetition frequency. B) Images of dye loaded microbubbles were obtained at 640x480 resolution at 32 frames per second. Images are shown before and after the ultrasound pulse is applied. Arrows indicate lipid shedding during ultrasound pulsing. Scale bar is 10 µm.
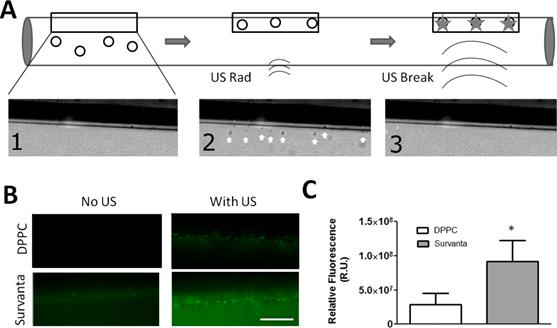
To further demonstrate this effect, we compared the ability of Survanta microbubbles to deposit drug under continuous flow (using fluorescent DiO as a model drug). In this experiment, biotinylated Survanta microbubbles were flowed through a capillary tube, and an ultrasound sequence (USRF followed by fragmentation, see methods) was used to deposit shell-loaded dye material onto the wall of an avidinated cellulose capillary tube (Fig. 8A). Survanta microbubbles demonstrated higher levels of drug deposition compared to biotinlyated-DPPC microbubbles with matched size and concentration (Fig. 8B). Survanta microbubbles without US application also showed higher background levels of dye deposition on the capillary wall, whereas DPPC microbubbles without US stimulation showed no detectable dye deposition on the capillary tube. This result indicates that a small amount of excess lipid was shed from Survanta microbubbles, perhaps by folds that were sheared off in flow. Normalizing by the background fluorescence, Survanta microbubbles demonstrated an ~3-fold increase in dye intensity on the capillary wall ((3.1 ± 1.2)x107 R.U. for DPPC microbubbles and (9.1 ± 3.0)x107 R.U. for Survanta microbubbles, P<0.05).
Targeted ultrasound deposition of the model drug DiO. A) (1) Biotinilayted microbubbles were perfused through an avidin coated capillary tube at 1 mL/hr. (2) Ultrasound radiation forces (1.5 MHz, 50 kPa PNP, 100% duty cycle for 5 seconds) were applied to force the microbubbles against the capillary tube wall. (3) Higher intensity fragmentation pulses (1.5 MHz, 250 kPa PNP) were applied 5 times to fragment the microbubbles in the field of view, depositing the fluorescent lipid dye onto the capillary tube surface. B) Fluorescent microscopy images of shell loaded dye deposited on the capillary tube wall with or without US application. Images were taken at the mid-plane of the capillary tube. C) Overall fluorescence accumulation on the capillary wall. Z-stacks were acquired at 10-µm spacing throughout (10 frames total) and the pixel intensities were summed (N=3 trials per group). *Indicates P<0.05 between DPPC and Survanta groups using a student's t-test.
Conclusion
The goal of this work was to examine the capability of lung surfactant extract to generate microbubbles with both acoustic responsiveness and enhanced drug payload. We demonstrated with fluorescence and bright-field microscopy, phosphorous assay and flow cytometry that Survanta microbubbles have roughly twice the lipid surface density of DPPC microbubbles. Following Rychak et al. [49], we further demonstrated that the lipid surface density can be increased on Survanta microbubbles through a simple pressurization technique, whereas the method is ineffective for DPPC microbubbles. DPPC is the primary lipid constituent of Survanta, and we therefore attributed the enhanced lipid payload to the surfactant proteins SP-B and SP-C, which are known to facilitate lipid shuttling back and forth between the monolayer and bilayer folds of the alveolar lining as it expands and contracts during breathing. We further showed that lung surfactant microbubbles are echogenic, they can be targeted to a surface by ligand-receptor binding and application of ultrasound radiation force and they can be fragmented by ultrasound to locally deliver a lipoid fluorescent dye. Overall, lung surfactant microbubbles show promise as a potential theranostic agent for ultrasound image-guided drug delivery of lipophilic drugs.
Study Limitations
In this study, we demonstrated that drug loading content on microbubbles can be improved by the presence of surface-active proteins that stabilize lipid folds on the monolayer shell. One drawback to the approach used in this study is that the ratio of surface-active proteins SP-B and SP-C to lipid DPPC was not well controlled (these ratios are variable from batch to batch, as specified by the manufacturer). Recent studies have demonstrated that supplementing DPPC with truncated SP-B or SP-B analogous peptides may facilitate folding during monolayer collapse [56, 57]. The use of recombinant peptides with synthetic lipids may provide better control over the degree of lipid folding, thus improving drug-loading efficiency. The use of recombinant peptides may also avoid premature lipid-fold shedding and immunogenic issues compared to bovine-derived lung surfactant used in this study. Another limitation of this study is that we estimated drug payload by measuring the fluorescence of a lipoid dye, rather than a hydrophobic/lipophilic drug compound (such as paclitaxel). We expect that the influence of lipid folds on drug loading will be similar, since hydrophobicity drives the association in both cases, but this requires further experimental confirmation. A final limitation to this study is that Survanta microbubbles as ultrasound contrast agents and drug delivery vehicles have yet to be investigated in vivo.
Supplementary Material
Fig.S1 - S4.
Acknowledgements
The authors wish to thank Dr. Cherry Chen for initial flow cytometry experiments, such as those shown in Supplementary Material: Figure S3. Funding for this work was provided by NSF grant CMMI-1100335 to MAB.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Wei K. Future applications of contrast ultrasound. J Cardiovasc Ultrasound. 2010;19:107-14
2. Suzuki R, Oda Y, Utoguchi N, Maruyama K. Progress in the development of ultrasound-mediated gene delivery systems utilizing nano- and microbubbles. J Control Release. 2010;149:36-41
3. Geis NA, Katus HA, Bekeredjian R. Microbubbles as a vehicle for gene and drug delivery: current clinical implications and future perspectives. Curr Pharm Des. 2012;18:2166-83
4. Ferrara K, Pollard R, Borden M. Ultrasound microbubble contrast agents: fundamentals and application to gene and drug delivery. Annu Rev Biomed Eng. 2007;9:415-47
5. Sheffield P, Trehan A, Boyd B, Wong OL. Microbubbles as ultrasound contrast agents and in targeted drug delivery. Crit Rev Biomed Eng. 2008;36:225-55
6. Sirsi S, Borden M. Microbubble Compositions, Properties and Biomedical Applications. Bubble Sci Eng Technol. 2009;1:3-17
7. Schmitz G. Ultrasonic imaging of molecular targets. Basic Res Cardiol. 2008;103:174-81
8. Lindner JR. Molecular imaging with contrast ultrasound and targeted microbubbles. J Nucl Cardiol. 2004;11:215-21
9. Stride E. Physical principles of microbubbles for ultrasound imaging and therapy. Cerebrovasc Dis. 2009;27(Suppl 2):1-13
10. Stride EP, Coussios CC. Cavitation and contrast: the use of bubbles in ultrasound imaging and therapy. Proc Inst Mech Eng H. 2010;224:171-91
11. Azmin M, Harfield C, Ahmad Z, Edirisinghe M, Stride E. How do microbubbles and ultrasound interact? Basic physical, dynamic and engineering principles. Curr Pharm Des. 2012;18:2118-34
12. Qin S, Caskey CF, Ferrara KW. Ultrasound contrast microbubbles in imaging and therapy: physical principles and engineering. Phys Med Biol. 2009;54:R27-57
13. Dayton PA, Morgan KE, Klibanov ALS, Brandenburger G, Nightingale KR, Ferrara KW. A preliminary evaluation of the effects of primary and secondary radiation forces on acoustic contrast agents. IEEE Trans Ultrason Ferroelectr Freq Control. 1997;44:1264-77
14. Dayton P, Klibanov A, Brandenburger G, Ferrara K. Acoustic radiation force in vivo: a mechanism to assist targeting of microbubbles. Ultrasound Med Biol. 1999;25:1195-201
15. Dayton PA, Allen JS, Ferrara KW. The magnitude of radiation force on ultrasound contrast agents. J Acoust Soc Am. 2002;112:2183-92
16. Zhao S, Borden M, Bloch SH, Kruse D, Ferrara KW, Dayton PA. Radiation-force assisted targeting facilitates ultrasonic molecular imaging. Mol Imaging. 2004;3:135-48
17. Gessner RC, Streeter JE, Kothadia R, Feingold S, Dayton PA. An in vivo validation of the application of acoustic radiation force to enhance the diagnostic utility of molecular imaging using 3-D ultrasound. Ultrasound Med Biol. 2012;38:651-60
18. Thomas DH, Butler M, Anderson T, Emmer M, Vos H, Borden M. et al. The "quasi-stable" lipid shelled microbubble in response to consecutive ultrasound pulses. Appl Phys Lett. 2012 101
19. Stieger SM, Caskey CF, Adamson RH, Qin SP, Curry FRE, Wisner ER. et al. Enhancement of vascular permeability with low-frequency contrast-enhanced ultrasound in the chorioallantoic membrane model. Radiology. 2007;243:112-21
20. Bekeredjian R, Chen S, Grayburn PA, Shohet RV. Augmentation of cardiac protein delivery using ultrasound targeted microbubble destruction. Ultrasound Med Biol. 2005;31:687-91
21. Frenkel PA, Chen S, Thai T, Shohet RV, Grayburn PA. DNA-loaded albumin microbubbles enhance ultrasound-mediated transfection in vitro. Ultrasound Med Biol. 2002;28:817-22
22. Lentacker I, Geers B, Demeester J, De Smedt SC, Sanders NN. Design and evaluation of doxorubicin-containing microbubbles for ultrasound-triggered doxorubicin delivery: cytotoxicity and mechanisms involved. Mol Ther. 2010;18:101-8
23. Lentacker I, De Smedt SC, Sanders NN. Drug loaded microbubble design for ultrasound triggered delivery. Soft Matter. 2009;5:2161-70
24. Panje CM, Wang DS, Pysz MA, Paulmurugan R, Ren Y, Tranquart F. et al. Ultrasound-mediated gene delivery with cationic versus neutral microbubbles: effect of DNA and microbubble dose on in vivo transfection efficiency. Theranostics. 2012;2:1078-91
25. Sirsi S, Borden M. Advances in Ultrasound Mediated Gene Therapy Using Microbubble Contrast Agents. Theranostics. 2012;2(12):1208-1222
26. Borden MA, Caskey CF, Little E, Gillies RJ, Ferrara KW. DNA and polylysine adsorption and multilayer construction onto cationic lipid-coated microbubbles. Langmuir. 2007;23:9401-8
27. Lentacker I, De Geest BG, Vandenbroucke RE, Peeters L, Demeester J, De Smedt SC. et al. Ultrasound-responsive polymer-coated microbubbles that bind and protect DNA. Langmuir. 2006;22:7273-8
28. Hernot S, Klibanov AL. Microbubbles in ultrasound-triggered drug and gene delivery. Adv Drug Deliv Rev. 2008;60:1153-66
29. Eisenbrey JR, Huang P, Hsu J, Wheatley MA. Ultrasound triggered cell death in vitro with doxorubicin loaded poly lactic-acid contrast agents. Ultrasonics. 2009;49:628-33
30. Eisenbrey JR, Burstein OM, Kambhampati R, Forsberg F, Liu JB, Wheatley MA. Development and optimization of a doxorubicin loaded poly(lactic acid) contrast agent for ultrasound directed drug delivery. J Control Release. 2010;143:38-44
31. Cochran MC, Eisenbrey J, Ouma RO, Soulen M, Wheatley MA. Doxorubicin and paclitaxel loaded microbubbles for ultrasound triggered drug delivery. Int J Pharm. 2011;414:161-70
32. Bloch SH, Wan M, Dayton PA, Ferrara KW. Optical observation of lipid- and polymer-shelled ultrasound microbubble contrast agents. Appl Phys Lett. 2004;84:631-3
33. Zhang S, Zong Y, Wan M, Yu X, Fu Q, Ding T. et al. Compare ultrasound-mediated heating and cavitation between flowing polymer- and lipid-shelled microbubbles during focused ultrasound exposures. J Acoust Soc Am. 2012;131:4845-55
34. Perez-Gil J. Structure of pulmonary surfactant membranes and films: the role of proteins and lipid-protein interactions. Biochim Biophys Acta. 2008;1778:1676-95
35. Perez-Gil J. Lipid-protein interactions of hydrophobic proteins SP-B and SP-C in lung surfactant assembly and dynamics. Pediatr Pathol Mol Med. 2001;20:445-69
36. Lee KY. Collapse mechanisms of Langmuir monolayers. Annu Rev Phys Chem. 2008;59:771-91
37. Discher BM, Schief WR, Vogel V, Hall SB. Phase separation in monolayers of pulmonary surfactant phospholipids at the air-water interface: Composition and structure. Biophys J. 1999;77:2051-61
38. Gomez-Gil L, Schurch D, Goormaghtigh E, Perez-Gil J. Pulmonary surfactant protein SP-C counteracts the deleterious effects of cholesterol on the activity of surfactant films under physiologically relevant compression-expansion dynamics. Biophys J. 2009;97:2736-45
39. Sirsi S, Pae C, Oh DKT, Blomback H, Koubaa A, Papahadjopoulos-Sternberg B. et al. Lung surfactant microbubbles. Soft Matter. 2009;5:4835-42
40. Stenger PC, Isbell SG, St Hillaire D, Zasadzinski JA. Rediscovering the Schulze-Hardy Rule in Competitive Adsorption to an Air-Water Interface. Langmuir. 2009;25:10045-50
41. Bernhard W, Mottaghian J, Gebert A, Rau GA, von der Hardt H, Poets CF. Commercial versus native surfactants - Surface activity, molecular components, and the effect of calcium. American Journal of Respiratory and Critical Care Medicine. 2000;162:1524-33
42. Blanco O, Perez-Gil J. Biochemical and pharmacological differences between preparations of exogenous natural surfactant used to treat Respiratory Distress Syndrome: Role of the different components in an efficient pulmonary surfactant. European Journal of Pharmacology. 2007;568:1-15
43. Braun A, Stenger PC, Warriner HE, Zasadzinski JA, Lu KW, Taeusch HW. A freeze-fracture transmission electron microscopy and small angle X-ray diffraction study of the effects of albumin, serum, and polymers on clinical lung surfactant microstructure. Biophys J. 2007;93:123-39
44. Walther FJ, Waring AJ, Sherman MA, Zasadzinski JA, Gordon LM. Hydrophobic surfactant proteins and their analogues. Neonatology. 2007;91:303-10
45. Unger EC, McCreery DC, Yellowhair D, Barrett T. Apparatus and method of making gas-filled vesicles of optimal size. Office USP, editor. USA. 1997
46. Feshitan JA, Chen CC, Kwan JJ, Borden MA. Microbubble size isolation by differential centrifugation. J Colloid Interface Sci. 2009;329:316-24
47. Chen CC, Borden MA. Ligand conjugation to bimodal poly(ethylene glycol) brush layers on microbubbles. Langmuir. 2010;26:13183-94
48. Chen PS, Toribara TV, Warner H. Microdetermination of Phosphorus. Anal Chem. 1956;28:1756-8
49. Rychak JJ, Lindner JR, Ley K, Klibanov AL. Deformable gas-filled microbubbles targeted to P-selectin. J Control Release. 2006;114:288-99
50. Kwan JJ, Borden MA. Microbubble dissolution in a multigas environment. Langmuir. 2010;26:6542-8
51. Lum AF, Borden MA, Dayton PA, Kruse DE, Simon SI, Ferrara KW. Ultrasound radiation force enables targeted deposition of model drug carriers loaded on microbubbles. J Control Release. 2006;111:128-34
52. Borden MA, Martinez GV, Ricker J, Tsvetkova N, Longo M, Gillies RJ. et al. Lateral phase separation in lipid-coated microbubbles. Langmuir. 2006;22:4291-7
53. Kwan JJ, Borden MA. Lipid monolayer collapse and microbubble stability. Adv Colloid Interface Sci. 2012;183-184:82-99
54. Kwan JJ, Borden MA. Lipid monolayer dilatational mechanics during microbubble gas exchange. Soft Matter. 2012;8:4756-66
55. Borden MA, Kruse DE, Caskey CF, Zhao S, Dayton PA, Ferrara KW. Influence of lipid shell physicochemical properties on ultrasound-induced microbubble destruction. IEEE Trans Ultrason Ferroelectr Freq Control. 2005;52:1992-2002
56. Ding J, Takamoto DY, von Nahmen A, Lipp MM, Lee KY, Waring AJ. et al. Effects of lung surfactant proteins, SP-B and SP-C, and palmitic acid on monolayer stability. Biophys J. 2001;80:2262-72
57. Holten-Andersen N, Michael Henderson J, Walther FJ, Waring AJ, Ruchala P, Notter RH. et al. KL(4) peptide induces reversible collapse structures on multiple length scales in model lung surfactant. Biophys J. 2011;101:2957-65
Author contact
![]() Corresponding author: Mark Andrew Borden, PhD. Department of Mechanical Engineering, University of Colorado, 1111 Engineering Drive, Boulder, CO 80309-0427 Phone: 303.492.7750 Fax: 303.492.3498 Email: mark.bordenedu.
Corresponding author: Mark Andrew Borden, PhD. Department of Mechanical Engineering, University of Colorado, 1111 Engineering Drive, Boulder, CO 80309-0427 Phone: 303.492.7750 Fax: 303.492.3498 Email: mark.bordenedu.