Impact Factor
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
The Pretargeting Alternative
Theranostics: Imaging Enhancement
Theranostics: Improving...
Summary
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2012; 2(5):523-540. doi:10.7150/thno.3582 This issue Cite
Review
Pretargeted Molecular Imaging and Radioimmunotherapy
David M. Goldenberg ![]() , Chien-Hsing Chang, Edmund A. Rossi, William J. McBride, Robert M. Sharkey
, Chien-Hsing Chang, Edmund A. Rossi, William J. McBride, Robert M. Sharkey
Center for Molecular Medicine and Immunology/Garden State Cancer Center, Morris Plains, NJ; Immunomedics, Inc., Morris Plains, NJ; IBC Pharmaceuticals, Inc., Morris Plains, NJ, USA.
Received 2011-10-2; Accepted 2011-10-31; Published 2012-5-17
Abstract

Pretargeting is a multi-step process that first has an unlabeled bispecific antibody (bsMAb) localize within a tumor by virtue of its anti-tumor binding site(s) before administering a small, fast-clearing radiolabeled compound that then attaches to the other portion of the bsMAb. The compound's rapid clearance significantly reduces radiation exposure outside of the tumor and its small size permits speedy delivery to the tumor, creating excellent tumor/nontumor ratios in less than 1 hour. Haptens that bind to an anti-hapten antibody, biotin that binds to streptavidin, or an oligonucleotide binding to a complementary oligonucleotide sequence have all been radiolabeled for use by pretargeting. This review will focus on a highly flexible anti-hapten bsMAb platform that has been used to target a variety of radionuclides to image (SPECT and PET) as well as treat tumors.
Keywords: bispecific antibody, cancer detection, pretargeting, radioimmunodetection, radioimmunotherapy.
Introduction
The exquisite specificity afforded by antibodies has long been recognized for their ability to serve as carriers of other substances. Studies in the 1950's were the first to show that radiolabeled antibodies directed against tissue antigens could specifically localize in these tissues, and shortly thereafter, selective tumor targeting was demonstrated [1, 2]. It was not until the early 1970's that suitable human tumor-associated antigens were identified that could be used for targeting radionuclides for tumor visualization and later for therapy [3-5]. Thus, radiolabeled antibodies were the first “theranostic” agents, capable of both detection and therapy.
Antibodies do not have any selective ability to home to tumors, but need to come into contact with cancer cells after being distributed through the fluid highways of the body, the blood and lymphatic channels, to enter the tumor's vascular supply. The imperfect vascular supply of a tumor creates an environment that is more permissive for macromolecules to enter their extravascular space than most normal tissues [6-9]. Thus, even a non-specific IgG and other macromolecules have an increased accretion in tumor compared to most normal tissues [10, 11]. Because tumors lack lymphatic drainage, backpressure inside builds, creating a physiological barrier that impedes a macromolecule's diffusion into the tumor; however, smaller molecules permeate better. As the antibody molecules leave the blood and encounters antigen on tumor cells in the extravascular compartment, they will be retained for variable lengths of time, dictated mainly by their affinity and valency (avidity), while the remaining antibody molecules in the body eventually clear by normal physiological mechanisms that remove proteins and foreign substances [12]. The selective binding actually impedes the antibody from migrating too far from the perivascular space where it first encounters the antigen, with antibodies that have a lower binding affinity being released more quickly and better able to continue penetrating deeper into the tumor than those with a higher affinity [13].
Since antibodies are designed to be retained in the serum, their re-circulation aids in building the concentration in the tumor to a maximum level over 1-2 days. However, even with delayed blood clearance, only a very small fraction of the injected dose will localize to the tumor. The slow blood clearance maintains background activity at high levels, and thus tumor discrimination can require several days. Reducing the molecular size or removing/altering the neonatal receptor-binding site, which is responsible for sustained IgG levels in the blood (by enzymatic digestion or molecular engineering), can accelerate blood clearance, but this reduces re-circulation, leading to decreased tumor accretion [14-18]. Because optimal targeting is a combination of high specific uptake and low retention in normal tissues, often the gains made in hastening clearance are offset by lower tumor uptake, and thus the net effect can be modest. This is particularly problematic for an agent that is intended for both imaging and therapy, where certain modifications that might improve imaging properties can compromise or place certain restrictions on the therapeutic application. For example, molecular engineering has created a wide variety of antibody forms with different valencies and molecular sizes. At a molecular size of ~25 kD, the smallest antibody fragment from an IgG, a scFv, has monovalent binding and clears exceptionally fast from the blood and tissues, creating much higher tumor/nontumor ratios much more quickly than an IgG (160 kD). This antibody form, and its divalent variant, a diabody, could be used for imaging, but tumor uptake and retention is reduced so significantly for both that they have virtually no therapeutic value as directly-labeled radioconjugates, at least for systemic applications. Other larger divalent forms, such as minibodies and (scFv)2-Fc constructs, have somewhat slower blood clearance with higher tumor retention, enhancing their use for imaging, but studies have suggested their therapeutic application would likely be restricted to radioiodinated forms [16, 19-21].
Indeed, one of the major issues facing investigators seeking radioconjugates for therapeutic use is isotope selection, which is often dictated by the pharmacokinetics and biodistribution of the directly-radiolabeled antibody. Radioiodine is commonly coupled to an antibody's tyrosine residues, and when catabolized, iodotyrosine is released from the cells, which is quickly and efficiently removed from the body with some retention by the thyroid [22-26]. 123I, 131I, 125I, and 124I all have been used for imaging and/or therapy, and since they can all be coupled to antibodies with the same procedure, radioiodine is often an attractive radionuclide for theranostic applications. Nevertheless, there are many more radionuclides of interest for imaging and therapy, particularly radiometals.
Radiometals are usually coupled to antibodies through an intermediate, a chelating agent. Chelating agents often have differing affinities for various metals, and thus the in vivo stability of one radiometal bound to a particular antibody-chelate conjugate might not be the same as another radiometal bound to the same conjugate. Radiometals that are brought into cells by an antibody are retained for long periods, because cells tend to retain metals, but metals held by chelates also are inhibited from being expelled [27-29]. Thus, radiometal-labeled antibodies will have significantly higher uptake in the liver and kidneys for IgG or smaller fragments, respectively. This retention also will occur in the tumor, with radiometal-labeled antibody accretion in tumor gradually increasing over time, reflecting the cumulative deposition of the radiometal in the cells, particularly for antibodies that readily internalize. Although radiometals become trapped in the liver, elevated uptake in the liver has not been problematic for directly-radiolabeled IgG, since dose-limiting hematologic toxicity occurs well before radiation doses to the liver approach critical levels. When using smaller antibody fragments that clear through the kidneys in an attempt to reduce red marrow exposure, renal uptake then can far exceed that of the tumor. Since kidneys have an upper threshold of tolerance of perhaps ~2500 cGy, while solid tumors may be eradicated by external beam irradiation with ~5000 cGy [30], it is difficult to envision a successful radiotherapeutic when renal exposure is so much higher than tumor. One way to circumvent these problems has been to explore compartmental treatment strategies, such as direct injection into surgical cavities in the cerebrum for brain cancers or the peritoneum for carcinomatosis [31]. In these examples, a larger fraction of the injected product will remain in the local compartment, which reduces the potential exposure of other tissues.
Thus, for optimal targeting, systemically-administered, directly-radiolabeled antibodies might need to use 2 different forms, e.g., a fragment for imaging and an IgG for therapy. However, imaging is rarely performed solely for the purpose of confirming uptake in known tumor sites, but instead to derive dosimetry estimates for the therapeutic or to ensure that the product does not have an altered biodistribution. For these purposes, the agent needs to have similar biodistribution and clearance properties as the therapeutic. This requirement often compromises its imaging quality and in turn optimal tumor detection.
The Pretargeting Alternative
Early development of bsMAb pretargeting. Pretargeting was first suggested by investigators who were developing chelates for radiochemistry. It was well known that, unlike radiolabeled antibodies that were slowly cleared from the body, chelated radiometals were quickly and efficiently eliminated [32]. Chelates are more easily radiolabeled than an antibody and often yield higher specific activities.
While most researchers were coupling chelates to antibodies for direct radiolabeling, this group had developed antibodies to EDTA derivatives and designed a new targeting agent, a bsMAb that bound both the tumor and separately to the radiolabeled chelate. The bsMAb was not to be radiolabeled, but instead would be injected and given time to pretarget to the tumor. Once in the tumor and cleared from the blood and tissues, the radiolabeled chelate would be injected, with the understanding that its small size would allow it to escape the vascular system quickly and then clear rapidly from all sites in the body, but when passing into the tumor, it would be captured by the pre-localized bsMAb. By separating the targeting of a slow clearing antibody from the fast clearing radionuclide, the procedure aimed to increase tumor/nontumor ratios and allow for more rapid imaging. Over time, the concept was refined, eventually leading to clinical testing, starting with an 111In-labeled benzyl-EDTA containing cobalt-bleomycin (BLEDTA IV), and later with a hydroxyethylthiourido-derivative of benzyl EDTA that gave better tumor uptake in animal testing, 111In-labeled EOTUBE [33-35]. The clinical procedure settled on the administration of 20 to 40 mg of a bsMAb, which was a chemically conjugated F(ab')2 composed of an anti-carcinoembryonic antigen (CEA) Fab' coupled to an anti-EOTUBE Fab'. Four days later, the patient received 111In-EOTUBE. This clinical investigation reported a detection sensitivity of 95% for known tumor lesions, some detected within 4 h of the 111In-EOTUBE injection, including hepatic metastases, a site where other clinical studies using an intact 111In-anti-CEA IgG frequently had difficulty because of the high background activity in the liver [36, 37].
It is perhaps not coincidental that CEA has been one of the most widely studied tumor antigens for pretargeting. Besides the fact that CEA targeting by directly radiolabeled antibodies was well established for many years, such as in the first preclinical and clinical studies of radioimmunodetection [3, 4], antibodies bound to CEA do not internalize readily. Since all pretargeting procedures have some lag time between the injection of the primary targeting agent and the radiolabeled effector, it is important for the antibody to remain accessible until the effector is given. In addition to CEA, we have examined several other pretargeting systems using antibodies that are not readily internalized: one against a colonic mucin (antibody Mu-9 [38]), another against a pancreatic mucin (based on the humanized antibody hPAM4 [39, 40]), and a third against CD20 found on many B-cell lymphomas (based on the humanized antibody veltuzumab [41, 42]). More recently, we have reported pretargeting results with a new tri-Fab bsMAb (TF12) [43] that is based on the humanized antibody designated hRS7, which binds to Trop-2 (trophoblast-2; also known as EGP-1 or epithelial glycoprotein-1), an antigen found in increased expression by many different types of epithelial cancers [44]. Successful pretargeting with the TF12 bsMAb was surprising, since early studies had reported rapid internalization of the murine RS7 IgG [45-47]. Recently we found that while ~10 to 20% of the antibody is rapidly internalized over 1-2 h, the remaining antibody remains accessible on the surface of the cell, with ~40-50% of the antibody internalized over 24 h (unpublished results). Thus, there was sufficient bsMAb remaining on the surface for highly successful pretargeting of a variety of human tumor xenografts, including prostate and ovarian cancers [43]. Other antibodies, such as against CD22, that are more readily and more completely internalized, do not make good pretargeting agents [48]. However, the TF12 anti-Trop-2 experience illustrates the importance of carefully reviewing how much antibody is retained on the surface of tumor cells before dismissing an antibody as a useful pretargeting agent.
Although the initial pretargeting concept was based on a bispecific antibody, other dual-binding systems have been employed successfully. Hnatowich et al. [49] described systems using avidin-biotin, and later, this group developed another pretargeting system based on oligomer binding by complementary oligomers [50-54]. The avidin-biotin pretargeting approach eventually split into 2 basic procedures, one using a streptavidin-IgG conjugate, which later evolved to using a molecularly-engineered multivalent scFv-streptavidin-fusion protein for binding radiolabeled biotin, and another that pursued a pretargeted biotinylated IgG conjugate that bound radiolabeled biotin after being bridged by streptavidin. These other pretargeting procedures have been reviewed previously [55-57] and will not be discussed further. While each of these other procedures has special merits, the avidin-biotin systems have to contend with the immunogenicity of avidin/streptavidin, since these are foreign proteins [58]. Hence, we have favored the bsMAb approach because the primary targeting agent can be humanized to reduce its immunogenicity.
Despite having encouraging clinical results with the first bsMAb pretargeting system, this was not pursued, perhaps in part because clinical studies with 123I- and 99mTc-labeled fragments of anti-CEA antibodies being developed at the same time were successful, with tumor localization possible within a few hours and an excellent ability to disclose hepatic metastases [59-64]. The lower cost and ease of preparation of 99mTc-Fab' displaced future development of 123I-labeled fragments. Ultimately 4 radiolabeled antibodies received FDA approval for imaging, but over time, all but one of these [Indium (111In) capromab pendetide; EUSA, Langhorne, PA) have been withdrawn, because 18F-fluor-deoxyglucose (18F-FDG) used with positron-emission tomography (PET) imaging systems provided better sensitivity at lower expense than the radiolabeled antibodies, and could also have high specificity when used with hybrid PET/CT cameras.
Instead of competing with directly-radiolabeled antibodies for imaging, all pretargeting procedures focused on their therapeutic prospects. With bsMAb pretargeting, investigators in France discovered they could enhance the binding of a hapten (e.g., DTPA loaded with indium) to a tumor pretargeted by a bsMAb by attaching 2 haptens to a short peptide composed of 2 amino acids, tyrosine and lysine [TL-(In)DTPA]. This localized-binding enhancement was termed the “affinity enhancement system” (AES) [65]. Others later confirmed that a structure containing 2 haptens improved uptake and retention in the tumor [66, 67]. With the TL-(In)DTPA hapten-peptide, tyrosine could be radiolabeled with 131I for therapy and imaging, while the DTPA haptens could chelate 111In. D-amino acids were used to reduce the risk of enzymatic cleavage [68]. Initial clinical studies examined the 111In-labeled agent to define the best conditions for pretargeting in patients, again using a chemically-conjugated Fab' x Fab' bsMAb targeting CEA, with the anti-hapten binding arm being an anti-(In)DTPA antibody [69, 70], while preclinical studies were focusing on the development of the 131I-labeled hapten-peptide [71-73].
Innovations in anti-hapten/hapten binding systems. One of the limitations of bsMAb-pretargeting system was the anti-hapten binding arm, which was commonly directed to the chelate loaded with the radiometal of interest. As Reardon et al. [32] had shown with the anti-EDTA antibodies, binding affinity changed, sometimes remarkably, based on which metal was loaded in the chelate. The 734 anti-(In)DTPA antibody used by the French investigators had an affinity of 10-9 M for (In)DTPA, but with (Ca)DTPA, the affinity dropped to 10-3 M [74]. Thus, that antibody was so specific for the indium-loaded DTPA that it would not bind as strongly to DTPA loaded with other metals. Additionally, the DTPA derivative used as the initial immunogen did not bind as avidly as other radionuclides of therapeutic interest, such as 90Y, which would lead to high bone uptake. Thus, while 131I is very suitable for therapy, other radionuclides would be of interest for a variety of reasons. However, if a different radionuclide were to be used, it appeared that a new antibody directed against the specific anti-chelate-metal complex might be required. This limitation was partially circumvented by inserting a ligand-binding agent that would bind 99mTc for imaging or rhenium (e.g., 188Re) for therapy. To do this, the peptide length was increased to 4 amino acids, placing the 2 DTPA moieties on the first and third amino acids, thereby keeping the spatial proximity of the 2 haptens in a similar configuration as the TL-(In)DTPA peptide, and inserting the 3-thiosemicarbazonyl)glyoxylcysteinyl (Tscg-Cys) moiety on a terminal Lys (fourth amino acid) [75]. While this manipulation successfully allowed the use of another series of radionuclides, this particular pretargeting system could not be expanded easily to other radionuclides.
Le Doussal et al. [65] first mentioned an anti-hapten antibody, 679, that bound to histamine-succinyl-glycine (HSG), and Janevik-Ivanovska et al. [76] then examined this antibody-hapten system more thoroughly, reporting various di-HSG peptide derivatives used with 131I. The 679 antibody was prepared originally as part of an effort to develop an immunoassay to detect histamine, binding HSG with nanomolar affinity, but not to histamine. Since HSG was not involved in binding the radionuclide, peptides could be crafted that maintained 2 HSG moieties with an appropriate spacing for AES, but other compounds suitable for binding any radionuclide of interest could be added (Figure 1).
The HSG-hapten binding system for binding a variety of radionuclides. (A) The initial bsMAb pretargeting systems utilized antibodies that bound to a specific chelate-metal complex, e.g., indium-loaded DTPA. As such, the anti-(In)DTPA antibody had a much lower binding affinity for DTPA loaded with another metal, such as 90Y. Thus, to bind another radiometal, like 90Y, a new anti-chelate antibody would need to be prepared. An antibody against a hapten that does not bind the radionuclide, such as the anti-HSG (histamine-succinyl-glycine) antibody, would open the possibility for using different radionuclides or other compounds with one system. (B) In the HSG-binding system, a peptide core of D-amino acids (aa) has 2 HSG haptens to enhance tumor-binding stability. The remaining structure can be modified to accommodate any radionuclide-binding agent. A tyrosine in the peptide core could be radioiodinated. We have also modified the carboxyl end of the peptide to allow for binding of fluorescent dyes. (C) For pretargeting, the bsMAb is first localized in the tumor, and after clearing from the blood and tissues, the radiolabeled divalent hapten-peptide is given. The divalent hapten structure enhances its binding avidity to the bsMAb, and it can potentially enhance the binding avidity of the bsMAb to the tumor as well. Trivalent bsMAbs with divalent binding to the tumor are preferred.
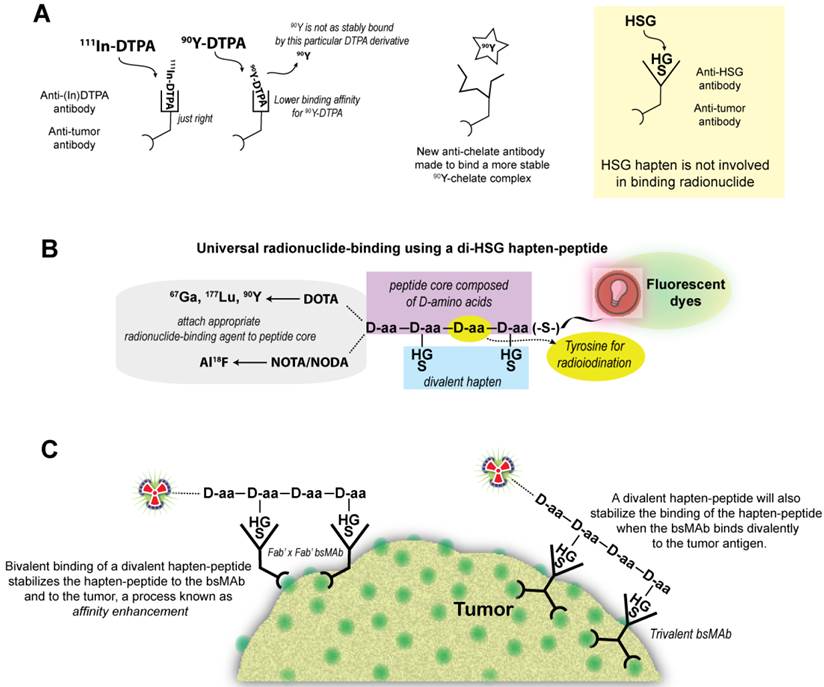
We showed the potential universality of this anti-hapten binding system for pretargeting using 3 different peptides, one having the Tscg-Cys moiety for binding 99mTc or 188Re, another with a single DOTA (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid) moiety, which was capable of binding 111In, 177Lu, and 90Y, and a third having a DOTA and Tscg-Cys moiety [38]. When radiolabeled with 99mTc, the hapten-peptide bearing Tscg-Cys and DOTA, designated IMP245, had significantly lower uptake in the tissues, such as liver and kidneys, than the hapten-peptide with just the Tscg-Cys moiety. This finding illustrates another important aspect of the pretargeting methodology; namely, the hapten-peptide structure can be modified in various ways to alter its biodistribution and clearance. As long as the structure contains 2 haptens, it will have higher uptake and retention in tumors than a peptide bearing a single hapten. We typically strive to ensure the hapten-peptide structure encourages renal over hepatobiliary elimination, with minimal renal retention, but the structure could be modified to encourage hepatic uptake if an indication required less background around the kidneys or urinary bladder. The tumor/kidney ratios for the various radiometal-labeled hapten-peptide structures that we have investigated have been ≥2:1, usually within 1 h, whereas tumor/kidney ratios for renally-filtered, directly-radiolabeled, antibody fragments/constructs can achieve this only when radioiodinated. In addition to harboring a single DOTA moiety, the di-HSG hapten-peptide IMP288 also contains a tyrosine, and has been radiolabeled successfully with124I in addition to 90Y, 177Lu, 111In, and 68Ga [77-80]. For clinical development, we showed the di-HSG hapten-peptide, IMP288, did not bind to or block the binding of histamine to H1, H2, H3, or H4; it could be given safely to guinea pigs (highly sensitive to histamine) in molar concentrations more than 1000-fold higher than required for human dosing with 90Y, and the anti-HSG antibody showed no binding to human tissues by immunohistology [81]. Thus, the anti-HSG/HSG system was compatible for human use and it offered the possibility for pretargeting virtually any radionuclide that could be bound stably to a chelate or chelate-like structure.
Innovation in bsMAb structure. The first bsMAbs were prepared chemically by combining Fab' fragments from each of the IgG antibodies of interest (Figure 2). The resulting F(ab')2 conjugate was smaller in size than an IgG lacking the Fc, so it was cleared effectively from the blood over several days. Animal studies with chemical conjugates using IgG, F(ab')2 and Fab' fragments revealed divalent binding to the tumor antigen yields higher and longer retention in the tumor [82], and thus we focused on developing humanized bispecific constructs that have divalent binding to tumor [83-85].
Bispecific antibody conjugates/constructs. Chemically conjugated bsMAb were prepared from Fab' fragments of the anti-tumor (α-T) and the anti-hapten (α-H) antibodies. The anti-tumor Fab' was coupled first to the bifunctional N,N”-o-phenylene-dimaleimide (PDM), and then the anti-hapten Fab' was added to form a stable Fab' x Fab' bsMAb. Molecularly engineered bsMAb used by our group include the hBS14 (anti-CEACAM5 x anti-679 HSG) and several tri-Fab (TF) constructs, each binding to a different tumor antigen, but all using the anti-HSG hapten-binding antibody. Whereas the hBS14 construct is prepared in a single antibody-producing clone, the tri-Fab Dock-and-Lock constructs are formed from 2 separate clones. One produces the anti-tumor fusion protein, using the docking and dimerization domain (DDD) peptide sequence that has a strategically placed cysteine (designated DDD2). The Fab-DDD2 forms a dimer, which has a docking domain that will bind to the anchoring domain sequence, which is also modified to include 2 cysteines (AD2). Thus, all these constructs have bivalent binding to the tumor and monovalent binding to the hapten.
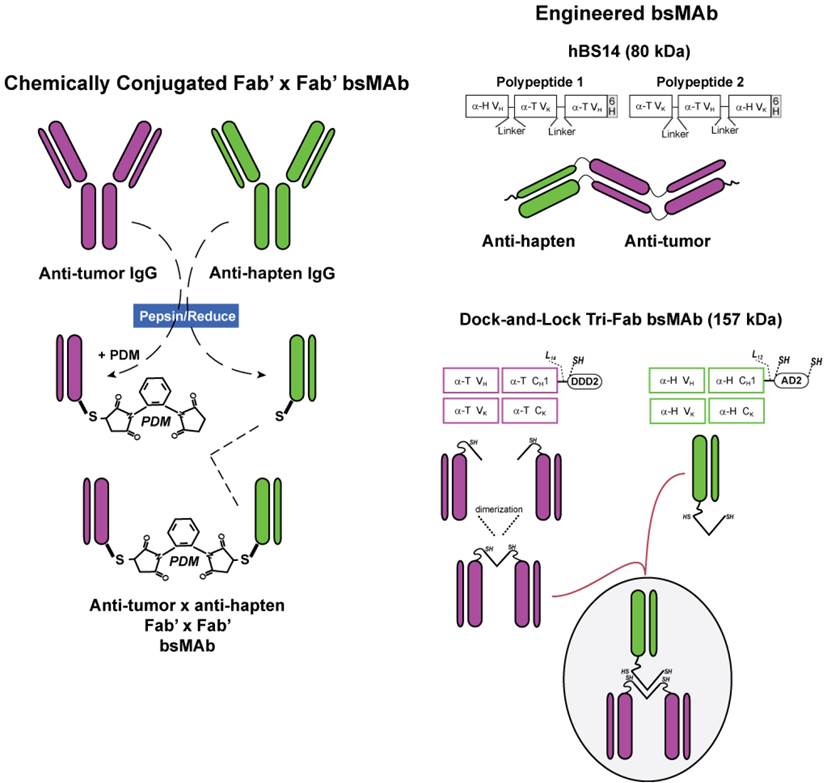
Clinical investigations have begun with a bsMAb prepared using the Dock-and-Lock (DNL) method (Figure 2). This procedure combines 3 Fabs, 2 directed against the tumor antigen and one against the hapten (HSG) in a unique manner, utilizing the natural binding interaction of peptides derived from two human proteins [85]. The dimerization and docking domain (or DDD) peptide consists of a 44 amino acid sequence derived from the regulatory subunit of human type II A-kinase. It forms a stable homodimer, which also acts as a docking site for the second peptide, the anchor domain (or AD), which consists of a 17-amino acid sequence derived from an interactive human A-kinase anchor protein. Once docked, the 3 Fabs have a specific orientation allowing them to each bind at their native affinity. These peptides were modified by inserting cysteines in strategic locations so that they will interact when the DDD and AD are docked together, thereby strengthening their binding by forming covalent disulfide bonds. Animal and patient studies found that these constructs are stable in serum [81]. This novel technology has allowed easy production of a number of different tri-Fab bsMAb, as well as many other multivalent, mono- or bispecific structures [57, 86, 87].
Principles of bsMAb pretargeting. AES dictates that the divalent hapten structure encourages retention in the tumor, where the concentration of the bsMAb is higher than in the blood or in the tissues. This enhancement occurs because the binding avidity of a divalent hapten is better than the affinity derived by a monovalent form. However, conceptually, the divalent hapten-peptide could cross-link 2 adjacent Fab' x Fab' bsMAbs, which would enhance the binding of the bsMAb to the tumor, particularly if the bsMAbs were themselves only monovalently-bound to the tumor. Hapten binding could thus cross-link 2 bsMAbs, stabilizing not only its hold on the bsMAb, but also the bsMAb binding to the tumor. An oligomer-pretargeting system is also enhanced by divalent binding [88, 89], whereas avidin-biotin approaches rely on their ultra-high affinity (10-15 M) for stable binding. However, with all of these approaches, the radiolabeled compound will be retained in the tumor only as long as the antibody remains bound to the antigen expressed on the tumor cell.
The principles of AES often focus on the divalent hapten for enhanced retention in the tumor, but the principle also relates to how the bsMAb and hapten-peptide interact in the serum. As long as the concentration of the bsMAb is low in the serum when the divalent hapten-peptide is given, it would most likely bind a single bsMAb. This monovalent binding allows the hapten-peptide to release readily in the serum and continue to be cleared rapidly, while in the tumor, where presumably the bsMAb concentration is higher, the divalent hapten-peptide will be held more tightly. The bsMAb concentration required for favorable release in the serum will vary based on the affinity of the anti-hapten antibody. With the anti-HSG/HSG system, we found that that as long as the concentration of the bsMAb in the blood was ≥10-fold lower than the concentration of the hapten-peptide the instant it was injected (assuming instantaneous distribution in the vascular volume), >95% would be cleared within 1 h [81].
As the concentration of the bsMAb increases relative to the hapten-peptide, the hapten-peptide's clearance slows, but it still clears much faster than most directly-radiolabeled antibody fragments. If the bsMAb in the blood is too high, the fraction of hapten-peptide available for effective tumor localization would be decreased significantly, resulting in low tumor uptake. It is tempting to assume that the formation of bsMAb-hapten-peptide complexes in the blood may be beneficial, with the prospect that these complexes would still localize in the tumor, but the important tenet behind pretargeting is rapid uptake in the tumor and clearance from the blood; formation of a large complex in the blood does not encourage rapid clearance of radiolabeled hapten-peptide, and in vivo studies have shown tumor uptake can be reduced significantly if the amount of complexation is unchecked [81, 90]. Thus, with a bsMAb pretargeting system, reducing the interval between a fixed amount of bsMAb and hapten-peptide will slow the blood clearance of the hapten-peptide. An acceptable clearance rate for the radiolabeled hapten-peptide may need to be determined empirically. Bispecific antibody pretargeting using a chemically-conjugated F(ab')2 found an interval of 4 days to be optimal with a 40 mg/m2 bsMAb dose [91, 92]. Using an anti-CEACAM5 x anti-HSG tri-Fab bsMAb (TF2), clinical studies have found that changing the interval from 5 days to 1 day in patients given 75 mg of the bsMAb had only modest effects on the hapten-peptide clearance (5-day interval, 96% and 99.9% cleared within 6 and 24 h; a 1-day interval had 83 and 98.9% cleared within 6 and 24 h) [93]. These results reflect the initial clinical finding that most of the TF2 bsMAb had cleared within 1 day [81, 93]. Ongoing studies are continuing to assess the effects of varying bsMAb and hapten-peptide dose, and interval, on hapten-peptide clearance and tumor localization with this system. It should be mentioned that with avidin-biotin methods, the ultra-high affinity makes it imperative that the primary targeting agent be cleared as thoroughly as possible, and therefore these methods have utilized a clearing step so the radiolabeled biotin can be given 1 day after the primary targeting agent injection.
There are 2 other important aspects that all pretargeting systems have in common. First, the dose of the primary targeting agent, in our case the bsMAb, has to be sufficient to optimize the capture of the radiolabeled hapten-peptide, and second, the dose of the hapten-peptide should be low, and therefore it should be radiolabeled at a high specific activity. We optimized the labeling of the di-HSG-DOTA hapten-peptide, IMP288, to a specific activity of ~2.8 mCi/nmole. At this level, radiolabeling yields are >97%, and therefore purification is not required (excess DTPA is added at the end of the labeling procedure to ensure no free 90Y is present). Higher specific activities are possible, but then purification also would be required.
Although the tri-Fab constructs have the same molecular size and divalent tumor-binding properties as an IgG, because they clear quickly from the blood, the percent uptake in the tumor is lower than a directly-radiolabeled IgG. Estimates based on radioiodinated tri-Fab given to tumor-bearing mice have found maximum accretion occurs within ~6 h, but blood concentrations are too high at this time for the hapten-peptide to be given with optimal targeting results. Thus, the hapten-peptide injection is delayed in animals for 16-24 h, and at this time, ~2 to 4% of the tri-Fab' is in the tumor.
Various models have indicated that it is best to administer enough of the pretargeting agent to nearly saturate antigen [94-96]. This is logical, since the pretargeted agent is acting as a binding site for the radiolabeled compound. However, while this might be true in theory, and perhaps even in xenograft models, this is just not practical clinically, where tumor burden and antigen content (and accessibility) are highly variable. One could elect to give exceeding high loading doses, but this would likely require a clearing agent to lower concentrations of the primary targeting agent in the blood to reasonable levels in a reasonable amount of time. We take a somewhat different view, suggesting that the optimal bsMAb dose does not have to be adjusted based on antigen content in the tumor, but rather on the amount of the hapten-peptide that will be given, with the hapten-peptide dose dependent on its specific activity and the maximum tolerated dose (MTD), if using a therapeutic. In our system, tumor uptake of the radiolabeled hapten-peptide is reasonably high when 10- to 20-fold more moles of the bsMAb are injected as compared to the moles of hapten-peptide dose planned to be given [90, 97]. While in xenograft models there may be incremental improvements in tumor uptake as the bsMAb dose is increased beyond this amount, increasing the bsMAb dose also introduces more moles of bsMAb in the serum, which can impact the interval used. While extending the interval will reduce bsMAb concentrations in the serum, supplies in the tumor may diminish as well, which could reduce the amount of hapten-peptide that could be captured [91]. Following the lead of how pretargeting is optimized in animals will provide a good starting point for clinical investigations, but because there are many factors that are beyond our control clinically, optimization often becomes a compromise to achieve a reasonably high tumor uptake and tumor/nontumor ratios.
While it may not be necessary to inject enough of the primary targeting agent to saturate the antigen in the tumor, other studies have shown there is a critical mass of bsMAb required for optimal uptake of the radiolabeled hapten-peptide (i.e., an appropriate dose of bsMAb is not governed only by the bsMAb/peptide molar ratio). For example, while a 10:1 bsMAb/hapten-peptide ratio gave excellent targeting results at a given amount of bsMAb and hapten-peptide, if the concentration of each agent was reduced, yet this proportionality kept the same, eventually a level would be reached where the amount of bsMAb in the tumor was too small to offer a reasonable capture of the hapten-peptide [97]. These preclinical results suggest that in situations where the specific activity of the hapten-peptide is very high, it may be necessary to maintain a minimum bsMAb dose to encourage optimal tumor uptake with high tumor/nontumor ratios. Early clinical studies with the anti-CEA x anti-(In)DTPA Fab' x Fab' bsMAb had indicated that the minimum bsMAb for acceptable tumor uptake should be 0.1 mg/kg using 0.1 nmole/kg of the hapten-peptide [69, 70]. While pretargeting procedures do require more adjustments than direct methods, preclinical studies provide considerable insights that help narrow the choices that will lead to excellent pretargeting results.
Theranostics: Imaging Enhancement
Pretargeting was first conceived as a procedure to improve imaging by reducing background activity, thereby enhancing tumor/nontumor ratios. Thus, it is not surprising that in preclinical testing, pretargeting has provided an exceptional ability to detect small lesions in xenograft models.
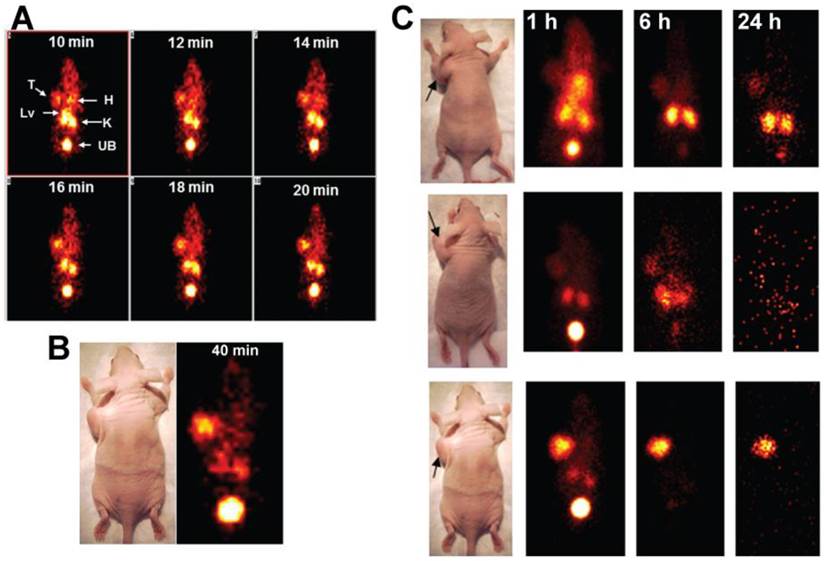
Initial testing of our pretargeting systems focused on the utility of a 99mTc-labeled hapten-peptide pretargeted with a humanized recombinant anti-CEACAM5 x anti-HSG bsMAb for detecting subcutaneous xenografts of a human colonic cancer cell line, GW-39 [97]. Arcitumomab (CEAScan®, Immunomedics, Inc., Morris Plains, NJ), a 99mTc-Fab' that also binds CEACAM5 and was approved by the FDA and EMA for detecting colonic cancer metastases was the primary comparator [98-100]. Dynamic imaging revealed uptake of a 99mTc-hapten-peptide could be appreciated within 10 min of its injection in the animals given the bsMAb, although there was still similar concentrations in the heart and liver, and somewhat higher uptake in the kidneys (Figure 3) [97]. Over the next 10 min, cardiac, hepatic, and renal uptake decreased and tumor uptake intensified, yielding clear images of the tumor within 20 to 40 min. In contrast, over this same period, the 99mTc-Fab' was seen only in the blood pool and kidneys. Although the dynamic imaging study used animals with large tumors, subcutaneously-implanted tumors as small as 0.1 g were easily discerned with pretargeting within 1 h [97]. Even after 24 h, pretargeting continued to be superior to the 99mTc-Fab' fragment.
Pretargeted 99mTc-hapten-peptide vs. 99mTc-Fab'. Nude mice bearing large (1.3 to 1.5 g) human colonic tumor xenografts (arrows in photos) were used to assess the dynamics of a 99mTc-hapten-peptide alone or pretargeted with an anti-CEA bsMAb given 48 h earlier, and compared to an 99mTc-anti-CEA Fab' fragment. Dynamic imaging was performed over 60 min, taking images at 2-min intervals. Static images of these same animals were then also taken at 1, 6, and 24 h. Panel A shows the dynamic images taken from 10 to 20 minutes that illustrate the pretargeted 99mTc-hapten-peptide with uptake in the tumor (T) as early as 10 min after its injection, but at this time, tumor uptake was similar to the heart (H), with more intense uptake in the kidneys (K) and liver (Lv). Substantial activity had already been eliminated in the urinary bladder (UB). Over the next 10 minutes, uptake in heart and liver diminished so that they were not visible, and the tumor's intensity was now similar to the kidneys. In Panel B, the 2-min dynamic ending at 40 min showed tumor uptake had exceeded the kidneys. The static images shown in Panel C illustrate the targeting seen with the 99mTc-anti-CEA Fab' fragment (top), the 99mTc-hapten peptide alone (no bsMAb was given; middle), and the pretargeted 99mTc-hapten-peptide (bottom). The 99mTc-anti-CEA Fab' showed only a minor blush in the tumor at 1 h, with most of the activity residing in the blood, liver and kidneys, with some elimination in the urinary bladder. The follow-up images show tumor localization, but renal uptake is much more prevalent. Even the 99mTc-hapten-peptide alone showed some uptake in the tumor over the first 6 h, but by 24 h, all activity was eliminated from the body. In contrast, the animal pretargeted with the anti-CEA bsMAb show strong tumor localization that persisted over time as the normal tissues continued to wash out.
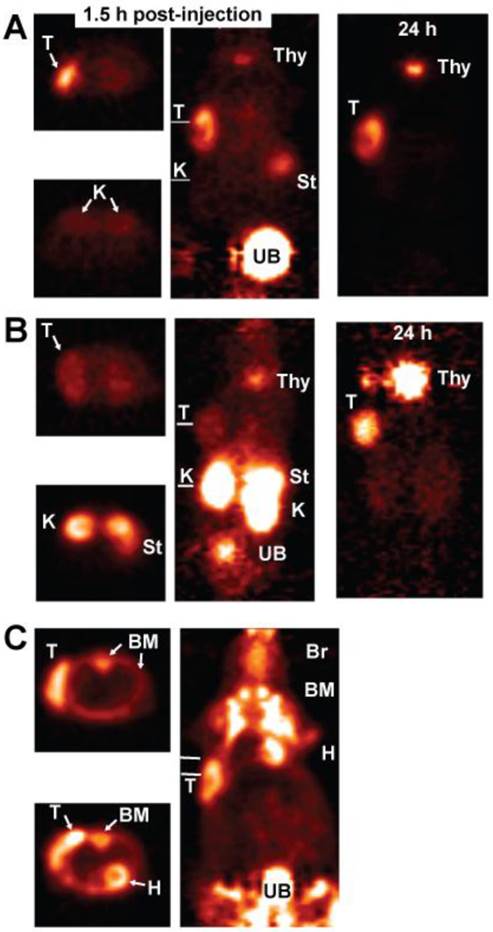
The rapidity and high sensitivity of pretargeting with the 99mTc-hapten-peptide suggested that this procedure could compete favorably with 18F-fluordeoxyglucose (18F-FDG), the most commonly used imaging agent in oncology. In order to compare the 2 procedures, we needed a hapten-peptide labeled with a PET-imaging radionuclide. One of the hapten-peptides being used for 90Y-labeling also had a tyrosine as one of the amino acids in its peptide core, and with the commercial availability of 124I for human use, a procedure was developed to radioiodinate it with 124I [77]. Animals bearing subcutaneous LS-174T human colonic cancer xenografts were pre-localized with the anti-CEACAM5 bsMAb and 1 day later given the 124I-hapten-peptide. Another group was fasted overnight and then given 18F-FDG, and a third group was injected with 124I-labeled Fab' of the same anti-CEACAM5 antibody used in the bsMAb. This was an important control, because directly-radiolabeled antibody fragments are often reported to have the best imaging properties when radioiodinated. This study found pretargeting provided less ambiguous localization of the tumor. Images of animal taken ~1.5 h after each injection showed similar uptake with the pretargeted 124I-hapten-peptide as 18F-FDG, but the pretargeted image had lower tissue uptake (Figure 4). The bone marrow of mice had exceptionally high uptake of 18F-FDG, along with the expected uptake in the heart wall and brain, and diffuse uptake in the abdomen. As in the earlier study with 99mTc-Fab', the 124I-Fab' was unable to demonstrate tumor localization over the first few hours, and there was much higher renal uptake and evidence of higher dehalogenation, with strong uptake in the stomach and later in the thyroid. While tumor/nontumor ratios improved for the 124I-Fab' by 24 h, with clear tumor localization, tumor uptake for the pretargeting procedure was nearly 15-fold higher. This was the first demonstration that pretargeting could improve image quality over 18F-FDG, while also showing it could provide much improved images than a directly radioiodinated Fab' fragment.
PET imaging of nude mice bearing a s.c. human colonic tumor (T). A (top panel) shows an animal given an anti-CEACAM5 bsMAb pretargeted 124I-hapten-peptide, B (middle panel) an 124I-labeled anti-CEACAM5 Fab', and C (bottom panel) was given 18F-FDG. The far left side of each panel shows a transverse section taken through a plane that includes the tumor, with white lines in the adjacent coronal sections indicating the approximate location of the plane. For A and B, another transverse section through the abdomen in the region of the kidneys (K) is also shown, whereas C shows a transverse section that thorough a plane that includes the heart and another lower plane below the heart. The coronal slices are on the posterior side of the animal in order to highlight the tumor that is in on left side of the image. All images are adjusted to the same intensity without background subtraction to give a better appreciation of the relative uptake in the tissues. The animals were not pre-medicated to ameliorate radioiodine uptake in the thyroid (Thy) or stomach (St). 18F-FDG has extensive uptake in the bone marrow (BM) of mice, as well as in the brain (Br) and heart wall (H).
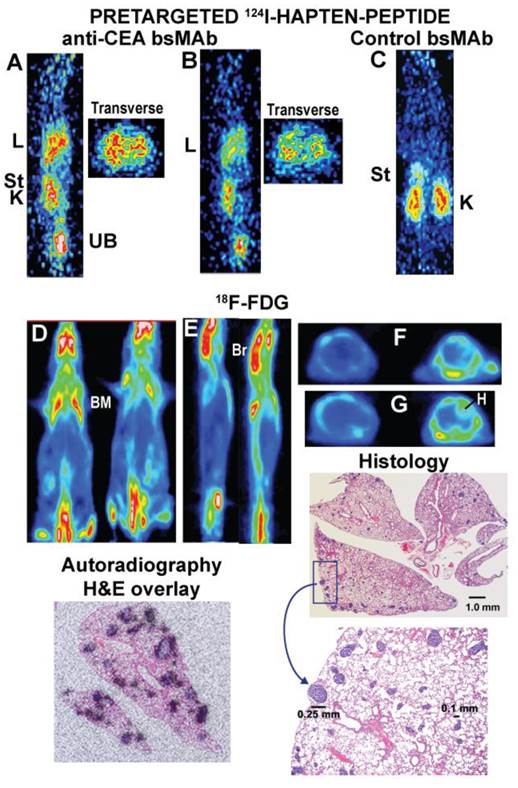
In order to provide a greater challenge for pretargeting, the GW-39 human colonic tumor cell line was injected intravenously, allowing it to develop as small metastatic colonies scattered within the lungs [101]. These animals were pretargeted using a tri-Fab anti-CEACAM5 bsMAb and one day later received the 124I-hapten-peptide, while a separate group of animals was given 18F-FDG (Figure 5) [79]. PET imaging showed a remarkable ability of the pretargeting procedure to detect uptake in the lungs, while there was no indication of tumor in the lungs of animals given 18F-FDG, even though biodistribution studies showed GW-39 was equally avid for FDG as LS-174T. Indeed, transverse sections of the chest in animals given the pretargeting procedure appeared to show discrete uptake in small nodules, but no evidence of targeting was seen with 18F-FDG. After completing the imaging study, a histological evaluation of the lungs revealed multiple nodules scattered throughout the lungs, with no single nodule exceeding 0.3 mm in diameter (Figure 5). Autoradiography studies in this same model using an 111In-labeled hapten-peptide showed that localization of the hapten-peptide was restricted to uptake around the tumor nodules, but not in the normal lung. Collectively, these data established the superior sensitivity afforded by the pretargeting procedure over 18F-FDG, disclosing pulmonary metastases that were smaller than metastases detected to-date by any other imaging procedure in such xenograft models. Further studies in a mouse model that bore a human tumor xenograft in one leg and a focal inflammation in the opposite one again confirmed the improved specificity of pretargeting over 18F-FDG, when the focal inflammation was not detected in the pretargeted animal, but did localize with 18F-FDG [102]. Thus, pretargeting has superior sensitivity and specificity over 18F-FDG in small animal imaging studies.
Localization of micrometastatic tumors the lungs of nude mice injected intravenously with a human colon cancer cell line. Nude mice were given a specific anti-CEACAM5 trivalent bsMAb or a control, non-binding trivalent bsMAb (anti-CD22 x anti-HSG). They then later received an 124I-hapten peptide. A separate group of animals received 18F-FDG. Images A and B are sagittal sections of 2 separate mice and corresponding transverse slices through the chest taken ~1 h after the 124I-hapten-injection. The lungs (L) of both animals showed increased uptake, and the transverse sections appear to show distinct uptake in small clusters. The sections were cut in a plane to include one of the kidneys (K) to illustrate uptake in this organ, as well as some activity in the stomach (St) immediately above the kidney. The activity is cleared in the urine (urinary bladder, UB). C shows a coronal section of a tumor-bearing mouse given the control bsMAb that did not find tumor in the chest. D shows coronal slices of 2 animals given 18F-FDG and imaged ~1 h later. E shows sagittal slices of these same 2 animals, and F and G are transverse sections through 2 different regions of the chest (F is higher than G). Even in the transverse sections, there was no indication of tumor involvement in lungs these animals, but there was intense uptake in the brain (Br), bone marrow (BM; e.g., ribs, scapula, spine, pelvis, femurs) and heart wall (H). The histology sections show lungs taken from one of the 18F-FDG imaged animals (animal to the right in panel D) that had multiple foci of tumor scattered in several lobes. A portion of the section highlighted in the rectangle is magnified, showing even the largest lesion in the field was only ~0.25 mm in diameter. A separate autoradiography study using animals with more advanced disease that were pretargeted with an anti-CEACAM5 bsMAb followed by an 111In-hapten-peptide showed uptake was specifically targeted to the tumor nodules in the lungs.
These imaging studies were initiated with 124I primarily because of the limited availability of PET-imaging radionuclides suitable for clinical use. 124I is commonly used with directly- radiolabeled antibodies because its ~4-day half-life allows for delayed imaging, but it is expensive and high-energy gamma emissions are problematic. 64Cu also has been used for small antibody fragments, peptides, and even for pretargeting, but it has limited availability and cost is a concern [103]. 68Ge/68Ga generators are becoming more widely available [104, 105]. 68Ga, which is essentially carrier-free with a physical half-life is ~1 h, could be an important agent for PET imaging with compounds that give an acceptable image within 1-2 h. The generators can be used for >6 months, which can significantly reduce the overall cost if multiple doses are prepared over this time. We found a DOTA, di-HSG hapten-peptide can be prepared at very high specific activities (e.g., starting at 48 mCi/nmole) and provide excellent images with 68Ga [102].
The preferred radionuclide for PET imaging remains 18F, but 18F chemistry is challenging and often laborious. McBride et al. [106] recently described a novel and unique method for binding 18F to peptides. Historically, while most radiofluorination methods have relied on direct binding of fluorine to carbon atoms, these authors took advantage of fluorine's high binding affinity to metals, particularly aluminum, where they were able to capture the highly stable aluminum-fluoride complex using a chelate. These efforts identified a NOTA structure with favorable binding properties to hold Al18F complexes stably in serum and in vivo. The initial compound inserted into the hapten-peptide, IMP449, had a radiolabeling of 5-20% with a specific activity of 0.5 to 1.3 mCi/nmol, but subsequent refinements in the chelate structure and labeling conditions have raised the radiolabeling yields to ≥80%, with specific activities as high as 4 mCi/nmol [107, 108]. The fluorination method is not applicable just for pretargeting peptides, but has been applied to other peptides as well [108-110]. The procedure is simple and quick, and while the general procedure will likely perform well with a number of peptides, others may require certain adaptations or purification processes to optimize yields or specific activities. However, the process for 18F-labeling of pretargeting peptides has been reduced to a single lyophilized vial that only requires the addition of 18F in saline, as used for bone imaging, and a brief cartridge purification procedure that can be completed within 20-30 min [110]. The method requires heating the chelate with the Al18F at high temperatures for 15 minutes, which might not be suitable for all peptides, but McBride et al. [111] recently described an adaptation of the procedure that allows even heat-sensitive compounds to be 18F-labeled easily and rapidly.
Theranostics: Improving Therapeutic Outcomes
Pretargeting procedures easily improved the problematic radiation exposure to the red marrow that has been dose-limiting for directly-radiolabeled IgG and even some fragments. However, achieving therapeutic results also requires the targeting agent to deliver a high enough concentration to tumor for a sufficient duration to elicit a significant response. As mentioned earlier, compounds that clear quickly from the blood, such as a pretargeted peptide, likely have only one chance of achieving maximum accretion in the tumor, since the concentration in the blood is depleted so quickly. Indeed, the faster a directly-radiolabeled antibody clears from the blood, the more the concentration in the tumor usually decreases. However, pretargeting appears to avoid this trend. Gautherot et al. found tumor uptake of a radioiodinated hapten-peptide pretargeted with an anti-CEA bsMAb could achieve similar tumor accretion as an 131I-F(ab')2 [71, 72]. They predicted and then showed a bsMAb pretargeting system using an 131I-labeled hapten-peptide would be a more effective therapeutic procedure for targeting radionuclides than an 131I-labeled IgG or F(ab')2 [71-73, 112, 113]. However, an even more surprising result was a report by Axworthy et al. [114], who found the uptake of radiolabeled biotin in a tumor pretargeted with a streptavidin-IgG conjugate could rival that of the directly-radiolabeled IgG, yet with much lower blood and tissue concentrations. This group later published a series of preclinical studies that showed a pretargeting approach using 90Y-biotin could cure tumor xenografts in mice more effectively than 90Y-labeled IgG [115]. All of these studies essentially revealed the therapeutic advantage for pretargeting is obtained by (a) the rapid uptake of the radiolabeled compound (biotin or hapten-peptide) that reaches a maximum accretion within 1-2 h, as compared with an IgG that requires 1-3 days, and (b) the exceptionally rapid elimination of radioactivity from the blood, significantly reducing red marrow exposure [57, 72, 115, 116]. These 2 factors combine to increase the radiation dose rate in the tumor, as well as providing better tumor/blood ratios.
In animal testing, we found that renal toxicity was dose-limiting for an optimized pretargeting procedure [116]. Thus, in the animals, blood clearance was sufficiently rapid to minimize marrow exposure, but because the vast majority of the activity was filtered in the kidneys and eliminated in the urine, the kidneys were the next organ of concern. Renal uptake in mice was relatively low, with a first generation hapten-peptide having a maximum uptake of just about 3% injected dose per gram (ID/g), while a second generation hapten-peptide that is now being used clinically was somewhat lower, being ~2% ID/g, with renal concentrations decreasing by about 20-40% per day [40]. Importantly, renal uptake of the radiolabeled hapten-peptide was not affected over a wide range of pretargeting conditions [81, 90], and thus we expect that the radiation-absorbed dose to the kidneys in patients will be relatively constant as adjustments are made. Early clinical data have suggested renal doses for the 90Y-hapten-peptide pretargeted with an anti-CEACAM5 bsMAb will be < 3 cGy/mCi [81]. In animals, tumor/kidney ratios easily exceed 2:1, which would give pretargeting an advantage over directly-radiolabeled antibody fragments that often have as much as 10-fold higher uptake in the kidneys than in tumor. Therapy studies with radiolabeled peptides have indicated kidney tolerance up to ~2700 cGy based on conventional MIRDOSE [117, 118], and thus if similar ratios in animals can be achieved in patients, tumors could receive ≥ 5000 cGy at a maximum tolerated dose. Historically, dosimetry data for solid tumors have shown a <2000 cGy delivery potential, with most tumors receiving <1000 cGy [119]. While this radiation dose delivered to lymphomas is highly effective, meaningful anti-tumor responses in solid tumors have been rare. However, one additional advantage for pretargeting is the manner in which the radiation is delivered; namely, very quickly. As we and others have emphasized [57, 72, 115, 116], pretargeting could potentially deliver the same radiation dose to tumor as a directly radiolabeled IgG, but because the hapten-peptide reaches its maximum uptake within 1 h, the dose rate that the radiation is delivered is significantly higher than with an IgG. This is expected to provide an added boost to the therapeutic potential for pretargeted radionuclide therapy, but we also have to keep in mind that other tissues are also being exposed at higher dose rate, and even if it is for a short duration, the toxicity could also be affected.
Being limited by renal toxicity has its drawbacks, because the clinical manifestation of these effects often takes many months to appear and years to reach full effect. Therefore, Phase I dose escalation trials will need to rely on imaging and dosimetry to guide patient treatment. While there have been some advances in renal dosimetry in an attempt to improve the correlation of exposure to toxicity by taking into account the biological effective dose, there is still a need for a more comprehensive data set before these estimates can fully account for long-term loss in renal function [120-125]. Interestingly, except for one trial where gastrointestinal toxicity was dose-limiting because of the specificity of the primary anti-EpCAM conjugate, other pretargeting trials using 90Y-biotin or an 131I-hapten-peptide have been limited by hematologic toxicity [55, 56]. Such toxicities likely reflect a diminished capacity of the red marrow because of prior treatments, but the radiation sensitivity threshold of the red marrow is so much lower than the kidneys that even when radiation is cleared rapidly from the blood, severe hematologic toxicity can occur. Medullary thyroid cancer patients were found to have a lower tolerance to a pretargeted 131I-hapten-peptide, which was attributed in part to unappreciated bone/bone marrow metastases [92, 126]. Indeed, these investigators found pretargeting could enhance the overall survival of a subpopulation of medullary thyroid cancer patients who had a short calcitonin doubling time (serum biomarker for this indication), attributing this result to the successful elimination of these metastases [127].
By reducing the risk of myelotoxicity, the possibility of combining this pretargeted radionuclide therapy with other treatment modalities is more feasible than with a directly- radiolabeled conjugate. Several reports have shown therapeutic improvements in animal models using various chemotherapeutic, as well as other agents, combined with radioimmunotherapy [40, 128-131]. There is even the possibility that pretargeted radionuclides can be combined with improved efficacy with antibody-drug conjugates, based on the fact that an antibody-SN-38 conjugate (SN-38 is the active ingredient in irinotecan) could be combined with a maximum tolerated dose of a directly radiolabeled 90Y-IgG [132]. Moving forward, we also need to keep in mind, as studies in animals have shown [40], that fractionating a pretargeting procedure may be the best course of action.
Summary
Pretargeting approaches are designed to have superior targeting properties over directly-targeted antibodies. Unlike directly-radiolabeled antibodies, where a different form of antibody might be required for optimal imaging than used for therapy, in pretargeting, the conditions that favor imaging will likely be identical to those for therapy. Even if a different hapten-peptide were necessary to allow a more favorable imaging radionuclide to be used, such as a positron-emitter, we often are able to modify the peptide structure in a manner that minimizes differences in biodistribution. Preclinical studies have already shown pretargeting can be more specific than conventional 18F-FDG, but specificity will be governed by the binding properties of the primary targeting antibody. Our initial clinical experience with an 111In-labeled hapten-peptide used primarily for dosimetry in a therapy trial encouragingly has found that pretargeting can localize 18F-FDG-positive lesions, but more studies aimed at optimizing the pretargeting conditions are necessary. In addition, an 18F-labeled hapten-peptide will soon be made available so that images can be acquired with similar equipment.
Pretargeting procedures do require more advanced investigations to determine suitable conditions for delivering the radiolabeled compound, but preclinical studies can provide useful insights to guide which starting conditions and adjustments will likely lead to the best localization results clinically. Pretargeting does require multiple injections, but the tri-Fab bsMAb pretargeting procedure has minimized this process to 2 injections, with clinical data suggesting the spacing will likely range within 1-3 days. Because the radiolabeled compound clears so quickly from the blood and body, substantially more radionuclide is required for pretargeted therapy than for direct targeting. Preclinical testing certainly indicates that improved anti-tumor responses with less toxicity occur in solid tumor and hematologic malignancies, possibly justifying these additional requirements. Clinical studies with the new generation tri-Fab bsMAb are assessing the prospects for this procedure. Fortunately, results to date confirm that pretargeted radioimmunodetection can be at least comparable to FDG-PET in terms of disclosing known sites of cancer. Whether the improved specificity noted and superiority to FDG-PET preclinically can be confirmed in patients must await clinical trials.
Acknowledgements
We thank Dr. T.M. Cardillo (Immunomedics, Inc.) for animal studies, Prof F. Kraeber-Bodéré, Prof J-F. Chatal, Dr. J. Barbet, and their colleagues (Nantes, France), as well as Prof O.C. Boerman, Prof. W.J. Oyen, and Dr. R. Schoffelen and their colleagues (Nijmegen, The Netherlands) for their clinical studies, for their collaboration, and advice.
Conflict of Interest
DM Goldenberg, C-H Chang, EA Rossi, and WJ McBride have financial interests in Immunomedics, Inc. or IBC Pharmaceutical, Inc. RM Sharkey declares no conflicts.
References
1. Pressman D, Keighley G. The zone of activity of antibodies as determined by the use of radioactive tracers. Fed Proc. 1948;7:308
2. Pressman D, Korngold L. The in vivo localization of anti-Wagner-osteogenic-sarcoma antibodies. Cancer. 1953;6:619-23
3. Goldenberg DM, DeLand F, Kim E, Bennett S, Primus FJ, van Nagell JR Jr. et al. Use of radiolabeled antibodies to carcinoembryonic antigen for the detection and localization of diverse cancers by external photoscanning. N Engl J Med. 1978;298:1384-6
4. Goldenberg DM, Preston DF, Primus FJ, Hansen HJ. Photoscan localization of GW-39 tumors in hamsters using radiolabeled anticarcinoembryonic antigen immunoglobulin G. Cancer Res. 1974;34:1-9
5. Goldenberg DM, Gaffar SA, Bennett SJ, Beach JL. Experimental radioimmunotherapy of a xenografted human colonic tumor (GW-39) producing carcinoembryonic antigen. Cancer Res. 1981;41:4354-60
6. Baxter LT, Jain RK. Transport of fluid and macromolecules in tumors. I. Role of interstitial pressure and convection. Microvasc Res. 1989;37:77-104
7. Sands H, Jones PL. Physiology of monoclonal antibody accretion by tumors. Cancer Treat Res. 1990;51:97-122
8. Jain RK. Physiological barriers to delivery of monoclonal antibodies and other macromolecules in tumors. Cancer Res. 1990;50:814s-9s
9. Reilly RM, Sandhu J, Alvarez-Diez TM, Gallinger S, Kirsh J, Stern H. Problems of delivery of monoclonal antibodies. Pharmaceutical and pharmacokinetic solutions. Clin Pharmacokinet. 1995;28:126-42
10. Primus FJ, Wang RH, Goldenberg DM, Hansen HJ. Localization of human GW-39 tumors in hamsters by radiolabeled heterospecific antibody to carcinoembryonic antigen. Cancer Res. 1973;33:2977-82
11. Fand I, Sharkey RM, Primus FJ, Cohen SA, Goldenberg DM. Relationship of radioantibody localization and cell viability in a xenografted human cancer model as measured by whole-body autoradiography. Cancer Res. 1987;47:2177-83
12. Rudnick SI, Adams GP. Affinity and avidity in antibody-based tumor targeting. Cancer Biother Radiopharm. 2009;24:155-61 doi:10.1089/cbr.2009.0627
13. Weinstein JN, van Osdol W. Early intervention in cancer using monoclonal antibodies and other biological ligands: micropharmacology and the "binding site barrier". Cancer Res. 1992;52:2747s-51s
14. Colcher D, Pavlinkova G, Beresford G, Booth BJ, Choudhury A, Batra SK. Pharmacokinetics and biodistribution of genetically-engineered antibodies. Q J Nucl Med. 1998;42:225-41
15. Ferl GZ, Wu AM, DiStefano JJ 3rd. A predictive model of therapeutic monoclonal antibody dynamics and regulation by the neonatal Fc receptor (FcRn). Ann Biomed Eng. 2005;33:1640-52 doi:10.1007/s10439-005-7410-3
16. Kenanova V, Wu AM. Tailoring antibodies for radionuclide delivery. Expert Opin Drug Deliv. 2006;3:53-70 doi:10.1517/17425247.3.1.53
17. Olafsen T, Wu AM. Antibody vectors for imaging. Semin Nucl Med. 2010;40:167-81 doi:10.1053/j.semnuclmed.2009.12.005
18. Suzuki T, Ishii-Watabe A, Tada M, Kobayashi T, Kanayasu-Toyoda T, Kawanishi T. et al. Importance of neonatal FcR in regulating the serum half-life of therapeutic proteins containing the Fc domain of human IgG1: a comparative study of the affinity of monoclonal antibodies and Fc-fusion proteins to human neonatal FcR. J Immunol. 2010;184:1968-76 doi:10.4049/jimmunol.0903296
19. Kenanova V, Olafsen T, Williams LE, Ruel NH, Longmate J, Yazaki PJ. et al. Radioiodinated versus radiometal-labeled anti-carcinoembryonic antigen single-chain Fv-Fc antibody fragments: optimal pharmacokinetics for therapy. Cancer Res. 2007;67:718-26 doi:10.1158/0008-5472.can-06-0454
20. Olafsen T, Kenanova VE, Sundaresan G, Anderson AL, Crow D, Yazaki PJ. et al. Optimizing radiolabeled engineered anti-p185HER2 antibody fragments for in vivo imaging. Cancer Res. 2005;65:5907-16 doi:10.1158/0008-5472.can-04-4472
21. Wu AM, Olafsen T. Antibodies for molecular imaging of cancer. Cancer J. 2008;14:191-7 doi:10.1097/PPO.0b013e31817b07ae
22. Khaw BA, Cooney J, Edgington T, Strauss HW. Differences in experimental tumor localization of dual-labeled monoclonal antibody. J Nucl Med. 1986;27:1293-9
23. Sands H, Jones PL. Methods for the study of the metabolism of radiolabeled monoclonal antibodies by liver and tumor. J Nucl Med. 1987;28:390-8
24. Geissler F, Anderson SK, Press O. Intracellular catabolism of radiolabeled anti-CD3 antibodies by leukemic T cells. Cell Immunol. 1991;137:96-110
25. Mattes MJ, Griffiths GL, Diril H, Goldenberg DM, Ong GL, Shih LB. Processing of antibody-radioisotope conjugates after binding to the surface of tumor cells. Cancer. 1994;73:787-93
26. Press OW, Shan D, Howell-Clark J, Eary J, Appelbaum FR, Matthews D. et al. Comparative metabolism and retention of iodine-125, yttrium-90, and indium-111 radioimmunoconjugates by cancer cells. Cancer Res. 1996;56:2123-9
27. Duncan JR, Welch MJ. Intracellular metabolism of indium-111-DTPA-labeled receptor targeted proteins. J Nucl Med. 1993;34:1728-38
28. Rogers BE, Franano FN, Duncan JR, Edwards WB, Anderson CJ, Connett JM. et al. Identification of metabolites of 111In-diethylenetriaminepentaacetic acid-monoclonal antibodies and antibody fragments in vivo. Cancer Res. 1995;55:5714s-20s
29. Stein R, Govindan SV, Mattes MJ, Chen S, Reed L, Newsome G. et al. Improved iodine radiolabels for monoclonal antibody therapy. Cancer Res. 2003;63:111-8
30. O'Donoghue J. Relevance of external beam dose-response relationships to kidney toxicity associated with radionuclide therapy. Cancer Biother Radiopharm. 2004;19:378-87
31. Sharkey RM, Goldenberg DM. Use of antibodies and immunoconjugates for the therapy of more accessible cancers. Adv Drug Deliv Rev. 2008;60:1407-20 doi:10.1016/j.addr.2008.04.011
32. Reardan DT, Meares CF, Goodwin DA, McTigue M, David GS, Stone MR. et al. Antibodies against metal chelates. Nature. 1985;316:265-8
33. Stickney DR, Slater JB, Kirk GA, Ahlem CN, Chang CH, Frincke JM. Bifunctional antibody: ZCE/CHA 111indium-BLEDTA-IV clinical imaging in colorectal carcinoma. Antibody Immunoconjug Radiopharm. 1989;2:1-13
34. Stickney DR, Anderson LD, Slater JB, Ahlem CN, Kirk GA, Schweighardt SA. et al. Bifunctional antibody: a binary radiopharmaceutical delivery system for imaging colorectal carcinoma. Cancer Res. 1991;51:6650-5
35. Chang CH, Sharkey RM, Rossi EA, Karacay H, McBride W, Hansen HJ. et al. Molecular advances in pretargeting radioimunotherapy with bispecific antibodies. Mol Cancer Ther. 2002;1:553-63
36. Divgi CR, McDermott K, Johnson DK, Schnobrich KE, Finn RD, Cohen AM. et al. Detection of hepatic metastases from colorectal carcinoma using indium-111 (111In) labeled monoclonal antibody (mAb): MSKCC experience with mAb 111In-C110. Int J Rad Appl Instrum B. 1991;18:705-10
37. Abdel-Nabi H, Doerr RJ, Evans NH, Farrell EE, Chan HW, Balu D. et al. Radioimmunodetection of colorectal carcinoma with 111In-labelled monoclonal antibody IVP ZCE 025 (a tissue culture-produced anti-CEA MAb). Nucl Med Commun. 1994;15:81-93
38. Sharkey RM, McBride WJ, Karacay H, Chang K, Griffiths GL, Hansen HJ. et al. A universal pretargeting system for cancer detection and therapy using bispecific antibody. Cancer Res. 2003;63:354-63
39. Gold DV, Goldenberg DM, Karacay H, Rossi EA, Chang CH, Cardillo TM. et al. A novel bispecific, trivalent antibody construct for targeting pancreatic carcinoma. Cancer Res. 2008;68:4819-26 doi:10.1158/0008-5472.can-08-0232
40. Karacay H, Sharkey RM, Gold DV, Ragland DR, McBride WJ, Rossi EA. et al. Pretargeted radioimmunotherapy of pancreatic cancer xenografts: TF10-90Y-IMP-288 alone and combined with gemcitabine. J Nucl Med. 2009;50:2008-16 doi:10.2967/jnumed.109.067686
41. Sharkey RM, Karacay H, Johnson CR, Litwin S, Rossi EA, McBride WJ. et al. Pretargeted versus directly targeted radioimmunotherapy combined with anti-CD20 antibody consolidation therapy of non-Hodgkin lymphoma. J Nucl Med. 2009;50:444-53 doi:10.2967/jnumed.108.058602
42. Sharkey RM, Karacay H, Litwin S, Rossi EA, McBride WJ, Chang CH. et al. Improved therapeutic results by pretargeted radioimmunotherapy of non-Hodgkin's lymphoma with a new recombinant, trivalent, anti-CD20, bispecific antibody. Cancer Res. 2008;68:5282-90 doi:10.1158/0008-5472.can-08-0037
43. Karacay H, Sharkey R, Rossi E, McBride B, Chang C-H, Goldenberg D. A new tri-Fab recombinant bispecific antibody (bsMAb) for pretargeting epithelial cancers: Studies with TF12 and 111In-labeled hapten-peptide (IMP 288) in ovarian cancer. J Nucl Med. 2010;51:abst1148
44. Cubas R, Li M, Chen C, Yao Q. Trop2: a possible therapeutic target for late stage epithelial carcinomas. Biochim Biophys Acta. 2009;1796:309-14 doi:10.1016/j.bbcan.2009.08.001
45. Ripani E, Sacchetti A, Corda D, Alberti S. Human Trop-2 is a tumor-associated calcium signal transducer. Int J Cancer. 1998;76:671-6
46. Stein R, Basu A, Goldenberg DM, Lloyd KO, Mattes MJ. Characterization of cluster 13: the epithelial/carcinoma antigen recognized by MAb RS7. Int J Cancer Suppl. 1994;8:98-102
47. Stein R, Chen S, Sharkey RM, Goldenberg DM. Murine monoclonal antibodies raised against human non-small cell carcinoma of the lung: specificity and tumor targeting. Cancer Res. 1990;50:1330-6
48. Pagel JM, Orgun N, Hamlin DK, Wilbur DS, Gooley TA, Gopal AK. et al. A comparative analysis of conventional and pretargeted radioimmunotherapy of B-cell lymphomas by targeting CD20, CD22, and HLA-DR singly and in combinations. Blood. 2009;113:4903-13 doi:10.1182/blood-2008-11-187401
49. Hnatowich DJ, Virzi F, Rusckowski M. Investigations of avidin and biotin for imaging applications. J Nucl Med. 1987;28:1294-302
50. Hnatowich DJ. Antisense and nuclear medicine. J Nucl Med. 1999;40:693-703
51. Wang Y, Chang F, Zhang Y, Liu N, Liu G, Gupta S. et al. Pretargeting with amplification using polymeric peptide nucleic acid. Bioconjug Chem. 2001;12:807-16
52. Liu G, Liu C, Zhang S, He J, Liu N, Gupta S. et al. Investigations of 99mTc morpholino pretargeting in mice. Nucl Med Commun. 2003;24:697-705 doi:10.1097/01.mnm.0000075200.18521.39
53. He J, Liu G, Gupta S, Zhang Y, Rusckowski M, Hnatowich DJ. Amplification targeting: a modified pretargeting approach with potential for signal amplification-proof of a concept. J Nucl Med. 2004;45:1087-95
54. Zhou J, Soontornworajit B, Snipes MP, Wang Y. Development of a novel pretargeting system with bifunctional nucleic acid molecules. Biochem Biophys Res Commun. 2009;386:521-5 doi:10.1016/j.bbrc.2009.06.090
55. Goldenberg DM, Sharkey RM, Paganelli G, Barbet J, Chatal JF. Antibody pretargeting advances cancer radioimmunodetection and radioimmunotherapy. J Clin Oncol. 2006;24:823-34 doi:10.1200/jco.2005.03.8471
56. Goldenberg DM, Chatal JF, Barbet J, Boerman O, Sharkey RM. Cancer imaging and therapy with bispecific antibody pretargeting. Update Cancer Ther. 2007;2:19-31 doi:10.1016/j.uct.2007.04.003
57. Sharkey RM, Rossi EA, Chang CH, Goldenberg DM. Improved cancer therapy and molecular imaging with multivalent, multispecific antibodies. Cancer Biother Radiopharm. 2010;25:1-12 doi:10.1089/cbr.2009.0690
58. Goldenberg DM, Chang CH, Sharkey RM, Rossi EA, Karacay H, McBride W. et al. Radioimmunotherapy: is avidin-biotin pretargeting the preferred choice among pretargeting methods? Eur J Nucl Med Mol Imaging. 2003;30:777-80 doi:10.1007/s00259-002-1089-6
59. Goldenberg DM. Future role of radiolabeled monoclonal antibodies in oncological diagnosis and therapy. Semin Nucl Med. 1989;19:332-9
60. Swayne LC, Goldenberg DM, Diehl WL, Macaulay RD, Derby LA, Trivino JZ. SPECT anti-CEA monoclonal antibody detection of occult colorectal carcinoma metastases. Clin Nucl Med. 1991;16:849-52
61. Goldenberg DM, Larson SM. Radioimmunodetection in cancer identification. J Nucl Med. 1992;33:803-14
62. Goldenberg DM, Wlodkowski TJ, Sharkey RM, Silberstein EB, Serafini AN. et al. Colorectal cancer imaging with iodine-123-labeled CEA monoclonal antibody fragments. J Nucl Med. 1993;34:61-70
63. Patt YZ, Podoloff DA, Curley S, Smith R, Badkhamkar VA, Lamki LM. et al. Monoclonal antibody imaging in patients with colorectal cancer and increasing levels of serum carcinoembryonic antigen. Experience with ZCE-025 and IMMU-4 monoclonal antibodies and proposed directions for clinical trials. Cancer. 1993;71:4293-7
64. Podoloff DA, Patt YZ, Curley SA, Kim EE, Bhadkamkar VA, Smith RE. Imaging of colorectal carcinoma with technetium-99m radiolabeled Fab' fragments. Semin Nucl Med. 1993;23:89-98
65. Le Doussal JM, Martin M, Gautherot E, Delaage M, Barbet J. In vitro and in vivo targeting of radiolabeled monovalent and divalent haptens with dual specificity monoclonal antibody conjugates: enhanced divalent hapten affinity for cell-bound antibody conjugate. J Nucl Med. 1989;30:1358-66
66. Goodwin DA, Meares CF, Watanabe N, McTigue M, Chaovapong W, Ransone CM. et al. Pharmacokinetics of pretargeted monoclonal antibody 2D12.5 and 88Y-Janus-2-(p-nitrobenzyl)-1,4,7,10-tetraazacyclododecanetetraacetic acid (DOTA) in BALB/c mice with KHJJ mouse adenocarcinoma: a model for 90Y radioimmunotherapy. Cancer Res. 1994;54:5937-46
67. Boerman OC, Kranenborg MH, Oosterwijk E, Griffiths GL, McBride WJ, Oyen WJ. et al. Pretargeting of renal cell carcinoma: improved tumor targeting with a bivalent chelate. Cancer Res. 1999;59:4400-5
68. Le Doussal JM, Barbet J, Delaage M. Bispecific-antibody-mediated targeting of radiolabeled bivalent haptens: theoretical, experimental and clinical results. Int J Cancer Suppl. 1992;7:58-62
69. Le Doussal JM, Chetanneau A, Gruaz-Guyon A, Martin M, Gautherot E, Lehur PA. et al. Bispecific monoclonal antibody-mediated targeting of an indium-111-labeled DTPA dimer to primary colorectal tumors: pharmacokinetics, biodistribution, scintigraphy and immune response. J Nucl Med. 1993;34:1662-71
70. Chetanneau A, Barbet J, Peltier P, Le Doussal JM, Gruaz-Guyon A, Bernard AM. et al. Pretargetted imaging of colorectal cancer recurrences using an 111In-labelled bivalent hapten and a bispecific antibody conjugate. Nucl Med Commun. 1994;15:972-80
71. Gautherot E, Bouhou J, Le Doussal JM, Manetti C, Martin M, Rouvier E. et al. Therapy for colon carcinoma xenografts with bispecific antibody-targeted, iodine-131-labeled bivalent hapten. Cancer. 1997;80:2618-23
72. Gautherot E, Le Doussal JM, Bouhou J, Manetti C, Martin M, Rouvier E. et al. Delivery of therapeutic doses of radioiodine using bispecific antibody-targeted bivalent haptens. J Nucl Med. 1998;39:1937-43
73. Gautherot E, Rouvier E, Daniel L, Loucif E, Bouhou J, Manetti C. et al. Pretargeted radioimmunotherapy of human colorectal xenografts with bispecific antibody and 131I-labeled bivalent hapten. J Nucl Med. 2000;41:480-7
74. Le Doussal JM, Gruaz-Guyon A, Martin M, Gautherot E, Delaage M, Barbet J. Targeting of indium 111-labeled bivalent hapten to human melanoma mediated by bispecific monoclonal antibody conjugates: imaging of tumors hosted in nude mice. Cancer Res. 1990;50:3445-52
75. Karacay H, McBride WJ, Griffiths GL, Sharkey RM, Barbet J, Hansen HJ. et al. Experimental pretargeting studies of cancer with a humanized anti-CEA x murine anti-[In-DTPA] bispecific antibody construct and a 99mTc-/188Re-labeled peptide. Bioconjug Chem. 2000;11:842-54
76. Janevik-Ivanovska E, Gautherot E, Hillairet de Boisferon M, Cohen M, Milhaud G, Tartar A. et al. Bivalent hapten-bearing peptides designed for iodine-131 pretargeted radioimmunotherapy. Bioconjug Chem. 1997;8:526-33
77. McBride WJ, Zanzonico P, Sharkey RM, Noren C, Karacay H, Rossi EA. et al. Bispecific antibody pretargeting PET (immunoPET) with an 124I-labeled hapten-peptide. J Nucl Med. 2006;47:1678-88
78. Sharkey RM, Karacay H, McBride WJ, Rossi EA, Chang CH, Goldenberg DM. Bispecific antibody pretargeting of radionuclides for immuno single-photon emission computed tomography and immuno positron emission tomography molecular imaging: an update. Clin Cancer Res. 2007;13:5577s-85s doi:10.1158/1078-0432.ccr-07-1087
79. Sharkey RM, Karacay H, Vallabhajosula S, McBride WJ, Rossi EA, Chang CH. et al. Metastatic human colonic carcinoma: molecular imaging with pretargeted SPECT and PET in a mouse model. Radiology. 2008;246:497-507 doi:10.1148/radiol.2462070229
80. Karacay H, Sharkey RM, McBride WJ, Rossi EA, Chang CH, Goldenberg DM. Optimization of hapten-peptide labeling for pretargeted immunoPET of bispecific antibody using generator-produced 68Ga. J Nucl Med. 2011;52:555-9 doi:10.2967/jnumed.110.083568
81. Sharkey RM, Rossi EA, McBride WJ, Chang CH, Goldenberg DM. Recombinant bispecific monoclonal antibodies prepared by the dock-and-lock strategy for pretargeted radioimmunotherapy. Semin Nucl Med. 2010;40:190-203 doi:10.1053/j.semnuclmed.2009.12.002
82. Karacay H, Sharkey RM, McBride WJ, Griffiths GL, Qu Z, Chang K. et al. Pretargeting for cancer radioimmunotherapy with bispecific antibodies: role of the bispecific antibody's valency for the tumor target antigen. Bioconjug Chem. 2002;13:1054-70
83. Rossi EA, Sharkey RM, McBride W, Karacay H, Zeng L, Hansen HJ. et al. Development of new multivalent-bispecific agents for pretargeting tumor localization and therapy. Clin Cancer Res. 2003;9:3886S-96S
84. Rossi EA, Chang CH, Losman MJ, Sharkey RM, Karacay H, McBride W. et al. Pretargeting of carcinoembryonic antigen-expressing cancers with a trivalent bispecific fusion protein produced in myeloma cells. Clin Cancer Res. 2005;11:7122s-9s doi:10.1158/1078-0432.ccr-1004-0020
85. Rossi EA, Goldenberg DM, Cardillo TM, McBride WJ, Sharkey RM, Chang CH. Stably tethered multifunctional structures of defined composition made by the dock and lock method for use in cancer targeting. Proc Natl Acad Sci U S A. 2006;103:6841-6 doi:10.1073/pnas.0600982103
86. Chang CH, Rossi EA, Goldenberg DM. The dock and lock method: a novel platform technology for building multivalent, multifunctional structures of defined composition with retained bioactivity. Clin Cancer Res. 2007;13:5586s-91s doi:10.1158/1078-0432.ccr-07-1217
87. Goldenberg DM, Rossi EA, Sharkey RM, McBride WJ, Chang CH. Multifunctional antibodies by the Dock-and-Lock method for improved cancer imaging and therapy by pretargeting. J Nucl Med. 2008;49:158-63 doi:10.2967/jnumed.107.046185
88. He J, Liu X, Zhang S, Liu G, Hnatowich DJ. Affinity enhancement bivalent morpholinos for pretargeting: surface plasmon resonance studies of molecular dimensions. Bioconjug Chem. 2005;16:1098-104 doi:10.1021/bc050061s
89. He J, Wang Y, Dou S, Liu X, Zhang S, Liu G. et al. Affinity enhancement pretargeting: synthesis and testing of a 99mTc-labeled bivalent MORF. Mol Pharm. 2010;7:1118-24 doi:10.1021/mp9002909
90. Sharkey RM, Karacay H, Richel H, McBride WJ, Rossi EA, Chang K. et al. Optimizing bispecific antibody pretargeting for use in radioimmunotherapy. Clin Cancer Res. 2003;9:3897S-913S
91. Kraeber-Bodere F, Faivre-Chauvet A, Ferrer L, Vuillez JP, Brard PY, Rousseau C. et al. Pharmacokinetics and dosimetry studies for optimization of anti-carcinoembryonic antigen x anti-hapten bispecific antibody-mediated pretargeting of Iodine-131-labeled hapten in a phase I radioimmunotherapy trial. Clin Cancer Res. 2003;9:3973S-81S
92. Kraeber-Bodere F, Rousseau C, Bodet-Milin C, Ferrer L, Faivre-Chauvet A, Campion L. et al. Targeting, toxicity, and efficacy of 2-step, pretargeted radioimmunotherapy using a chimeric bispecific antibody and 131I-labeled bivalent hapten in a phase I optimization clinical trial. J Nucl Med. 2006;47:247-55
93. Schoffelen R, Boerman O, van der Graaf W, van Herpen C, Sharkey R, McBride W. et al. Phase I clinical study of the feasibility of pretargeted radioimmunotherapy (PT-RAIT) in patients with colorectal cancer (CRC): First results. J Nucl Med. 2011;52:abst358
94. Liu G, Dou S, Rusckowski M, Hnatowich DJ. An experimental and theoretical evaluation of the influence of pretargeting antibody on the tumor accumulation of effector. Mol Cancer Ther. 2008;7:1025-32 doi:10.1158/1535-7163.mct-07-2203
95. Liu G, Hnatowich DJ. A semiempirical model of tumor pretargeting. Bioconjug Chem. 2008;19:2095-104 doi:10.1021/bc8002748
96. Breitz HB, Weiden PL, Beaumier PL, Axworthy DB, Seiler C, Su FM. et al. Clinical optimization of pretargeted radioimmunotherapy with antibody-streptavidin conjugate and 90Y-DOTA-biotin. J Nucl Med. 2000;41:131-40
97. Sharkey RM, Cardillo TM, Rossi EA, Chang CH, Karacay H, McBride WJ. et al. Signal amplification in molecular imaging by pretargeting a multivalent, bispecific antibody. Nat Med. 2005;11:1250-5 doi:10.1038/nm1322
98. Moffat FL Jr, Pinsky CM, Hammershaimb L, Petrelli NJ, Patt YZ, Whaley FS. et al. Clinical utility of external immunoscintigraphy with the IMMU-4 technetium-99m Fab' antibody fragment in patients undergoing surgery for carcinoma of the colon and rectum: results of a pivotal, phase III trial. The Immunomedics Study Group. J Clin Oncol. 1996;14:2295-305
99. Hughes K, Pinsky CM, Petrelli NJ, Moffat FL, Patt YZ, Hammershaimb L. et al. Use of carcinoembryonic antigen radioimmunodetection and computed tomography for predicting the resectability of recurrent colorectal cancer. Ann Surg. 1997;226:621-31
100. Erb DA, Nabi HA. Clinical and technical considerations for imaging colorectal cancers with technetium-99m-labeled antiCEA Fab' fragment. J Nucl Med Technol. 2000;28:12-8
101. Sharkey RM, Weadock KS, Natale A, Haywood L, Aninipot R, Blumenthal RD. et al. Successful radioimmunotherapy for lung metastasis of human colonic cancer in nude mice. J Natl Cancer Inst. 1991;83:627-32
102. Schoffelen R, Sharkey RM, Goldenberg DM, Franssen G, McBride WJ, Rossi EA. et al. Pretargeted immuno-positron emission tomography imaging of carcinoembryonic antigen-expressing tumors with a bispecific antibody and a 68Ga- and 18F-labeled hapten peptide in mice with human tumor xenografts. Mol Cancer Ther. 2010;9:1019-27 doi:10.1158/1535-7163.mct-09-0862
103. Lewis MR, Wang M, Axworthy DB, Theodore LJ, Mallet RW, Fritzberg AR. et al. In vivo evaluation of pretargeted 64Cu for tumor imaging and therapy. J Nucl Med. 2003;44:1284-92
104. Pagou M, Zerizer I, Al-Nahhas A. Can gallium-68 compounds partly replace 18F-FDG in PET molecular imaging? Hell J Nucl Med. 2009;12:102-5
105. Rice SL, Roney CA, Daumar P, Lewis JS. The next generation of positron emission tomography radiopharmaceuticals in oncology. Semin Nucl Med. 2011;41:265-82 doi:10.1053/j.semnuclmed.2011.02.002
106. McBride WJ, Sharkey RM, Karacay H, D'Souza CA, Rossi EA, Laverman P. et al. A novel method of 18F radiolabeling for PET. J Nucl Med. 2009;50:991-8 doi:10.2967/jnumed.108.060418
107. McBride WJ, D'Souza CA, Sharkey RM, Karacay H, Rossi EA, Chang CH. et al. Improved 18F labeling of peptides with a fluoride-aluminum-chelate complex. Bioconjug Chem. 2010;21:1331-40 doi:10.1021/bc100137x
108. D'Souza CA, McBride WJ, Sharkey RM, Todaro LJ, Goldenberg DM. High-yielding aqueous 18F-labeling of peptides via Al18F chelation. Bioconjug Chem. 2011 doi:10.1021/bc200175c
109. Laverman P, McBride WJ, Sharkey RM, Eek A, Joosten L, Oyen WJ. et al. A novel facile method of labeling octreotide with 18F-fluorine. J Nucl Med. 2010;51:454-61 doi:10.2967/jnumed.109.066902
110. McBride WJ, D'Souza CA, Cardillo TM, Goldenberg DM. A facile kit for rapid radiofluorination of peptides for PET. J Nucl Med. 2011;52:313-4
111. McBride WJ, D'Souza CA, Sharkey RM, Goldenberg DM. The radiolabeling of proteins by the [18F]AlF method. Appl Radiat Isot. 2011 in press
112. Kraeber-Bodere F, Faibre-Chauvet A, Sai-Maurel C, Gautherot E, Fiche M, Campion L. et al. Bispecific antibody and bivalent hapten radioimmunotherapy in CEA-producing medullary thyroid cancer xenograft. J Nucl Med. 1999;40:198-204
113. Kraeber-Bodere F, Faivre-Chauvet A, Sai-Maurel C, Campion L, Fiche M, Gautherot E. et al. Toxicity and efficacy of radioimmunotherapy in carcinoembryonic antigen-producing medullary thyroid cancer xenograft: comparison of iodine 131-labeled F(ab')2 and pretargeted bivalent hapten and evaluation of repeated injections. Clin Cancer Res. 1999;5:3183s-9s
114. Axworthy DB, Fritzberg AR, Hylarides MD, Mallet RW, Theodore LJ, Gustavson LM. et al. Preclinical evaulation of an anti-tumor monoclonal antibody/streptavidin conjugate for pretargeted 90Y radioimmunotherapy in a mouse xenograft model. J Immunother. 1994;16:158
115. Axworthy DB, Reno JM, Hylarides MD, Mallett RW, Theodore LJ, Gustavson LM. et al. Cure of human carcinoma xenografts by a single dose of pretargeted yttrium-90 with negligible toxicity. Proc Natl Acad Sci U S A. 2000;97:1802-7
116. Karacay H, Brard PY, Sharkey RM, Chang CH, Rossi EA, McBride WJ. et al. Therapeutic advantage of pretargeted radioimmunotherapy using a recombinant bispecific antibody in a human colon cancer xenograft. Clin Cancer Res. 2005;11:7879-85 doi:10.1158/1078-0432.ccr-05-1246
117. Barone R, Borson-Chazot F, Valkema R, Walrand S, Chauvin F, Gogou L. et al. Patient-specific dosimetry in predicting renal toxicity with 90Y-DOTATOC: relevance of kidney volume and dose rate in finding a dose-effect relationship. J Nucl Med. 2005;46(Suppl 1):99S-106S
118. Valkema R, Pauwels SA, Kvols LK, Kwekkeboom DJ, Jamar F, de Jong M. et al. Long-term follow-up of renal function after peptide receptor radiation therapy with 90Y-DOTA0,Tyr3-octreotide and 177Lu-DOTA0, Tyr3-octreotate. J Nucl Med. 2005;46(Suppl 1):83S-91S
119. Sharkey RM, Goldenberg DM. Perspectives on cancer therapy with radiolabeled monoclonal antibodies. J Nucl Med. 2005;46(Suppl 1):115S-27S
120. Bouchet LG, Bolch WE, Blanco HP, Wessels BW, Siegel JA, Rajon DA. et al. MIRD Pamphlet No 19: absorbed fractions and radionuclide S values for six age-dependent multiregion models of the kidney. J Nucl Med. 2003;44:1113-47
121. Bodei L, Cremonesi M, Ferrari M, Pacifici M, Grana CM, Bartolomei M. et al. Long-term evaluation of renal toxicity after peptide receptor radionuclide therapy with 90Y-DOTATOC and 177Lu-DOTATATE: the role of associated risk factors. Eur J Nucl Med Mol Imaging. 2008;35:1847-56 doi:10.1007/s00259-008-0778-1
122. Wessels BW, Konijnenberg MW, Dale RG, Breitz HB, Cremonesi M, Meredith RF. et al. MIRD pamphlet No. 20: the effect of model assumptions on kidney dosimetry and response--implications for radionuclide therapy. J Nucl Med. 2008;49:1884-99 doi:10.2967/jnumed.108.053173
123. Siegel JA, Stabin MG, Sharkey RM. Renal dosimetry in peptide radionuclide receptor therapy. Cancer Biother Radiopharm. 2010;25:581-8 doi:10.1089/cbr.2010.0805
124. Stabin MG, Sharkey RM, Siegel JA. RADAR commentary: Evolution and current status of dosimetry in nuclear medicine. J Nucl Med. 2011;52:1156-61 doi:10.2967/jnumed.111.088666
125. Imhof A, Brunner P, Marincek N, Briel M, Schindler C, Rasch H. et al. Response, survival, and long-term toxicity after therapy with the radiolabeled somatostatin analogue [90Y-DOTA]-TOC in metastasized neuroendocrine cancers. J Clin Oncol. 2011;29:2416-23 doi:10.1200/jco.2010.33.7873
126. Mirallie E, Vuillez JP, Bardet S, Frampas E, Dupas B, Ferrer L. et al. High frequency of bone/bone marrow involvement in advanced medullary thyroid cancer. J Clin Endocrinol Metab. 2005;90:779-88 doi:10.1210/jc.2004-1500
127. Chatal JF, Campion L, Kraeber-Bodere F, Bardet S, Vuillez JP, Charbonnel B. et al. Survival improvement in patients with medullary thyroid carcinoma who undergo pretargeted anti-carcinoembryonic-antigen radioimmunotherapy: a collaborative study with the French Endocrine Tumor Group. J Clin Oncol. 2006;24:1705-11 doi:10.1200/jco.2005.04.4917
128. Kraeber-Bodere F, Sai-Maurel C, Campion L, Faivre-Chauvet A, Mirallie E, Cherel M. et al. Enhanced antitumor activity of combined pretargeted radioimmunotherapy and paclitaxel in medullary thyroid cancer xenograft. Mol Cancer Ther. 2002;1:267-74
129. Graves SS, Dearstyne E, Lin Y, Zuo Y, Sanderson J, Schultz J. et al. Combination therapy with pretarget CC49 radioimmunotherapy and gemcitabine prolongs tumor doubling time in a murine xenograft model of colon cancer more effectively than either monotherapy. Clin Cancer Res. 2003;9:3712-21
130. Kraeber-Bodere F, Bodet-Milin C, Niaudet C, Sai-Maurel C, Moreau A, Faivre-Chauvet A. et al. Comparative toxicity and efficacy of combined radioimmunotherapy and antiangiogenic therapy in carcinoembryonic antigen-expressing medullary thyroid cancer xenograft. J Nucl Med. 2010;51:624-31 doi:10.2967/jnumed.109.070714
131. Salaun PY, Bodet-Milin C, Frampas E, Oudoux A, Sai-Maurel C, Faivre-Chauvet A. et al. Toxicity and efficacy of combined radioimmunotherapy and bevacizumab in a mouse model of medullary thyroid carcinoma. Cancer. 2010;116:1053-8 doi:10.1002/cncr.24792
132. Sharkey RM, Karacay H, Govindan SV, Goldenberg DM. Combination radioimmunotherapy and chemoimmunotherapy involving different or the same targets improves therapy of human pancreatic carcinoma xenograft models. Mol Cancer Ther. 2011;10:1072-81 doi:10.1158/1535-7163.mct-11-0115
Author contact
![]() Corresponding author: DM Goldenberg, CMMI/GSCC, 300 American Road, Morris Plains, NJ 07950; Phone: 973-844-7000; Fax: 973-844-7020; E-mail: dmg.gscancernet.
Corresponding author: DM Goldenberg, CMMI/GSCC, 300 American Road, Morris Plains, NJ 07950; Phone: 973-844-7000; Fax: 973-844-7020; E-mail: dmg.gscancernet.