Theranostics
13.3
Impact Factor
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Methods
Results
Discussion
Conclusion
Abbreviations
Acknowledgements
Supplementary Material
References
Introduction
Methods
Results
Discussion
Conclusion
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(14):3781-3796. doi:10.7150/thno.24821 This issue Cite
Research Paper
Nano-delivery of fraxinellone remodels tumor microenvironment and facilitates therapeutic vaccination in desmoplastic melanoma
Lin Hou1,2,3,4,#, Qi Liu1,#, Limei Shen1, Yun Liu1, Xueqiong Zhang1, Fengqian Chen5, Leaf Huang1, ![]()
1. Division of Pharmacoengineering and Molecular Pharmaceutics and Center for Nanotechnology in Drug Delivery, Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA.
2. School of Pharmaceutical Sciences, Zhengzhou University, 100 Kexue Avenue, Zhengzhou 450001, China.
3. Collaborative Innovation Center of New Drug Research and Safety Evaluation, Henan Province, China.
4. Key Laboratory of Targeting and Diagnosis for Critical Diseases, Henan Province, China.
5. Department of Environmental Toxicology, The Institute of Environmental and Human Health (TIEHH) and the Center for Biotechnology & Genomics, Texas Tech University, Lubbock, TX 79416, USA.
# These authors contributed equally
Received 2018-1-8; Accepted 2018-3-16; Published 2018-6-13
Citation:
Hou L, Liu Q, Shen L, Liu Y, Zhang X, Chen F, Huang L. Nano-delivery of fraxinellone remodels tumor microenvironment and facilitates therapeutic vaccination in desmoplastic melanoma. Theranostics 2018; 8(14):3781-3796. doi:10.7150/thno.24821. https://www.thno.org/v08p3781.htm
Other stylesAbstract

Rationale: Tumor-associated fibroblasts (TAFs) play a critical role in the suppressive immune tumor microenvironment (TME), compromising the efficacy of immunotherapy. To overcome this therapeutic hurdle, we developed a nanoemulsion (NE) formulation to deliver fraxinellone (Frax), an anti-fibrotic medicine, to TAFs, as an approach to reverse immunosuppressive TME of desmoplastic melanoma.
Methods: Frax NE was prepared by an ultrasonic emulsification method. The tumor inhibition effect was evaluated by immunofluorescence staining, masson trichrome staining and western blot analysis. Immune cell populations in tumor and LNs were detected by flow cytometry.
Results: This Frax NE, with a particle size of around 145 nm, can efficiently accumulate in the tumor site after systemic administration and was taken up by TAFs and tumor cells. A significant decrease in TAFs and stroma deposition was observed after intravenous administration of Frax NE, and Frax NE treatment also remolded the tumor immune microenvironment, as was reflected by an increase of natural-killer cells, cytotoxic T cells (CTLs) as well as a decrease of regulatory B cells, and myeloid-derived suppressor cells in the TME. In addition, after treatment by Frax NEs, T helper 1 (Th1) cytokines of interferon gamma (IFN-γ), which effectively elicit anti-tumor immunity, were enhanced. Transforming growth factor-β (TGF-β), chemokine (C-C motif) ligand 2 (CCL2) and interleukin 6 (IL6), which inhibit the development of anti-tumor immunity, were reduced. Although Frax NE demonstrated an inhibitory effect on tumor growth, this mono-therapy could only achieve partial antitumor efficacy, and the tumor growth effect was not maintained long-term after dosing stopped. Therefore, a tumor-specific peptide vaccine was combined with Frax NEs. The combination led to enhanced tumor-specific T-cell infiltration, activated death receptors on the tumor cell surface, and induced increased apoptotic tumor cell death.
Conclusion: Collectively, Frax NE combined with tumor-specific peptide vaccine might be an effective and safe strategy to remodel fibrotic TME, thereby enhancing immune response activation, resulting in a prolonged efficiency for advanced desmoplastic melanoma.
Keywords: fraxinellone, nanoemulsion, desmoplastic melanoma, tumor microenvironment, tumor-associated fibroblasts
Introduction
Melanoma, the most lethal skin cancer, has an incremental incidence, few durable therapies, and a low survival rate (< 10%) [1]. Its progression is often associated with mutations, and 50-60% of melanomas harbor activating BRAF mutations (over 90% are V600E) [2, 3], rendering this tumor much more desmoplastic and difficult to treat [4]. In the past few years, BRAF mutant inhibitors like vemurafenib and dabrafenib produced desirable clinical responses in the short term [5, 6], but tumors rapidly acquired resistance, which still remains a challenge to effective therapy [7]. Notably, besides cancer cells, the stroma of the desmoplastic melanoma includes tumor-associated fibroblasts (TAFs), T cells, B cells and immunosuppressive cells (Figure S1A). In particular, TAFs are one of the most prominent stromal cell types, which reach up to 17% of the total cell population (Figure S1A).
It has been reported that TAFs are the receivers as well as the inducers of tumorigenic activation signals. Emerging evidence suggests that TAFs can modulate the immunosuppression of tumor microenvironment (TME) through diverse mechanisms, thereby supporting tumor progression [8, 9]. For example, TAFs can suppress cytotoxic T lymphocyte (CTL)-driven antitumor immunity and mediate immune suppression by modulating myeloid cells, such as tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), and tolerogenic dendritic cells (DCs) [10]. TAFs can also mediate epithelial-mesenchymal transition (EMT) of carcinoma cells, thereby contributing to the progression of cancer. Additionally, some growth factors secreted by TAFs like transforming growth factor-β (TGF-β) play an important role in promoting the carcinogenic process [11]. As a result, a bi-directional activation between cancer cells and TAFs has been identified as the leading cause in the formation of the malignant phenotype of cancer [8]. Taken together, TAFs are the potential target for treatment of desmoplastic melanoma and targeting TAFs will render both malignant and stromal compartments more responsive to immunotherapies. Encouragingly, our previous investigation on modifying TAFs through delivery of apoptosis-reducing ligand has proved effective to treat desmoplastic cancers [12, 13].
In recent years, we have studied natural products that target TAFs, especially on the interaction loop between TAFs and cancer cells. The focus of this work is on TGF-β, one of the key mediators for fibroblast activation and tissue fibrosis [14]. Fraxinellone (Frax), a compound isolated from the root bark of Dictamnus dasycarpus, is reported to resolve liver fibrosis by reducing CUG-binding protein 1 (CUGBP1) expression and consequently regulating TGF-β and interferon gamma (IFN-γ) signaling [15]. Other studies have examined Frax for its actions such as anti-inflammatory, neuroprotective, antinociceptive, and vasorelaxation activities [16-18]. However, the effect of Frax on TAFs in TME has not been studied. Therefore, as a part of our research on TAFs modification, we investigated the anti-fibrotic properties of Frax in TME. In order to enhance the targeting ability, aminoethyl anisamide (AEAA), a targeting ligand of sigma receptor used in our laboratory, was added on the surface of the formulation [19].
As accumulating investigations have proved the importance of TME modulation in alleviating the offensive behavior of melanoma [20, 21], we hypothesize that targeted delivery of Frax to the tumor site will lead to deactivation of TAFs and reduce tumor load. Nevertheless, remodeling TME alone might affect tumor growth partially. To further improve anti-cancer activity, vaccination aiming at the tumor-specific antigen, BRAFV600E, which is specifically overexpressed in BRAF-mutant melanoma, is introduced herein [22]. In our previous studies, a mannose-modified lipid calcium phosphate (LCP) nanoparticles-based vaccine, including both tumor-specific antigen (modified BRAFV600E peptide (pSpSSFGLANEKSI)) and CpG oligodeoxynucleotides adjuvant, was observed to be potent in triggering an antigen-specific CTL response and remarkably suppressed initial tumor growth. However, we also found that the immunotherapeutic efficiency of the vaccine was compromised at the later stage of tumor growth due to increased immune suppression in the TME. Accordingly, in this study, a synergistic therapy combining Frax and tumor-specific peptide vaccine was hypothesized to regulate the TME and negate its suppressive surroundings, thus increasing the anti-tumor immune response, inhibiting tumor growth and prolonging the survival duration.
Methods
Materials
Fraxinellone was purchased from Shanghai Tauto Biotech Co. Ltd (Shanghai, China). Lecithin from soybean was purchased from Santa Cruz Biotechnology, Inc (Dallas, Texas). Dioleoylphosphatydic acid (DOPA), 1,2-Dioleoyl-3-trimethylammonium-propane chloride salt (DOTAP), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(polyethylene glycol)-2000 (DSPE-PEG-2000), and the 3-(N-succinimidyloxyglutaryl)aminopropyl, polyethyleneglycol-carbamyl distearoylphosphatidyl-etha-nolamine (DSPE-PEG-NHS) were purchased from Avanti Polar Lipids (Alabaster, AL). Pluronic F68 was provided by BASF (Florham Park, NJ). DSPE-PEG-aminoethyl anisamide (DSPE-PEG-AEAA) was synthesized based on the previously reported methods [23]. Briefly, 4-methoxybenzoyl chloride and 2-bromoethylamine hydrobromide were mixed at room temperature for 6 h. Then, DSPE-PEG-NH2 was added into the above solvent and stirred in an oil bath at 65-70 ℃ for 24 h. Finally, the reactant was washed and lyophilized for further use. PEG-DSPE-mannose was synthesized from DSPE-PEG-NHS and 4-amino phenyl-mannopyranoside. CpG ODN 1826 (5'-TCCATGACGTTCCTGACGTT-3') were obtained from Sigma-Aldrich (Darmstadt, Germany).
Cell lines and animals
Murine BRAF-mutant melanoma cell line BPD6 (BRAFV600EPTEN-/-, syngeneic with C57BL/6) was provided by Brent Hanks (Duke Cancer Institute, Durham, NC) and cultivated in DMEM Medium (Invitrogen, Carlsbad, CA) containing 1% Penicillin/Streptomycin (Invitrogen) and 10% bovine calf serum at 37 °C with 5% CO2. Female C57BL/6 mice (6-8 weeks old) were ordered from Charles River Laboratories (Wilmington, MA). All animal handling procedures were approved by the University of North Carolina at Chapel Hill's Institutional Animal Care and Use Committee.
Preparation and characterization of Frax-loaded AEAA-modified NE (Frax NE)
Frax NE was prepared by an ultrasonic emulsification method. In brief, Frax was firstly dissolved in dimethyl sulfoxide (DMSO) (no more than 1% in total formulation) and mixed with lecithin from soybean and sesame oil. Subsequently, Pluronic F68 solution (100 mg/mL) containing targeting ligand DSPE-PEG-AEAA was added into the drug mixture above drop by drop under stirring. After stirring for 5 min at room temperature, the resultant mixture was ultrasonicated on an ice bath for 5 min to produce NE. The particle size and morphology of Frax NE was determined by a Malvern ZetaSizer Nano series (Westborough, MA) and a JEOL 100CX II TEM (JEOL, Japan), respectively. The encapsulation efficiency of AEAA-modified NE was measured using HPLC (Shimadzu LC-20AT, Kyoto, Japan). In vitro stability was evaluated by determining the diameter size by DLS (Malvern, United Kingdom) at room temperature.
To investigate the targeting ability of this NE, DiI-labeled NE with or without AEAA were prepared by the same method as above without addition of Frax but with 0.5% DiI added. After intravenous injection of DiI-labeled NE for 24 h, mice were euthanized, and tumors as well as major organs (heart, liver, spleen, lung and kidney) were collected. The bio-distribution was visualized and quantitatively measured with IVIS® Kinetics Optical System (Perkin Elmer, CA). The excitation wavelength was set at 520 nm, while the emission wavelength was 570 nm.
Additionally, intra-tumoral cellular uptake by cells of interest (tumor cells and TAFs) was evaluated by flow cytometry. Briefly, tumor tissues were dissociated with 1 mg/mL collagenase (Invitrogen), and 200 μg/mL DNAase (Invitrogen) in DMED/2% FBS for 40 min to generate a single-cell suspension. Tumor cells were stained with PE-conjugated MART1 antibody (Melan-A antibody, sc-20032 PE, Santa Cruz Biotechnology), and TAFs were stained with FAP antibody (anti-Fibroblast activation protein antibody, abT28244, Abcam). The cells were then subjected to flow cytometric analysis, and the ratios of DiI-loaded NE distributed in different cell populations were calculated.
Furthermore, a LC/MS instrument (Shimadzu LCMS-2020, Kyoto, Japan) was also utilized to quantitatively analyze the accumulation of Frax NE in the tumor site at predetermined times (1, 3, 8, 12, 24 h) and study the pharmacokinetics profile. Separation of analytes was carried out on a Thermo Scientific C18 column (100 mm × 4.6 mm, 2.6 μm) (Thermo Fisher Scientific, Waltham, MA USA); the flow rate was set to 0.2 mL/min, and the column temperature was 35 ℃.
Tumor growth inhibition
The stroma-rich desmoplastic melanoma model was established as previously reported [22]. Mice were inoculated subcutaneously with 1×106 BPD6 cells on their lower flank. When the tumor volume reached about 200 mm3, mice were separated into the following groups (n = 6): Untreated group (PBS), Frax oral suspension group (Frax oral, 120 mg/kg), and Frax NE group (Frax NE, 30 mg/kg). As the control, Frax oral was prepared by suspending Frax directly in a 0.5% carboxymethylcellulose (CMC) solution with grinding. Frax was administrated p.o. or i.v. every other day 5 times, and the tumor volumes were monitored by caliper every 2 days and calculated as (a×b2)/2, where 'a' represents the larger diameter and 'b' represents the smaller one. At the endpoint of the tumor inhibition study, we sacrificed the mice, and tumors were harvested and weighed. The inhibition ratio (IR) was defined as IR (%) = ((Wc-Wt)/Wc) × 100, where Wc and Wt are the average tumor weights for the control group and each treatment group, respectively.
To evaluate the combination therapy with BRAF peptide vaccine, BPD6 tumor-bearing mice (tumor volume reached about 200 mm3) were randomly divided into four groups (n = 8-10): Untreated group (PBS), Frax NE group (Frax NE, 30 mg/kg), BRAF peptide vaccine group (Vaccine, (BRAF peptide + CpG) 100 μg/mice) and Frax NE combined with BRAF peptide vaccine group (Combo). BRAF peptide vaccine was prepared as described previously [22]. For the single vaccine and combo therapy groups, vaccination was administrated on day 9 and boosted on day 15 subcutaneously. Intravenous injections of Frax NE were also given every 2 days for a total of 5 doses. Tumor volume was measured as above, and mice were sacrificed before tumor volume reached 2000 mm3 under the animal safety protocol.
Long-term survival was also monitored on BPD6-bearing mice with different treatments (n = 8, in each treatment group). Kaplan-Meier curves and median survival were quantified and calculated using GraphPad.
Immunofluorescence staining and Masson trichrome staining
Tissue section staining was executed following the procedure of deparaffinization, antigen retrieval, permeabilization, and 1% bovine serum albumin (BSA) blocking. Primary antibodies with or without fluorophore conjugation were incubated at 4 ℃ overnight. The samples using non-conjugated primary antibodies were treated with secondary antibodies for 1 h at room temperature on the next day. Nuclei were counterstained with DAPI (Vector Laboratories Inc., Burlingame, CA). Images were acquired using fluorescence microscopy (Nikon, Tokyo, Japan) and five fields were selected at random for quantitative analysis by ImageJ software.
The Masson trichrome assay was performed to detect collagen among tumor tissue. Tumor slides were stained using a Masson Trichrome Kit by the UNC Tissue Procurement Core.
Flow cytometry analysis
Immune cell populations in tumor and LNs were detected by flow cytometry. In brief, tumors were treated with collagenase A and DNAase for 40 min at 37 ℃. Then, single cells in treated tumors and LNs were collected in FACs buffer. For intracellular staining, the cells were treated with penetration buffer (BD, Franklin Lakes, NJ) as the manufacturer's instruction suggested. Different kinds of immune lymphocytes were stained with different kinds of fluorescein-conjugated antibodies.
Western blot analysis
Western blot was performed on proteins extracted from the tumor tissues after treatment or cells in the lysis buffer. The extracted proteins were separated by 4-12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE electrophoresis) (Invitrogen) and transferred onto polyvinylidene difluoride (PVDF) membranes (Bio-Rad, Hercules, California). The membranes were incubated with the indicated antibodies at 4 ℃ overnight. Primary antibodies were directed against P-SMAD2 (Cell signaling, 3108S), α-smooth muscle actin (α-SMA) (Abcam, ab124964), CUGBP1 (Abcam, ab129115) and GAPDH (Santa Cruz Biotechnology, sc-25778). Membranes were then incubated with a horseradish peroxidase-coupled secondary antibody, and signals were observed using the Pierce ECL Western Blotting Substrate (Thermo, Rockford, IL). The relative expression level of protein was quantified with ImageJ software.
Quantitative real-time PCR assay
Whole RNA was obtained from tumor tissues using an RNeasy kit (Qiagen, Valencia, CA), and cDNA was reverse-transcribed using the SuperScript First-Strand Synthesis System for RT-PCR (Invitrogen, Grand Island, NY). 100 ng of cDNA was amplified with Taqman Universal Probes Supermix System (Bio-rad, Hercules, CA) and mouse-specific primers. Primers for mouse TGF-β, CUGBP1, chemokine (C-C motif) ligand 2 (CCL2), Interleukin 6 (IL6), C-X-C motif chemokine 13 (CXCL13), Insulin-like Growth Factors (IGF-1), Fibroblast Growth Factor (FGF-2) and C-X-C motif chemokine 12 (CXCL12) were purchased from ThermoFisher Scientific (Waltham, MA USA). The GAPDH RNA expression was used as internal control. Reactions were conducted using a 7500 Real-Time PCR System, and the data were analyzed with the 7500 Software.
In vivo CTL assay and ELISPOT assay
In vivo CTL and ELISPOT assay were conducted as in the previously published protocol [24]. For the CTL assay, splenocytes from healthy mice were collected and pulsed by BRAFV600E peptide (10 μM) or OVA peptide (10 μM). The BRAFV600E peptide-pulsed and OVA peptide-pulsed cells were then labeled with 4 μM carboxyfluorescein succinimidyl ester (CFSE) or 0.4 μM CFSE, respectively. The mice treated with different formulations, according to the methods in the tumor growth inhibition section, were injected with equal amounts of CFSEhigh (BRAFV600E-pulsed cells) and CFSElow (OVA-pulsed cells). 18 h later, splenocytes were collected and analyzed by flow cytometry. The number of CFSEhigh as well as CFSElow cells was calculated, and BRAFV600E-specific lysis was enumerated [25].
For ELISPOT assay, spleen and draining LNs of mice treated with different formulations were collected, and single cells from these tissues were seeded on the capture antibody-coated 96-well plate. The production of IFN-γ was detected by testing antibody addition followed by enzyme conjugate magnification, and measured with BDTM ELISPOT assay system (BD Pharmingen, San Diego, CA) as the manufacturer's instructions advised.
TUNEL assay
Slides were deparaffinized and stained using a TUNEL assay kit (Pierce, Madison, WI) according to the manufacturer's instruction. Cell nuclei that were stained with FITC (green) were defined as TUNEL-positive nuclei. The images were acquired by fluorescence microscopy (Nikon, Tokyo, Japan) and quantitatively analyzed using ImageJ.
Safety evaluation
Body weights of mice were measured every other day starting from the treatment. At the endpoint, the mice were sacrificed, blood was collected, and plasma was obtained by centrifugation at 4500 rcf for 5 min. Blood urea nitrogen (BUN), creatinine, aspartate aminotransferase (AST) and alanine aminotransferase (ALT) levels were detected as indicators of renal and hepatic function. Whole blood was also gathered for the measurement of myelosuppression by counting the Red blood cells (RBC), white blood cell (WBC), platelets (PLT), hemoglobin (HGB) and hematocrits (HCT). Major organs, such as heart, liver, spleen, lung and kidney were fixed and used for H&E staining by UNC histology facility to evaluate the organ-specific toxicity.
Statistical analysis
Results were expressed as mean ± SD, and statistically evaluated by Student's test or one-way analysis of variance (ANOVA). P values smaller than 0.05 were considered to be significant.
Results
Preparation and characterization of Frax NE
Frax is so hydrophobic that it is very difficult to be loaded into traditional drug delivery systems. It is well-known that nanoemulsion (NE) is a colloidal particulate system, which is manufactured to improve drug solubilization and enhance therapeutic efficacy. As a result, Frax was formulated in the NE [26, 27], the preparation process of which is shown in Figure S1B. In NEs, the combination of surfactants with oils offers a superior advantage over a co-solvent system or other nanocarriers in terms of drug-loading capacity for hydrophobic compounds [28]. To avoid the toxicity of traditional small molecular surfactants, we used the biocompatible lecithin from soybean as the emulsifier herein. To achieve tumor targeting ability, AEAA was used as the targeting group on the NE, as our previous studies have confirmed that AEAA is the sigma receptor ligand, which is overexpressed on cancer cells and TAFs [19, 29]. Moreover, the preparation procedure of Frax NE was much simpler than that of other nano-systems, and thus endows it with translational potential.
Frax NE was prepared by the method of ultrasonic emulsification, which is very efficient in constructing this formulation. The Frax NE concentrated solution appeared opalescent with a yellow color, and the average particle size was 148.1 ± 1.3 nm. The morphology of the nanoemulsion by TEM analysis is shown in Figure S2A, revealing a spherical shape and uniform droplet. The concentration of Frax in the NE was 2 mg/mL, the encapsulation efficiency was about 90% and Frax NE was found to be stable for about 20 days of storage at room temperature. There was no significant difference in diameter size, appearance and dilution ability, which indicated that Frax NE was chemically and physically stable.
To investigate the accumulation of NE in tumors, the biodistribution of DiI-loaded NE with or without AEAA modification was recorded using IVIS imaging (Figure S2B). 24 h post-injection, the higher fluorescence signal in tumors was observed even without targeting ligand. This enhanced distribution of DiI-labeled NE at the tumor site was attributed to the enhanced permeability and retention (EPR) effect. By contrast, AEAA-modified DiI-loaded NE demonstrated higher tumor targeting ability, and semi-quantitative biodistribution analysis in major organs was also performed. The ratio of fluorescence intensity to tissue weights of AEAA-modified NE was significantly increased at the tumor region and decreased at other organs as compared to that of non-targeted NE.
Furthermore, the plasma concentration-time and tissue distribution profiles of Frax were characterized after intravenous administration of Frax NE (30 mg/kg) and oral administration of Frax suspension, respectively. As shown in Figure S2C-D, data fitting results displayed that the pharmacokinetics behavior of Frax NE fitted a two-compartment model, the value of the total area-under-the-curve (AUC) was 139.88 ± 4.5 μg·h/mL, and t1/2 was 6.03 ± 0.67 h. This suggested that Frax NE can circulate for a longer time in the blood than Frax oral suspension. Notably, the Frax accumulated in the tumor was dramatically greater than that of the oral control. The MRT (0→∞) values of Frax for Frax NE was 2.9-fold compared with the control, which indicated that the injected NE were targeted to and stayed in the tumor tissue for an extended time.
After confirming the targeting ability of Frax NE, especially with the AEAA-modification, the DiI-loaded NE accumulation in various cell populations within the tumor was further performed by flow cytometry. Based on the results (Figure S2E), we found that approximately 22.3% of NE in the TME was taken up by the tumor cells (MART1 positive) and 20.4% was absorbed by TAFs (FAP positive) 24 h post-injection. In most cases, uptake of nanoparticles results from binding with the cell surface, and thus more AEAA-modified NE entered cells with overexpressed sigma receptor regardless of size compared to non-targeted NE, as expected.
Evaluation of therapeutic efficacy and changes in TME after Frax NE treatment
The antitumor efficacy of Frax was investigated after we confirmed the tumor-targeting ability of Frax NE. Therapy began when tumor sizes reached 200 mm3 in order to form the stromal-vessel structure. The tumor volume curve (Figure 1A) demonstrated that Frax treatments can significantly inhibit tumor growth compared with the PBS group. In addition, Frax NE exhibited higher antitumor effect, even though the dosage of Frax oral suspension was 4 times higher than that of Frax NE. Moreover, the inhibition ratios of these two Frax formulations were calculated based on the tumor weight at the endpoint (Figure 1B). IRs for Frax NE and Frax oral were 35.3±2.5% and 51.0±3.5%, respectively, which agreed with the results of tumor inhibition measurements.
Because Frax was reported to treat liver fibrosis [15], we initially examined the changes of TAFs as well as the CUGBP1 levels in tumor tissue samples. CUGBP1, standing for CUG-binding protein 1, was reported to be involved in posttranscriptional regulatory networks, TGF-β/IFN-γ balancing, fibrogenesis and tumorigenesis [30]. It is also the target of Frax. As can be seen in Figure 1C, α-SMA-positive TAFs were significantly reduced in the Frax NE group compared with the untreated PBS group, and the morphology of TAFs also changed from compact ribbons to small dots. Of note, the CUGBP1 expression was decreased with downregulating fibrosis.
We have shown that Frax NE could suppress tumor growth and formation of TAFs, but the reason for these effects needed to be further studied. Firstly, we confirmed that empty NE without Frax had no influence on tumor growth by using MTT and tumor volume observation (data not shown). Therefore, we looked for alterations of immune cell populations in the TME. In tumor-bearing hosts, the immune suppressive cells such as MDSC as well as regulatory B cells (Bregs), and the checkpoint of programmed death ligand-1 (PD-L1) play crucial roles in immune suppression, and countering their function is important for immunotherapeutic treatment [31]. As seen in Figure 1D, the percentages of MDSC (CD11b+Gr1+), Bregs (CD1d+CD19+) and PD-L1 in leukocytes in the Frax NE group were much lower than those in the PBS group, measured by flow cytometry of whole tumor tissue. In contrast, CTLs and natural-killer (NK) cells increased significantly, which suggested that the change of the TME morphology might facilitate T cell infiltration and innate immune response. There was no significant difference for memory T cells between the PBS group and the Frax NE group. Although we have found Frax NE could inhibit tumor growth, possibly due to the remodeling of TAFs and TME as aforementioned, the survival duration was not prolonged, and tumors grew back after drug withdrawal (Figure 1E).
Frax NE improves the antitumor effect and reprograms TAFs when combined with BRAF peptide vaccine in stroma-rich melanoma
To improve the antitumor efficacy of Frax NE and increase the survival rate, combination therapy was taken into consideration. As Frax NE could remodel TAFs and reduce intra-tumor suppressive cells in the TME, we hypothesized that combination therapy with a vaccine that induces antigen-specific CTL response would be successful, especially in advanced BRAF-mutant melanoma. Our group developed a BRAF peptide vaccine previously [22], and it was introduced into the combo group herein.
Compared with the PBS group, all treatment groups showed reduced tumor growth rates (Figure 2A). As expected, the combo group exhibited the best anti-cancer effects, suggesting the advantages of combination therapy. Meanwhile, in an overall survival analysis after the final day of treatment, median survival was also elevated in the combo group (Figure 2B), conveying not only a potent therapeutic effect but also a long-lasting overall response [32].
To investigate the mechanism of anti-tumor effects, we firstly used Masson's trichrome staining to study the morphology and collagen content of tumors after treatment. In Figure 2C, collagen deposition and fibrosis were observed abundantly in tumor sections of the untreated group. By contrast, Frax NE and Combo treatment significantly ameliorated the pathological changes. Simultaneously, the percentages of α-SMA and CUGBP1 were quantitatively analyzed by Image J under confocal imaging (Figure 2D), which displayed similar trends as aforementioned. However, α-SMA percentages in the vaccine only and PBS groups were alike, while CUGBP1 in the whole tumor increased partially. Moreover, the relative mRNA expression of CUGBP1 was a further evidence of our staining analyses (Figure 2E, left panel).
Figure 1
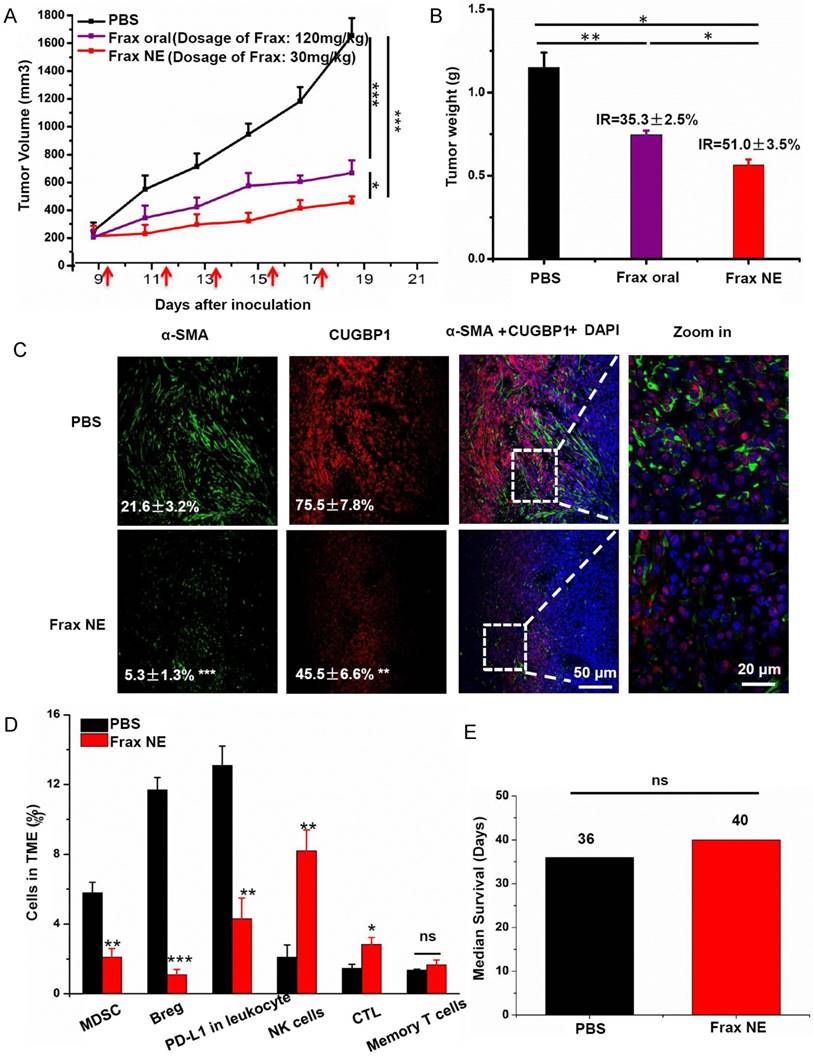
Tumor inhibition effects and TME changes in vivo after treatment with Frax. (A) Tumor volume change as a function of time. The dosage of Frax by oral administration is 4 times higher than that of Frax NE by intravenous injection. Frax was administrated p.o. or i.v. every other day 5 times (small arrows under the axis represent the day of dosing). (B) Tumor weight at the end of the experiment (day 23). Inhibition ratio (IR) was calculated. (C) Confocal analysis for α-SMA and CUGBP1 from tumor tissue sections. (D) Comparison of different immune cells in the TME between BPD6 tumor-bearing mice with and without treatment using flow cytometric analysis. (E) The survival data from treatment and no-treatment groups. Numbers shown in white indicate the average % of each cell type in the tumor. The statistical analyses were calculated by comparison with the control group if not specifically mentioned. Data are shown as mean ± SD. (n = 5-8, * P < 0.05, ** P < 0.01, *** P < 0.001)
Figure 2
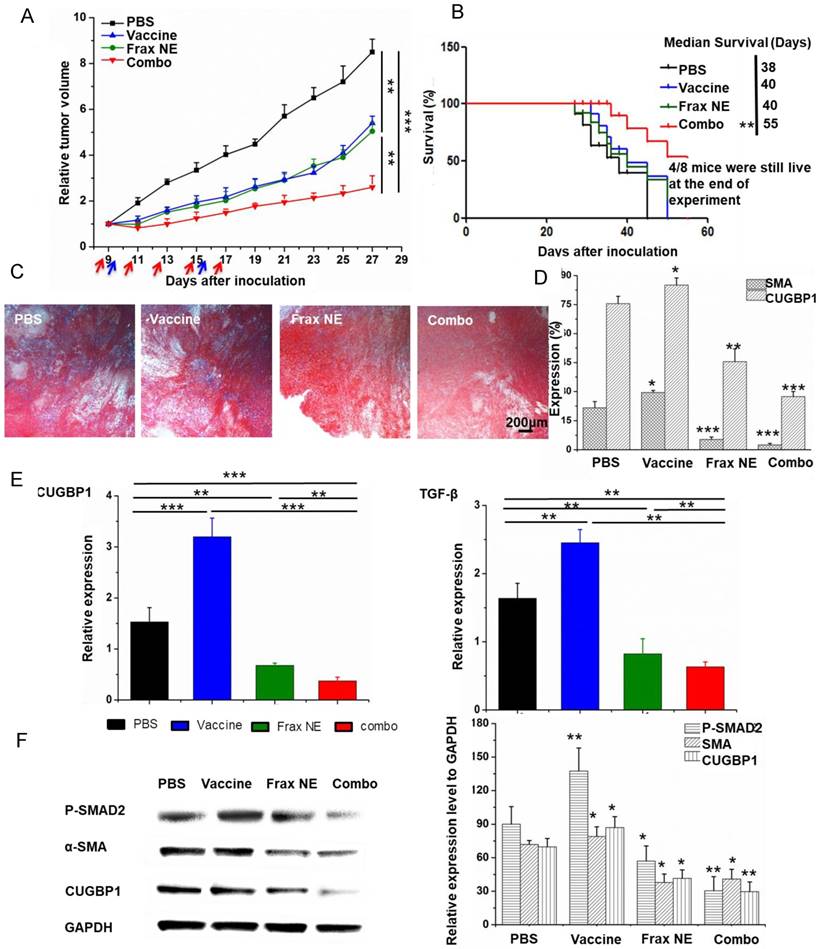
Tumor inhibition effects and TME changes in vivo after treatment with Frax NE combined with vaccine. (A) Tumor inhibition curve of BPD6 tumor-bearing mice using different formulation treatments (PBS, Vaccine, Frax NE and Combo). Frax (red arrow) was administrated i.v. every other day 5 times at a dose of 30 mg/kg. For the vaccine alone and combo therapy groups, vaccination (blue arrow) was administrated on day 9 and boosted on day 15 subcutaneously. (B) The survival proportions of the treated groups. (C) Masson's trichrome stain for collagen. (D) Quantitative analysis of α-SMA and CUGBP1 to evaluate the effects of different treatments on the inhibition of fibroblasts by confocal microscopy. (E) Changes of cytokines in the TME evaluated using quantitative RT-PCR. (F) Western blot analysis of BPD6 tumor protein levels after different treatments. The statistical analyses were calculated by comparison with the control group if not specifically mentioned. All data are shown as mean ± SD. (n = 8-10, * P < 0.05, ** P < 0.01, *** P < 0.001)
We wondered about the underlying relationship between treatment and TAFs after confirming that Frax might have an influence on changes of TAFs. As it is commonly accepted that the majority of TAFs are transdifferentiated from resident fibroblasts in response to TGF-β [33, 34], the TGF-β expression and downstream portions of TGF-β signaling pathway, involving P-SMAD2 and α-SMA, were examined. Data demonstrated that treatment with Frax alone or combined with vaccine resulted in reduced TGF-β expression, but it was also noteworthy that vaccination significantly increased the expression of TGF-β in the TME on a RNA level (Figure 2E, right panel), which would inhibit the development of anti-tumor immunity [35].
Western blotting (Figure 2F) revealed that P-SMAD2 level dramatically reduced in tumors in the Frax NE and Combo groups, but vaccine monotherapy increased this protein partially compared to the PBS-treated group. Decreased α-SMA and CUGBP1 expression after treatment agreed with the previous results.
Apoptosis of neighboring tumor cells caused by combination therapy of Frax NE and BRAF peptide vaccine induces an antigen-specific immune response
In order to study the potential effects from the histological cross-sections, tumor cell apoptosis was quantified via a TUNEL assay (Figure 3A). All three treatment groups displayed a greater number of apoptotic cells than the PBS group, and the combo group exhibited the highest level of cell apoptosis (46.6±2.7%). These findings correlated with the tumor inhibition data above, which was possibly due to the immune cells killing inducing potent cell death within the TME.
To further determine whether the antitumor potency was caused by a robust immune response, antigen-specific CTL response and IFN-γ production ELISPOT assay were performed. ELISPOT assay results in Figure 3B confirmed the eliciting IFN-γ release capacity of the vaccine, as in our published paper [22]. Moreover, Frax NE also moderately boosted efficacy, and the combo group exhibited the most sufficient stimulation to secrete IFN-γ. For the CTL assay (Figure 3C), mice immunized with BRAF peptide showed partial (~43.6%) efficacy, whereas mice receiving combination therapy proved the most effective (~57.2%), indicating that combination therapy can induce a potent in vivo CTL response compared to monotherapy.
It is well known that DCs, as an essential component of vaccination, are required to home to secondary lymphoid organs to prime T cell responses [36-38]. We detected the DCs and T cell population by flow cytometric analysis (Figure 3D), and the data revealed that the three treatment groups all promoted DC activation, with an increase of 0.5-1-fold compared with the PBS group. Among these groups, the combo group possessed the greatest capacity to facilitate DC activation and induced the highest level of CD8+ T cells within LNs. Memory T cells and activated NKs, which might play a critical role in the initiation of T-cell responses by contributing to DC maturation [39], demonstrated the similar trend as above within LNs (Figure S3B). These results confirmed that manipulating the DCs could induce T-cell activation and proliferation.
Along with the increase of active DCs and T cells in LNs, immune boosting cells such as CD8+ T cells, memory T cells and NK cells were found to be increased in tumors, which was determined by both immunofluorescence staining and flow cytometry (Figure 3E). As can be seen from the tumor slices, small numbers of T cells were observed in the tumor region, but they were located in the border of tumor tissue. In comparison, both the vaccine and Frax NE groups showed boosted T cells penetration in the TME, but the most extensive T-cell infiltration was found in the combo group. Interestingly, memory T cells in the tumor region were not altered much in the Frax NE group, while marked enhancement was found in the other two groups, especially in the combo group. Significantly, NKs that participated in the early immune response against the tumor and contributed to the adaptive immune response were elevated 3-8-fold after treatment, and the combo group had nearly 14% of NKs within the TME. In agreement with the staining results, flow cytometry analysis confirmed our observation. It was found that IFN-γ within the whole tumor also increased at the mRNA level (Figure S3A) and the leukocytes cell level (Figure S3C, upper left panel).
Remodeling of the TME and enhanced immune cell infiltration resulted in the superior antitumor effect of the combination therapy
Collectively, we observed that Frax NE combined with BRAF peptide vaccine triggered the best immunotherapeutic efficacy, including improved tumor inhibition, T-cell penetration, NKs activation and IFN-γ secretion. Together with results that collagen deposition and TAFs decreased remarkably in the TME, these antitumor effects were probably due to remodeling of the immunosuppressive TME. Therefore, immunosuppressive cells within the TME such as MDSCs, Bregs and TAMs, which were the dominating myeloid infiltrates, were examined by immunostaining of tumor sections and flow cytometry.
As shown in Figure 4A, the percentages of MDSCs in the Frax NE and combo groups were much lower than in the control group, whereas more MDSCs were found in the vaccine-only group (measured by immunostaining and flow cytometry). Meanwhile, the ratio of TAMs exhibiting M1 signatures (tumor-suppressing) to M2 signatures (tumor-promoting) significantly increased, which was modulated by TAFs in the TME (Figure S3C, lower left panel, measured by flow cytometry). In addition, PD-L1 immune checkpoint on leukocytes accordingly decreased (Figure S2).
Figure 3
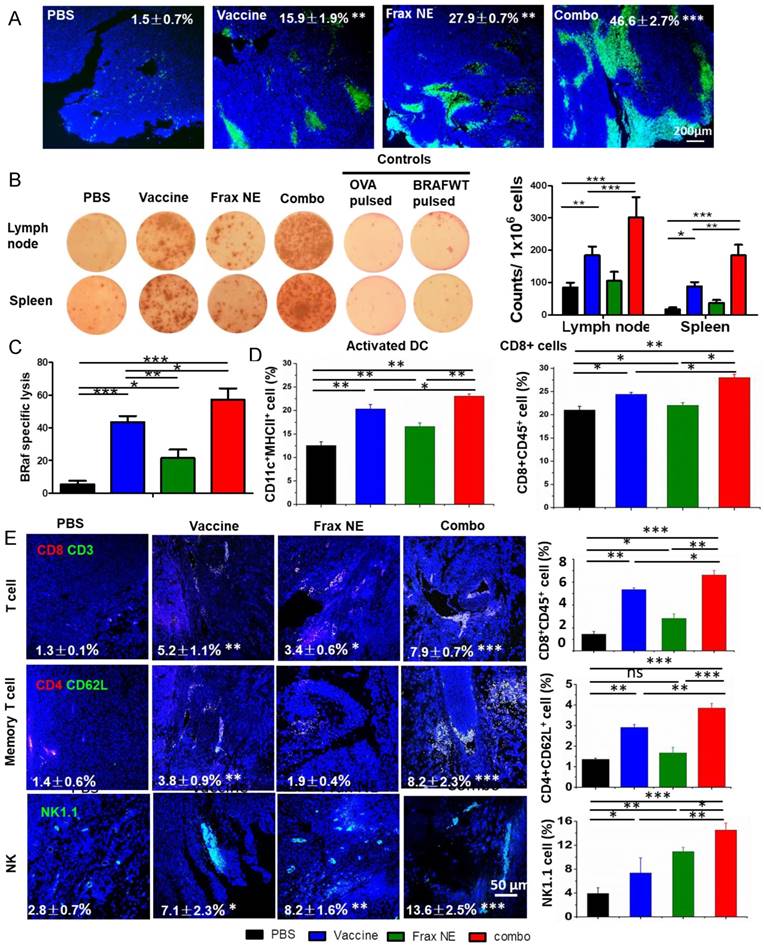
Enhanced T-cell infiltration into the TME induced potent CTL killing. (A) TUNEL staining of tumor sections after different treatments. (B) IFN-γ production after treatment was measured with ELISPOT assay. (C) In vivo CTL response after treatment with either Frax NE, vaccine or combo was measured through CFSE high/low staining of splenocytes collected from naïve mice, which were pulsed with BRAFV600E peptide (CFSEhigh cells) or with OVA control peptide (CFSElow cells), respectively. An equal mixture of both pulsed cells was injected into the vaccinated mice via tail vein. 18 h after injection, mice were euthanized and splenocytes were collected, washed and analyzed via flow cytometry. (D) Changes of immune cells quantified by flow cytometric analysis of lymph nodes. (E) Confocal and flow cytometric analysis of immune cells infiltration in the TME. Numbers shown in white indicate the average % of each cell type in the tumor. The statistical analyses were calculated by comparison with the control group if not specifically mentioned. All data are shown as mean ± SD. (n = 6, * P < 0.05, ** P < 0.01, *** P < 0.001)
Figure 4
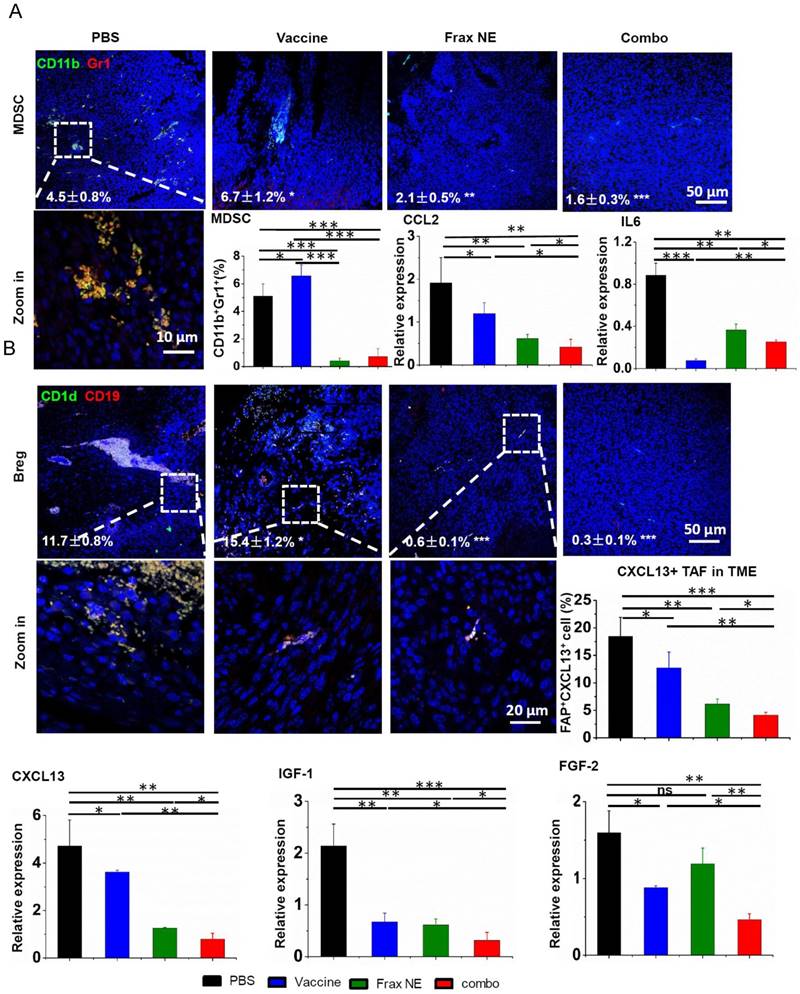
Changes of tumor-infiltrating immune cells and cytokines in the TME. C57BL/6 mice were inoculated with 1×106 BPD6 cells on day 0. Vaccine was injected on day 9 and 15; Frax NE were i.v. administered on days 9, 11, 13, 15 and 17 at a dose of 30 mg/kg alone or combined with vaccine, respectively. Mice were sacrificed on day 23 and tumors were harvested for immunostaining evaluation, flow cytometry and quantitative RT-PCR assay to detect MDSC (A) and Bregs (B). CCL2 and IL6 mediate MDSC recruitment. CXCL13, IGF-1 and FGF-2 facilitate B cells within the TME to differentiate into Bregs. Numbers shown in white indicate the average % of each cell type in the tumor. The statistical analyses were calculated by comparison with the control group if not specifically mentioned. All data are shown as mean ± SD. (n = 6, * P < 0.05, ** P < 0.01, *** P < 0.001).
According to reports that IL-6 and CCL2 produced by TAFs mediate MDSC recruitment and differentiation of macrophages into pro-tumor M2 phenotype [10, 40-42], the mRNA expressions of IL6 and CCL2 were checked by quantitative RT-PCR (Figure 4A). Treatment resulted in reduction of these two T helper 2 (Th2) cytokines, which are critical for immunosuppression, thus inhibiting tumor progression. Although IL6 in the Frax NE and combo groups exhibited a little higher expression than in the vaccine group, it still did not change the overall tendency compared to the PBS group. It is worth mentioning that IFN-γ, the T helper 1 (Th1) cytokine that was more effective in eliciting anti-tumor immunity, was also dramatically elevated at the mRNA level, especially in the combo group (Figure S3A).
Figure 5
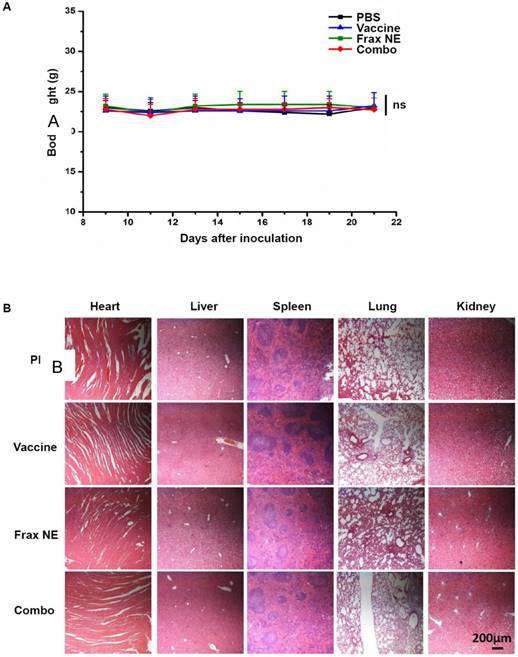
Safety evaluation of vaccine, Frax NE and combo treatment. (A) Body weight change. (B) H&E morphology evaluation of major organs after treatment. ns: P > 0.05.
Interestingly, we noticed that a large number of Bregs appeared in the TME. Bregs originated from normal B cells, which were attracted by tumor cells and converted into Bregs by high expression of TGF-β within the TME. Bregs can induce the generation of MDSCs and promote tumor cells to form a suppressive milieu [43]. However, Frax NE and combo treatment significantly downregulated the Bregs, indicating that Frax NE could remodel the immunosuppressive TME in favor of therapy (Figure 4B). As CXCL13, which is predominantly secreted by TAFs and cancer cells, played a vital role in attracting B cells into the TME [44], we detected this chemokine through flow cytometry and RT-PCR analysis. As expected, CXCL13 levels within TME were much lower in the Frax NE and combo groups, compared with the PBS group, and after vaccine treatment it was also partially reduced. Furthermore, IGF-1 (produced by tumor stroma-derived cells) and FGF-2 (produced by tumor cells) expressions, two important growth factors involved in tumor-associated B cells crosstalk with tumor cells [11], were both suppressed compared with that of the PBS group. As reported, FGF-2 plays a key part in converting normal B cells to tumor-associated B cells and could induce B cells to generate inflammatory factors and cytokines, most notably IGF-1. On the other hand, IGF-1 can form heterogeneous tumor subpopulations possessing cancer stem cell-like properties [4]. Therefore, Frax might have an important part in disturbing this interaction.
Meanwhile, CXCL12, known as stromal-derived factor 1 (SDF-1), is a key chemokine inhibiting T-cell infiltration [45]. Inhibiting of the CXCL12/CXCR4 axis has become a promising TME modulation strategy that improves checkpoint inhibitor efficacy [32]. We found that CXCL12 was significantly decreased at the mRNA transcriptional level (Figure S3C, lower right panel), thus further facilitating effective immune killing of cancer cells.
Safety evaluation of the different treatments
Safety evaluation is an important aspect in the development of immunotherapies. The body weights of mice treated with the above regimens did not reduce throughout the tumor inhibition experiment (Figure 5A). Administration of all formulations showed no significant changes in ALT, AST, creatinine, or BUN levels, suggesting that there was no severe damage to renal and hepatic functions. Further analysis of blood cell levels demonstrated no signs of change compared with control healthy mice (Table 1-2). Moreover, the H&E staining results also indicated no morphological differences in major organs after treatment (Figure 5B).
Table 1
Whole cell counts of tumor-bearing mice.
| Sample# | WBC (103/μL) | HCT (%) | RBC (106/μL) | HGB (g/dL) | PLT (105/μL) |
|---|---|---|---|---|---|
| Healthy | 2.8±0.1 | 46.8±3.1 | 10.1±1.0 | 15.7±1.3 | 11±0.7 |
| PBS | 5.5±0.7 | 35.0±10.3 | 8.0±2.7 | 10.5±2.9 | 14.2±3.7 |
| Vaccine | 4.8±0.6 | 47.4±1.8 | 10.1±0.1 | 15.4±0.7 | 14.4±5.1 |
| Frax NE | 5.5±1.1 | 46.7±2.7 | 10.0±0.3 | 14.7±0.9 | 13.5±4.7 |
| Combo | 4.65±0.2 | 36.0±8.1 | 8.0±1.6 | 12±2.1 | 13.5±1.5 |
| Normal range | 2.6-10.1 | 32.8-48 | 6.5-10.1 | 10.1-16.1 | 7.8-15.4 |
Table 2
Serum biochemical values of tumor-bearing mice.
| Sample# | BUN (mg/dL) | Creatinine (mg/dL) | AST (U/L) | ALT (U/L) |
|---|---|---|---|---|
| Healthy | 12.0±2.5 | 0.26±0.0 | 220.1±30.2 | 102.3±10.6 |
| PBS | 21.0±1.7 | 0.30±0.1 | 200.7±42.0 | 18.3±7.5 |
| Vaccine | 15±2.1 | 0.34±0.1 | 193.3±32.2 | 124±12 |
| Frax NE | 17.0±2.0 | 0.33±0.1 | 169.5±24.9 | 52.7±2.1 |
| Combo | 9±1.4 | 0.4±0.1 | 186.7±36.2 | 116.6±21.1 |
| Normal range | 8-33 | 0.2-0.9 | 54-298 | 17-132 |
Discussion
TAFs are believed to be essential for synthesis and deposition of the extracellular matrix (ECM) by producing various collagens as well as fibronectin [46], and can act like a mutagen that increases the tumorigenic ability of cancer cells. In addition, TAFs are a rich origin of different secreted factors such as cytokines, chemokines (e.g., IL6, CXCL12, CXCL13), and growth factors including TGF-β, FGF as well as VEGF [47], which mediate communication between cancer cells and TAFs. Nowadays, it has been generally accepted that TGF-β can transform normal fibroblasts into TAFs and regulate pivotal biological functions in cancers [48-50], rendering TGF-β more attractive in the field of cancer immunotherapy. Taken together, a natural product Frax, which was recently reported to treat liver fibrosis by inhibiting TGF-β signaling and triggering IFN-γ signaling [15], was considered in our investigation to remodel TME by targeting TAFs.
Notably, Frax NE herein indeed demonstrated antitumor efficacy in the desmoplastic BRAFV600E mutant melanoma model, which is extremely formidable to cure. Unfortunately, there are only a few reports about the pharmacology of Frax, most of which are focused on its anti-bacterial, anti-inflammatory and neuroprotective properties [16, 18, 51]. We noticed that, as a component of Dictamnus dasycarpus root bark, the anticancer activity was mentioned nebulously in some introductions of projects, books [52] or Chinese patents. But, the molecular mechanism of Frax in cancer has not been characterized. As aforementioned, Wu et al. reported that Frax could reduce the mRNA and protein expression of α-SMA by inhibiting CUGBP1, which balancing the TGF-β/IFN-γ signaling pathways, for the therapy of liver fibrosis [15]. Accumulating evidences have indicated that TGF-β/SMAD signaling is the most crucial pathway in the pathogenesis of fibrosis [53, 54]. Moreover, several studies showed that paracrine secretion of TGF-β can activate stromal fibroblast and produce immune-suppressive effects to modulate the TME for the benefit of melanoma growth [55]. Therefore, we questioned whether Frax inhibited tumor growth by such mechanism. We found that protein expressions of α-SMA and CUGBP1 in the NIH-3T3 cell line (which was activated with 10 ng/mL TGF-β-mimicking TAFs in vitro [56]) were both downregulated by Frax NE in a dose-dependent manner. But Frax NE only had a slight influence on BPD6 tumor cells even with a high dosage, which indicated that our formulation primarily focused on TAFs, not tumor cells (data not shown). Furthermore, Frax NE also reduced mRNA expression of TGF-β and phosphorylation of its downstream protein SMAD2 in BPD6 tumor-bearing mice after treatment, accompanied by decreased protein expression of α-SMA and CUGBP1. This was accordant with the process of the well-established TGF-β/SMADs signaling pathway. Briefly, TGF-β1 binds with its receptor II (TβRII) and activates the TGF-β receptor type II-kinase, resulting in phosphorylation of SMAD2 and SMAD3, which then associate with SMAD4 to form a heteromeric complex to regulate transcription of their target gene [57], which is related to fibrosis. These findings suggested that Frax NE might play a crucial role in inhibiting TGF-β signaling so as to suppress TAFs in the TME, possibly with regard to downregulation of CUGBP1. Recently, some studies revealed that CUGBP1 is overexpressed in cancer tissue and, accompanied by its binding target transcripts, functions to control cellular growth as well as homeostasis [58, 59]. So, we hypothesize that disruptions by Frax in this network might also interfere with the development of tumors, and the in-depth mechanism is being explored in our ongoing research.
Meanwhile, our results displayed that Frax NE triggered IFN-γ production and downregulated IL6 as well as TNF-α expression. IFN-γ, an immunomodulatory cytokine secreted by immune cells such as CD4+ Th1 cells, CD8+ T cells and NKs, can act on TAFs and change their promoting effects on tumor growth [60] by inhibiting activation and proliferation of fibroblasts [61]. In contrast, IL6 and TNF-α are pro-inflammatory cytokines that can induce generation of free radicals and damage DNA, potentially leading to tumor initiation and enhancement of tumor invasive properties [62]. Changes in these cytokines also elucidate the antitumor efficacy of Frax NE. It is worth mentioning that Kim et al. and Wu et al. both confirmed the anti-inflammatory effect (inhibition of the release of IL6 and TNF-α) of Frax by its association with the NF-κB signaling pathway, which regulates the transcription of most inflammatory factors [16, 18].
Many treatments for cancer, especially aiming at TAFs, continue to evolve, including TGF-β inhibitors (antisense oligonucleotides, monoclonal antibodies and small molecules) [63, 64] and IFN-γ. Anti-TGF-β therapy aims to treat not only tumor cells but also the TME, thus generating systemic effects on tumorigenesis. However, long-time use of these medications can cause severe side effects (vascular problems and multi-organ inflammatory disease) and its clinical failure is mainly due to its poor pharmacokinetics and low specificity. We found that Frax NE improved its pharmacokinetics profile and did not produce any adverse reactions at the tested dosage levels in mice, though administered for a long time. Therefore, our fibroblast-targeting Frax NE could be able to interrupt the interaction between TAFs and the tumor, thus resulting in modulation of the TME, and, if combined with BRAF peptide vaccine, suppression of tumor growth and prolongation of host survival.
Conclusion
In summary, we successfully developed Frax NE, a TAFs-targeted formulation of anti-fibrosis TCM, which could interfere with the crosstalk between TAFs and tumor cells, thus changing cytokine profiles as well as stromal structures, and dramatically decreasing MDSCs and Bregs in the TME, thereby remodeling the immunosuppressive microenvironment. After combination with BRAF peptide vaccine, enhanced antitumor efficacy was achieved through abrogating tumor-associated immune suppression and promoting infiltration of immune cells such as CTLs, NK cells and memory T cells. Hence, our investigations provided an immunotherapeutic strategy for the treatment of advanced BRAF-mutant melanoma.
Abbreviations
AEAA: aminoethyl anisamide; ALT: alanine aminotransferase; AST: aspartate aminotransferase; BUN: blood urea nitrogen; CCL2: chemokine (C-C motif) ligand 2; CMC: carboxymethylcellulose; CTL: cytotoxic T lymphocyte; CUGBP1: CUG-binding protein 1; CXCL12: C-X-C motif chemokine 12; CXCL13: C-X-C motif chemokine 13; DCs: tolerogenic dendritic cells; DMSO: dimethyl sulfoxide; DOPA: Dioleoylphosphatydic acid; DOTAP: 1,2-Dioleoyl-3-trimethylammonium-propane chloride salt; EMT: epithelial-mesenchymal transition; FGF-2: fibroblast growth factor; Frax: fraxinellone; HCT: hematocrits; HGB: hemoglobin; TGF-β: transforming growth factor-β; IFN-γ: interferon gamma; IGF-1: insulin-like growth factors; IL6: interleukin 6; LCP: lipid calcium phosphate; MDSCs: myeloid-derived suppressor cells; PLT: platelets; RBC: red blood cells; α-SMA: α-smooth muscle actin; TAF: tumor associated fibroblasts; TAMs: tumor associated macrophages; TME: tumor microenvironment; WBC: white blood cell.
Acknowledgements
This work was supported by NIH grant CA198999. Lin Hou was supported by China Scholarship Council. We thank Dr. K.H. Lee for the use of LC/MS instrument. We thank Dr. Rihe Liu, Dr. Yuhua Wang and Ryan Kramer for editing the manuscript.
Supplementary Material
Supplementary figures and tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Whipple CA, Brinckerhoff CE. BRAF(V600E) melanoma cells secrete factors that activate stromal fibroblasts and enhance tumourigenicity. Br J Cancer. 2014;111:1625-33
2. Guadarrama-Orozco JA, Ortega-Gomez A, Ruiz-Garcia EB, Astudillo-de la Vega H, Meneses-Garcia A, Lopez-Camarillo C. Braf V600E mutation in melanoma: translational current scenario. Clin Transl Oncol. 2016;18:863-71
3. Liu Q, Das M, Liu Y, Huang L. Targeted drug delivery to melanoma. Adv Drug Deliv Rev, in press. DOI: 10.1016/j.addr.2017.09.016
4. Liu Q, Zhu H, Tiruthani K, Shen L, Chen F, Gao K. et al. Nanoparticle-Mediated Trapping of Wnt Family Member 5A in Tumor Microenvironments Enhances Immunotherapy for B-Raf Proto-Oncogene Mutant Melanoma. ACS Nano. 2018;12:1250-261
5. Guerriero L, Palmieri G, De Marco M, Cossu A, Remondelli P, Capunzo M. et al. The anti-apoptotic BAG3 protein is involved in BRAF inhibitor resistance in melanoma cells. Oncotarget. 2017;8:80393-404
6. Ascierto PA, Kirkwood JM, Grob JJ, Simeone E, Grimaldi AM, Maio M. et al. The role of BRAF V600 mutation in melanoma. J Transl Med. 2012;10:85
7. Hartsough E, Shao Y, Aplin AE. Resistance to RAF inhibitors revisited. J Invest Dermatol. 2014;134:319-25
8. Tao LL, Huang GC, Song HZ, Chen YT, Chen LB. Cancer associated fibroblasts: An essential role in the tumor microenvironment. Oncol Lett. 2017;14:2611-20
9. Cohen N, Shani O, Raz Y, Sharon Y, Hoffman D, Abramovitz L. et al. Fibroblasts drive an immunosuppressive and growth-promoting microenvironment in breast cancer via secretion of Chitinase 3-like 1. Oncogene. 2017;36:4457-68
10. Jiang H, Hegde S, DeNardo DG. Tumor-associated fibrosis as a regulator of tumor immunity and response to immunotherapy. Cancer Immunol Immunother. 2017;66:1037-48
11. Somasundaram R, Zhang G, Fukunaga-Kalabis M, Perego M, Krepler C, Xu XW. et al. Tumor-associated B-cells induce tumor heterogeneity and therapy resistance. Nat Commun. 2017;8:607
12. Miao L, Liu Q, Lin CM, Luo C, Wang YH, Liu LN. et al. Targeting Tumor-Associated Fibroblasts for Therapeutic Delivery in Desmoplastic Tumors. Cancer Res. 2017;77:719-31
13. Miao L, Guo S, Lin CM, Liu Q, Huang L. Nanoformulations for combination or cascade anticancer therapy. Adv Drug Deliv Rev. 2017;115:1-2
14. Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6:392
15. Wu X, Wu X, Ma Y, Shao F, Tan Y, Tan T. et al. CUG-binding protein 1 regulates HSC activation and liver fibrogenesis. Nat Commun. 2016;7:13498
16. Kim JH, Park YM, Shin JS, Park SJ, Choi JH, Jung HJ. et al. Fraxinellone inhibits lipopolysaccharide-induced inducible nitric oxide synthase and cyclooxygenase-2 expression by negatively regulating nuclear factor-kappa B in RAW 264.7 macrophages cells. Biol Pharm Bull. 2009;32:1062-8
17. Sun Y, Qin Y, Gong FY, Wu XF, Hua ZC, Chen T. et al. Selective triggering of apoptosis of concanavalin A-activated T cells by fraxinellone for the treatment of T-cell-dependent hepatitis in mice. Biochem Pharmacol. 2009;77:1717-24
18. Wu XF, Ouyang ZJ, Feng LL, Chen G, Guo WJ, Shen Y. et al. Suppression of NF-kappaB signaling and NLRP3 inflammasome activation in macrophages is responsible for the amelioration of experimental murine colitis by the natural compound fraxinellone. Toxicol Appl Pharmacol. 2014;281:146-56
19. Goodwin TJ, Huang L. On the article "Findings questioning the involvement of Sigma-1 receptor in the uptake of anisamide-decorated particles" [J. Control. Release 224 (2016) 229-238]: Letter to the Editor 1 (September 14, 2016). J Control Release. 2016;243:382-5
20. Giavina-Bianchi MH, Giavina-Bianchi PF, Neto CF. Melanoma: tumor microenvironment and new treatments. An Bras Dermatol. 2017;92:156-66
21. Somasundaram R, Herlyn M, Wagner SN. The role of tumor microenvironment in melanoma therapy resistance. Melanoma Manag. 2016;3:23-32
22. Liu Q, Zhu H, Liu Y, Musetti S, Huang L. BRAF peptide vaccine facilitates therapy of murine BRAF-mutant melanoma. Cancer Immunol Immunother. 2017;67:299-310
23. Banerjee R, Tyagi P, Li S, Huang L. Anisamide-targeted stealth liposomes: a potent carrier for targeting doxorubicin to human prostate cancer cells. Int J Cancer. 2004;112:693-700
24. Xu Z, Ramishetti S, Tseng YC, Guo S, Wang Y, Huang L. Multifunctional nanoparticles co-delivering Trp2 peptide and CpG adjuvant induce potent cytotoxic T-lymphocyte response against melanoma and its lung metastasis. J Control Release. 2013;172:259-65
25. Liu L, Wang Y, Miao L, Liu Q, Musetti S, Li J. et al. Combination Immunotherapy of MUC1 mRNA Nano-vaccine and CTLA-4 Blockade Effectively Inhibits Growth of Triple Negative Breast Cancer. Mol Ther. 2018;26:45-55
26. Gupta A, Eral HB, Hatton TA, Doyle PS. Nanoemulsions: formation, properties and applications. Soft Matter. 2016;12:2826-41
27. Jaiswal M, Dudhe R, Sharma PK. Nanoemulsion: an advanced mode of drug delivery system. 3 Biotech. 2015:5 123-7
28. Wang Y, Wu KC, Zhao BX, Zhao X, Wang X, Chen S. et al. A novel paclitaxel microemulsion containing a reduced amount of Cremophor EL: pharmacokinetics, biodistribution, and in vivo antitumor efficacy and safety. J Biomed Biotechnol. 2011;2011:854872
29. Hu K, Miao L, Goodwin TJ, Li J, Liu Q, Huang L. Quercetin Remodels the Tumor Microenvironment To Improve the Permeation, Retention, and Antitumor Effects of Nanoparticles. ACS Nano. 2017;11:4916-25
30. Lu H, Yu Z, Liu S, Cui L, Chen X, Yao R. CUGBP1 promotes cell proliferation and suppresses apoptosis via down-regulating C/EBPalpha in human non-small cell lung cancers. Med Oncol. 2015;32:82
31. Lindau D, Gielen P, Kroesen M, Wesseling P, Adema GJ. The immunosuppressive tumour network: myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology. 2013;138:105-15
32. Miao L, Li J, Liu Q, Feng R, Das M, Lin CM. et al. Transient and Local Expression of Chemokine and Immune Checkpoint Traps To Treat Pancreatic Cancer. ACS Nano. 2017;11:8690-706
33. Hawinkels LJ, Paauwe M, Verspaget HW, Wiercinska E, van der Zon JM, van der Ploeg K. et al. Interaction with colon cancer cells hyperactivates TGF-beta signaling in cancer-associated fibroblasts. Oncogene. 2014;33:97-107
34. Liu C, Zhang Y, Lim S, Hosaka K, Yang Y, Pavlova T. et al. A Zebrafish Model Discovers a Novel Mechanism of Stromal Fibroblast-Mediated Cancer Metastasis. Clin Cancer Res. 2017;23:4769-79
35. Zhang Q, Liu XY, Zhang T, Zhang XF, Zhao L, Long F. et al. The dual-functional capability of cytokine-induced killer cells and application in tumor immunology. Hum Immunol. 2015;76:385-91
36. Zhou Q, Zhang Y, Du J, Li Y, Zhou Y, Fu Q. et al. Different-Sized Gold Nanoparticle Activator/Antigen Increases Dendritic Cells Accumulation in Liver-Draining Lymph Nodes and CD8+ T Cell Responses. ACS Nano. 2016;10:2678-92
37. Palucka K, Banchereau J. Dendritic-cell-based therapeutic cancer vaccines. Immunity. 2013;39:38-48
38. Ye Y, Wang C, Zhang X, Hu Q, Zhang Y, Liu Q. et al. A melanin-mediated cancer immunotherapy patch. Sci Immunol. 2017;2:5692
39. Walzer T, Dalod M, Robbins SH, Zitvogel L, Vivier E. Natural-killer cells and dendritic cells: "l'union fait la force". Blood. 2005;106:2252-8
40. Yang XG, Lin YL, Shi YH, Li BJ, Liu WR, Yin W. et al. FAP Promotes Immunosuppression by Cancer-Associated Fibroblasts in the Tumor Microenvironment via STAT3-CCL2 Signaling. Cancer Res. 2016;76:4124-35
41. Mace TA, Ameen Z, Collins A, Wojcik S, Mair M, Young GS. et al. Pancreatic Cancer-Associated Stellate Cells Promote Differentiation of Myeloid-Derived Suppressor Cells in a STAT3-Dependent Manner. Cancer Res. 2013;73:3007-18
42. Umansky V, Blattner C, Gebhardt C, Utikal J. The Role of Myeloid-Derived Suppressor Cells (MDSC) in Cancer Progression. Vaccines (Basel). 2016;4:36
43. Bodogai M, Moritoh K, Lee-Chang C, Hollander CM, Sherman-Baust CA, Wersto RP. et al. Immunosuppressive and Prometastatic Functions of Myeloid-Derived Suppressive Cells Rely upon Education from Tumor-Associated B Cells. Cancer Res. 2015;75:3456-65
44. Guy TV, Terry AM, Bolton HA, Hancock DG, Shklovskaya E, Fazekas de St. Groth B. Pro- and anti-tumour effects of B cells and antibodies in cancer: a comparison of clinical studies and preclinical models. Cancer Immunol Immunother. 2016;65:885-96
45. Liepelt A, Tacke F. Stromal cell-derived factor-1 (SDF-1) as a target in liver diseases. Am J Physiol Gastrointest Liver Physiol. 2016;311:G203
46. Xing F, Saidou J, Watabe K. Cancer associated fibroblasts (CAFs) in tumor microenvironment. Front Biosci (Landmark Ed). 2010;15:166-79
47. Augsten M. Cancer-associated fibroblasts as another polarized cell type of the tumor microenvironment. Front Oncol. 2014;4:62
48. Tiwari R, Bargmann W, Bose HR Jr. Activation of the TGF-beta/Smad signaling pathway in oncogenic transformation by v-Rel. Virology. 2011;413:60-71
49. Eyler CE, Rich JN. Looking in the miR-ror: TGF-beta-mediated activation of NF-kappaB in glioma. J Clin Invest. 2012;122:3473-5
50. Bierie B, Moses HL. TGF-β and cancer. Cytokine Growth Factor Rev. 2006;17:29-40
51. Roy A, Saraf S. Limonoids: overview of significant bioactive triterpenes distributed in plants kingdom. Biol Pharm Bull. 2006;29:191-201
52. Xu J-P. Cancer inhibitors from Chinese natural medicines. Boca Raton: CRC Press, Taylor & Francis Group. 2017
53. Xu F, Liu C, Zhou D, Zhang L. TGF-beta/SMAD Pathway and Its Regulation in Hepatic Fibrosis. J Histochem Cytochem. 2016;64:157-67
54. Lan HY. Diverse roles of TGF-beta/Smads in renal fibrosis and inflammation. Int J Biol Sci. 2011;7:1056-67
55. Perrot CY, Javelaud D, Mauviel A. Insights into the Transforming Growth Factor-beta Signaling Pathway in Cutaneous Melanoma. Ann Dermatol. 2013;25:135-44
56. Negmadjanov U, Godic Z, Rizvi F, Emelyanova L, Ross G, Richards J. et al. TGF-beta1-mediated differentiation of fibroblasts is associated with increased mitochondrial content and cellular respiration. PLoS One. 2015;10:e0123046
57. Lasfar A, Cohen-Solal KA. Resistance to transforming growth factor beta-mediated tumor suppression in melanoma: are multiple mechanisms in place? Carcinogenesis. 2010;31:1710-7
58. Wang X, Jiao W, Zhao Y, Zhang L, Yao R, Wang Y. et al. CUG-binding protein 1 (CUGBP1) expression and prognosis of brain metastases from non-small cell lung cancer. Thorac Cancer. 2016;7:32-8
59. Gao CH, Yu Z, Liu SH, Xin H, Li XM. Overexpression of CUGBP1 is associated with the progression of non-small cell lung cancer. Tumor Biol. 2015;36:4583-9
60. Lu Y, Yang W, Qin C, Zhang L, Deng J, Liu S. et al. Responsiveness of stromal fibroblasts to IFN-gamma blocks tumor growth via angiostasis. J Immunol. 2009;183:6413-21
61. Bansal R, Tomar T, Ostman A, Poelstra K, Prakash J. Selective Targeting of Interferon gamma to Stromal Fibroblasts and Pericytes as a Novel Therapeutic Approach to Inhibit Angiogenesis and Tumor Growth. Mol Cancer Ther. 2012;11:2419-28
62. Landskron G, De la Fuente M, Thuwajit P, Thuwajit C, Hermoso MA. Chronic inflammation and cytokines in the tumor microenvironment. J Immunol Res. 2014;2014:149185
63. Lahn M, Kloeker S, Berry BS. TGF-beta inhibitors for the treatment of cancer. Expert Opin Investig Drugs. 2005;14:629-43
64. Saunier EF, Akhurst RJ. TGF beta inhibition for cancer therapy. Curr Cancer Drug Targets. 2006;6:565-78
Author contact
![]() Corresponding author: leafhunc.edu
Corresponding author: leafhunc.edu
Citation styles
APA
Hou, L., Liu, Q., Shen, L., Liu, Y., Zhang, X., Chen, F., Huang, L. (2018). Nano-delivery of fraxinellone remodels tumor microenvironment and facilitates therapeutic vaccination in desmoplastic melanoma. Theranostics, 8(14), 3781-3796. https://doi.org/10.7150/thno.24821.
ACS
Hou, L.; Liu, Q.; Shen, L.; Liu, Y.; Zhang, X.; Chen, F.; Huang, L. Nano-delivery of fraxinellone remodels tumor microenvironment and facilitates therapeutic vaccination in desmoplastic melanoma. Theranostics 2018, 8 (14), 3781-3796. DOI: 10.7150/thno.24821.
NLM
Hou L, Liu Q, Shen L, Liu Y, Zhang X, Chen F, Huang L. Nano-delivery of fraxinellone remodels tumor microenvironment and facilitates therapeutic vaccination in desmoplastic melanoma. Theranostics 2018; 8(14):3781-3796. doi:10.7150/thno.24821. https://www.thno.org/v08p3781.htm
CSE
Hou L, Liu Q, Shen L, Liu Y, Zhang X, Chen F, Huang L. 2018. Nano-delivery of fraxinellone remodels tumor microenvironment and facilitates therapeutic vaccination in desmoplastic melanoma. Theranostics. 8(14):3781-3796.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.