Impact Factor
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusions
Supplementary Material
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2016; 6(12):2250-2266. doi:10.7150/thno.15710 This issue Cite
Research Paper
Combination Therapy for Ulcerative Colitis: Orally Targeted Nanoparticles Prevent Mucosal Damage and Relieve Inflammation
Bo Xiao1,2 ![]() , Zhan Zhang2, Emilie Viennois2,3, Yuejun Kang1, Mingzhen Zhang2, Moon Kwon Han2, Jiucun Chen1, Didier Merlin2,3
, Zhan Zhang2, Emilie Viennois2,3, Yuejun Kang1, Mingzhen Zhang2, Moon Kwon Han2, Jiucun Chen1, Didier Merlin2,3
1. Institute for Clean Energy and Advanced Materials, Faculty of Materials and Energy, Southwest University, Chongqing, 400715, P. R. China.
2. Institute for Biomedical Sciences, Center for Diagnostics and Therapeutics, Georgia State University, Atlanta, 30302, USA.
3. Atlanta Veterans Affairs Medical Center, Decatur, 30033, USA.
Received 2016-3-31; Accepted 2016-8-12; Published 2016-9-25
Abstract

Combination therapy is an emerging strategy that is under intensive preclinical investigation for the treatment of various diseases. CD98 is highly overexpressed on the surfaces of epithelial cells and macrophages in the colon tissue with ulcerative colitis (UC), which is usually associated with mucosal damage and inflammation. We previously proved that CD98 siRNA (siCD98)-induced down-regulation of CD98 in colitis tissue decreased the severity of UC to a certain extent. In an effort to further improve the therapeutic efficacy, we aim to simultaneously deliver siCD98 in combination with a potent anti-inflammatory agent, curcumin (CUR), using hyaluronic acid (HA)-functionalized polymeric nanoparticles (NPs). The resultant spherical HA-siCD98/CUR-NPs are featured by a desirable particle size (∼246 nm) and slightly negative zeta potential (∼-14 mV). The NPs functionalized with HA are able to guide the co-delivery of drugs to the targeted cells related to UC therapy (colonic epithelial cells and macrophages). Compared to either siCD98- or CUR-based monotherapy, co-delivery of siCD98 and CUR by HA-functionalized NPs can exert combinational effects against UC by protecting the mucosal layer and alleviating inflammation both in vitro and in vivo. This study shows the promising capability of the co-delivered siCD98 and CUR for boosting the conventional monotherapy via this novel nanotherapeutic agent, which offers a structurally simple platform for orally administered delivery of drugs to target cells in UC therapy.
Keywords: oral administration, combination therapy, mucosal protection, anti-inflammation, targeted nanoparticle, ulcerative colitis.
Introduction
Ulcerative colitis (UC) is a chronic relapsing idiopathic disease characterized by epithelial barrier damage and disruption of inflammatory homeostasis in colon [1-3]. The conventional treatments are mainly restricted to control inflammation [4, 5]. However, the main goal of clinical UC therapy is to not only control inflammation but also achieve mucosal healing. Therefore, novel therapeutic strategies are urgently needed to address the limitations of existing treatments.
CD98 is a transmembrane protein complex for neutral amino acid transportation, which also can regulate integrin signaling-mediated functions.[6, 7] The specific molecular ratio between CD98/LAT1 and β1 integrin might determine the polarity of colonic epithelial cells.[6] This ratio has been changed because of the up-regulation of CD98 in the colonic epithelial cells of mice with active colitis,[8, 9] inducing epithelial layer disintegration.[8] On the other hand, CD98 plays a crucial role in controlling homeostatic and innate immune responses in the colon. Thus, the down-regulation of CD98 expression in colonic cells could be a promising therapeutic strategy against UC, which might both prevent mucosal damage and control inflammation. Small-interfering RNA (siRNA)-mediated knockdown of certain protein has been shown to offer an alternative therapeutic strategy in UC therapy [4]. Indeed, our group recently demonstrated that CD98 siRNA (siCD98)-mediated down-regulation of colonic CD98 expression could decrease the severity of experimentally induced UC in mice. However, mice in the treatment group still exhibited obvious disease symptoms [10].
UC is generally expected to arise due to the combined effects of genetic background and disordered immune responses [11]. Therefore, the use of a single-target therapeutic strategy is suboptimal in UC therapy. Recently, a combination therapy that acts on multiple targets or a single target through different pathways simultaneously has shown the potential to address this concern in complex disease systems [12-14]. Specifically, the combined application of siRNAs and chemical drugs has proved to be more effective than the use of siRNAs or chemical drugs alone [15-17]. To the best of our knowledge, however, no studies have been reported to date employing a combination for UC disease. Meanwhile, curcumin (CUR), a hydrophobic molecule isolated from the rhizome of turmeric, is known for its numerous attractive pharmacological effects (e.g. anti-inflammatory and wound-healing activities).[18] In addition, CUR has the capacity to reduce mucosal neutrophil infiltration and control the signal transduction pathways involved in inflammatory responses.[19] Importantly, CUR is well tolerated by human with no toxicity up to 12 g/day for 3 months.[20] Herein, in the present study, we propose a combination therapy by taking advantage of both siCD98 and CUR for UC treatment.
To achieve combinational effect of siCD98 and CUR, they should be simultaneously delivered to the same cells with an optimal ratio.[17, 21] To date, a variety of carriers have been applied for the co-delivery of siRNAs and chemical drugs, such as block copolymer micelles, liposomes and nanoparticles (NPs).[15, 22-24] As one of the most widely used drug carriers, polymeric NP offers the benefits of high drug encapsulation capacity, controlled drug-release kinetics, and ability of surface functionalization.[25] However, application of NPs has been seriously restrained by their low selectivity to colitis tissue, especially the key cells related to UC therapy (colonic epithelial cells and macrophages).[26] In UC treatment, orally administered NP-based drug delivery can theoretically be achieved through either passive or active targeting mechanisms. Although NPs preferentially accumulate passively in colitis tissues via the epithelial enhanced permeability and retention (eEPR) effect [27, 28], the therapeutic efficacy of such passive colitis-targeting NPs is still far from optimum. Therefore, it is desirable to develop effective strategies for active targeting of colitis tissues, which can realize highly selective drug accumulation at colitis sites and efficient cellular internalization by the target cells.[26] Moreover, hyaluronic acid (HA) can specifically bind to glycoprotein CD44, which is over-expressed on the surface of colonic epithelial cells and macrophages in UC tissues.[29-31] The specific affinity of HA to CD44 inspires us to utilize HA-functionalized NPs for the targeted treatment of colitis.
In this study, we synthesized HA-functionalized siCD98/CUR-loaded polymeric NPs (HA-siCD98/CUR-NPs, as depicted in Scheme 1a); and characterized their physicochemical properties and targeted drug delivery capability. Particularly, we investigated their functions to trigger mucosal protection, anti-inflammation, and the synergistic effects both in vitro and in vivo (Scheme 1b).
The synergistic therapeutic effects of HA-siCD98/CUR-NPs against UC. (a) A schematic illustration for the fabrication of HA-siCD98/CUR-NPs. (b) Oral administration of HA-siCD98/CUR-NPs embedded in hydrogel (chitosan/alginate) confers synergistic therapeutic effects against UC by protecting mucosa and alleviating inflammation.
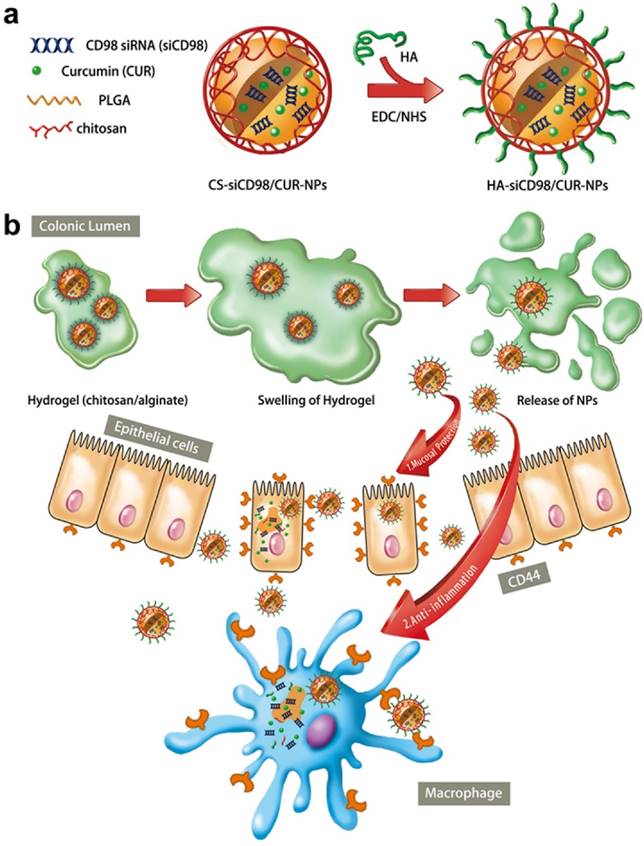
Materials and Methods
Materials
PLGA (lactide:glycolide = 50:50, Mw = 38-54 kg/mol), poly(vinyl alcohol) (PVA, 86-89% hydrolyzed, low molecular weight), chitosan, sodium nitrite, spermidine, CUR, 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDC), N-hydroxysuccinimide (NHS), 2-(N-morpholino) ethanesulfonic acid sodium salt (MES), lipopolysaccharide (LPS) from Salmonella enteric serotype typhimurium and fluorescein isothiocyanate-dextran (FITC-dextran, 3 kDa) were purchased from Sigma-Aldrich (St. Louis, MO). Chitosan was purified before its subsequent application, and its molecular weight was tailored by depolymerization using sodium nitrite following a reported method [32]. Viscosity-average molecular weights of the original chitosan and resulting chitosan were determined as respective 246 kDa and 18 kDa using a reported method [33]. The depolymerized chitosan was used in the NPs fabrication process. DNA Gel Loading Dye (6X) was from Thermo Fisher Scientific Inc. (Waltham, MA). HA (Mw = 20 kDa) was obtained from Lifecore Biomedical, LLC (Chaska, MN). Paraformaldehyde stock solution (16%) was from Electron Microscopy Science (Hatfield, PA). 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT), 4ʹ,6-diamidino-2-phenyl-indole dihydrochloride (DAPI), Alexa Fluor 568 phalloidin (Mw = 1590) and 1,1ʹ-dioctadecyl-3,3,3ʹ,3ʹ-tetramethylindotricarbocyanine iodide (DiR) were supplied from Invitrogen (Eugene, OR). Mouse siCD98, human siCD98 and scrambled siRNA were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Dextran sulphate sodium (DSS, 36-50 kDa) was obtained from MP Biomedicals (Aurora, OH). Buffered formalin (10%) was supplied from EMD Millipore (Billerica, MA). Hematoxylin and eosin were from Richard-Allan Scientific (Kalamazoo, MI). All commercial products were used without further purification.
Fabrication of HA-siCD98/CUR-NPs
siCD98/CUR-loaded NPs and siCD98-loaded NPs were prepared by a double emulsion solvent evaporation technique. Briefly, siRNA was reconstituted in RNase-free water, and was then complexed with spermidine at N/P ratio of 8:1. The resultant mixture was kept at room temperature for 15 min to form spermidine/siRNA complexes. PLGA (100 mg) and, optionally, CUR (total 6 mg) were dissolved in 2 mL of dichloromethane. The aqueous solution (0.1 mL) was then added dropwise to the oil phase to form the first emulsion. PVA solutions (5%) with chitosan (0.5%) was prepared in diluted hydrochloric acid solution (0.1%). Addition of the first emulsion to 4 mL PVA (5%)/chitosan (0.5%) solution and subsequent sonication (six times, 10 s each time) of the whole mixture formed a double emulsion. This double emulsion was immediately poured into 100 mL of aqueous acidic solution containing 0.3% PVA with 0.03% chitosan. After that, the organic solvent was evaporated under low vacuum conditions (Rotary evaporator, Yamato RE200, Santa Clara, CA). The NPs formed by this method were collected by centrifugation at 17,418 g for 20 min, washed three times with deionized water, and re-suspended in aqueous solution containing 5% threhalose. Finally, the resultant NPs were dried in a lyophilizer at a temperature below -50 °C and a vacuum level of 0.05 mbar, and stored at -20 oC in airtight container. In addition, CUR-loaded NPs were fabricated by a single emulsion solvent evaporation technique as we described previously [25, 34]. The depolymerized chitosan-coated NPs were herein designated as CS-coated NPs.
As for the fabrication of HA-functionalized NPs, the CS-coated NPs obtained above were dispersed in MES buffer (pH = 5.5). The carboxyl group of HA was activated for 2 h by EDC/NHS. The HA solution was added to CS-coated NPs suspension, and resultant mixture was allowed to react at ambient temperature with stirring for 4 h. The final NPs were collected by centrifugation at 17,418 g for 20 min, washed three times with deionized water, and re-suspended in aqueous solution containing 5% threhalose. Finally, the resultant NPs were dried in a lyophilizer, and stored at -20 oC in airtight container.
Characterization of NPs
Particle sizes (nm), polydispersity index (PDI) and zeta potential (mV) of NPs were measured by dynamic light scattering (DLS) using 90 Plus/BI-MAS (Multi-angle particle sizing) or DLS after applying an electric field using a ZetaPlus (Zeta potential analyzer, Brookhaven Instruments Corporation). The diameters (nm), PDI or zeta potential (mV) of NPs were measured using 3 runs. Each run is an average of 10 measurements. The average values were based on the measurement on repeated NPs.
The morphology of NPs was observed with a transmission electron microscopy (TEM, LEO 906E, Zeiss, Germany) at an 80 kV accelerating voltage. A drop of diluted NP suspension was mounted onto 400-mesh carbon-coated copper grids and dried before analysis.
To determine loading amount of siRNA in NPs, NPs (3 mg) were dissolved in 0.5 mL of dichloromethane. The released siRNA was extracted from the organic phase using 0.8 mL Tris-EDTA (TE) buffer (10 mM Tris-HCl, 1 mM EDTA, pH = 8.0). The TE buffer was added to the organic solution, and the resultant mixture was vortexed vigorously for 5 min and then centrifuged at 13,400 g for 5 min at 4 oC. The supernatant was collected and analyzed for double-stranded RNA content using the Quant-ITTM PicoGreenTM assay according to the procedures recommended by the manufacturer (Invitrogen).
The entrapped CUR in NPs was determined by measuring its intrinsic fluorescence on a Perkine Elmer EnSpire multimode plate reader (Perkin Elmer, Boston, MA). In a typical example, NPs (3 mg) were dissolved in dimethyl sulfoxide (DMSO). Then the solution (100 μL) was transferred to a black 96-well plate. The fluorescence intensity of CUR was measured at 528 nm emission wavelength (485 nm excitation wavelength). The drug loading and encapsulation efficiency were defined as follows [35, 36]:
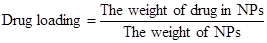
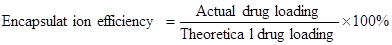
The morphology of hydrogel with NPs was examined using scanning electronic microscopy (SEM, LEO 1450VP, Zeiss, Germany). The hydrogel with NPs was freeze-dried, mounted on the carbon adhesive tape, and sputter-coated with a mixture of gold and palladium (60:40) in an argon atmosphere under low pressure.
To evaluate the integrity of siRNA in NPs, siRNA was extracted from HA-siCD98-NPs (3 mg) according to the method described in the section of quantification of siRNA above. Supernatant containing siRNA (300 μL) were dried in a lyophilizer, and then 20 μL TE buffer was added to dissolve siRNA. Agarose gel (4%, W/V) containing GelRed solution (0.5 μg/mL) was prepared, and the samples were electrophoresed at 100 V for 25 min. The resulting siRNA migration patterns were viewed under UV transilluminator.
Release profiles of siCD98 and CUR from HA-siCD98/CUR-NPs
NPs (3 mg) were suspended in PBS solution (1.0 mL) and incubated at 37 oC with gentle shaking (120 rpm). The amount of released siRNA from the NPs was measured at various time intervals over 72 h. During each measurement, the NPs suspension was centrifuged at 13,400 g for 5 min. The supernatant was removed and its siRNA content was analyzed using the same method as described above. The precipitated NPs were re-suspended in an equal volume of fresh PBS solution and incubated under the same conditions for continuous monitoring of siRNA release.
The release behavior of CUR from NPs was conducted by the dialysis method. Briefly, NPs were dispersed in PBS to form a suspension (equal to 250 μg of drug). The suspension was transferred into a regenerated cellulose dialysis tube (molecular weight cut-off = 10 kDa) and the sample-filled tube was closed tightly at both ends to keep each tube surface area equivalent. The closed bag was subsequently put into a centrifuge tube, and immersed in 20 mL PBS release medium containing 0.1% Tween-80. Tween-80 was employed in PBS to maintain the solubility of CUR in aqueous phase. The tube was put in a water bath shaking at 120 rpm at 37 oC. At appropriate time points, outer solution was taken for measurement and fresh release medium was added. The amount of CUR in the outer solution was measured using the same method as described above.
Cell culture
Colon-26 cell (murine colon carcinoma cell line), Raw 264.7 macrophage (murine leukaemic monocyte macrophage cell line) and Caco2-brush border expressing cell (Caco2-BBE, human colon carcinoma cell line) were maintained in respective RPMI 1640 medium, DMEM medium and DMEM medium supplemented with 100 U/mL penicillin, 100 μg/mL streptomycin and 10% fetal bovine serum (FBS). The cells were grown at 37 °C under 5% CO2, the growth medium was replaced every 48 h, and the cells were sub-cultured 1:10 twice a week. Colon-26 cells and Caco2-BBE cells were detached from the culture flasks by incubating the cells with respective accutase and trypsin for around 2 min at 37 °C. Raw 264.7 macrophages were gently detached with a sterile cell scraper.
Biocompatibility of NPs
For MTT assay, Colon-26 cells and Raw 264.7 macrophages were seeded at a respective density of 2×104 and 8×103 cells/well in 96-well plates and incubated overnight. After 5, 24 or 48 h of exposure to NP suspensions, the cells were then incubated with 100 μL MTT working solution at 37 °C for 4 h. This solution was prepared in serum-free medium with the MTT concentration of 0.5 mg/mL. Thereafter, the media were discarded and 50 μL DMSO was added to each well prior to spectrophotometric measurements at 570 nm. Untreated cells were used as a negative reference, whereas cells treated with 0.5% Triton X-100 were the positive control.
Intracellular NPs uptake visualization
Colon-26 cells and Raw 264.7 macrophages were seeded in eight-chamber tissue culture glass slide (BD Falcon, Bedford) at a respective density of 5×104 and 2×104 cells/well and incubated overnight. After 5 h of exposure to HA-CUR-NPs (CUR, 100 μM), the cells were thoroughly rinsed with PBS to eliminate excess of NPs, and then fixed in 4% paraformaldehyde for 20 min. To observe cellular uptake of NPs, DAPI was diluted 10,000 times and added to the wells for staining cells for 5 min. Images were acquired using the FITC channel and DAPI channel on an Olympus fluorescence microscopy equipped with a Hamamatsu Digital Camera ORCA-03G.
Quantification of cellular uptake using flow cytometry (FCM)
Colon-26 cells and Raw 264.7 macrophages were seeded in 12-well plates at a respective density of 4×105 and 1×105 cells/well and incubated overnight. The medium was exchanged to serum-free medium containing various NPs (equal to 25 and 50 μM CUR for Colon-26 cells and Raw 264.7 macrophages, respectively). Cell treated with blank NPs were used as a negative control. After incubation for different time periods (0, 1, 3 and 5 h), the cells were thoroughly rinsed with PBS to eliminate excess NPs, which were not taken up by cells. Subsequently, the treated cells were harvested using accutase or trypsin, transferred to centrifuge tubes, and centrifuged at 1,800 g for 5 min. Upon removal of the supernatant, the cells were re-suspended in FCM buffer, transferred to round-bottom polystyrene test tubes (BD Falcon, 12 × 75 mm), and kept at 4 oC until further analysis. Analytical FCM was performed using the FITC channel on the FCM CantoTM (BD Biosciences). A total of 5,000 ungated cells were analyzed. The competition assay was carried out to confirm the cellular uptake of HA-functionalized NPs. The uptake efficiency of Colon-26 cells and Raw 264.7 macrophages with these NPs in the presence of free HA (5 mg/mL) as a competitor was also tested by FCM.
In vitro mucosal protection assay
Since the transepithelial barrier is important for intestinal inflammation, we studied the protection effects of various NPs on intestinal barrier function in vitro. Mucosal protection capability was examined by measuring the flux of FITC-dextran across cell monolayer after the treatment of LPS to Raw 264.7 macrophages. Raw 264.7 macrophages were seeded into the 24-well plate and incubated overnight to completely adhere to the well. Confluent and polarized Caco2-BBE cells grown on filters (pore size, 0.4 μm) were treated with different NPs for 24 h. 100 μM of FITC-dextran was added to the apical side of Caco2-BBE monolayer. The transwell insert were added into multiple plate wells preloaded with Raw 264.7 macrophages. The LPS (5 μg/mL) was added the basolateral side in this model. After incubation for 3 h, the medium (100 μL) was collected from the basolateral chambers, and transferred to a black 96-well plate for measuring the FITC fluorescence on a Perkine Elmer Enspire multimode plate reader (Perkin Elmer, Boston, MA). The amount of FITC-dextran that had crossed the epithelial layer was read off a standard curve.
In vitro gene silencing efficiency test
Colon-26 cells and Raw 264.7 macrophages were seeded in 6-well plates at a respective density of 3×105 and 1×105 cells/well and incubated overnight. After co-culture with various NPs for 5 h, cells were incubated in medium containing 10% FBS for 19, 43, 67 or 91 h. Raw 264.7 macrophages were then stimulated with LPS (1 μg/mL, Sigma) for 3 h. Total RNA was extracted using RNeasy Plus Mini Kit (Qiagen). The cDNA was generated from the total RNAs isolated above using the Maxima first strand cDNA synthesis kit (Fermentas) according to the manufacturer's instruction. CD98 and TNF-α mRNA expression levels were quantified by RT-PCR using Maxima® SYBR Green/ROX qPCR Master Mix (Fermentas). The data were normalized to the internal control: 36B4. Relative gene expression levels were calculated using the 2-∆∆Ct method. Sequences of all the primers used for RT-PCR are given in Table S1 (Supplementary Information).
Induction of UC mice model and oral administration of NPs
FVB male mice (8 weeks of age, The Jackson Laboratory) were used in the animal experiments. Mice were group housed (25 °C), photoperiod (12:12-h light-dark cycle), and allowed unrestricted access to potables and standard mouse chow. All the animal experiments were approved by Georgia State University Institutional Animal Care and Use Committee. UC was induced by replacing their drinking water with a 2.8% (wt/vol) DSS solution for 7 days. To deliver various NPs to mice colonic lumen, we encapsulated them into a hydrogel comprised of chitosan and alginate at a ratio of 3/7 (wt/wt) and double-gavaged the mice for 6 days (a detailed protocol is available at http://www.natureprotocols.com/2009/09/03/a_method_to_target_bioactive_c.php). Mice receiving NPs were treated with 5 mg/kg of CUR and/or 16.55 μg/kg of siCD98 per day. Control mice were given water only. Mice were observed daily and evaluated for changes in body weight and development of clinical symptoms of colitis. Feces were collected during the DSS treatment and drug administration. Mice were sacrificed by CO2 euthanasia. Spleen weight and colon length were measured. A small piece (50 mg) of distal colon was taken for MPO and RNA analysis, and the remaining of the colon was used for histopathological analysis.
Ex vivo imaging
To track the HA-functionalized NPs in gastrointestinal tract (GIT) after oral administration, the near infrared dye DiR was employed as a fluorescence probe. The fabrication process of HA-DiR-NPs was exactly the same as that of HA-CUR-NPs, except that CUR was replaced by DiR. The amount of DiR in HA-DiR-NPs was examined by a UV-Vis spectra method. Colitis was induced in FVB male mice (8 weeks of age, The Jackson Laboratory) by replacing their drinking water with a 3.5% (wt/vol) DSS (36-50 kDa). HA-DiR-NP-embedded hydrogel was orally administered to mice at an equivalent DiR concentration (0.5 mg DiR/kg per mouse) after 6 days of DSS treatment. After oral administration for respective 8, 16, or 24 h, the mice were sacrificed to obtain GIT. The images were captured using the IVIS spectrum imaging system (PerkinElmer/Caliper LifeSciences, Hopkinton, MA).
In vivo cellular uptake of NPs
Colitis was induced in FVB male mice (8 weeks of age, The Jackson Laboratory) by replacing their drinking water with a 3.5% (wt/vol) DSS (36-50 kDa). Mice were orally administered with HA-CUR-NP-embedded hydrogel (5 mg CUR/kg per mouse) after 6 days of DSS treatment. After 12 h of oral administration, mice were sacrificed by CO2 euthanasia. Colon tissues were extracted and rinsed thoroughly with PBS and embedded in Optimal Cutting Temperature compound. Sections (6 μm) were stained with Alexa Fluor 568 phalloidin and DAPI. Images were acquired using an Olympus equipped with a Hamamatsu Digital Camera ORCA-03G.
Quantification of fecal Lcn-2 by ELISA
Fecal samples were reconstituted in PBS containing 0.1% Tween-20 (100 mg/mL) and vortexed for 20 min to get a homogenous fecal suspension. These samples were then centrifuged for 10 min at 13,400 g and 4 oC. Clear supernatants were collected and stored at -20 oC until analysis. Lcn-2 levels were estimated in the supernatants using Duoset murine Lcn-2 ELISA kit (R&D Systems, Minneapolis). Feces from healthy control mice were used as negative controls.
MPO activity
Neutrophil infiltration into the colon was quantified by measuring MPO activity. Briefly, a portion of colon was homogenized in 1:20 (w/v) of 50 mM phosphate buffer (pH 6.0) containing 0.5% hexadecyltrimethyl ammonium bromide (Sigma) on ice using a homogenizer (Polytron, Luzern, Switzerland). The homogenate was then sonicated for 10 s, freeze-thawed three times, and centrifuged at 16,000 g for 15 min. The supernatant was then added to 1 mg/mL O-dianisidine hydrochloride (Sigma) and 0.0005% hydrogen peroxide, and the change in absorbance at 460 nm was measured. MPO activity was expressed as units per g of colonic tissue, where one unit was defined as the amount that degrades 1 μmol of hydrogen peroxide per min at 25 °C.
Histological analysis
Tissue samples were evaluated for mucosal architecture change, cellular infiltration, inflammation, goblet cell depletion, surface epithelial cell hyperplasia, and signs of epithelial regeneration using light microscopy of hematoxylin and eosin (H&E) staining. These values were used to assess the degrees of mucosal damage and repair in various groups. Colon tissues were fixed in 10% buffered formalin and embedded in paraffin. Tissue sections with a thickness of 5 μm were stained with hematoxylin and eosin followed by imaging using bright-field microscopy. Histological scoring was performed on the basis of 3 parameters (the severity of inflammation, crypt damage and ulceration) as we described previously.[37] A minimum of three sections of the colon per animal were scored.
In vivo gene silencing efficiency by RT-PCR
Total RNA was extracted from the tissue samples using RNeasy Plus Mini Kit (Qiagen). The cDNA was generated from the total RNAs isolated above using the Maxima first strand cDNA synthesis kit (Fermentas) according to the manufacturer's instruction. The mRNA expression levels of CD98 and TNF-α were quantified by real-time RT-PCR using Maxima® SYBR Green/ROX qPCR Master Mix (Fermentas).
Statistical analysis
Statistical analysis was performed using ANOVA test followed by a Bonferroni post-hoc test (GraphPad Prism) or Student's t-test. Data were expressed as mean ± standard error of mean (S.E.M.). Statistical significance was represented by *P<0.05 and **P<0.01.
Results
Preparation and characterization of HA-functionalized NPs
The HA-siCD98/CUR-NPs were prepared using a double emulsion-solvent evaporation method, which is a well-established technique for fabricating drug-loaded NPs [38]. The inner aqueous phase (containing siRNA/spermidine complexes) was added to the organic phase (containing PLGA and CUR) to form a first water/oil emulsion under sonication. We then added this first emulsion to the aqueous phase (containing emulsifier) and performed sonication to form a water/oil/water emulsion based on the Gibbs-Marangoni effect and a capillary break-up mechanism.[39] Next, organic solvent (dichloromethane) was removed by evaporation under reduced pressure to permit CUR molecules to transfer to PLGA hydrophobic core through the “like dissolves like” principle.[40] This process could allow for the restriction of inner aqueous phase (containing siRNA/spermidine complexes) and CUR to the hydrophobic PLGA core simultaneously.
Emulsifiers at NP surface enable the separation of oil and water phases, thus preventing the aggregation of NPs [41]. PVA, a common amphiphilic copolymer, has been extensively used as an emulsifier for the preparation of PLGA NPs [42]. In the process of NP formation, the hydrophobic parts of PVA penetrate into the organic phase and remain entrapped in the polymeric matrix of NPs. Meanwhile, its hydrophilic parts form NP corona and further stabilize NP through steric hindrance. Our group has often used chitosan, a natural biocompatible polymer, for the surface modification of PLGA NPs [25]. The coating ability of chitosan could reflect the entangling of its chains with PVA and/or the adsorption of positively charged chitosan to the negatively charged surfaces of the NPs. Finally, HA was conjugated to the CS-coated NPs via the formation of an ester bond between the carboxyl group of HA and the amino group of chitosan at the distal end of NPs, using EDC/NHS chemistry in MES buffer. Based on this strategy, we successfully produced the HA-functionalized NPs (Table 1).
Characteristics of the siCD98/CUR-loaded nanoparticles (mean ± S.E.M.; n=3).
| Nanoparticles | Particle Size (nm) | PDI | Zeta-potential (mV) | Drug Loading | Encapsulation Efficiency (%) | ||
|---|---|---|---|---|---|---|---|
| siCD98 (ng/mg) | CUR (μg/mg) | siCD98 | CUR | ||||
| HA-siCD98-NPs | 240.5 ± 5.3 | 0.171 | -15.8 ± 3.2 | 142.6 ± 15.1 | - | 56.1 ± 4.8 | - |
| HA-CUR-NPs | 253.9 ± 9.1 | 0.149 | -17.9 ± 2.9 | - | 45.7 ± 11.2 | - | 61.7 ± 10.3 |
| HA-siCD98/CUR-NPs | 246.2 ± 7.8 | 0.221 | -13.7 ± 4.1 | 52.9 ± 3.9 | 39.4 ± 7.1 | 24.2 ± 1.5 | 53.2 ± 7.4 |
| CUL-CUR-NPs | 260.5 ± 11.4 | 0.247 | -15.3 ± 5.2 | - | 51.5 ± 9.6 | - | 64.5 ± 8.9 |
Particle size and zeta potential are important parameters that directly impact the stability and cellular uptake of NPs [43]. As indicated in Table 1, DLS measurements revealed that the average hydrodynamic diameter of the HA-functionalized NPs was in the range of 240.5 to 253.9 nm. The particle size of the control NPs (carboxymethyl cellulose-coated CUR-loaded NPs; CUL-CUR-NPs) was slightly larger than that of its counterpart HA-CUR-NPs. The PDI value less than 0.3 indicated the narrow size distribution for all the NPs. The CS-coated NPs were electropositive (data not shown), whereas all of the HA-functionalized NPs and CUL-CUR-NPs had negative zeta potentials ranging from -13 to -18 mV. This reflects the chemical conjugation of negatively charged HA or CUL to the NPs surfaces.
The NPs were subjected to morphological characterization by TEM. As shown in representative TEM images, CS-siCD98/CUR-NPs (Figure 1a) and HA-siCD8/CUR-NPs (Figure 1b) were spherical in shape and had mean diameters of approximately 128.2 and 199.7 nm, respectively. The modest discrepancies in the diameters measured by TEM and DLS were ascribed to differences in the surface states of the NPs under the different test conditions, as also described in our previous studies [44]. The NPs are in a fully hydrated (swollen) state when examined by DLS, whereas they have to be completely dehydrated for TEM characterization.
Physicochemical and morphological characterization of NPs. Representative TEM images of CS-siCD98/CUR-NPs (a) and HA-siCD98/CUR-NPs (b). The insert highlights the TEM of an individual NP. (c) Representative size distribution of HA-siCD98/CUR-NPs. (d) In vitro cumulative release of siCD98 and CUR from HA-siCD98/CUR-NPs at 37 oC. Data are presented as means ± S.E.M. (n=3). (e) Cellular uptake profiles of HA-CUR-NPs in Colon-26 cells and Raw 264.7 macrophages after treatment with a CUR concentration of 100 μM for 5 h. After co-incubation, the cells were processed for fluorescence staining. Fixed cells were stained with DAPI for visualization of nuclei (purple). Scale bar represents 10 μm.
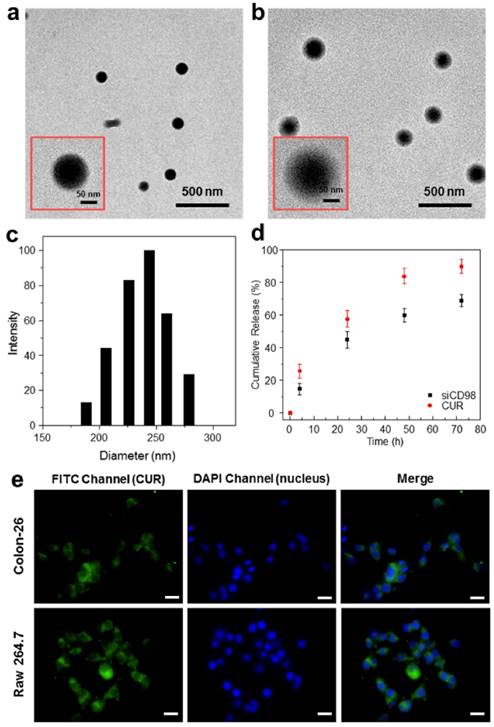
The NPs generated herein exhibited narrow size distribution (Figure 1c). As shown in Table 1, the siCD98-loading capacity ranged from 52.9 to 142.6 ng/mL, while the CUR-loading capacity ranged from 39 to 51 μg/mg. The encapsulation efficiencies depended on the numbers and types of the drugs used in the synthetic process, with single drug-loaded NPs exhibiting higher encapsulation efficiencies compared to dual drug-loaded NPs. We also found that the drug encapsulation efficiencies were lower than the data in our previous results [28, 29, 46]. The reason might be that drugs (siCD98 and CUR) close to the NPs surface released in the reaction solution during HA conjugation. Accordingly, no obvious initial burst release was detected in Figure 1d.
To investigate the integrity of siRNA extracted from NPs, electrophoresis was performed. As shown in Figure S1a, no breakage in siRNA fragment (Lane 2) was observed in comparison to control siRNA (Lane 1), indicating that siRNA was intact after extraction from NPs. In addition, we found that the band corresponding to siRNA extracted from NPs was slightly lagging behind the band of control siRNA. A reasonable explanation is that when the external electric field is applied to a fluid in agarose gel, electro-osmosis is expected to occur. As respect to siRNA extracted from NPs, it was mixed with large amount of constituents of TE buffer after lyophilization, and the concentrated ion distribution had a strong impact on the siRNA migration in electric field during electro-osmosis. It was also observed that dyes (bromophenol blue and xylene cyanol FF) from DNA Gel Loading Dye in Lane 2 were slightly lagging during electrophoresis (Figure S1b).
HA-functionalized NPs simultaneously release siCD98 and CUR in vitro
Controlled release of siCD98 and CUR from NPs is an important prerequisite for the synergistic therapy of UC. Here, the in vitro drug release profiles for HA-siCD98/CUR-NPs were monitored in vitro as a function of time (Figures 1d). At 24 h of incubation, HA-siCD98/CUR-NPs showed a release of about 44.9% and 57.6% of the siCD98 and CUR, respectively. This was followed by a more moderate drug release over the next 48 h. Cumulatively, approximately 68.8% and 89.7% of the siCD98 and CUR, respectively, were released from the NPs over 72 h. These results indicated that HA-siCD98/CUR-NPs were characterized by a slow release of the encapsulated drugs, which might be useful for simultaneous and sustained release of siRNAs and drugs.
Furthermore, the results revealed that the HA-functionalized NPs released siCD98 and CUR in two phases, each of which showed a constant rate. The initial release phase mainly represented siCD98 and CUR that were bound or adsorbed onto the surfaces of the NPs, while the subsequent phase was primarily dependent on the progressive erosion or degradation of the NP matrix, which allowed the siRNAs and drugs to escape from the NPs.
NPs do not affect cell viability
Cytotoxicity is a primary concern in efforts to develop novel formulations for UC therapy. To evaluate the cytotoxicity of the obtained NPs, we treated Colon-26 cells and Raw 264.7 macrophages with various NPs, and examined cell viability using the MTT assay (Supplementary Information: Figure S2). A low degree of cytotoxicity was observed in the groups treated with HA-CUR-NPs. However, neither HA-siCD98-NPs nor HA-siCD98/CUR-NPs caused obvious cytotoxicity in either cell line, even after 48 h of co-incubation. This indicates that these NPs have excellent biocompatibility. In addition, we examined the cytotoxicity of HA-CUR-NPs at higher CUR concentrations up to 125 µM after 5 h of co-incubation (Supplementary Information: Figure S3). It was found that HA-CUR-NPs exhibited slight cytotoxicity on Colon-26 and Raw 264.7 at a CUR concentration of 125 µM, under which the cells retained viability over 81%. However, at CUR concentrations below 100 uM, more than 88% of cells were viable. Therefore, HA-CUR-NPs were well tolerated by both cell lines under CUR concentration of 100 uM, and this concentration was used for further cellular uptake imaging test.
HA functionalization increases cellular uptake of NPs
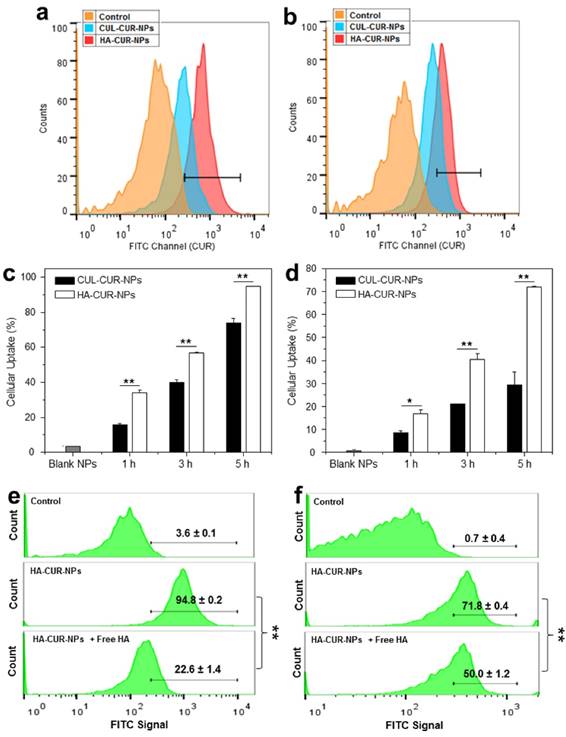
Efficient cellular uptake of NPs is critical for UC therapy. In this work, we used the intrinsic fluorescence of CUR to investigate the internalization of NPs into two cell types (colonic epithelial cells and macrophages), which were believed to be the key targets in UC treatment. Colon-26 cells and Raw 264.7 macrophages were treated for 5 h with HA-CUR-NPs (CUR, 100 μM). As expected, the untreated control cells showed no fluorescence (data not shown), whereas clear drug accumulation was detected in both cell lines treated with HA-CUR-NPs (Figure 1e).
To investigate whether the surface functionalization of NPs by HA could promote the cellular uptake efficiency, we treated Colon-26 cells and Raw 264.7 macrophages with HA-CUR-NPs or CUL-CUR-NPs (25 and 50 μM CUR for two cell lines, respectively), and investigated their cellular uptake profiles after 0, 1, 3, and 5 h of co-incubation. The fluorescence intensities of both HA-CUR-NP-treated cell lines (Figure 2a-2d) were remarkably higher than those in the corresponding CUL-CUR-NP-treated cells, indicating that surface modification with HA enhanced the cellular uptake of NPs. To confirm the HA receptor-mediated cellular uptake of HA-functionalized NPs, we also examined the cellular uptake efficiency of HA-CUR-NPs by both cell lines in medium supplemented with free HA, which competed with the HA groups of the HA-CUR-NPs. As shown in Figure 2e-2f, the cellular uptake of HA-CUR-NPs decreased substantially in the presence of free HA, suggesting that HA-functionalized NPs were internalized into Colon-26 cells and Raw 264.7 macrophages via HA receptor-mediated endocytosis.
HA functionalization increases the cellular uptake of NPs. Flow cytometry histogram overlays show the cellular uptake of NPs by Colon-26 cells (a) and Raw 267.4 macrophages (b) after 5 h of treatment. Analysis of the percentage of CUR fluorescence-positive Colon-26 cells (c) and Raw 267.4 macrophages (d) after treatment with a respective CUR concentration of 25 μM and 50μM. Competitive cellular uptake profiles of HA-functionalized NPs by Colon-26 cells (e) and Raw 264.7 macrophages (f). HA-CUR-NPs were incubated with cells in the presence of free HA (5 mg/mL) as a competitor. Each point represents the mean ± S.E.M. (n=3). Statistical significance was assessed using Studentʹs t-test (*P<0.05 and **P<0.01).
NPs protect against mucosal damage in vitro
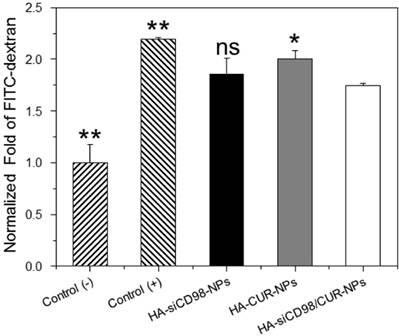
The inflamed tissues are usually characterized by numerous immune cells that produce excess pro-inflammatory cytokines [45]. In this study, we established an in vitro model of gut inflammation. Specifically, a monolayer of Caco2-BBE cells with tight junctions were formed on the apical side of a transwell apparatus, while Raw 264.7 macrophages were placed on the basolateral side. LPS (5 μg/mL) was then applied to the basolateral compartment to imitate gut inflammation. As shown in Figure 3, the FITC signal in the supernatant of the basolateral wells was significantly higher for LPS-stimulated Raw 264.7 macrophages compared to control (non-LPS-treated) cells, indicating that LPS treatment damaged the Caco2-BBE monolayer and induced the penetration of FITC-dextran through the monolayer. This finding was in good agreement with a previous report [46]. Moreover, we observed that the FITC signal was slightly lower for cells treated with HA-siCD98-NPs (siCD98, 10 ng/mL) or HA-CUR-NPs (CUR, 8.2 μM), suggesting that single drug-loaded NPs had minimal effect on mucosal protection. In contrast, the FITC signal was significantly decreased in the supernatants of HA-siCD98/CUR-NP-treated cells (siCD98, 10 ng/mL; CUR, 8.2 μM), indicating that siCD98 and CUR synergistically reinforced the Caco2-BBE monolayer and prevented mucosal damage.
HA-siCD98/CUR-NPs prevent mucosa damage. Cells were cultured in transwell plates, and Caco2-BBE cell layer was treated with various NPs for 24 h. Subsequently, FITC-labeled dextran (100 μM) was added to the apical side of Caco2-BBE monolayer and LPS (5 μg/mL) was added the basolateral side with Raw 264.7 macrophages. After 3 h of incubation, the fluorescent intensity of medium was tested. Each point represents the mean ± S.E.M. (n=3). Statistical significance was assessed using the Student's t-test (*P<0.05 and **P<0.01).
HA-functionalized NPs reduce expressions of CD98 and TNF-α in vitro
We next assessed whether HA-functionalized NPs had synergistic anti-inflammatory properties in vitro. Compared to untreated Colon-26 cells, cells treated with each of the HA-functionalized NPs exhibited remarkable decrease of CD98 after 24 h of co-incubation (Figure 4a). Furthermore, the CD98 gene expression levels in Colon-26 cells treated with HA-siCD98-NPs decreased as the concentration of siCD98 increased. Interestingly, HA-CUR-NPs also efficiently down-regulated CD98 in Colon-26 cells, but this effect decreased as the concentration of CUR increased. HA-siCD98/CUR-NPs showed the maximal ability to down-regulate CD98 in Colon-26 cells, indicating that there was a synergistic effect between siCD98 and CUR. Compared to LPS (1 μg/mL)-treated Raw 264.7 macrophages with or without NP treatment, all of the NP-treated cells exhibited significantly lower levels of CD98 mRNA expression levels (Figure 4b).
In vitro gene downregulation capacities of NPs against Colon-26 cells and RAW 267.4 macrophages. (a) CD98 mRNA expression levels of Colon-26 cells exposed to NPs for 24 h. The mRNA expression levels of CD98 (b) and TNF-α (c) for Raw 264.7 macrophages exposed to NPs for 24 h. After treatment by NPs, cells were treated with LPS (1 μg/mL) for 3 h. (d) CD98 mRNA expression levels of Colon-26 cells exposed to NPs for 48 h. The mRNA expression levels of CD98 (e) and TNF-α (f) for Raw 264.7 macrophages exposed to NPs for 48 h. After treatment by NPs, cells were treated with LPS (1 μg/mL) for 3 h. Each point represents the mean ± S.E.M. (n=3). Statistical significance was assessed using the Student's t-test (*P<0.05, **P<0.01; ns, non-significant).
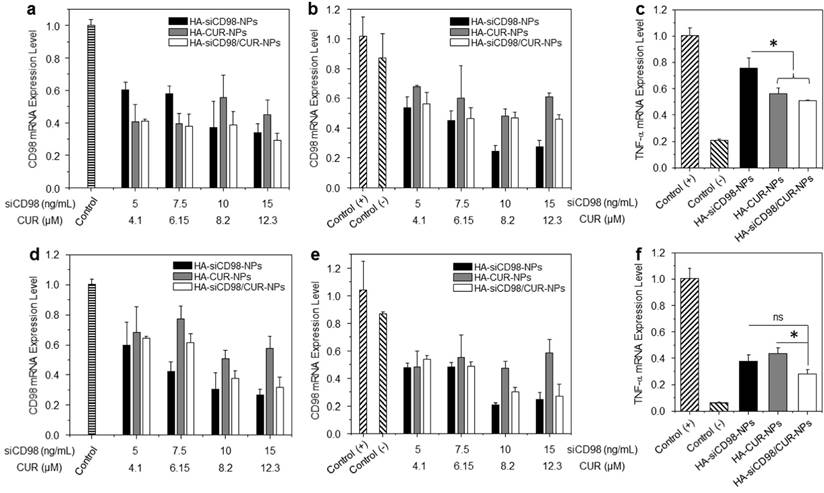
We further studied whether HA-functionalized NPs could attenuate inflammation in vitro. TNF-α is the major pro-inflammatory cytokine secreted by macrophages during inflammation. We measured the TNF-α mRNA expression level in Raw 264.7 macrophages after treatment of siCD98-loaded NPs. It was found that there was significant decrease of CD98 mRNA levels induced by NP treatment, which attenuated the activation of TNF-α in Raw 264.7 macrophages (Figure 4c). In the control experiments, blank HA-functionalized NPs and HA-scrambled siRNA-NPs did not produce any obvious knockdown of CD98 relative to the levels observed in LPS-stimulated macrophages (Supplementary Information: Figure S4a and Figure S4b). These results confirmed that CD98 is a critical factor during inflammation, and that the TNF-α mRNA expression level could potentially be ameliorated by CD98 knockdown. Moreover, among these three NP-treated groups, Raw 264.7 macrophages treated with HA-siCD98/CUR-NPs exhibited the lowest TNF-α mRNA expression level, suggesting a synergistic anti-inflammatory effect of siCD98 and CUR.
We also investigated the downregulation efficiencies of CD98 and TNF-α after 48 h of treatment in Colon-26 cells and Raw 264.7 macrophages. There was no synergistic effect observed with respect to CD98 gene knockdown efficiency (Figure 4d-4e). However, HA-siCD98/CUR-NPs showed a clear synergistic effect to down-regulate the TNF-α mRNA expression (Figure 4f). Finally, we tested the RNAi efficiency of NPs after 72 h and 96 h of co-incubation. It was found that HA-siCD98/CUR-NPs did not cause notable knockdown of CD98 in both cell lines, indicating the dilution effect of drugs (siCD98 and CUR) after cell proliferation (Supplementary Information: Figure S4c).
Hydrogel encapsulation of HA-functionalized NPs for oral administration
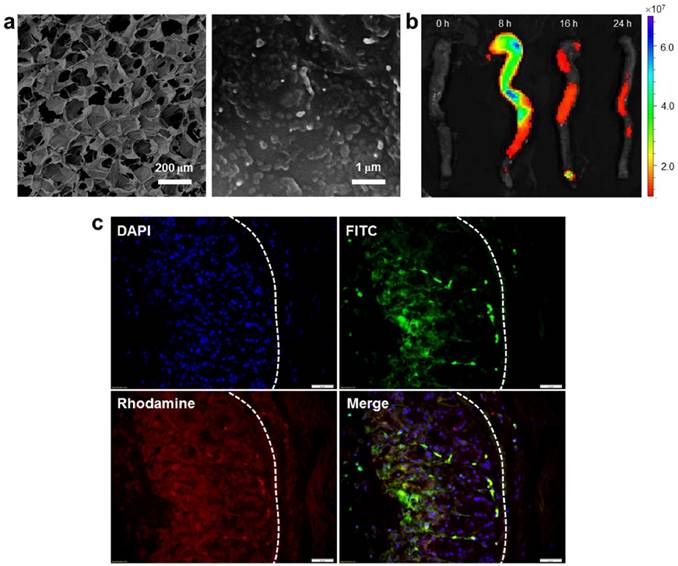
To enable the HA-functionalized NPs to be delivered to the colonic lumen, we encapsulated them in hydrogel comprised of chitosan and alginate at a weight ratio of 3:7. We first need to ensure that the NPs are uniformly dispersed in the chitosan/alginate hydrogel, which is essential for in vivo applications [47]. NPs were dispersed in the chitosan/alginate matrix, and the NH3+ of chitosan and the COO- of the alginate were chelated with a mixture of Ca2+ and SO42-. The NPs were encapsulated with the polysaccharide mixture and then freeze-dried. The obtained scaffold was cut transversely and processed for SEM. As shown in Figure 5a, SEM analysis revealed that the NPs were homogenously dispersed in the chitosan/alginate matrix without obvious cluster or agglomerate.
The uptake profiles of HA-functionalized NPs by colitis tissue. (a) Representative SEM images of the resulting hydrogel (chitosan and alginate) loaded with NPs. (b) Typical images of colon imaging showing accumulation of orally HA-functionalized NPs embedded in hydrogel in colon at four different time points (0, 8, 16 and 24 h). (c) Colitis tissue uptake of NPs after 12 h of oral administration of HA-functionalized NPs embedded in hydrogel (5 mg CUR/kg per mouse). Fixed colitis tissues were stained with Alexa Fluor 568 phalloidin and DAPI to visualize actin (red) and nuclei (purple), respectively. Scale bar represents 20 μm.
Colitis tissue uptake orally HA-functionalized NPs efficiently
To investigate the in vivo bio-distribution of HA-functionalized NPs in the GIT, we orally administered DSS-treated mice (a model of UC) with HA-DiR-NPs encapsulated in hydrogel, and examined the time-dependent passage and in vivo targeting efficacy of the formulations using near infrared fluorescence (NIRF) imaging. As shown in Figure. 5b and Figure S5 (Supplementary Information), a strong NIRF signal was observed in the small intestine and colon at 8 h after oral administration. This signal decreased gradually with time, indicating that rapid clearance occurred in the feces. As the fluorescence was relatively weak by 24 h post-administration, mice were gavaged daily during the treatment process.
Next, we evaluated the tissue bio-distribution of HA-functionalized NPs. HA-CUR-NPs encapsulated in hydrogel were orally administered to DSS-treated mice. Once the NPs were released from the hydrogel, they had to go through the mucus layer. As shown in Figure 5c, tissue cross sections revealed that almost all colonic epithelial cells had internalized the NPs (green) by 12 h after oral administration of HA-CUR-NPs. Importantly, we also found that large amounts of NPs penetrated deep into the mucosa and were taken up by the cells (e.g. macrophages).
Orally administered HA-functionalized NPs can help relieve DSS-induced UC in mice
The DSS-induced mouse model of UC is an easily generated and highly reproducible model that can be correlated with human UC [48, 49]. Here, we investigated whether oral administration of hydrogel-encapsulated HA-functionalized NPs could help relieve the clinical manifestation of DSS-induced UC in mice. Body weight loss is typically used as a main marker of the colitis phenotype. The changes of body weight among DSS-treated mouse groups were evaluated after administration of hydrogel loaded with different NPs. As seen in Figure 6a, all of the DSS-treated mouse groups exhibited body weight loss that peaked on day 11. The HA-siCD98/CUR-NP-treated group showed the smallest body weight loss on day 11, and thereafter showed the best body weight recovery among the three NP-treated groups. As shown in Figure 6b, fecal Lcn-2 levels significantly increased in all the mice groups after 6 days of DSS treatment. Furthermore, the fecal Lcn-2 levels in all the NP-treated groups were much lower than that of DSS-treated control group, and HA-siCD98/CUR-NPs-treated group exhibited the lowest fecal Lcn-2 level during the whole treatment process among the NP-treated groups.
Oral administration of HA-functionalized NPs embedded in hydrogel relieve DSS-induced UC in mice. (a) Mouse body weight over time, normalized as a percentage of day zero body weight and given as the mean of each treatment group. Arrows indicate the days of oral administration of drug-loaded NPs. (b) The real-time concentration of fecal Lcn-2. Fecal Lcn-2 levels were measured by ELISA. (c) Colonic MPO activity, spleen weight and colon length of mice in different treatment groups. The MPO results are expressed as units of MPO activity per gram of tissue. (d) The mRNA expression levels of CD98 and TNF-α in mice with different treatments. Each point represents the mean ± S.E.M. (n=5). Statistical significance was assessed using ANOVA test followed by a Bonferroni post-hoc test (*P<0.05 and **P<0.01).
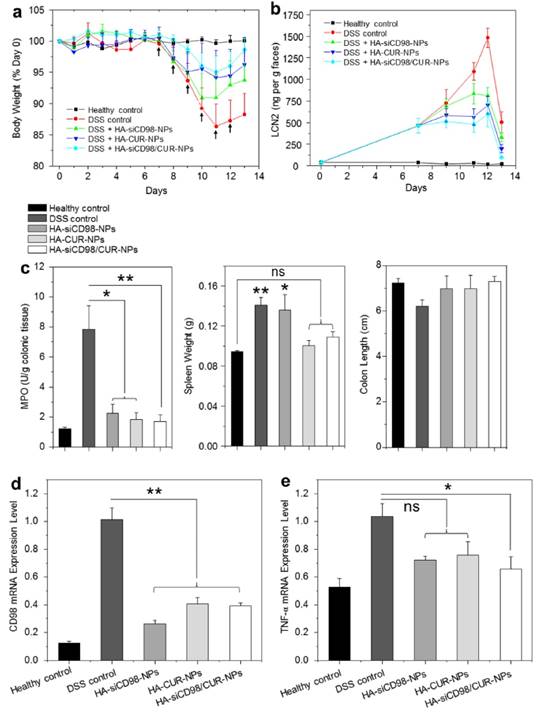
MPO activity is a direct indicator of the infiltration of neutrophils into the colonic mucosa, and is thus an important marker of inflammation during DSS treatment. As shown in Figure 6c, the colonic MPO activities were significantly lower in the three NP-treated groups than in the DSS-treated control group. DSS-treated control group has a significantly higher spleen weight compared to healthy control group. The spleen weights of the HA-CUR-NP- and HA-siCD98/CUR-NP-treated groups did not differ significantly from that in the healthy control group. Moreover, the colon lengths are not altered within the different groups. In terms of mRNA expression levels, we observed remarkable decrease of colonic CD98 expression (by 59.2% to 73.8%) in the treatment groups compared to the DSS-treated control group (Figure 6d). This was accompanied by notable suppression of the TNF-α mRNA expression levels (Figure 6e). Importantly, the levels of CD98 and TNF-α were much lower in the HA-siCD98/CUR-NP-treated group compared to the DSS-treated control group.
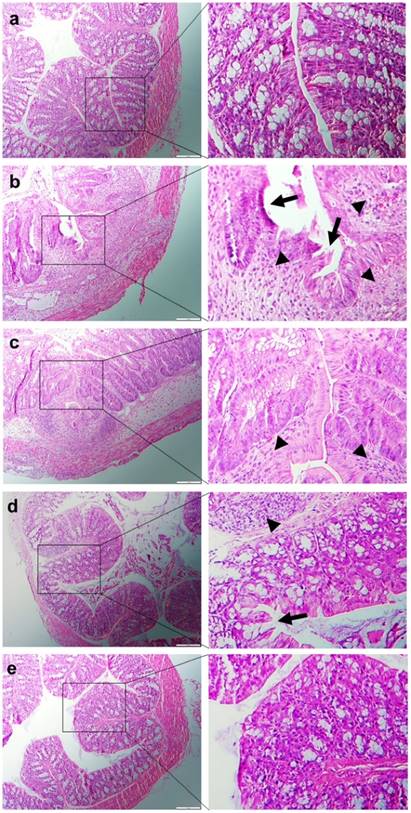
H&E-stained colon cryosections were evaluated for histological changes. Colon tissues from the healthy control group were characterized by normal colon histology, with no sign of inflammation or disruption of the healthy tissue morphology (Figure 7a). In contrast, colon tissues from the DSS-treated control group exhibited clear signs of inflammation, including epithelial disruption, goblet cell depletion, and significant infiltration of inflammatory cells into the mucosa (Figure 7b). However, tissues from the treatment groups showed much less inflammation (Figure 7c-e). Indeed, colon tissues from the HA-siCD98/CUR-NP-treated group exhibited almost the same tissue morphology as that observed in the healthy control group, especially with respect to the integration of the colonic epithelial layer and the infiltration of inflammatory cells (Figure 7e). The histological score of the DSS-treated control group was significantly higher than the other four mice groups (Supplementary Information: Figure S6). In addition, HA-siCD98/CUR-NP-treated group showed the lowest histological score among the three NP-treated groups. In summary, there results clearly demonstrated that HA-siCD98/CUR-NPs, which showed the highest therapeutic efficacy among the three tested NPs, could efficiently promote colon tissue recovery from UC.
Representative H&E-stained colon sections from DSS-treated mice administered with daily double gavages of hydrogel with different NPs for 6 days. Healthy control mice group (a), DSS-treated mice group (b), HA-siCD98-NPs-treated mice group (c), HA-CUR-NPs-treated mice group (d) and HA-siCD98/CUR-NPs-treated mice group (e). Arrow: erosion; arrowhead: lymphocyte infiltration. Scale bar represents 100 μm.
Discussion
To date, although several medications have proven to be effective in controlling UC, there is still no thorough and permanent curative [50]. Combination therapy based on a siRNA and a chemical drug in a single NP platform has recently shown synergistic therapeutic efficacy in the treatment of many important diseases, such as cancer, skin inflammation, and rheumatoid arthritis [51-54]. In this study, we developed an oral formulation (HA-functionalized NPs in hydrogel) for targeted co-delivery of a siRNA and a chemical drug to the key cells related to UC therapy (colonic epithelial cells and macrophages).
In the context of UC therapy via oral administration, drug-loaded NPs have to transport through GIT and be released into colonic lumen. In our previous studies, we have optimized the hydrogel (chitosan/alginate) matrix so that it can be administered via the oral route and collapse at colonic pH [10, 37, 55, 56]. Thus, NPs encapsulated into this hydrogel matrix could be primarily released in the colonic lumen rather than other segments of the GIT with harsh environment, such as the stomach or small intestine.
It is generally accepted that cells preferentially internalize NPs with the diameter less than 400 nm.[57, 58] Moreover, it has been shown that NPs with diameters less than 10 μm can potentially accumulate in colitis tissues based on the eEPR effect; this has been mainly ascribed to the histopathological abnormalities of colitis tissues, including the disrupted intestinal barrier function, increased epithelial permeability, and significant infiltration of inflammatory cells into the mucosa.[29] In addition, the inflamed mucosa is also characterized by accumulation of positively charged proteins. Langer and his co-workers developed a negatively charged hydrogel and demonstrated that their drug-loaded hydrogel preferentially adhered to the inflamed colonic mucosa [59]. Therefore, our HA-functionalized NPs with nanoscale particle size and negative surface charge (Figure 1a-c and Table 1) could faciliate their penetration into inflamed colon, adhesion to the positively charged inflamed tissue and further internalization into targeted cells.
To achieve active targeting, several receptor-specific ligands can drive colitis-targeted delivery, including lectin, transferrin and bacterial adhesins [29]. However, very few ligands have been developed that can simultaneously target colonic epithelial cells and macrophages (two types of key cells in UC therapy). Although we previously found that an antibody against CD98 could target NPs to these cell types [10] this strategy was proved to be relatively costly. Meanwhile, HA is a native ligand to CD44 which is a cell membrane glycoprotein highly overexpressed on the surface of colonic epithelial cells and macrophages in UC tissues.[29, 60] We have previously reported that surface functionalization with HA increases the cellular uptake efficiency of NPs through receptor-mediated interactions.[25] Our present results suggest that HA could be a good alternative for UC-targeted therapy, as it shows excellent targeting capability (Figure 2) and biocompatibility, and is relatively inexpensive. HA-siCD98/CUR-NPs were also found to accelerate mucosal healing and alleviate inflammation, which are the two main therapeutic purposes in UC therapy (Figure 3 and Figure 4). Most importantly, mice group treated with HA-siCD98/CUR-NPs-hydrogel (Figure 5) exhibited similar parameters (body weight, MPO activity, colon length, spleen weight, and histologic appearance) as healthy control mice group (Figure 6 and Figure 7), and even much better than the single drug-based treatment groups.
In comparison to the reported oral drug formulations for UC treatment, our HA-functionalized NP-hydrogel system has a number of beneficial properties as following. (1) Excellent biocompatibility. siRNA and CUR are well tolerated by GIT, and all the carrier polymers are generally recognized as safe materials, facilitating their rapid translation to clinical use; (2) Excellent stability. PLGA NPs allows their long-term storage and convenient use in the clinic; (3) Cell specificity. HA-functionalized NP-hydrogel system has the capacity to be delivered to the colon, accumulate in inflamed colonic sites, and further actively target to the key cells related to UC therapy (Scheme 1); (4) Easy production. The process for preparing this NP-hydrogel system is relatively simple and easy to scale up.
Conclusions
This study presented the first example of targeted and simultaneous delivery of a siRNA and a chemical drug via orally administered polymeric NPs for UC therapy. We conclude the following concerning the synergistic therapeutic effect of this combination therapy: (1) HA-functionalized NPs can realize targeted delivery of drugs to the key cells related to UC therapy (colonic epithelial cells and macrophages). (2) siCD98 and CUR can synergistically prevent mucosal damage and reduce inflammation. (3) Treatment with hydrogel-encapsulated HA-siCD98/CUR-NPs inhibits the DSS-induced over-expression of the genes encoding CD98 and TNF-α in the colon. (4) Orally administered hydrogel-encapsulated HA-siCD98/CUR-NPs exhibit a better therapeutic effect against UC compared to the single drug-based formulations. We believe that this effective and biocompatible formulation provides a promising approach for a synergistic combination therapy of UC.
Supplementary Material
Supplementary tables and figures.
Abbreviations
UC: ulcerative colitis; siCD98: CD98 siRNA; CUR: curcumin; HA: hyaluronic acid; NP: nanoparticle; LAT-1: L-type amino acid transporter 1; siRNA: small-interfering RNA; MPO: myeloperoxidase; eEPR: epithelial enhanced permeability and retention; PLGA: poly(lactic acid/glycolic acid); PVA: poly(vinyl alcohol); EDC: 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide; NHS: N-hydroxysuccinimide; MES: 2-(N-morpholino) ethanesulfonic acid sodium salt; LPS: lipopolysaccharide; MTT: 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide; FITC: fluorescein isothiocyanate; DAPI: 4ʹ,6-diamidino-2-phenyl-indole dihydrochloride; DiR: 1,1ʹ-diocta decyl-3,3,3ʹ,3ʹ-tetramethylindotricarbocyanine iodide; DSS: dextran sulphate sodium; DLS: dynamic light scattering; TEM: transmission electron microscopy; SEM: scanning electronic microscopy; FCM: flow cytometry; GIT: gastrointestinal tract; H&E: hematoxylin and eosin; NIRF: near infrared fluorescence.
Acknowledgements
This work was supported by grants from the Department of Veterans Affairs (BX002526), the National Institutes of Health of Diabetes and Digestive and Kidney by the grant (RO1-DK-071594), the Fellowship from Crohn's and Colitis Foundation of America (E.V and M.Z), the National Natural Science Foundation of China (51503172 and 81571807), the Fundamental Research Funds for the Central Universities (SWU114086 and XDJK2015C067) and the Scientific Research Foundation for the Returned Overseas Chinese Scholars (State Education Ministry). D.M. is a recipient of a Career Scientist Award from the Department of Veterans Affairs.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Gupta RB, Harpaz N, Itzkowitz S, Hossain S, Matula S, Kornbluth A. et al. Histologic inflammation is a risk factor for progression to colorectal neoplasia in ulcerative colitis: A cohort study. Gastroenterology. 2007;133:1099-105
2. Hang TTN, Dalmasso G, Torkvist L, Halfvarson J, Yan YT, Laroui H. et al. CD98 expression modulates intestinal homeostasis, inflammation, and colitis-associated cancer in mice. J Clin Invest. 2011;121:1733-47
3. Abraham C, Cho JH. Mechanisms of disease: Inflammatory Bowel Disease. New Engl J Med. 2009;361:2066-78
4. Xiao B, Laroui H, Ayyadurai S, Viennois E, Charania MA, Zhang YC. et al. Mannosylated bioreducible nanoparticle-mediated macrophage-specific TNF-alpha RNA interference for IBD therapy. Biomaterials. 2013;34:7471-82
5. Laroui H, Theiss AL, Yan Y, Dalmasso G, Nguyen HT, Sitaraman SV. et al. Functional TNFalpha gene silencing mediated by polyethyleneimine/TNFalpha siRNA nanocomplexes in inflamed colon. Biomaterials. 2011;32:1218-28
6. Yan YT, Vasudevan S, Nguyen HTT, Merlin D. Intestinal epithelial CD98: An oligomeric and multifunctional protein. Biochim Biophys Acta. 2008;1780:1087-92
7. Hang TTN, Merlin D. Homeostatic and innate immune responses: role of the transmembrane glycoprotein CD98. Cell Mol Life Sci. 2012;69:3015-26
8. Kucharzik T, Lugering A, Yan Y, Driss A, Charrier L, Sitaraman S. et al. Activation of epithelial CD98 glycoprotein perpetuates colonic inflammation. Lab Invest. 2005;85:932-41
9. Cantor JM, Ginsberg MH. CD98 at the crossroads of adaptive immunity and cancer. J Cell Sci. 2012;125:1373-82
10. Xiao B, Laroui H, Viennois E, Ayyadurai S, Charania MA, Zhang Y. et al. Nanoparticles with surface antibody against CD98 and carrying CD98 small interfering RNA reduce colitis in mice. Gastroenterology. 2014;146:1289-300
11. Pithadia AB, Jain S. Treatment of inflammatory bowel disease (IBD). Pharmacol Rep. 2011;63:629-42
12. Bang YJ, Van Cutsem E, Feyereislova A, Chung HC, Shen L, Sawaki A. et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet. 2010;376:687-97
13. Home PD, Pocock SJ, Beck-Nielsen H, Curtis PS, Gomis R, Hanefeld M. et al. Rosiglitazone evaluated for cardiovascular outcomes in oral agent combination therapy for type 2 diabetes (RECORD): a multicentre, randomised, open-label trial. Lancet. 2009;373:2125-35
14. Park JS, Yang HN, Jeon SY, Woo DG, Kim MS, Park KH. The use of anti-COX2 siRNA coated onto PLGA nanoparticles loading dexamethasone in the treatment of rheumatoid arthritis. Biomaterials. 2012;33:8600-12
15. Sun TM, Du JZ, Yao YD, Mao CQ, Dou S, Huang SY. et al. Simultaneous Delivery of siRNA and Paclitaxel via a "Two-in-One" Micelleplex Promotes Synergistic Tumor Suppression. ACS nano. 2011;5:1483-94
16. Deng ZJ, Morton SW, Ben-Akiva E, Dreaden EC, Shopsowitz KE, Hammond PT. Layer-by-Layer Nanoparticles for Systemic Codelivery of an Anticancer Drug and siRNA for Potential Triple-Negative Breast Cancer Treatment. ACS nano. 2013;7:9571-84
17. Xu XY, Xie K, Zhang XQ, Pridgen EM, Park GY, Cui DS. et al. Enhancing tumor cell response to chemotherapy through nanoparticle-mediated codelivery of siRNA and cisplatin prodrug. Proc Natl Acad Sci U S A. 2013;110:18638-43
18. Ali T, Shakir F, Morton J. Curcumin and inflammatory bowel disease: biological mechanisms and clinical implication. Digestion. 2012;85:249-55
19. Pu HL, Chiang WL, Maiti B, Liao ZX, Ho YC, Shim MS. et al. Nanoparticles with Dual Responses to Oxidative Stress and Reduced pH for Drug Release and Anti-inflammatory Applications. ACS nano. 2014;8:1213-21
20. Gupta SC, Patchva S, Aggarwal BB. Therapeutic Roles of Curcumin: Lessons Learned from Clinical Trials. AAPS J. 2013;15:195-218
21. Xiong XB, Lavasanifar A. Traceable Multifunctional Micellar Nanocarriers for Cancer-Targeted Co-delivery of MDR-1 siRNA and Doxorubicin. ACS nano. 2011;5:5202-13
22. Biswas S, Deshpande PP, Navarro G, Dodwadkar NS, Torchilin VR. Lipid modified triblock PAMAM-based nanocarriers for siRNA drug co-delivery. Biomaterials. 2013;34:1289-301
23. Dong DW, Xiang B, Gao W, Yang ZZ, Li JQ, Qi XR. pH-responsive complexes using prefunctionalized polymers for synchronous delivery of doxorubicin and siRNA to cancer cells. Biomaterials. 2013;34:4849-59
24. Zhao J, Mi Y, Feng SS. Targeted co-delivery of docetaxel and siPlk1 by herceptin-conjugated vitamin E TPGS based immunomicelles. Biomaterials. 2013;34:3411-21
25. Xiao B, Han MK, Viennois E, Wang L, Zhang M, Si X. et al. Hyaluronic acid-functionalized polymeric nanoparticles for colon cancer-targeted combination chemotherapy. Nanoscale. 2015;7:17745-55
26. Xiao B, Yang Y, Viennois E, Zhang YC, Ayyadurai S, Baker MT. et al. Glycoprotein CD98 as a receptor for colitis-targeted delivery of nanoparticles. J Mater Chem B Mater Biol Med. 2014;2:1499-508
27. Xiao B, Merlin D. Oral colon-specific therapeutic approaches toward treatment of inflammatory bowel disease. Expert Opin Drug Deliv. 2012;9:1393-407
28. Lamprecht A. IBD Selective nanoparticle adhesion can enhance colitis therapy. Nat Rev Gastro Hepat. 2010;7:311-2
29. Fromont Hankard G, Cezard JP, Aigrain Y, Navarro J, Peuchmaur M. CD44 variant expression in inflammatory colonic mucosa is not disease specific but associated with increased crypt cell proliferation. Histopathology. 1998;32:317-21
30. Farkas S, Hornung M, Sattler C, Anthuber M, Gunthert U, Herfarth H. et al. Short-term treatment with anti-CD44v7 antibody, but not CD44v4, restores the gut mucosa in established chronic dextran sulphate sodium (DSS)-induced colitis in mice. Clin Exp Immunol. 2005;142:260-7
31. Dreaden EC, Morton SW, Shopsowitz KE, Choi JH, Deng ZJ, Cho NJ. et al. Bimodal Tumor-Targeting from Microenvironment Responsive Hyaluronan Layer-by-Layer (LbL) Nanoparticles. ACS nano. 2014;8:8374-82
32. Lavertu M, Methot S, Tran-Khanh N, Buschmann MD. High efficiency gene transfer using chitosan/DNA nanoparticles with specific combinations of molecular weight and degree of deacetylation. Biomaterials. 2006;27:4815-24
33. Badawy MEI, Rabea EI. Potential of the biopolymer chitosan with different molecular weights to control postharvest gray mold of tomato fruit. Postharvest Biol Technol. 2009;51:110-7
34. Xiao B, Si X, Han MK, Viennois E, Zhang M, Merlin D. Co-delivery of camptothecin and curcumin by cationic polymeric nanoparticles for synergistic colon cancer combination chemotherapy. J Mater Chem B Mater Biol Med. 2015;3:7724-33
35. Cun D, Jensen DK, Maltesen MJ, Bunker M, Whiteside P, Scurr D. et al. High loading efficiency and sustained release of siRNA encapsulated in PLGA nanoparticles: quality by design optimization and characterization. Eur J Pharm Biopharm. 2011;77:26-35
36. Cun D, Foged C, Yang M, Frokjaer S, Nielsen HM. Preparation and characterization of poly(DL-lactide-co-glycolide) nanoparticles for siRNA delivery. Int J Pharm. 2010;390:70-5
37. Laroui H, Geem D, Xiao B, Viennois E, Rakhya P, Denning T. et al. Targeting intestinal inflammation with CD98 siRNA/PEI-loaded nanoparticles. Mol Ther. 2014;22:69-80
38. Cohen-Sela E, Chorny M, Koroukhov N, Danenberg HD, Golomb G. A new double emulsion solvent diffusion technique for encapsulating hydrophilic molecules in PLGA nanoparticles. J Control Release. 2009;133:90-5
39. Mora-Huertas CE, Fessi H, Elaissari A. Influence of process and formulation parameters on the formation of submicron particles by solvent displacement and emulsification-diffusion methods Critical comparison. Adv Colloid Interface Sci. 2011;163:90-122
40. Chen YF, Rosenzweig Z. Luminescent CdSe quantum dot doped stabilized micelles. Nano Lett. 2002;2:1299-302
41. Mu L, Feng SS. PLGA/TPGS nanoparticles for controlled release of paclitaxel: Effects of the emulsifier and drug loading ratio. Pharm Res. 2003;20:1864-72
42. Hassan CM, Peppas NA. Structure and applications of poly(vinyl alcohol) hydrogels produced by conventional crosslinking or by freezing/thawing methods. Adv Polym Sci. 2000;153:37-65
43. Mora-Huertas CE, Fessi H, Elaissari A. Influence of process and formulation parameters on the formation of submicron particles by solvent displacement and emulsification-diffusion methods critical comparison. Adv Colloid Interface Sci. 2011;163:90-122
44. Xiao B, Zhang M, Viennois E, Zhang Y, Wei N, Baker MT. et al. Inhibition of MDR1 gene expression and enhancing cellular uptake for effective colon cancer treatment using dual-surface-functionalized nanoparticles. Biomaterials. 2015;48:147-60
45. MacDermott RP. Chemokines in the inflammatory bowel diseases. J Clin Immunol. 1999;19:266-72
46. Lewis K, Caldwell J, Phan V, Prescott D, Nazli A, Wang A. et al. Decreased epithelial barrier function evoked by exposure to metabolic stress and nonpathogenic E. coli is enhanced by TNF-alpha. Am J Physiol Gastrointest Liver Physiol. 2008;294:G669-78
47. Theiss AL, Laroui H, Obertone TS, Chowdhury I, Thompson WE, Merlin D. et al. Nanoparticle-based therapeutic delivery of prohibitin to the colonic epithelial cells ameliorates acute murine colitis. Inflamm Bowel Dis. 2011;17:1163-76
48. Perse M, Cerar A. Dextran sodium sulphate colitis mouse model: traps and tricks. J Biomed Biotechnol. 2012;2012:718617
49. Grisham MB. Do different animal models of IBD serve different purposes? Inflamm Bowel Dis. 2008;14:S132-3
50. Patel MV, Patel KB, Gupta SN. Effects of Ayurvedic treatment on forty-three patients of ulcerative colitis. Ayu. 2010;31:478-81
51. Yang T, Li B, Qi S, Liu Y, Gai Y, Ye P. et al. Co-delivery of doxorubicin and Bmi1 siRNA by folate receptor targeted liposomes exhibits enhanced anti-tumor effects in vitro and in vivo. Theranostics. 2014;4:1096-111
52. Yu H, Guo C, Feng B, Liu J, Chen X, Wang D. et al. Triple-Layered pH-Responsive Micelleplexes Loaded with siRNA and Cisplatin Prodrug for NF-Kappa B Targeted Treatment of Metastatic Breast Cancer. Theranostics. 2016;6:14-27
53. Park JS, Yang HN, Jeon SY, Woo DG, Kim MS, Park KH. The use of anti-COX2 siRNA coated onto PLGA nanoparticles loading dexamethasone in the treatment of rheumatoid arthritis. Biomaterials. 2012;33:8600-12
54. Desai PR, Marepally S, Patel AR, Voshavar C, Chaudhuri A, Singh M. Topical delivery of anti-TNFalpha siRNA and capsaicin via novel lipid-polymer hybrid nanoparticles efficiently inhibits skin inflammation in vivo. J Control Release. 2013;170:51-63
55. Laroui H, Dalmasso G, Nguyen HT, Yan Y, Sitaraman SV, Merlin D. Drug-loaded nanoparticles targeted to the colon with polysaccharide hydrogel reduce colitis in a mouse model. Gastroenterology. 2010;138:843-53
56. Laroui H, Viennois E, Xiao B, Canup BS, Geem D, Denning TL. et al. Fab'-bearing siRNA TNFalpha-loaded nanoparticles targeted to colonic macrophages offer an effective therapy for experimental colitis. J Control Release. 2014;186:41-53
57. Kim TH, Ihm JE, Choi YJ, Nah JW, Cho CS. Efficient gene delivery by urocanic acid-modified chitosan. J Control Release. 2003;93:389-402
58. Liu Y, Reineke TM. Hydroxyl stereochemistry and amine number within poly(glycoamidoamine)s affect intracellular DNA delivery. J Am Chem Soc. 2005;127:3004-15
59. Zhang S, Ermann J, Succi MD, Zhou A, Hamilton MJ, Cao B. et al. An inflammation-targeting hydrogel for local drug delivery in inflammatory bowel disease. Sci Transl Med. 2015;7:300ra128
60. Farkas S, Hornung M, Sattler C, Anthuber M, Gunthert U, Herfarth H. et al. Short-term treatment with anti-CD44v7 antibody, but not CD44v4, restores the gut mucosa in established chronic dextran sulphate sodium (DSS)-induced colitis in mice. Clin Exp Immunol. 2005;142:260-7
Author contact
![]() Corresponding author: Bo Xiao, Ph.D., Institute for Clean Energy and Advanced Materials, Faculty of Materials and Energy, Southwest University, Chongqing, 400715, P. R. China. Email: hustboxiaocom or bxiaoedu; Tel: +86-23-6825-4762; Fax: +86-23-6825-4969.
Corresponding author: Bo Xiao, Ph.D., Institute for Clean Energy and Advanced Materials, Faculty of Materials and Energy, Southwest University, Chongqing, 400715, P. R. China. Email: hustboxiaocom or bxiaoedu; Tel: +86-23-6825-4762; Fax: +86-23-6825-4969.