Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
2. Materials and Methods
3. Results and Discussion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2011; 1:18-27. doi:10.7150/thno/v01p0018 This volume Cite
Research Paper
Real-Time Video Imaging of Protease Expression In Vivo
Lei Zhu1,2, Jin Xie1, Magdalena Swierczewska1,3, Fan Zhang1, Qimeng Quan1, Ying Ma1, Xuexun Fang2, Kwangmeyung Kim4, Seulki Lee1, ![]() , Xiaoyuan Chen1,
, Xiaoyuan Chen1, ![]()
1. Laboratory of Molecular Imaging and Nanomedicine, National Institute of Biomedical Imaging and Bioengineering, National Institutes of Health, Bethesda, Maryland 20892, USA
2. Key Laboratory of Molecular Enzymology and Enzyme Engineering of Ministry Education, Jilin University, Changchun 130023, P.R. China
3. Department of Biomedical Engineering, Stony Brook University, Stony Brook, NY 11794, USA
4. Biomedical Research Center, Korea Institute of Science and Technology, Seoul 136-791, Korea
Published 2011-1-12
Abstract
We demonstrate the first true real-time in vivo video imaging of extracellular protease expression using an ultrafast-acting and extended-use activatable probe. This simple, one-step technique is capable of boosting fluorescent signals upon target protease cleavage as early as 30 minutes from injection in a small animal model and is able to sustain the strong fluorescent signal up to 24 hours. Using this method, we video imaged the expression and inhibition of matrix metalloproteinases (MMPs) in a tumor-bearing mouse model. The current platform can be universally applied to any target protease of interest with a known peptide substrate and is adaptable to a wide range of real-time imaging applications with high throughputs such as for in vivo drug screening, examinations of the therapeutic efficacy of drugs, and monitoring of disease onset and development in animal models.
Keywords: matrix metalloproteinases (MMPs), activatable probes, near-infrared fluorescence imaging, real-time in vivo video imaging, peptide substrate, PEGylation.
Introduction
Most new drug candidates generated during in vitro screening turn out to be invalid after time-consuming and costly testing in animal models. Therefore, there is an urgent need for development of noninvasive, real-time, sensitive, and cost-effective tools with high throughput for monitoring and early detection of drug efficacy in vivo. Optical molecular imaging provides many advantages over other imaging modalities, including high sensitivity and safe detection with non-radioactive materials, using readily available instruments, at moderate cost [1]. Further improvements in performance of in vivo optical imaging are dependent on the development of sophisticated imaging probes that exhibit high sensitivity and low background noise. Among the diverse applications, peptide-based molecular beacons, so called protease activatable optical probes, have enabled in vivo imaging of protease activity and demonstrated promising results in the field of protease research and protease-targeted drug development [2-3].
Proteases are known as exceptionally critical signaling proteins that are involved in numerous processes, such as inflammation, as well as cancer, neurological disorders, and cardiovascular diseases [4]. It is of no surprise, therefore, that several small-molecule inhibitors targeting proteases are already on the market, and a significant number of new therapies based on protease inhibition are under vigorous clinical investigation. The unique differential expression of proteases in diseases enable particular types of protease to be used as a specific biomarkers for diagnosis and as target molecules for protease-activated prodrugs [5]. Considerable efforts have been made to identify the role of certain proteases in given biological processes and to screen specific molecules that can regulate protease expression. Most experimental methods are based on the use of protease reporters or molecular beacons. Moreover, they are limited to in vitro applications.
With the development of hydrophilic near-infrared (NIR) dyes and quenchers, it is now possible to use conventional molecular beacon constructs as in vivo imaging agents [6-7]. These probes are optically silent (quenched) in their native state and are activated in the presence of a specific protease, thereby generating an NIR fluorescence signal. However, the inherent instability, short half-life, and nonspecific activation of peptides and small compounds are still major obstacles to their in vivo application by systemic administration. Conjugation of macromolecules, such as high molecular weight poly(amino acids) and poly(ethylene) glycol (PEG), efficiently increases the in vivo stability [8], but decreases the sensitivity and specificity of the probes. This is because conjugated macromolecules require longer circulation times to produce high contrast images through accumulation at tumor sites by the enhanced permeability and retention effect (EPR), and a consequence can be non-specific signal activation by proteases present in the blood. For example, commercially available VisEn's protease activatable probes targeting matrix metalloproteinases (MMPs) (MMPSense™, VisEn, Bedford, MA, USA) have been widely used [9]. However, the probes typically take a long time (~ 24 hr) to be fully activated in vitro and in vivo, which may be due to their conjugated, high-molecular-weight polymer backbone. Delayed activation hampers real-time and high-throughput in vivo applications. Therefore, it is important to strike a balance between stability and sensitivity of the probes in vivo to enable quick screening and true real-time imaging of enzyme activity in live animals, and to achieve superior target-to-background contrast. Recently, we and others have developed various novel activatable imaging probes that can provide high-resolution imaging and low background signals [3, 10-13]. Although these reported systems are sensitive, they have limited applications due to the modest and delayed fluorescent changes of the probes, thus allowing only snap-shot in vivo images, rather than real-time video images of the whole-body.
In this study, we present a simple method to upgrade a conventional molecular beacon to an ultrafast-acting and extended-use activatable probe. We found that the PEGylation of a molecular beacon with a specific size PEG can significantly alter the activation properties of the probe and permit real-time video imaging of protease expression in vivo. We chose MMPs as our initial target proteases. MMPs are a family of zinc-dependent endopeptidases and represent the most well-known proteases associated with tumorigenesis [14].
2. Materials and Methods
Cell culture and animal models. Squamous cell carcinoma SCC-7 were cultured in RPMI 1640 medium containing 10% (v/v) fetal bovine serum (Invitrogen) supplemented with penicillin (100 μg/ml) and streptomycin (100 μg/ml) at 37 ºC with 5% CO2. SCC-7 tumor model was developed by subcutaneous injection of 1×106 cells into the right front flank of female athymic nude mice (Harlan Laboratories). The mice were used for optical studies when the tumor volume reached about 300 mm3. All animal studies were conducted in accordance with the principles and procedures outlined in the NRC Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of the Clinical Center, NIH.
Synthesis of the MMP-Pns. MMP-Pn is composed of i) Cy5.5 as a NIR dye (ex/em: 675/695 nm), ii) MMP substrate GPLGVRGKGG as a recognition site indicated by italics with the cleavage site between Gly and Val, iii) Cy5.5 quencher BHQ-3 (maximum abs. 650 nm), and iv) PEG as a backbone. The peptide sequence was synthesized by standard solid-phase Fmoc peptide chemistry. MMP-Pn was synthesized through serial conjugation and deprotection of peptide in solution followed by preparative reversed-phase high-performance liquid chromatography (RP-HPLC) purifications. The final products were confirmed by analytical RP-HPLC and LC-mass spectroscopy. Cy5.5-NHS was purchased from GE Healthcare (Piscataway, NJ) and BHQ-3 NHS ester was obtained from Biosearch Technology (Navato, CA). Amino PEG analogs NH2-R1 were purchased from Quanta Biodesign (Powell, OH) and Sigma-Aldrich (St-Louis, MO). All other chemicals were of analytical grade and were purchased from Sigma-Aldrich.
In vitro enzyme test. The activity of the MMP-Pns was investigated in vitro by incubating the probe (15 nM) in the reaction buffer (50 mM Tris·HCl, 10 mM CaCl2·2H2O, 0.15 M NaCl, 0.05% Brij35, pH 7.8, TCNB buffer) containing an appropriate amount of activated MMPs. Recombinant human MMPs were activated with 2.5 mM p-aminophenylmercuric acetate (APMA) in the reaction buffer for 1 to 2 hrs at 37 ºC before use. A homophenylalanine-hydroxamin acid based broad-spectrum MMP inhibitor MMP-I (MMP Inhibitor III, Merck KGaA, Darmstadt, Germany) was used for the MMP inhibition test. Fluorescence intensity was monitored using a spectrofluorometer equipped with micro-well plate reader (F-7000 Fluorescence Spectrophotometer, Hitach, Tokyo, Japan) every 10 min at 37 °C. The excitation wavelength was set at 675 nm and emission spectra were recorded from 680 to 800 nm.
In vivo imaging. Optical image aquisition and analysis were done using a Maestro 2 imaging system (Cri, Woburn, MA, USA). The MMP-Pns (100 μL in PBS pH 7.4, 300 nM) and MMPSense 680™ (150 μL, as recommended by the manufacturer, VisEn, Bedford, MA, USA) were injected via tail vein. Imaging was performed for 24 hrs using Maestro 2 configured for Cy5.5 detection at the indicated time points after the injection of the probes. For video imaging, 100 μL of the MMP-P12 was injected via tail vein catheter and imaging was performed every 10 sec for the first 5 min, 15 sec for 5 to 30 min, and 20 sec for 30 to 60 min, respectively, using Maestro's DyCE function. To inhibit the MMP activity, the MMP-I (1 mM) was injected intratumorally 30 min before the intravenous injection of the probe. Images were normalized and analyzed by using Maestro software. For quantitative comparison, the regions of interest (ROI) were drawn over tumor (T) and normal tissue (N, muscle), and the average signal (×106 photons×cm-2×s-1) for each area was measured. Results were presented as means ± s.d. for a group of 3-6 animals.
Ex vivo biodistribution. At 4 h after injection of the probes, mice were sacrificed and tumors and major organs were carefully excised and rinsed with saline. Excised samples were immediately imaged and analyzed by using Maestro 2. Results were presented as means ± s.d. for a group of 3 animals.
Histological analysis. Excised tumor tissue slices (4 μm) were fixed with cold acetone for 20 min and dried in air for 30 min at room temperature. After blocking with 10% BSA for 30 min, the sections were incubated with Rabbit anti-MMP-2, -9 and MMP-13 antibodies (10 μg/mL) for 60 min at room temperature in the dark, and then visualized with FITC-conjugated donkey anti-rabbit secondary antibody. Finally, the slices were mounted with DAPI-containing mounting medium under an epifluorescence microscope (Olympus, X81). For NIR fluorescence imaging, tumor slices were viewed by fluorescence microscopy equipped with a Cy5.5 filter.
3. Results and Discussion
We hypothesized that PEGylation of molecular beacons by PEGs of different size would induce different probe specificity and sensitivity effects, since PEG can alter the stability, half-life, and enzyme susceptibility of the probe dependent on the steric hindrance and molecular weight (MW) of the PEG [15]. In this study, we found that the addition of small MW PEG as a backbone to a fluorescently-quenched molecular beacon can significantly alter the beacon's activation profiles under physiological conditions. We synthesized a series of PEGylated molecular beacons, screened their activity in vitro and in vivo, and assessed their suitability for video imaging of protease expression in an MMP-overexpressing tumor-bearing mouse model. We began by preparing MMP activatable molecular beacons without a PEG backbone, termed MMP-P0.. The MMP-P0 consisted of Cy5.5 and BHQ-3 as a near-infrared dye/dark quencher pair, Cy5.5-GPLGVRGK(BHQ-3)GG[10]. Next, PEGs of different sizes were conjugated to the C-termini of the MMP-P0 (MMP-Pn, where “n” represents the number of repeating ethylene glycol units, -CH2CH2O-) (Table 1 and Fig. 1). MMP-13 was used as a model MMP for in vitro screening since MMP-P0 showed highest specificity against MMP-13 (Fig. 2a). The activity of each MMP-Pn (n = 0, 4, 12, 24, and 67) was tested by incubating 15 nM of each probe in a 96-well microplate containing the reaction buffer and 40 nM of activated MMP-13, with and without a broad spectrum hydroxamate-type MMP inhibitor (MMP-I). Fluorescence spectrometry clearly demonstrated that MMP-Pns can be significantly and fully activated (> 25-fold vs. without MMP) in the presence of MMP-13 within an hour (Fig. 2b and Fig. 2c). All MMP-Pns showed a linear relationship between the activated fluorescent signals and MMP-13 concentrations and were able to detect nanomolar concentrations of enzyme (Fig. 2d). The rate of activation was slightly enhanced for MMP-P12, which may be due to the improved solubility of the probe by PEG, but decreased when higher MW PEG was conjugated because of a steric-hindrance effect. However, no significant differences among the MMP-Pns were observed under the tested in vitro conditions.
Chemical structures of the MMP-Pns. Molecular weight (MW) of the MMP-P0 is 2324.95.
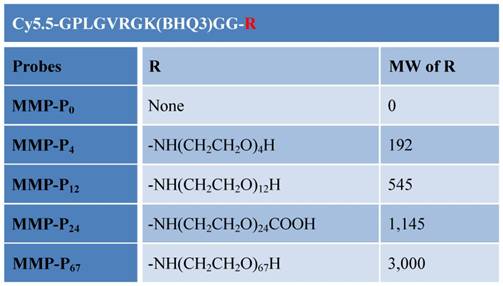
(a) Synthesis of the MMP-Pns. Reagents: a) HSPyU, 2% DIPEA/DMF, NH2-R1; b) 20% piperdine/DMF; c) Cy5.5-NHS ester, 2% DIPEA/DMF; d) TFA/water/TIS (95/2.5/2.5, v/v/v); e) BHQ-3 NHS ester, 2% DIPEA/DMF.
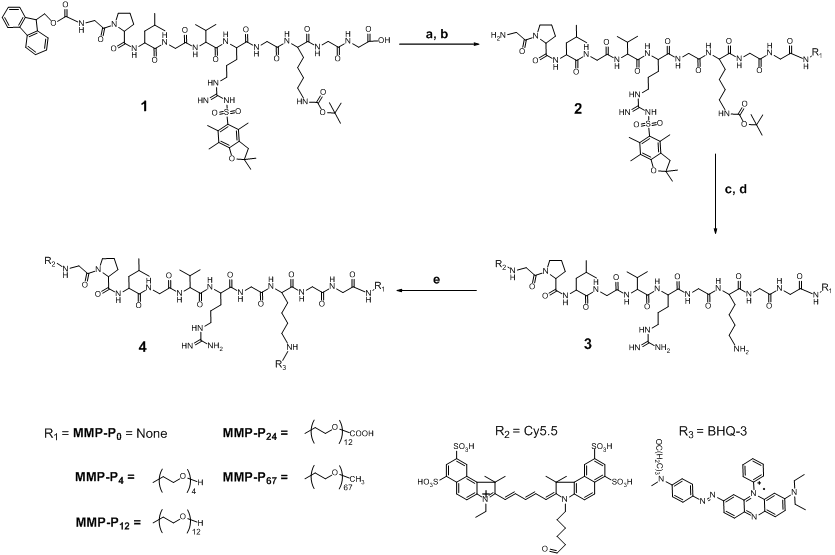
(a) Recovery of fluorescence of the MMP-P0 (15 nM) in the presence of various activated MMPs (40 nmol/L) following 80 min incubation at 37 ºC. (b, c) Fluorescence activation of the MMP-Ps (20 nM) in a solution containing activated MMP-13 (40 nmol/L) following 80 min incubation at 37 ºC. (b) Fluorescence emission spectra of the MMP-Ps before (quenched state, low fluorescence intensity, signals enlarged in inset) and after (activated state, increased fluorescence intensity) addition of activated MMP-13. (c) Fluorescence emission kinetic spectra of the activated MMP-Ps in the presence of MMP-13 with and without a broad spectrum MMP inhibitor (MMP-I). (d) Fluorescence intensities of the MMP-Pns in the presence of various concentrations of activated MMP-13 (1, 2, 5, 10, 20, and 40 nM) following a 20 min incubation. Means ± s.d. (n = 3).
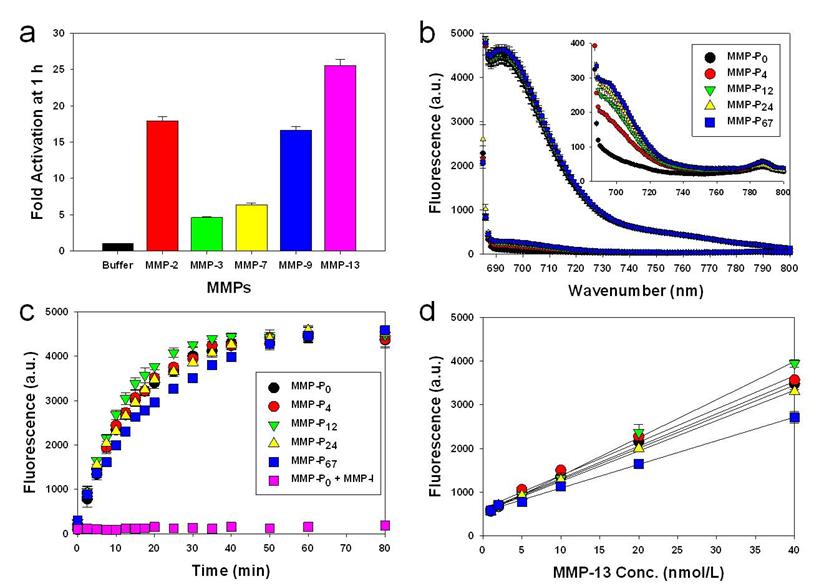
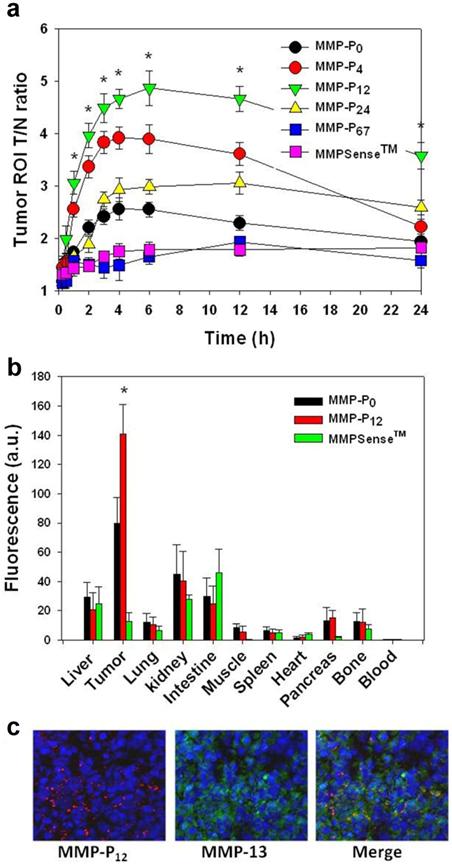
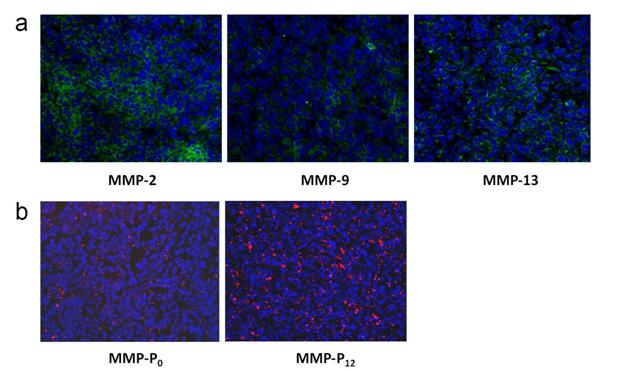
To evaluate whether a particular type of MMP-Pn can improve the visualization of overexpressed MMPs in vivo, the MMP-Pns and MMPSense 680™ were administered intravenously into separate MMP-positive SCC-7 tumor-bearing mice. In vivo imaging was performed for 24 hrs using a small-animal imaging system (Maestro 2, Cri, Woburn, MA, USA). Surprisingly, modification of MMP-Pn with small MW PEG showed significantly reduced activation time (ultrafast-acting and extended-use) in vivo. Fig. 3 shows representative serial images of mice at selected time points. In contrast to the in vitro activity, MMP-P4 and MMP-P12 clearly showed early onset of activation in vivo and provided high NIR fluorescence signals in the MMP-positive tumor region for a longer time compared to other MMP-Pns and MMPSense 680™. The activation of fluorescence signals in the tumor region was enhanced when the MW of the PEG backbone was increased up to 500 Da and gradually decreased with further increases of PEG MW. MMPSense 680™ showed a slightly positive fluorescent signal in the tumor region 24 hrs post injection (Fig. 4), however, the normalized signal was significantly lower than all MMP-Pns, except for MMP-P48. The ratio of the signal in the regions of interest (ROI) in the tumor compared to the normal region (T/N) of MMP-P12 was up to 3-fold higher than any other tested compounds at any time point (Fig. 5a). The biodistributions of MMP-P0, MMP-P12 and MMPSense 680™ were evaluated ex vivo after careful removal of tumors and other organs, after in vivo imaging, at 4 hrs post injection. As shown in Fig. 5b, MMP-P12 was activated predominantly in the tumors. Tumor sections were also analyzed. Fluorescence microscopy showed strong fluorescence signal consistent with in vivo imaging and anti-MMP antibody staining (for MMP-2, 9, 13), revealing strong expression of MMPs in SCC7 tumors (Fig. 5c and Fig. 6). These results indicate that molecular beacon targeting of specific proteases can be tested efficiently and optimized by simple small-MW PEG bioconjugation for in vivo applications in disease models.
Whole-mouse imaging with Maestro 2 (CRI). (a) Representative serial fluorescence images of MMPs-positive SCC7 tumor-bearing mice injected intravenously with the MMP-Ps and MMPSense 680™. Images were acquired at the indicated time points and were normalized by the maximum average value. The color bar indicates radiant efficiency (low, 0; high, 2.62×105).
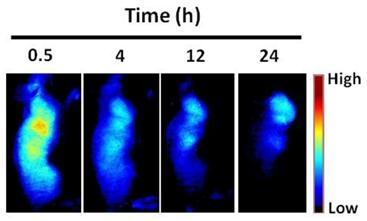
Representative serial fluorescence images of MMP-positive SCC7 tumor bearing mice injected intravenously with 150 μL of MMPSense 680™ (2 nmol). The color bar indicates radiant efficiency (low, 0; high, 4.7×104).
(a) Region of interest (ROI) tumor-to-normal tissue (T/N) ratio analysis of fluorescence intensity of SCC7 tumor in vivo. (b) Biodistribution of the MMP-P0, MMP-P12 and MMPSense 680™ 4 h post-injection. (c) NIR fluorescence microscopy of SCC7 tumor injected with MMP-P12 (left), tumor section counterstained with primary antibody for MMP-13 (middle) and their merged images (right). Means ± s.d. (n = 3-6/group). *P < 0.05 (Student's t-test) for MMP-P12 relative to other probes.
(a) Immunohistochemistry of MMP expression in a SCC7 tumor section counterstained with primary antibodies for MMP-2, 9, and 13, respectively. (b) NIR fluorescence microscopy of SCC7 tumor injected with MMP-P0 and MMP-P12. The slices were mounted with DAPI-containing mounting medium and examined under an epifluorescence microscope using Cy5.5 filter (Olympus X81). Original magnification was ×200. Exposure time: 1 sec.
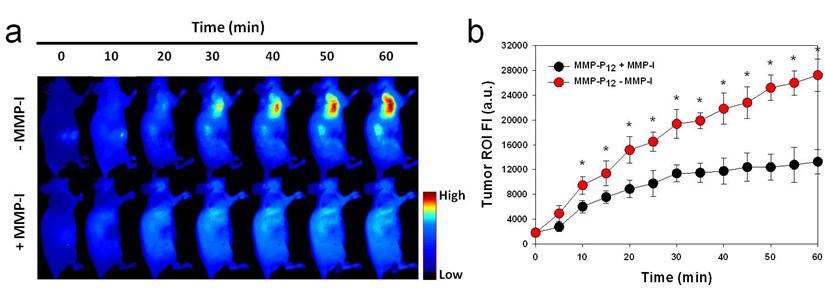
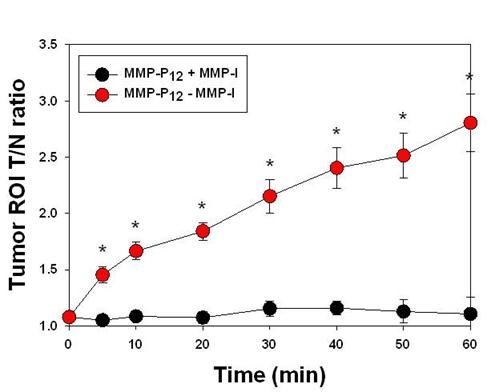
After systemic validation of MMP-Pns, we investigated whether we could video image the activity of MMPs in vivo. To inhibit the activity of MMPs in SCC-7 tumor-bearing mice, MMP-I was intratumorally injected 30 min before the probe injection. MMP-P12 was injected into mice either directly or following MMP-I treatment, using a tail vein catheter during continuous imaging procedures. The animals were imaged every 10 to 20 sec for 1 hr and video images were generated by using imaging software (DyCE, Cri, Woburn, MA, USA). Fig. 7a shows a typical series of video images. The MMP-P12 generated strong NIR fluorescence signals as early as 20 to 30 min after the probe injection and enabled clear visualization of MMPs in the tumor region thereafter (Supplementary Video 1). In contrast, the overall NIR fluorescent signals were significantly decreased when the activity of MMPs was suppressed by the MMP inhibitor (Supplementary Video 2). The mean fluorescent intensity of the tumor ROI and the T/N ratios analyzed from normalized snapshot images clearly confirm that the fluorescent activation of MMP-P12 can be inhibited significantly in the presence of MMP-I (Fig. 7b and Fig. 8). These results demonstrate the possibility of using our probe to monitor expression and inhibition of MMPs in vivo in real-time.
(a) Real-time video imaging of MMP expression in SCC7 tumor-bearing mice. MMP-P12 was injected into mice with or without MMP-I treatment via tail vein catheter during the imaging procedures. Whole-mouse images were obtained every 10 to 20 sec for 1 hr. Images were normalized by the maximum average value. The color bar indicates radiant efficiency (low, 0; high, 4.8×104). (b) ROI analysis of fluorescence intensity in tumor. The original video images (Supplemental Video 1 and Supplemental Video 2) are presented in the Supplemental Information. Means ± s.d. (n = 3). *P < 0.05 (Student's t-test).
ROI T/N ratio analysis of fluorescence intensity in SCC7 tumor with or without MMP-I. Means ± s.d. (n = 3). *P < 0.05 (Student's t-test).
In summary, we've presented a screening platform of activatable imaging probes to target extracellular proteases of interest in vivo. Any known peptide-based molecular beacon construct designed for in vitro application could be successfully tuned to become an in vivo imaging probe to visualize target proteases non-invasively and spatiotemporally in a rapid and efficient fashion. Optimized probes can be applied to a wide array of in vivo applications, such as drug screening with higher throughput, examinations of therapeutic efficacy, and monitoring of disease onset and development in animal models. The advantages over protease reporters or other polymer-based agents, are greater flexibility and tunability, and easier synthesis, scale-up, and handling. In this study, we did not attempt to quantify protease expression. We anticipate that current development of a real-time quantitative imaging system using advanced imaging software algorithms will improve the capabilities of our probes in the near future.
Supplementary Material
Supplementary Video 1Acknowledgements
This work was supported by the Intramural Research Program (IRP) of the National Institute of Biomedical Imaging and Bioengineering (NIBIB), NIH. J.X. is partially supported by an NIH pathway to independence grant (K99/R00). S.L. acknowledges a National Research Council Research Associateship Award funded by the National Institute of Standards and Technology (NIST) and the IRP of NIBIB, NIH.
Conflict of Interest
The authors have declared that no conflict of interest exists.
References
1. Weissleder R, Pittet MJ. Imaging in the era of molecular oncology. Nature. 2008;452:580-9
2. Lee S, Park K, Kim K, Choi K, Kwon IC. Activatable imaging probes with amplified fluorescent signals. Chem Commun (Camb). 2008:4250-60
3. Lee S, Xie J, Chen X. Activatable molecular probes for cancer imaging. Curr Top Med Chem. 2010;10:1135-44
4. Turk B. Targeting proteases: successes, failures and future prospects. Nat Rev Drug Discov. 2006;5:785-99
5. Law B, Tung CH. Proteolysis: a biological process adapted in drug delivery, therapy, and imaging. Bioconjug Chem. 2009;20:1683-95
6. Lee S, Xie J, Chen X. Peptides and peptide hormones for molecular imaging and disease diagnosis. Chem Rev. 2010;110:3087-111
7. Lee S, Park K, Lee SY, Ryu JH, Park JW, Ahn HJ. et al. Dark quenched matrix metalloproteinase fluorogenic probe for imaging osteoarthritis development in vivo. Bioconjug Chem. 2008;19:1743-7
8. Bremer C, Tung CH, Weissleder R. In vivo molecular target assessment of matrix metalloproteinase inhibition. Nat Med. 2001;7:743-8
9. In vivo Technology. PerkinElmer Inc. http://www.visenmedical.com/index.html
10. Lee S, Ryu JH, Park K, Lee A, Lee SY, Youn IC. et al. Polymeric nanoparticle-based activatable near-infrared nanosensor for protease determination in vivo. Nano Lett. 2009;9:4412-6
11. Nahrendorf M, Waterman P, Thurber G, Groves K, Rajopadhye M, Panizzi P. et al. Hybrid in vivo FMT-CT imaging of protease activity in atherosclerosis with customized nanosensors. Arterioscler Thromb Vasc Biol. 2009;29:1444-51
12. Barnett EM, Zhang X, Maxwell D, Chang Q, Piwnica-Worms D. Single-cell imaging of retinal ganglion cell apoptosis with a cell-penetrating, activatable peptide probe in an in vivo glaucoma model. Proc Natl Acad Sci U S A. 2009;106:9391-6
13. Nguyen QT, Olson ES, Aguilera TA, Jiang T, Scadeng M, Ellies LG. et al. Surgery with molecular fluorescence imaging using activatable cell-penetrating peptides decreases residual cancer and improves survival. Proc Natl Acad Sci U S A. 2010;107:4317-22
14. Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141:52-67
15. Kang JS, Deluca PP, Lee KC. Emerging PEGylated drugs. Expert Opin Emerg Drugs. 2009;14:363-80
Author contact
![]() Corresponding author: X.C. (Shawn.Chengov) or S.L. (Seulki.Leegov).
Corresponding author: X.C. (Shawn.Chengov) or S.L. (Seulki.Leegov).