Impact Factor
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results and Discussion
Conclusions
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2013; 3(3):181-189. doi:10.7150/thno.5984 This issue Cite
Research Paper
Lewis Acid-Assisted Isotopic 18F-19F Exchange in BODIPY Dyes: Facile Generation of Positron Emission Tomography/Fluorescence Dual Modality Agents for Tumor Imaging
Shuanglong Liu1, Tzu-Pin Lin2, Dan Li1,3, Lauren Leamer2, Hong Shan3, Zibo Li1 ![]() , François P. Gabbaï2
, François P. Gabbaï2 ![]() , Peter S. Conti1
, Peter S. Conti1
1. Molecular Imaging Center, Department of Radiology, University of Southern California, Los Angeles 90033, USA.
2. Department of Chemistry, Texas A&M University, College Station, Texas 77843, USA.
3. Department of Radiology, The Third Affiliated Hospital of Sun Yat-sen University, Guangzhou 510630, China.
Received 2013-1-30; Accepted 2013-2-9; Published 2013-2-21
Abstract
Positron emission tomography (PET) is a powerful technique for imaging biological pathways in vivo, particularly those that are key targets in disease processes. In contrast, fluorescence imaging has demonstrated to be a superior method for image-guided surgery, such as tumor removal. Although the integration of PET and optical imaging could provide an attractive strategy for patient management, there is a significant shortage of established platforms/methods for PET/optical probe construction. In this study, various reaction conditions were explored to develop a simple and fast method allowing for the introduction of [18F]-fluoride into BODIPY dyes. Through a systematic optimization of the reaction conditions, we found that BODIPY dyes, including commercial amine-reactive BODIPY succinimidyl esters, may be converted into their radioactive analogues in the matter of minutes via a 18F-19F isotopic exchange reaction promoted by a Lewis acid such as SnCl4. An integrin-targeting RGD peptide was also conjugated with [18F]BODIPY® R6G , derived from the commercially available BODIPY® R6G fluorescent tag, to provide a [18F]-RGD conjugate in 82% yield. In vivo evaluation of this imaging probe showed a discernible tumor uptake in the U87MG xenograft model. The dual modality imaging properties of the probe was confirmed by ex vivo fluorescence and microPET imaging experiments. In summary, in the matter of minutes, BODIPY dyes were converted into their “hot” radioactive analogues via a 18F-19F isotopic exchange reaction promoted by a Lewis acid. This approach, which can be applied to commercial BODIPY dyes, provides easy access to positron emission tomography/fluorescence dual modality imaging agents.
Keywords: PET, fluorsence, dual modality, BODIPY, 18F-19F exchange.
Introduction
Molecular imaging is a fast growing research area involving the development and evaluation of novel tools, reagents and methods to image specific molecular pathways in vivo; particularly those that are key targets in disease processes [1-3]. Within this area of research, positron emission tomography (PET) has emerged as one of the most powerful clinical imaging techniques because it can provide critical in vivo information on the distribution of radiolabeled biomolecules for non-invasive diagnosis [4]. Fluorescence imaging is another valuable technique that has been used for intraoperative tumor detection [1]. Since both PET and fluorescence imaging have unique features suitable for clinical application, a system that integrates these two imaging modalities could greatly benefit patient management, for example by providing complimentary diagnosis information during surgery in a non-invasive manner. Stimulated by the potential that such dual modality imaging agent may present in the clinical field [5, 6], we have recently investigated the radiofluorination of BODIPY dyes. Inspired by the work of Perrin [7-13], Blower [14-16], and Tsien [17], we used the boron center as a site for radiofluorination [18, 19] and prepared the first example of a [18F]BODIPY as a dual modality imaging agent [20]. This new approach, which has been recently adopted by Weissleder and Mazitschek [21, 22], is attractive because the positron emitting and fluorescence properties of the imaging agent are confined to the same molecular compartment. It is also based on the use of 18F, a radionuclide of choice for PET [23-30].
The successful implementation of this approach is non-trivial because of the stability of the B-F bond in the parent BODIPY dye. After an initial series of studies on the activation of the B-F bond with Lewis acidic reagents [31] such as trimethylsilyl triflate (TMSOTf) [19], we found that introduction of 18F could be achieved from the corresponding hydroxide derivative in aqueous media at low pH or in MeCN with TMSOTf as a B-OH bond activator (no-carrier added method, Scheme 1/Figure A) [20], Weissleder and Mazitschek showed that radiofluorination could be carried out in MeCN by converting the BODIPY dye into a boron-triflate derivative and by allowing it to react with 18Fˉ (Figure A) [21, 22]. These two approaches rest on the intermediacy of an activated BODIPY dye with a labile hydroxide or a triflate anion bound to the boron atom. Despite the undisputed success of both approaches, some problems remain. The first approach requires the synthesis of a BODIPY hydroxide as well as acidic conditions that may lead to decomposition reactions. The second approach necessitates the synthesis of a boron triflate derivative.
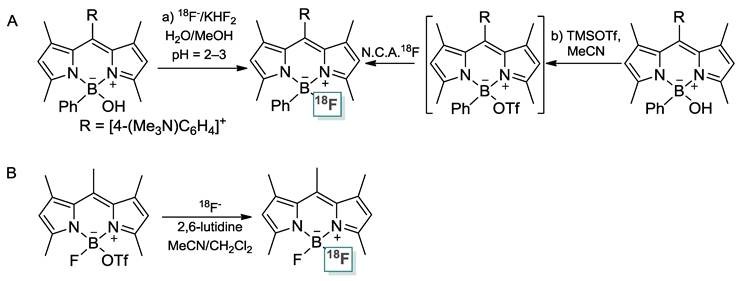
As part of our continuing interest in this field of research, we have decided to broaden the scope of our studies and test Lewis acidic reagents that would promote fluoride exchange reactions, thus circumventing the need for an activated hydroxide or triflate derivative. In this report, we describe the results of this effort and introduce a simple and highly efficient approach toward integrated PET/fluorescence imaging agents, based on BODIPY dyes. We also demonstrate the potential of our approach for tumor imaging.
Materials and Methods
1,3,5,7-tetramethyl-8-[4-(N,N-dimethylamino)phenyl]-4,4-difluoro-4-bora-3a,4a-diaza-s-indacene were prepared according to the reported procedures [32]. Solvents were dried by passing through an alumina column (toluene and CH2Cl2) or refluxing under N2 over Na/K (Et2O and THF). All other solvents and chemicals were used as received. Ambient temperature NMR spectra were recorded on a Varian Unity Inova 400 FT NMR (399.59 MHz for 1H, 128.20 MHz for 11B, 100.45 MHz for 13C, and 375.89 MHz for 19F) spectrometer. Chemical shifts (δ) are given in ppm and are referenced against residual solvent signals (1H, 13C) or external BF3-Et2O (11B, 19F). Elemental analyses were performed at Atlantic Microlab (Norcross, GA). Electrospray mass spectra were obtained with a SciexQstarr Pulsar and a Protana Nanospray ion source. The radiosynthetic work was carried out with the following equipment and methods. The syringe filter and polyethersulfone membranes (pore size, 0.22 μm; diameter, 13 mm) were obtained from Nalge Nunc International (Rochester, NY). Analytical reversed-phase high-performance liquid chromatography (HPLC) was on a Waters 515 chromatography system with a Waters 2487 dual λ absorbance detector and model 2200 scaler-ratemeter radiation detector from Ludlum Measurements, Inc. (Sweetwater, TX). Empower 2 software from Waters Corporation (Milford, MA) was used to record the chromatograms. HPLC was performed on a phenomenex Luna 5µ C18 column (250 × 4.6 mm). The flow was 1 mL/min, with the mobile phase starting from 95% solvent A (0.1% TFA in water) and 5% solvent B (0.1% TFA in MeCN) (0-2 min) to 5% solvent A and 95% solvent B at 22 min.
Synthesis of 1+OTf-
To a CH2Cl2 (5 mL) solution of 1,3,5,7-tetramethyl-8-[4-(N,N-dimethylamino)phenyl]-4,4-difluoro-4-bora-3a,4a-diaza-s-indacene (150 mg, 0.408 mmol) was added neat methyl trifluoromethanesulfonate (80 mg, 0.490 mmol) dropwise at ambient temperature. The formation of an orange precipitate was observed after stirring for 15 min. This solid was collected by filtration and washed with n-pentane (20 mL) and Et2O (5 mL) to afford a pure sample of 1+OTf- (147 mg, 68%). 1H NMR (399.59 MHz, DMSO-d6): δ 1.33 (s, 6H, dipyrrin-CH3), 2.45 (s, 6H, dipyrrin-CH3), 3.65 (s, 9H, N-CH3), 6.21 (s, 2H, dipyrrin-CH), 7.71 (d, 2H, 3JH-H = 8.9 Hz, phenyl-CH), 8.13 (d, 2H, 3JH-H = 8.9 Hz, phenyl-CH). 13C NMR (100.45 MHz, DMSO-d6): δ 14.6, 56.9, 121.9, 122.1, 130.3, 30.8, 136.2, 140.1, 143.0, 148.4, 155.9. 19F NMR (375.89 MHz, DMSO-d6): δ -77.1 (s, 3F, OTf), -142.9 (q, 2F, 1JB-F = 32.2 Hz, BF2). 11B NMR (128.20 MHz, DMSO-d6): δ 0.89 (t, 1JB-F = 32.9 Hz). HRMS (ESI+) calcd for 1+ (C22H27BF2N3+): 382.2265, found: 382.2247.
Synthesis of 2-RGD
Compound 2 (BODIPY® R6G) (1.5 µmol) in 50 µL DMSO and c(RGDyK) (400 µg, 0.66 µmol, denoted as RGD) were mixed together and treated with N,N-diisopropylethylamine (5 µL). After 2 h incubation, the reaction mixture was subjected to HPLC purification. 2-RGD (Rt = 11.9 min) was obtained in 80% yield. ESI-MS for 2-RGD: m/z 942.4 for [M+H]+ (Chemical formula: C45H55BF2N11O9, calculated m/z value: 942.4).
Radiolabeling
Fluorine-18 was produced as [18F]fluoride ion by the 18O(p,n)18F reaction in [18O]water using a CTI/Siemens RDS112 11MeV cyclotron. [18F]fluoride was trapped by a QMA cartridge and eluted off with tetrabutylammonium bicarbonate (TBAB) into a 5-mL V-vial (Type I Borosilicate), followed by azeotropic drying. [18F]-fluoride was then dissolved in anhydrous MeCN for the labeling reactions.
Radiofluorination of 1+: In a typical experimental, 1+OTf- (0.37 µmol) was mixed with SnCl4 (3.0 µmol) in MeCN (20 µL). The resulting solution was then combined with a MeCN solution (50 µL) of [18F]fluoride (10 ± 3 mCi). After shaking at room temperature for 10 min, an aliquot of the reaction mixture (50-100 µCi) was collected for HPLC analysis. Integration of the radio-chromatogram indicated a conversion with a RCY > 95%. For animal studies, 3 ± 1 mCi of [18F]1+ was purified by HPLC. The HPLC mobile phase was then removed by rotary evaporation and the activity was reconstituted in 1 mL phosphate-buffered saline (PBS) and passed through a 0.22 µm syringe filter for in vivo animal experiments. Counted from the end of bombardment, the drying step was 25 min. Other steps include the reaction time (10 min), the sample preparation time (approx. 5 min), HPLC purification (20 min), and the rotary evaporation of the solvent (10-15 min).
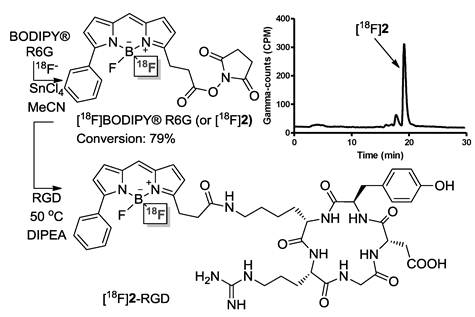
Radiofluorination of 2: Compound 2 (130 µg, 0.23 µmol) was mixed with SnCl4 (3.0 µmol) in MeCN (20 µL). The resulting solution was then combined with a MeCN solution (50 µL) of [18F]fluoride (10 ± 3 mCi). After shaking at room temperature for 10 min, a portion of the reaction mixture (3 ± 1 mCi) was loaded on the HPLC for purification. [18F]2 was obtained in a 79% labeling yield with an estimated specific activity of 35 ± 10 mCi/µmol.
Synthesis of [18F]2-RGD: [18F]2 was azeotropically dried twice at 80 °C using anhydrous MeCN. Then RGD (200 µg, 0.33 µmol) in DMSO (100 µL) was added to [18F]2 (2 mCi, 0.07 µmol), followed by addition of 2 µL diisopropylethylamine. The reaction remained at 50 °C under shaking for 15 min. After quenching the reaction with a 5% acetic acid solution (1 mL), a portion of the reaction mixture (1 ± 0.2 mCi) was loaded on the HPLC for purification. The HPLC solvents were removed by rotary evaporation and the activity was reconstituted in 1 mL PBS and passed through a 0.22 µm syringe filter for in vivo animal experiments. [18F]2-RGD was obtained in an 82% yield with an estimated specific activity of 19 ± 4 mCi/µmol.
MicroPET Imaging
Animal procedures were performed according to a protocol approved by the University of Southern California Institutional Animal Care and Use Committee. The detailed procedure was published previously [33]. In brief, each mouse was injected with 50 ± 10 μCi of the [18F] probe via the tail vein. The imaging data were achieved with the mice under anesthesia using isoflurane (5% for induction and 2% for maintenance in 100% O2). The regions of interest (ROIs) were converted to counts per gram per min based on the assumption of 1 g/mL tissue density. Dividing counts per gram per minute by injected dose gave the image ROI derived %ID/g values.
Ex vivo fluorescence imaging
Ex vivo fluorescence imaging was performed using the Xenogen Lumina XR Imaging System and analyzed using the IVIS Living Imaging 3.0 software (Caliper Life Sciences, Alameda, CA, USA). The detailed information was reported previously in our publication [34].
Results and Discussion
In our initial effort, we decided to investigate the radiofluorination of 1+, a BODIPY dye decorated by a peripheral trimethylammonium group and isolated as a triflate salt. The fluoride exchange reaction was initially studied in aqueous media under acidic condition at pH 2.0 (Table 1, entry 1), a set of conditions that has proven effective for the radiolabeling of fluoroborate species [15, 16, 35]. Unfortunately, this reaction only afforded a trace amount of [18F]1+ (< 2% yield), with most of the BODIPY dye succumbing to the acidic conditions. This result led us to investigate the radiofluorination of 1+ in acetonitrile (MeCN), using TMSOTf as an activator (entry 2). Under optimized conditions, this reaction afforded [18F]1+ with a yield approaching 20%. Increasing the amount of activator did not improve the yield because of the competitive formation of [18F]TMSF [20].
Radiosynthetic results for the 18F-labeling of 1+OTf-.
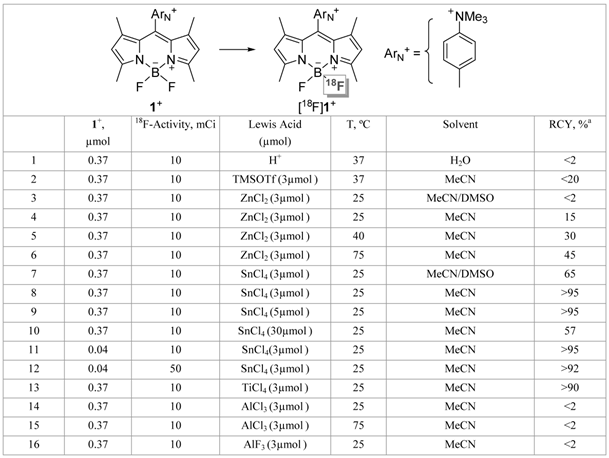
a % RCY calculated from the radio HPLC analysis by dividing the area of the product peak by the sum of all the peak areas. The RCYs were decay-corrected. The final reaction volume was kept at 70 µL for entry 3-15.
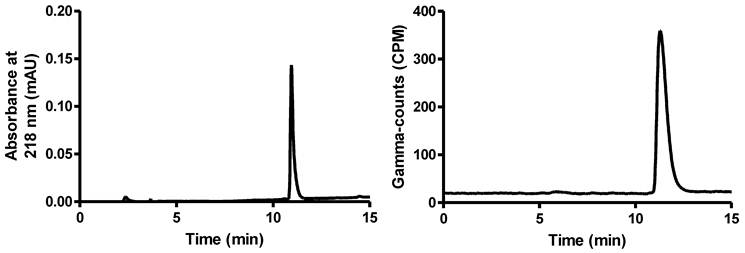
Faced with these limitations, we decided to investigate the use of alternative Lewis acid activators. Realizing that strong Lewis acids may hamper 18F-19F isotopic exchange by irreversible fluoride anion binding, we decided to focus on Lewis acids of intermediate strength. Using the anion accepting scale established by Gutmann in his seminal studies [36], we chose to start with a relatively weak Lewis acid such as ZnCl2. The results obtained with this Lewis acid provided an initial validation of our approach, with a non-negligible radiochemical yield (RCY) observed when the reaction was carried out in MeCN with an excess of ZnCl2 at elevated temperature (Table 1, entries 3-6). However, these high temperature reactions were complicated by decomposition of the dye and formation of hydrophobic byproducts. Next, we decided to focus on the more Lewis acidic SnCl4. This Lewis acid proved remarkably efficient at promoting 18F-19F isotopic exchange, even at room temperature (entries 7-12). As illustrated by the crude HPLC traces shown in Figure 1, an almost quantitative yield was obtained when an 8-fold or 14-fold molar excess of SnCl4 was employed with respect to the starting BODIPY dye 1+ (entries 8 and 9). We observed a decrease in yield at very high molar excess (80-fold excess, entry 10), a phenomenon most likely resulting from the sequestration of fluoride by the large excess of the tin reagent. The yield was not compromised with lower loading of 1+ (entry 11). This approach can also be implemented with higher amounts of starting activity (50 vs. 10 mCi), affording the [18F]1+ with a specific activity close to 1 Ci/µmol (calculated based on starting activity and RCY, entry 12). We also noticed that more polar solvents such as DMSO compromised the yield, a phenomenon that we assign to increasing solvent-Lewis acid interactions (entry 3 and 7). The isotopic exchange reaction is also efficiently promoted by TiCl4 (entry 13) but not AlCl3 and AlF3 (entries 14-16) which presumably sequester [18F]fluoride rather than promoting exchange [37, 38].
Left: UV trace of 1+ as the standard reference. Right: Crude radio-HPLC profile for the 18F-labeling of 1+ from entry 8 in Table 1.
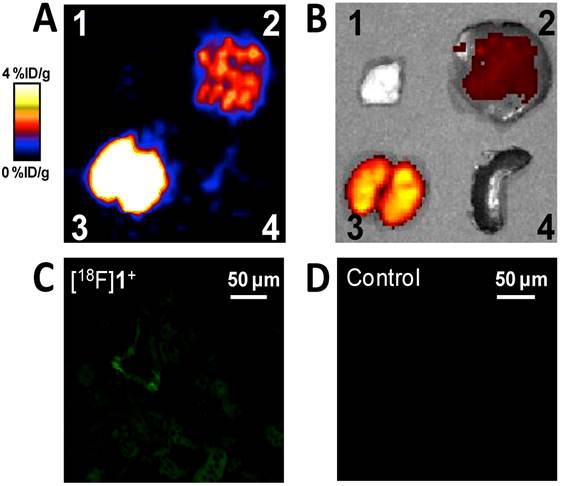
We then studied the stability of [18F]1+ in a PBS buffer at pH 7.5 over a period of several hours. The HPLC profile showed that 97% of [18F]1+ remained after a 6 h incubation, thus indicating this derivative's remarkable resistance to hydrolysis at physiological pH (supporting information). In order to further validate our compound and demonstrate its potential for in vivo PET imaging, [18F]1+ was injected into normal nude mice that were imaged using a microPET scanner at 0.5 h, 1 h, and 3 h post injection (supporting information). As expected, we did not observe obvious bone uptake even at 3 h post injection, which is consistent with the reported high stability of BODIPY dyes [20]. Next, we endeavoured to confirm the dual modality potential of the probe. The animal was euthanized and the liver, the kidneys, and a muscle sample were harvested for ex vivo PET/fluorescence imaging. As shown in Figure 2, the ex vivo microPET and fluorescence imaging correlate well with each other. We also sectioned a kidney of the animal and observed the natural green fluorescence of [18F]1+ using a fluorescence microscope (Figure 2C). By comparison, only weak tissue auto-fluorescence was observed in the kidney of the control mouse (a mouse without [18F]1+ injection) (Figure 2D). These fluorescence images confirm the dual modality properties of the probe and also suggest that fluorescence microscopy can be used to study the localization of the radioactive probes within the tissue (for example, the distribution of a PET probe within the tumor tissue).
Representative ex vivo microPET (A) and fluorescence (B) imaging of dissected organs of a nude mouse. The observation of [18F]1+ in mouse kidneys (C) and the kidney of a control animal (D). The animal was sacrificed after the microPET scan 3 h post injection. 1. Muscle. 2. Liver. 3. Kidneys. 4. Spleen.
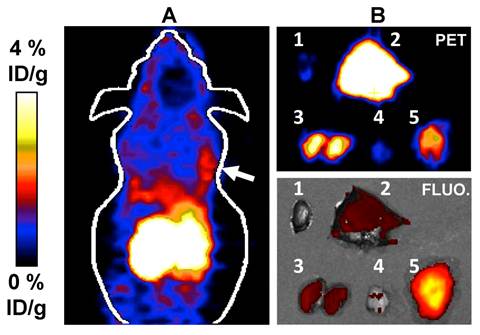
The clinical relevance of our approach to cancer imaging was established using human glioblastoma (U87MG) tumor-bearing mice as an animal model [39, 40]. Since it has been firmly established that the Arg-Gly-Asp (RGD) peptide binds to integrin αvβ3 [41-44], a receptor protein that is over-expressed in U87MG cells, we considered the construction of a radiolabeled BODIPY/RGD peptide conjugate. In our proof of principle study, we found that the conditions used for the radiolableling of 1+ could also be applied to 2, a commercially available BODIPY N-succinimide ester (BODIPY® R6G), which was converted into [18F]2 with a yield of 79% (Figure 3). Further, we found that the resulting radiolabeled dye [18F]2 could be easily conjugated with the Arg-Gly-Asp (RGD) peptide [39] in MeCN and in the presence of DIPEA to afford [18F]2-RGD in 82% RCY. In vivo evaluation of [18F]2-RGD using U87MG tumor-bearing nude mice showed a strong PET signal from the liver and kidneys of the animal, in accordance with the lipophilic nature of the conjugate and its clearance via the urinary tract, respectively. More importantly, in vivo PET imaging showed a discernible tumor uptake of the conjugate. The dual modality imaging properties of the probe was confirmed by ex vivo fluorescence and microPET imaging experiments (Figure 3). The harvested liver, kidneys, and tumor showed a strong PET signal thus corroborating the in vivo imaging results. In a somewhat fortuitous fashion, we observed a much stronger fluorescence signal from the tumor than from the kidneys and liver. We rationalized this differences by the high vascularization of the kidneys and liver whose hemoglobin absorbs much of the excitation light (λ = 535 nm). The emission band of the BODIPY at λmax 550 nm also coincides with the strong absorption bands of hemoglobin, leading to a further reduction of the fluorescence signal. These interferences are greatly diminished in the less vascularized tumor, which gives rise to a much stronger fluorescence signal. These images, and in particular those of the tumor, provide an original demonstration of the dual modality properties of this novel BODIPY-based cancer imaging probe ([18F]2-RGD). Such results bear no precedent, since previously reported cancer specific [18F]BODIPY conjugates are yet to be studied in vivo and imaged in or ex vivo [21, 22].
Top: Scheme for the synthesis of [18F]2 and [18F]2-RGD and HPLC trace showing [18F]2 in the crude reaction mixture. Bottom: microPET imaging (A, the white arrow indicates the tumor location) and Ex vivo PET/fluorescence imaging (B) of major organs and tumor 0.5 h after injection of [18F]2-RGD into a U87MG tumor bearing nude mouse. The fluorescence images were obtained by irradiation of the organs at λ= 535 nm. The fluorescence image was reconstructed based on the emission intensity measured at λ= 580 nm. 1: Heart, 2: Liver, 3: Kidneys, 4:Muscle, 5: Tumor.
Conclusions
We have discovered that Lewis acids such as SnCl4 can be used to promote the 18F-labeling of BODIPY dyes with high efficiency. We also provide evidence that our new approach can be applied to the preparation of PET/fluorescence BODIPY-based cancer imaging probes. We are currently testing the generality of our approach with a particular focus to extend it to BODIPY fluorophores and disease-specific conjugates that emit in the NIR region.
Supplementary Material
Characterization of the BODIPY compounds and conjugates, their radiofluorination and their use for animal imaging.
Acknowledgements
We acknowledge support from the National Science Foundation (CHE-0952912), the Welch Foundation (A-1423), the USC Department of Radiology; the Department of Energy (DE-SC0002353), the American Cancer Society (121991-MRSG-12-034-01-CCE), Robert E. and May R. Wright Foundation and the Southern California Clinical Translational Science Institute (To S. Liu), NNSF of Guangdong (no. U1032002), NNSF of China (no.81071206), and the National Cancer Institute (P30CA014089).
Competing interests
The authors have declared that no competing interest exists.
References
1. Margolis DJ, Hoffman JM, Herfkens RJ, Jeffrey RB, Quon A, Gambhir SS. Molecular imaging techniques in body imaging. Radiology. 2007;245:333-56
2. Baum RP, Kulkarni HR. THERANOSTICS: From Molecular Imaging Using Ga-68 Labeled Tracers and PET/CT to Personalized Radionuclide Therapy - The Bad Berka Experience. Theranostics. 2012;2:437-47
3. Velikyan I. Molecular imaging and radiotherapy: theranostics for personalized patient management. Theranostics. 2012;2:424-6
4. Hoffman JM, Gambhir SS. Molecular imaging: the vision and opportunity for radiology in the future. Radiology. 2007;244:39-47
5. Loudet A, Burgess K. BODIPY dyes and their derivatives: Syntheses and spectroscopic properties. Chem Rev. 2007;107:4891-932
6. Ulrich G, Ziessel R, Harriman A. The chemistry of fluorescent Bodipy dyes: versatility unsurpassed. Angew Chem Int Ed. 2008;47:1184-201
7. Li Y, Ting R, Harwig CW, auf dKU, Bellac CL, Lange PF. et al. Towards kit-like 18F-labeling of marimastat, a noncovalent inhibitor drug for in vivo PET imaging cancer associated matrix metalloproteases. Med Chem Comm. 2011;2:942-9
8. auf dem Keller U, Bellac CL, Li Y, Lou Y, Lange PF, Ting R. et al. Novel Matrix Metalloproteinase Inhibitor [18F]Marimastat-Aryltrifluoroborate as a Probe for In vivo Positron Emission Tomography Imaging in Cancer. Cancer Res. 2010;70:7562-9
9. Harwig CW, Ting R, Adam MJ, Ruth TJ, Perrin DM. Synthesis and characterization of 2,6-difluoro-4-carboxyphenylboronic acid and a biotin derivative thereof as captors of anionic aqueous [18F]-fluoride for the preparation of [18F/19F]-labeled aryltrifluoroborates with high kinetic stability. Tetrahedron Lett. 2008;49:3152-6
10. Ting R, Lo J, Adam MJ, Ruth TJ, Perrin DM. Capturing aqueous [18F]-fluoride with an arylboronic ester for PET: Synthesis and aqueous stability of a fluorescent [18F]-labeled aryltrifluoroborate. J Fluorine Chem. 2008;129:349-58
11. Ting R, Adam MJ, Ruth TJ, Perrin DM. Arylfluoroborates and Alkylfluorosilicates as Potential PET Imaging Agents: High-Yielding Aqueous Biomolecular 18F-Labeling. J Am Chem Soc. 2005;127:13094-5
12. Ting R, Harwig CW, Lo J, Li Y, Adam MJ, Ruth TJ. et al. Substituent Effects on Aryltrifluoroborate Solvolysis in Water: Implications for Suzuki-Miyaura Coupling and the Design of Stable 18F-Labeled Aryltrifluoroborates for Use in PET Imaging. J Org Chem. 2008;73:4662-70
13. Ting R, Harwig C, auf dem Keller U, McCormick S, Austin P, Overall CM. et al. Toward [18F]-Labeled Aryltrifluoroborate Radiotracers: In Vivo Positron Emission Tomography Imaging of Stable Aryltrifluoroborate Clearance in Mice. J Am Chem Soc. 2008;130:12045-55
14. Smith GE, Sladen HL, Biagini SCG, Blower PJ. Inorganic approaches for radiolabelling biomolecules with fluorine-18 for imaging with Positron Emission Tomography. Dalton Trans. 2011;40:6196-205
15. Jauregui-Osoro M, Sunassee K, Weeks AJ, Berry DJ, Paul RL, Cleij M. et al. Synthesis and biological evaluation of [18F]tetrafluoroborate: a PET imaging agent for thyroid disease and reporter gene imaging of the sodium/iodide symporter. Eur J Nucl Med Mol Imaging. 2010;37:2108-16
16. Weeks AJ, Jauregui-Osoro M, Cleij M, Blower JE, Ballinger JR, Blower PJ. Evaluation of [18F]-tetrafluoroborate as a potential PET imaging agent for the human sodium/iodide symporter in a new colon carcinoma cell line, HCT116, expressing hNIS. Nucl Med Commun. 2011;32:98-105
17. Ting R, Aguilera TA, Crisp JL, Hall DJ, Eckelman WC, Vera DR. et al. Fast 18F Labeling of a Near-Infrared Fluorophore Enables Positron Emission Tomography and Optical Imaging of Sentinel Lymph Nodes. Bioconjug Chem. 2010;21:1811-9
18. Hudnall TW, Lin T-P, Gabbaï FP. Substitution of hydroxide by fluoride at the boron center of a BODIPY dye. J Fluorine Chem. 2010;131:1182-6
19. Hudnall TW, Gabbaï FP. A BODIPY boronium cation for the sensing of fluoride ions. Chem Commun. 2008:4596-7
20. Li Z, Lin T-P, Liu S, Huang C-W, Hudnall TW, Gabbaï FP. et al. Rapid aqueous [18F]-labeling of a bodipy dye for positron emission tomography/fluorescence dual modality imaging. Chem Commun. 2011;47:9324-6
21. Hendricks JA, Keliher EJ, Wan D, Hilderbrand SA, Weissleder R, Mazitschek R. Corrigendum: Synthesis of [18F]BODIPY: Bifunctional Reporter for Hybrid Optical/Positron Emission Tomography Imaging. Angew Chem Int Ed. 2012;51:6813
22. Hendricks JA, Keliher EJ, Wan D, Hilderbrand SA, Weissleder R, Mazitschek R. Synthesis of [18F]BODIPY: Bifunctional Reporter for Hybrid Optical/Positron Emission Tomography Imaging. Angew Chem Int Ed. 2012;51:4603-6
23. Purser S, Moore PR, Swallow S, Gouverneur V. Fluorine in medicinal chemistry. Chem Soc Rev. 2008;37:320-30
24. Miller PW, Long NJ, Vilar R, Gee AD. Synthesis of C-11, F-18, O-15, and N-13 Radiolabels for Positron Emission Tomography. Angew Chem Int Ed. 2008;47:8998-9033
25. Dolle F, Roeda D, Kuhnast B, Lasne M-C. Fluorine-18 chemistry for molecular imaging with positron emission tomography. In: Tressaud A, editor. Fluorine and Health: Elsevier, Amsterdam. 2008:3-65
26. Smith TAD. F-18 Fluoride labelling of macromolecules in aqueous conditions: silicon and boroaryl-based F-18 fluorine acceptors, F-18 FDG conjugation and (AlF)-F-18 chelation. J Labelled Compd Radiopharm. 2012;55:281-8
27. Gu YH, Huang D, Liu ZG, Huang JG, Zeng WB. Labeling Strategies with F-18 for Positron Emission Tomography Imaging. Med Chem. 2011;7:334-44
28. Olberg DE, Hjelstuen OK. Labeling Strategies of Peptides with F-18 for Positron Emission Tomography. Curr Top Med Chem. 2010;10:1669-79
29. Li ZB, Conti PS. Radiopharmaceutical chemistry for positron emission tomography. Adv Drug Deliver Rev. 2010;62:1031-51
30. Hooker JM. Modular strategies for PET imaging agents. Curr Opin Chem Biol. 2010;14:105-11
31. Bonnier C, Piers WE, Parvez M, Sorensen TS. Borenium cations derived from BODIPY dyes. Chem Commun. 2008:4593-5
32. Hoogendoorn S, Blom AE, Willems LI, van der Marel GA, Overkleeft HS. Synthesis of pH-activatable red fluorescent BODIPY dyes with distinct functionalities. Org Lett. 2011;13:5656-9
33. Huang CW, Li Z, Cai H, Shahinian T, Conti PS. Biological stability evaluation of the alpha2beta1 receptor imaging agents: diamsar and DOTA conjugated DGEA peptide. Bioconjug Chem. 2011;22:256-63
34. Li Z, Lin TP, Liu S, Huang CW, Hudnall TW, Gabbai FP. et al. Rapid aqueous [18F]-labeling of a bodipy dye for positron emission tomography/fluorescence dual modality imaging. Chem Commun (Camb). 2011;47:9324-6
35. Li Z, Chansaenpak K, Liu S, Wade CR, Zhao H, Conti PS. et al. Harvesting [18F]-fluoride ions in water via direct 18F-19F isotopic exchange: Radiofluorination of zwitterionic aryltrifluoroborates and in vivo stability studies. Med Chem Comm. 2012;3:1305-8
36. Gutmann VHT. The solvent benzoyl chloride. I. The formation of anhydrous chlorides and their behavior in benzoyl chloride. Monatsh Chem. 1957;88:216-27
37. McBride WJ, Sharkey RM, Karacay H, D'Souza CA, Rossi EA, Laverman P. et al. A Novel Method of 18F Radiolabeling for PET. J Nucl Med. 2009;50:991-8
38. McBride WJ, D'Souza CA, Karacay H, Sharkey RM, Goldenberg DM. New Lyophilized Kit for Rapid Radiofluorination of Peptides. Bioconjug Chem. 2012;23:538-47
39. Wu Z, Li ZB, Chen K, Cai W, He L, Chin FT. et al. microPET of Tumor Integrin αvβ3 Expression Using 18F-Labeled PEGylated Tetrameric RGD Peptide (18F-FPRGD4). J Nucl Med. 2007;48:1536-44
40. Li ZB, Cai W, Cao Q, Chen K, Wu Z, He L. et al. 64Cu-labeled tetrameric and octameric RGD peptides for small-animal PET of tumor αvβ3 integrin expression. J Nucl Med. 2007;48:1162-71
41. Xiong J-P, Stehle T, Zhang R, Joachimiak A, Frech M, Goodman SL. et al. Crystal Structure of the Extracellular Segment of Integrin αvβ3 in Complex with an Arg-Gly-Asp Ligand. Science. 2002;296:151-5
42. Beer AJ, Kessler H, Wester HJ, Schwaiger M. PET Imaging of Integrin αvβ3 Expression. Theranostics. 2011;1:48-57
43. Chen K, Chen X. Integrin targeted delivery of chemotherapeutics. Theranostics. 2011;1:189-200
44. Morrison M, Cuthbertson A. Integrin imaging to evaluate treatment response. Theranostics. 2011;1:149-53
Author contact
![]() Corresponding author: Zibo Li, PhD. Assistant Professor, Email: ziboliedu Molecular Imaging Center, Department of Radiology, University of Southern California. François P. Gabbaï, Email: francoisedu Department of Chemistry, Texas A&M University.
Corresponding author: Zibo Li, PhD. Assistant Professor, Email: ziboliedu Molecular Imaging Center, Department of Radiology, University of Southern California. François P. Gabbaï, Email: francoisedu Department of Chemistry, Texas A&M University.