Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Results
Discussion
Materials and Methods
Abbreviations
Supplementary Material
Acknowledgements
References
Introduction
Results
Discussion
Materials and Methods
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(10):2754-2767. doi:10.7150/thno.33395 This issue Cite
Research Paper
The Circadian Clock Gene Bmal1 Controls Intestinal Exporter MRP2 and Drug Disposition
Fangjun Yu1, Tianpeng Zhang1,2, Cui Zhou1, Haiman Xu1, Lianxia Guo1, Min Chen1, Baojian Wu1 ![]()
1. Research Center for Biopharmaceutics and Pharmacokinetics, College of Pharmacy, Jinan University, 601 Huangpu Avenue West, Guangzhou, China.
2. Integrated Chinese and Western Medicine Postdoctoral research station, Jinan University, 601 Huangpu Avenue West, Guangzhou, China
Received 2019-1-22; Accepted 2019-3-31; Published 2019-4-13
Citation:
Yu F, Zhang T, Zhou C, Xu H, Guo L, Chen M, Wu B. The Circadian Clock Gene Bmal1 Controls Intestinal Exporter MRP2 and Drug Disposition. Theranostics 2019; 9(10):2754-2767. doi:10.7150/thno.33395. https://www.thno.org/v09p2754.htm
Other stylesAbstract

The intestinal exporter MRP2 plays an important role in disposition and elimination of a wide range of drugs. Here, we aimed to clarify the impact of circadian clock on intestinal MRP2, and to determine the molecular mechanisms for generation of diurnal MRP2 expression.
Methods: The regulatory effects of Bmal1 on intestinal MRP2 expression were assessed using intestine-specific Bmal1 knockout (Bmal1iKO) mice and colon cancer cells. The relative mRNA and protein levels were determined by qPCR and Western blotting, respectively. Everted gut sac, cell viability and in situ intestinal perfusion experiments were performed to evaluate intestinal efflux of the MRP2 substrate methotrexate (MTX). Toxicity and pharmacokinetic experiments were performed with Bmal1iKO mice and control littermates (Bmal1fl/fl mice) after oral gavage of MTX. Transcriptional gene regulation was investigated using luciferase reporter, mobility shift and chromatin immunoprecipitation (ChIP) assays.
Results: Bmal1iKO mice were generated by inter-crossing the mice carrying a Bmal1 exon 8 floxed allele (Bmal1fl/fl) with Villin-Cre mice. Intestinal MRP2 expression exhibited a diurnal oscillation in Bmal1fl/fl mice with a zenith value at ZT6. Bmal1 ablation caused reductions in Mrp2 mRNA and protein levels [as well as in transport activity (measured by MTX)], and blunted their diurnal rhythms. Intestinal ablation of Bmal1 abrogated circadian time-dependency of MTX pharmacokinetics and toxicity. Bmal1/BMAL1 regulation of rhythmic Mrp2/MRP2 expression was also confirmed in the colon cancer CT26 and Caco-2 cells. Based on a combination of luciferase reporter, mobility shift and ChIP assays, we found that Dbp activated and E4bp4 repressed Mrp2 transcription via specific binding to a same D-box (-100/-89 bp) element in promoter region. Further, Bmal1 directly activated the transcription of Dbp and Rev-erbα through the E-boxes, whereas it negatively regulated E4bp4 via the transcriptional repressor Rev-erbα. Positive regulation of Mrp2 by Rev-erbα was also observed, and attained through modulation of E4bp4.
Conclusion: Bmal1 coordinates temporal expressions of DBP (a MRP2 activator), REV-ERBα (an E4BP4 repressor) and E4BP4 (a MRP2 repressor), generating diurnal MRP2 expression.
Keywords: Bmal1, MRP2, Dbp, E4bp4, MTX, Chronotoxicity
Introduction
Mammalian circadian clock drives the expressions of thousands of genes (i.e., ''clock-controlled genes'') and regulates circadian rhythms in many biochemical and physiological processes [1,2]. Defects in circadian clock have been associated with various diseases such as diabetes, obesity, and cancers [3,4]. At the molecular level, circadian clock essentially is a transcriptional regulatory loop system [5]. The central loop consists of BMAL1/CLOCK as the positive limb (activates gene transcription) and PER/CRY as the negative limb (inhibits gene transcription) [6,7,8]. In addition to regulating circadian gene expression, BMAL1 plays important roles in regulation of many other physiological processes such as lipogenesis, fatty acid oxidation, amino acid metabolism, and immune responses [9, 10, 11, 12]. Several studies have shown that circadian clock is present in small intestine and colon, where it regulates cell growth and proliferation [13,14,15,16].
Small intestine represents the first line of oral drug detoxification (but the first barrier to oral drug absorption). Intestinal detoxification system mainly consists of xenobiotic-metabolizing enzymes [XMEs, e.g., cytochromes P450 (CYPs)] and efflux transporters (also known as exporters) [17]. The former converts drug molecules into inactive metabolites (note that the pro-drug is an exception), whereas the latter blocks intestinal uptake to the body by expelling the drug substrates to the gut lumen. The multidrug resistance proteins (MRPs/ABCCs) are the C subfamily proteins of the superfamily of ATP-binding cassette (ABC) exporters [18]. MRP2 is an important MRP protein best known for its apical membrane location in enterocytes and hepatocytes [19]. This unique location supports its critical roles in intestinal defense against xenobiotic threats and in biliary drug elimination [20,21].
Circadian clock has been implicated in regulation of drug metabolism and detoxification [22,23]. This regulation results in dosing time-dependency of drug tolerance/toxicity and efficacy [22,23,24,25]. For instance, the hepatotoxicity of acetaminophen was more severe during the night than the daytime [22]. The circadian knowledge can be exploited to optimize drug treatment outcomes (an area of chronotherapeutics) [26]. Circadian clock regulates drug detoxification through modulation of expressions of XMEs (e.g., CYP2B10 and CYP2E1) and efflux transporters (e.g., P-glycoprotein/P-gp) [23,27]. Several clock output genes [e.g., the three proline- and acid-rich (PAR) basic leucine zipper (bZIP) proteins HLF (hepatic leukemia factor), TEF (thyrotroph embryonic factor) and DBP (D-site binding protein) as well as E4BP4 (E4 promoter-binding protein 4)] have been shown to play a role in generation of circadian expression of CYP2B10 and P-gp [23,28]. However, whether and how the central clock genes such as BMAL1 and CLOCK regulate XMEs and transporters remain largely unknown.
Methotrexate (MTX) is an inhibitor of dihydrofolate reductase (DHFR) and used to treat neoplastic cancers and autoimmune diseases (e.g., rheumatoid arthritis and psoriasis) [29,30]. However, the dose and duration of MTX treatment are severely limited by hepatic and renal toxicities [30,31]. Therefore, it is of great interest to identify the influencing factors for MTX-induced toxicity. An early study shows circadian rhythms in MTX toxicity and pharmacokinetics despite the lack of mechanistic explorations [32]. This circadian time differences might be applied to down-regulate MTX-induced toxicity and to improve the therapeutic efficacy [32]. Multiple transporters are involved in intestinal disposition of MTX. Proton-coupled folate transporter (PCFT/SLC46A1) and reduced folate carrier-1 (RFC-1/SLC19A1) participate in intestinal uptake of MTX, whereas MRP2 limits MTX absorption by blocking entrance into enterocytes [33,34]. Of note, intestinal expression of MRP2 exhibits a circadian oscillation [35]. However, whether and how intestinal circadian MRP2 contributes to the rhythms in MTX toxicity and kinetics remain unknown.
To clarify the impact of circadian clock on intestinal MRP2, we established intestine-specific Bmal1 knockout (Bmal1iKO) mice in this study. Intestinal ablation of Bmal1 disrupted diurnal expression of MRP2, and abrogated the circadian time-dependency of MTX (a MRP2 substrate) absorption and toxicity. Cell-based assays confirmed positive regulation of MRP2 by Bmal1. Mechanistic studies revealed that Bmal1 regulated intestinal MRP2 expression through DBP and REV-ERBα/E4BP4 axis. Our study resolved the molecular links between circadian clock and intestinal MRP2, thereby providing an avenue for management of MTX toxicity.
Results
Generation of genetic mice lacking intestinal Bmal1 (Bmal1iKO mice)
Bmal1iKO mice were generated by inter-crossing the mice carrying a Bmal1 exon 8 floxed allele (Bmal1fl/fl) with Villin-Cre mice. The genetic mice were first validated by PCR genotyping using mouse tails (Figure 1A). Bmal1iKO mice showed recombination of loxP sites and Villin-Cre recombinase (Figure 1A). qPCR and Western blot assays confirmed the absence of intestinal Bmal1 in Bmal1iKO mice (Figure 1B/C & Supplementary Figure 1A). However, the mRNA expression of Bmal1 were unaffected in other tissues such as the liver, kidney and brain (Figure 1B). Bmal1iKO mice were phenotypically normal, with no signs of defects in morphology, proliferation, and apoptosis of intestinal epithelial cells (Figure 1D and E). The mice also have normal expressions of the tight junction proteins zonula occludens-1 (ZO-1) and occludin, suggesting intact intestinal barrier structure (Figure 1F).
Disrupted diurnal expression of intestinal MRP2 in Bmal1iKO mice
Intestinal Mrp2 mRNA exhibited a diurnal oscillation in Bmal1fl/fl mice with a zenith value at ZT6 (Figure 2A). Similarly, a diurnal time-dependent expression oscillation was observed for intestinal MRP2 protein (Figure 2B). Bmal1 ablation caused reductions in Mrp2 mRNA and protein levels, and blunted their diurnal rhythms (Figure 2A/B). The immunofluorescence assays confirmed reduced MRP2 protein in Bmal1iKO mice as compared to the control littermates (Figure 2C). In everted gut sac experiments, the mucosal-to-serosal transport of and intestinal accumulation of methotrexate (MTX, a well-known MRP2 substrate) were significantly enhanced because of Bmal1 deletion (Figure 2D). In situ intestinal perfusion showed higher levels of MTX absorption and of tissue accumulation in Bmal1iKO mice than in control littermates (Figure 2E). These data indicated regulation of MRP2 expression and activity in the intestine by Bmal1.
Figure 1
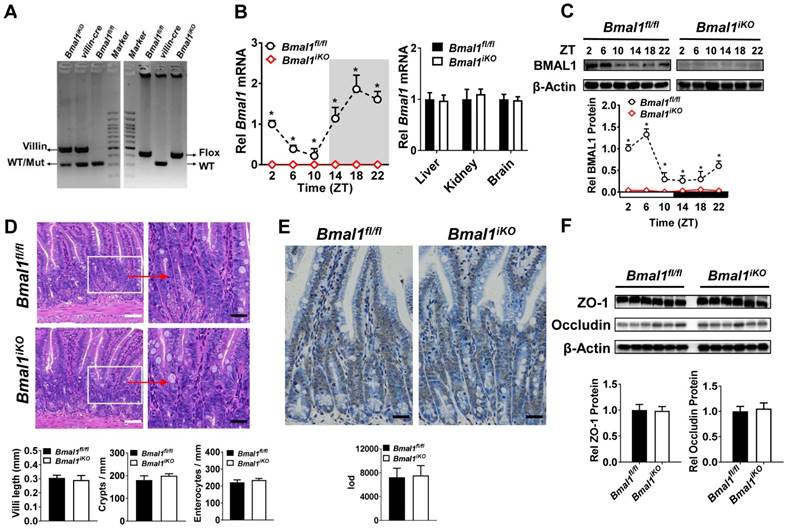
Establishment of genetic mice lacking intestinal Bmal1. (A) Mouse genotyping results generated from agarose gel electrophoresis. Bmal1iKO mice showed recombination of loxP sites and Villin-Cre recombinase. (B) qRT-PCR analysis of Bmal1 transcript abundance in small intestine and other control tissues of Bmal1fl/fl and Bmal1iKO mice. Data are presented as the fold change in Bmal1 expression normalized to Hmbs and relative to ZT2 of Bmal1fl/fl mice. Data are mean ± SD (n = 6). *p < 0.05 (Two-way ANOVA with Bonferroni post hoc test). (C) Western bolt analysis of BMAL1 protein in small intestine of Bmal1fl/fl and Bmal1iKO mice. Data are mean ± SD (n = 6). *p < 0.05 (Two-way ANOVA with Bonferroni post hoc test). (D) Representative hematoxylin and eosin (H&E) staining of small intestine from Bmal1fl/fl and Bmal1iKO mice. Bmal1iKO mice have normal morphology in small intestine. Scale bar (black) = 100 μm, scale bar (white) = 500 μm. (E) Apoptosis was determined by TUNEL staining in small intestine of Bmal1fl/fl and Bmal1iKO mice. The positive apoptotic nuclei are indicated by integral optical density (IOD). Scale bar (black) = 100 μm. (F) Western blot analysis of tight junction proteins zonula occludens-1 (ZO-1) and occludin in small intestine of Bmal1fl/fl and Bmal1iKO mice. Data are mean ± SD (n = 6).
Intestinal ablation of Bmal1 abrogates circadian time-dependency of MTX pharmacokinetics and toxicity
We further preformed in vivo MTX pharmacokinetic and toxicity experiments to clarify the impact of Bmal1 on intestinal MRP2 expression and activity. As expected, pharmacokinetic behavior of MTX (oral gavage, 50 mg/kg) was dependent on circadian dosing time in Bmal1fl/fl mice (Figure 3A). The AUC (area under the curve, representing systemic exposure and the extent of drug absorption) value and tissue (intestinal and extraintestinal) accumulation were higher at dosing time of ZT14 than at ZT2 (Figure 3B/C and Supplementary Figure 2A/B). This was accounted for by a lower intestinal MRP2 (an efflux pump that limits MTX absorption) expression at ZT14 than at ZT2 (Figure 2A/B). Intestinal ablation of Bmal1 led to enhancement of MTX absorption (increased AUC values) and abrogated dosing time differences in its absorption (Figure 3A/B). Likewise, intestinal and extraintestinal accumulation of MTX were increased and their dosing time-dependency were lost in Bmal1iKO mice (Figure 3C and Supplementary Figure 2A/B).
Oral gavage of MTX (1000 mg/kg) induced hepatoxicity [reflected by alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels] and nephrotoxicity [reflected by blood urea nitrogen (BUN) and serum creatinine (Scr) levels] in Bmal1fl/fl mice (pilot experiments were conducted initially to ascertain the effect of the toxin in control mice) and the toxicities were dependent on circadian dosing time (more severe toxicity at ZT14 than at ZT2) (Figure 3D-G, left panels). Intestinal ablation of Bmal1 exacerbated MTX-induced toxicities and abrogated the dosing time differences in MTX toxicities (Figure 3D-G, right panels). In addition, Bmal1fl/fl mice showed differential responses (dependent on circadian time of dosing) in the survival rate after MTX administration (Figure 3H). A higher mortality (but not statistically significant) was observed at dosing time of ZT14 (Figure 3H). Intestinal ablation of Bmal1 increased the mortality rates and abrogated the time differences in mortality (Figure 3H). Taken together, Bmal1 regulates the diurnal oscillation of MRP2, thereby impacting the circadian time-dependent pharmacokinetic and toxicity of the MRP2 substrate MTX.
Figure 2
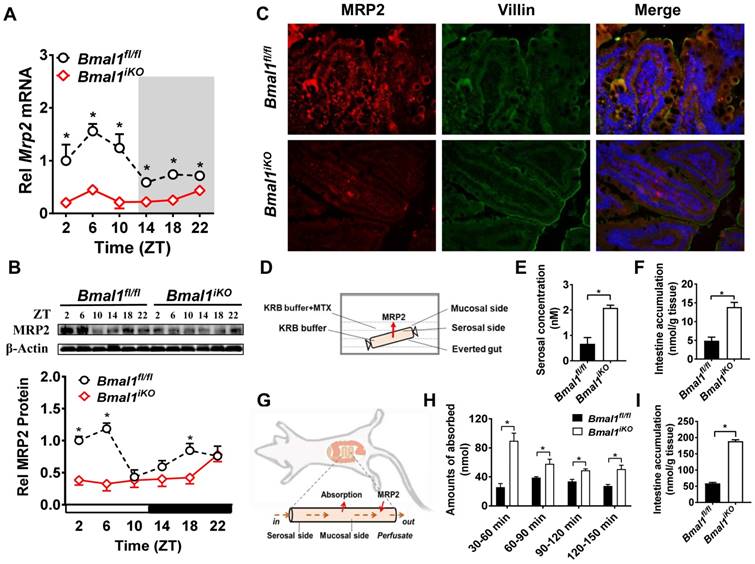
Intestine-specific deletion of Bmal1 disrupted diurnal expression of Mrp2. (A) qRT-PCR analysis of Mrp2 mRNA in small intestine of Bmal1fl/fl and Bmal1iKO mice. Data are presented as the fold change in Mrp2 expression normalized to Hmbs and relative to ZT2 of Bmal1fl/fl mice. Data are mean ± SD (n = 6). *p < 0.05 (Two-way ANOVA with Bonferroni post hoc test). (B) Western blot analysis of MRP2 protein in small intestine of Bmal1fl/fl and Bmal1iKO mice. Data are mean ± SD (n = 6). *p < 0.05 (Two-way ANOVA with Bonferroni post hoc test). (C) Double immunofluorescence staining of MRP2 (red) and Villin (green) in small intestine of Bmal1fl/fl and Bmal1iKO mice at ZT2. (D) Schematic diagram illustrating the everted intestinal sac experiment. (E) Bmal1 ablation increases MTX transport from mucosal to serosal in everted intestinal sac model with 20 μM MTX. Data are mean ± SD (n = 3). *p < 0.05 (t test). (F) Bmal1 ablation increases intestine MTX accumulation in everted intestinal sac model with 20 μM MTX. Data are mean ± SD (n = 3). *p < 0.05 (t test). (G) Schematic diagram for in situ single-pass intestinal perfusion experiments. (H) The absorption of MTX in in situ single-pass intestinal perfusion with 20 μM MTX in Bmal1fl/fl and Bmal1iKO mice. Data are mean ± SD (n = 5). *p < 0.05 (Two-way ANOVA with Bonferroni post hoc test). (I) MTX accumulation in small intestine of Bmal1fl/fl and Bmal1iKO mice in in situ single-pass intestinal perfusion with 20 μM MTX. Data are mean ± SD (n = 5). *p < 0.05 (t test).
Bmal1 (BMAL1) regulates Mrp2 (MRP2) expression in colon cancer cells
Overexpression of Bmal1 (BMAL1) led to elevations in Mrp2 (MRP2) mRNA and protein in CT26 and Caco-2 cells (Figure 4A/B). Knockdown of Bmal1 (BMAL1) by siRNA resulted in decreased levels of Mrp2 (MRP2) mRNA and protein in the cells (Figure 4C/D). In line with down-regulated MRP2 expression, Bmal1 knockdown led to a significant increase in cellular uptake of MTX at different time points (Figure 4E). Increased cellular uptake was also observed at a wide range of MTX concentrations (25-100 μM) (Figure 4E). Overexpression of Bmal1 increased the resistance of CT26 cells to MTX toxicity, whereas Bmal1 knockdown sensitized the cells to MTX toxicity (Figure 4F). Furthermore, a serum shock induced rhythmic expression of Mrp2 in CT26 cells (Figure 4G). Bmal1 silencing decreased Mrp2 expression, and blunted its rhythmicity in serum-shocked CT26 cells (Figure 4G). In addition, Dbp and Rev-erbα were downregulated and their rhythmicities were blunted. E4bp4 was upregulated and its rhythmicity was dampened (Supplementary Figure 3). Taken together, the cell data supported that Bmal1 positively regulates circadian Mrp2 expression.
Figure 3
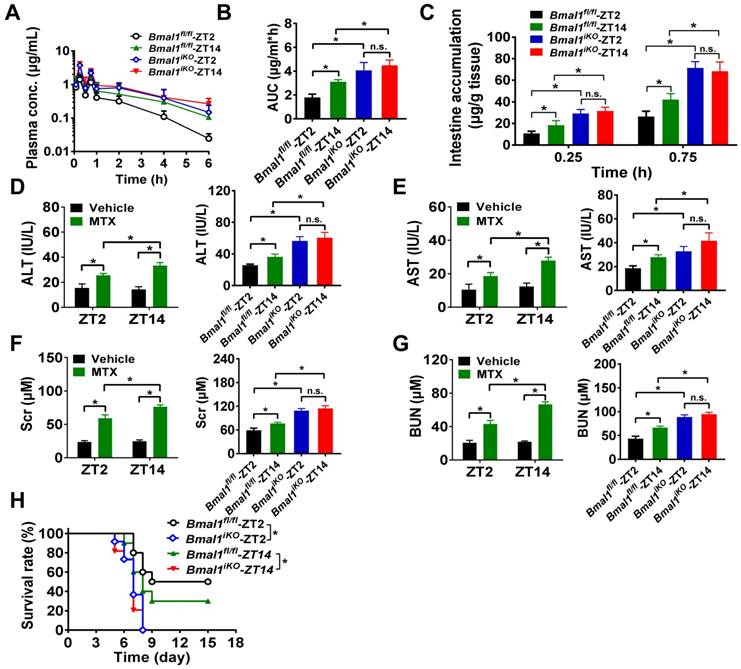
Intestinal ablation of Bmal1 abrogates time-dependency of MTX pharmacokinetics and toxicity. (A) Time profiles of the plasma concentration of MTX after oral administration (50 mg/kg) at ZT2 and ZT14 in Bmal1fl/fl and Bmal1iKO mice. Data are mean ± SD (n = 5). (B) The AUC value after oral administration of MTX (50 mg/kg) at dosing time of ZT2 and ZT14 in Bmal1fl/fl and Bmal1iKO mice. Data are mean ± SD (n = 5). (C) The intestinal accumulation of MTX after oral administration (50 mg/kg) at dosing time of ZT2 and ZT14 in Bmal1fl/fl and Bmal1iKO mice. Data are mean ± SD (n = 5). (D) Serum ALT levels in Bmal1fl/fl and Bmal1iKO mice 24 h after vehicle or MTX administration (1000 mg/kg, i.g., n = 5) at ZT2 and ZT14. (E) Serum AST levels in Bmal1fl/fl and Bmal1iKO mice 24 h after vehicle or MTX administration (1000 mg/kg, i.g., n = 5) at ZT2 and ZT14. (F) Serum Scr levels in Bmal1fl/fl and Bmal1iKO mice 24 h after vehicle or MTX administration (1000 mg/kg, i.g., n = 5) at ZT2 and ZT14. (G) Serum BUN levels in Bmal1fl/fl and Bmal1iKO mice 24 h after vehicle or MTX administration (1000 mg/kg, i.g., n = 5) at ZT2 and ZT14. (H) Survival rates of Bmal1fl/fl and Bmal1iKO mice after MTX administration (1000 mg/kg, i.g., n = 10) at ZT2 and ZT14. Statistics for panels B-G were performed with Two-way ANOVA and Bonferroni post hoc test (*p < 0.05). Statistical analyses for survival in panel H were performed with the logrank test (*p < 0.05).
Dbp and E4bp4 are transcriptional regulators of Mrp2
Dbp (a PAR bZip transcription factor) overexpression led to an increase in Mrp2 mRNA in CT26 cells, whereas E4bp4 overexpression resulted in reduced Mrp2 expression (Figure 5A). Mrp2 expression was increased in E4bp4-/- mice (Figure 5B). These data suggested Dbp as an activator and E4bp4 as a repressor of Mrp2. Moreover, Dbp dose-dependently induced, whereas E4bp4 dose-dependently inhibited Mrp2-Luc reporter activity (Figure 5C/D). Interestingly, E4bp4 repressed Dbp-mediated transactivation of Mrp2, suggesting competing roles of the two transcriptional factors in gene regulation (Figure 5E). Sequence analysis identified four potential D-box (a motif for DBP and E4BP4 binding) elements in Mrp2 promoter region, namely, D1, D2, D3, and D4. Truncation and mutation experiments revealed D4 as the actual site for DBP and E4BP4 binding (Figure 5F/G). Through binding to D4, DBP activates but E4BP4 inactivates Mrp2 transcription (Figure 5F/G). Direct interactions between DBP/E4BP4 proteins and the D4 element were confirmed by using EMSA assays (Figure 5H). Furthermore, intestinal DBP and E4BP4 proteins were recruited to the D4 element of Mrp2 promoter in Bmal1fl/fl mice based on ChIP assays (Figure 5I/J). By contrast, recruitment of E4BP4 to Mrp2 promoter was not observed in E4bp4 knockout mice (Figure 5K). Taken together, DBP activated and E4BP4 repressed Mrp2 transcription via specific binding to a same D-box in promoter region.
Bmal1 regulates intestinal Mrp2 via Dbp and Rev-erbα/E4bp4 axis
Sequence analysis revealed one putative E-box (-1293/-1277 bp) in Mrp2 promoter region. To determine whether this putative E-box element is functional, we performed Mrp2 luciferase reporter assays using HEK293T, CT26 and NIH3T3 cells. Overexpression or knockdown of Bmal1 had no effect on Mrp2 promoter activity (Figure 6A), suggesting that an indirect rather than direct mechanism is involved in regulation of Mrp2 by Bmal1. We next investigated the role of circadian transcription factors in Bmal1 regulation of Mrp2. Bmal1 ablation led to downregulation of Dbp and Rev-erbα expression, and upregulation of E4bp4 expression in the small intestine (Figure 6B/C). By contrast, the expressions of Tef and Hlf were unaffected (two additional PAR bZip transcription factors, Supplementary Figure 4A). Positive regulation of Dbp/Rev-erbα and negative regulation of E4bp4 was also confirmed in CT26 cells (Figure 6D). Bmal1 is a transcriptional activator that has been shown to directly regulate Dbp and Rev-erbα through E-boxes [36,37]. By contrast, an indirect mechanism involving a negative regulator is necessary for Bmal1 regulation of E4bp4. We found intestinal E4bp4 is under the control of Rev-erbα as Rev-erbα knockout upregulates E4bp4 in mice (Figure 6E). This was supported by the fact that Rev-erbα trans-represses E4bp4 through direct binding to the RevRE element in the promoter [38]. Thus, Bmal1 negatively regulates E4bp4 via the transcriptional repressor Rev-erbα.
Figure 4
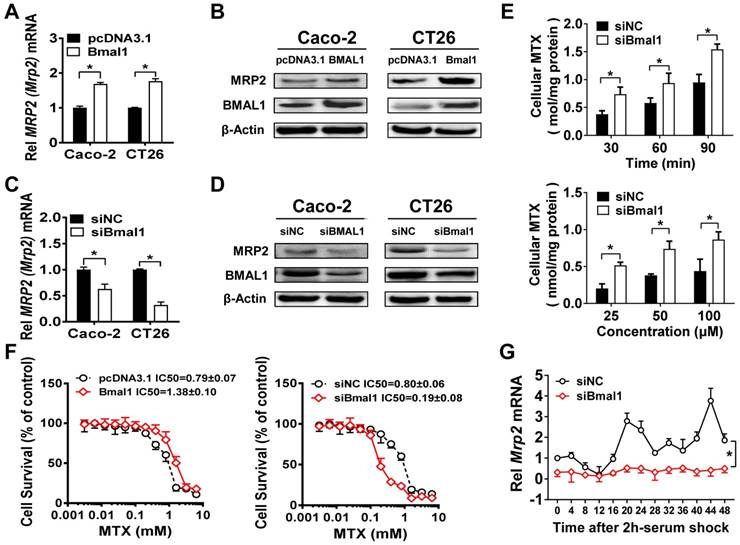
Bmal1(BMAL1) regulates Mrp2(MRP2) expression in colon cancer cells. (A) Mrp2 (MRP2) mRNA levels in CT26 or Caco-2 cells transfected with Bmal1 (BMAL1) expression plasmids for 36 h. Data are presented as the fold change in gene expression normalized to Hmbs. Data are mean ± SD (n = 5). *p < 0.05 (t-test). (B) Western blot analysis of MRP2 and BMAL1 protein levels in CT26 or Caco-2 cells transfected with Bmal1 (BMAL1) expression plasmids for 48 h. (C) Mrp2 (MRP2) mRNA levels in CT26 or Caco-2 cells transfected with siBmal1 (siBMAL1) for 36 h. Data are presented as the fold change in gene expression normalized to Hmbs. Data are mean ± SD (n = 5). *p < 0.05 (t-test). (D) Western blot analysis of MRP2 and BMAL1 protein levels in CT26 or Caco-2 cells transfected with siBmal1 (siBMAL1) for 48 h. (E) Effects of Bmal1 knockdown on cellular uptake of MTX at different time points (30, 60 and 90 min) and different concentrations (25, 50 and 100 μM) in CT26 cells. Data are mean ± SD (n = 5). *p < 0.05 (Two-way ANOVA with Bonferroni post hoc test). (F) Effects of Bmal1 knockdown or overexpression on CT26 cell viability with exposure to indicated concentrations of MTX. Data are mean ± SD (n = 6). (G) Bmal1 knockdown attenuates Mrp2 rhythmicity in serum-shocked CT26 cells. Data are mean ± SD (n = 6). *p < 0.05 (Two-way ANOVA).
Figure 5
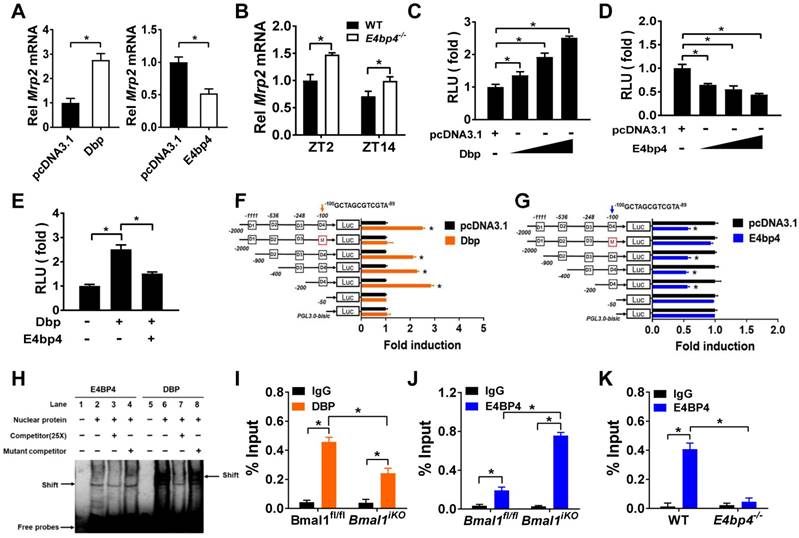
Dbp and E4bp4 are transcriptional regulators of Mrp2. (A) Mrp2 mRNA expressions in CT26 cells transfected with Dbp or E4bp4 expression plasmid for 36 h. Data are presented as the fold change in Mrp2 expression normalized to Hmbs. (B) Intestinal Mrp2 mRNA expressions at ZT2 and ZT14 in WT and E4bp4-/- mice. Data are presented as the fold change in Mrp2 expression normalized to Hmbs and relative to ZT2 of WT mice. (C) Dbp dose-dependently activated Mrp2 transcription in luciferase reporter assays. NIH3T3 cells were transfected with Mrp2-luc reporter and different amounts of Dbp plasmids. After 24 h transfection, luciferase reporter activities were measured. (D) E4bp4 dose-dependently inhibited Mrp2 promoter activity. (E) E4bp4 repressed Dbp-mediated transactivation of Mrp2. (F) Influence of transactivation by Dbp on deleted or mutated potential D-box in Mrp2 promoter region. (G) Influence of transrepression by E4bp4 on deleted or mutated potential D-box in Mrp2 promoter region. (H) EMSA assays showed direct interactions between DBP/E4BP4 proteins and the D4 element. HEK293T cells were transfected with Dbp or E4bp4 plasmid. After 48 h treatment, EMSA assays were performed with nuclear extracts from the transfected cells. (I) ChIP assay showing the relative occupancy of DBP at Mrp2 promoter region in Bmal1fl/fl and Bmal1iKO mice. (J) ChIP assay showing the relative occupancy of E4BP4 at Mrp2 promoter region in Bmal1fl/fl and Bmal1iKO mice. (K) ChIP assay showing the relative occupancy of E4BP4 at Mrp2 promoter region in WT and E4bp4-/- mice. All data except EMSA assays are shown as mean ± SD (n = 6). Statistics for panels A and C-G were performed with Student's t test (*p < 0.05). Statistics for panels B, I, J and K were performed with Two-way ANOVA and Bonferroni post hoc test (*p < 0.05).
Rev-erbα overexpression led to an increase in Mrp2 mRNA in CT26 cells (Figure 6F). Consistently, Rev-erbα ablation caused a decrease in Mrp2 expression in mice and in CT26 cells (Figure 6G/H). This positive regulation of Mrp2 by Rev-erbα was achieved through modulation of E4bp4 (Figure 6E/F/H). Rev-erbα ablation led to enhanced binding of E4BP4 to Mrp2 promoter (Figure 6I). Knockdown of Rev-erbα or E4bp4 attenuated the activation effects of Bmal1 on Mrp2 expression (Figure 6J). In addition, Bmal1 ablation led to reduced binding of DBP and enhanced binding of E4BP4 to Mrp2 promoter (Figure 5I and J). All these data supported critical roles of Dbp and Rev-erbα/E4bp4 axis in mediating Bmal1 regulation of intestinal Mrp2.
Discussion
The exporter MRP2 plays an important role in disposition and elimination of a wide range of drugs, including antineoplastics (e.g., methotrexate, cisplatin, doxorubicin and irinotecan), antihypertensives (e.g., olmesartan and temocaprilate), antiretrovirals (e.g., adefovir, lopinavir and saquinavir), and antibiotics (e.g., ampicillin and azithromycin) [39]. In this study, we identified a critical role for the central clock gene Bmal1 in generation of diurnal expression and activity of intestinal MRP2 (Figures 1 & 2). Moreover, we elucidated the molecular mechanisms for Bmal1 control of MRP2. BMAL1 regulates Mrp2 transcription and expression through DBP and REV-ERBα/E4BP4 axis (Figure 7). To be specific, through its own rhythmic expression, BMAL1 induces circadian expressions of the target genes Dbp and Rev-erbα (Figure 7). The former directly activates Mrp2 transcription, whereas the latter represses Mrp2 transcription via E4bp4, a negative regulator of Mrp2 (Figure 7). DBP and E4BP4 coordinately act on Mrp2 transcription in a time-dependent manner (Figure 7). DBP action (induction of Mrp2 expression) dominates in the daytime, whereas E4BP4 action (inhibition of Mrp2 expression) dominates in the night, thereby generating a diurnal oscillation in MRP2 expression (Figure 7).
Figure 6
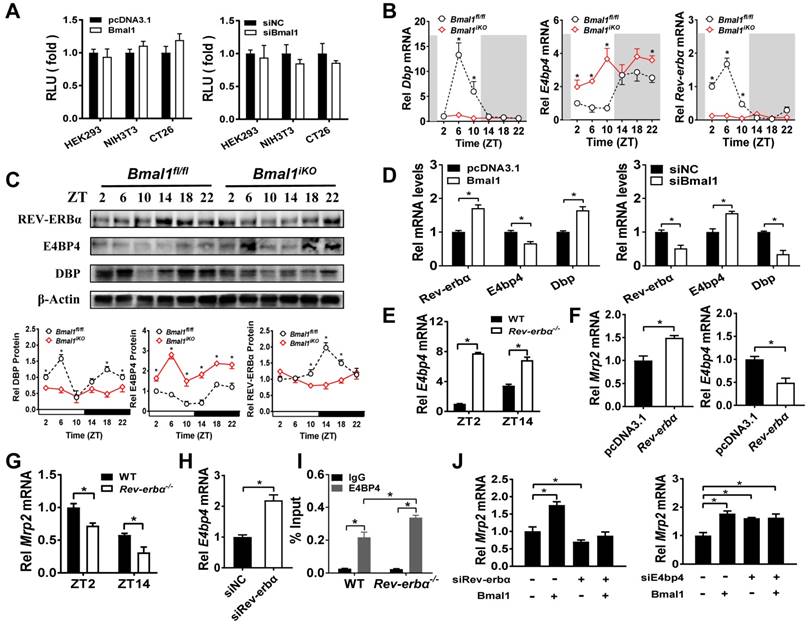
Bmal1 regulates intestinal Mrp2 via Dbp and Reverbα-E4bp4 axis (A) Overexpression or knockdown of Bmal1 had no effect on Mrp2 promoter activity. HEK293T, CT26 or NIH3T3 cells were transfected with Mrp2-Luc reporter and indicated expression plasmids. After 24 h treatment, luciferase reporter activities were measured. (B) qRT-PCR analysis of Dbp, E4bp4 and Rev-erbα transcript abundance in small intestine of Bmal1fl/fl and Bmal1iKO mice. Data are presented as the fold change in gene expression normalized to Hmbs and relative to ZT2 of Bmal1fl/fl mice. Data are mean ± SD (n = 6). (C) Western blot analysis of DBP, E4BP4 and REV-ERBα protein in small intestine of Bmal1fl/fl and Bmal1iKO mice. Data are mean ± SD (n = 6). (D) Effects of Bmal1 overexpression or knockdown on mRNA expressions of Dbp, E4bp4 and Rev-erbα in CT26 cells. (E) Intestinal E4bp4 mRNA levels in WT and Rev-erbα-/- mice at ZT2 and ZT14. (F) Effects of Reverbα overexpression on mRNA expressions of Mrp2 and E4bp4 in CT26 cells. (G) Intestinal Mrp2 mRNA levels in WT and Rev-erbα-/- mice at ZT2 and ZT14. (H) Effects of Reverbα knockdown on Mrp2 mRNA expression in CT26 cells. (I) ChIP assay showing the relative occupancy of E4BP4 at Mrp2 promoter region in WT and Rev-erbα-/- mice. (J) Knockdown of Rev-erbα or E4bp4 attenuated the activation effects of Bmal1 on Mrp2 expression. CT26 cells were transfected with indicated expression plasmids. After 36 h treatment, mRNA was extracted and quantified by qRT-PCR. All data are shown as mean ± SD (n = 6). Statistics for panels B, C, E, G, I and J were performed with Two-way ANOVA and Bonferroni post hoc test (*p < 0.05). Statistics for panels D, F and H were performed with Student's t test (*p < 0.05).
Since CLOCK protein is a partner of BMAL1 in regulation of circadian gene expression by acting on E-box elements [40]. We attempted to validate the mechanisms for circadian clock-controlled MRP2 expression using Clock-/- mice. Clock ablation resulted in reduced expression of Mrp2 in the small intestine (Supplementary Figure 5A). This was accompanied by down-regulated Dbp and Rev-erbα, and up-regulated E4bp4 (Supplementary Figure 5B-D). The data support the antagonistic roles of DBP and E4BP4 in generation of circadian MRP2 expression, and the mediating role of REV-ERBα in BMAL1/CLOCK regulation of E4BP4/MRP2 (Figure 7).
Like MRP2, P-gp (Mdr1a) is an apical membrane exporter and rhythmically expressed in the intestine [41]. A previous study shows that HLF (a PAR bZip transcription factor) and E4BP4 proteins consist of a reciprocating mechanism in which HLF activates the transcription of the Mdr1a gene, whereas E4BP4 periodically suppresses transcription when E4BP4 is abundant [27]. Thereby, HLF and E4BP4 control the rhythm in Mdr1a expression although the actual roles of central clock genes such as Bmal1 in this exporter's expression was unexplored [27]. We determined the relative expressions of Hlf and Tef (another PAR bZip transcription factor) in Bmal1iKO mice versus control littermates (Supplementary Figure 4A). The roles of Hlf and Tef in Bmal1 regulation of intestinal MRP2 were excluded because their expressions were unaffected in Bmal1iKO mice and Hlf/Tef had no effects on Mrp2 expression (Supplementary Figure 4A-D). This was also supported by the study of Oishi K et al in which Dbp rather than Tef and Hlf was down-regulated in Clock-deficient mice [42]. Therefore, Bmal1 controls the rhythm in Mrp2 expression through its output circadian proteins DBP (rather than HLF as shown for Mdr1a) and REV-ERBα/E4BP4 (Figure 7).
Figure 7
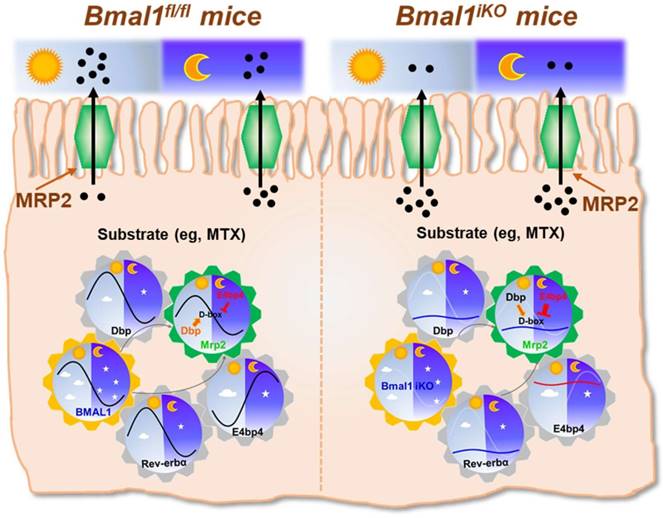
Schematic summarizing major findings in this study. Bmal1 regulates diurnal expression of intestinal Mrp2 via Dbp and Rev-erbα/E4bp4 axis, thereby altering the disposition of MRP2 substrates.
We observed more severe toxicity of MTX in the early dark period (ZT14) than in the early morning period (ZT2) (Figure 3). This appears to contradict with previous observation that MTX is more toxic in the early light period [43]. The apparent “discrepancy” stems from a difference in the route of drug administration. MTX is orally administered to mice herein, but was intraperitoneally injected to mice in previous chronotoxicity studies [43]. Intestinal efflux (by MRP2) is a key determinant to MTX pharmacokinetics and systemic exposure in the former case, whereas renal clearance is in the latter case [43]. Therefore, the chronotoxicity of oral MTX was mainly dependent on the circadian rhythm of MRP2 expression/activity. A lower toxicity at ZT2 was associated with a higher MRP2 expression/activity (and a lower drug absorption), and vice versa (Figures 2&3). A previous study suggests circadian rhythms of DNA synthesis and DHFR activity (i.e., the sensitivity of living organisms to MTX; increased activities in the light phase and decreased activities in the dark phase) as additional contributors to MTX chronotoxicity [43]. However, we argue for negligible or no contributions from diurnal DNA synthesis and DHFR activity because their rhythms are inconsistent with the observed rhythm of oral MTX toxicity (Figure 3).
It was noteworthy that MTX is not a specific substrate of MRP2 [34]. It can be also transported by other MRP proteins such as MRP1 (ABCC1) and MRP3 (ABCC3), located in the basolateral membrane of enterocytes [44]. In addition, uptake transporters such as SLC19A1 and SLC46A1 are involved in intestinal disposition of MTX [34]. However, intestinal expressions of Mrp1/3 and the two uptake transporters were unaffected in Bmal1iKO mice (Supplementary Figure 6A). Therefore, altered MTX pharmacokinetics and toxicity in the genetic mice were solely ascribed to down-regulated MRP2 activity (Figure 3). Although MTX undergoes minimal metabolism in the body, we assessed the effects of Bmal1 on intestinal XMEs. Bmal1 appears to regulate a very limited number of XMEs such as Cyp2c29 and Cyp2c55 (Supplementary Figure 6B). Our study lends a strong support to the notion that circadian clock regulates xenobiotic/drug detoxification [45]. Individuals with disrupted circadian clock (e.g., shift workers) are expected to have impaired drug detoxification, leading to altered drug toxicity and efficacy. Therefore, circadian clock may be an important factor in the design of drug dosing regimen.
In summary, we generated intestine-specific Bmal1 knockout (Bmal1iKO) mice and identified Bmal1 as a key regulator of circadian intestinal MRP2 expression. Bmal1 coordinated temporal expressions of DBP (a MRP2 activator) and REV-ERBα/E4BP4 (a MRP2 repressor), realizing a diurnal oscillation in MRP2 expression.
Materials and Methods
Chemicals and reagents
Methotrexate (MTX) and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) were purchased from Sigma-Aldrich (St. Louis, MO). The antibodies used for Western blotting were as follows: anti-MRP2 antibody (OriGene TA313641), anti-REV-ERBα antibody (Sigma-Aldrich WH0009572M2), anti-E4BP4 antibody (MBL M225-3), anti-BMAL1 antibody (Abcam ab3350), anti-DBP antibody (Abcam ab22824), anti-ZO-1 antibody (Invitrogen 40-2200), anti-Occludin antibody (abcam ab31721) and anti-β-Actin (Abcam ab8226). The antibodies used for immunofluorescence assay were as follows: anti-MRP2 antibody (OriGene TA313641) and anti-Villin antibody (Proteintech 66096-1-Ig). For ChIP assays, normal rabbit IgG was purchased from Cell Signaling Technology (Beverly, MA); Antibody against DBP (sc-32899) was obtained from Santa Cruz Biotechnology (Santa Cruz, CA); Antibody against E4BP4 (11773-1-AP) was purchased from Proteintech (Chicago, IL). Plasmids: pGL3.0 and pRL-TK vectors were purchased from Promega (Madison, WI). Mrp2 (2000)-Luc, Mrp2 (900)-Luc, Mrp2 (400)-Luc, Mrp2 (200)-Luc, Mrp2 (50)-Luc, Bmal1/BMAL1, Rev-erbα, E4bp4, Dbp, Hlf and Tef were synthesized by Biowit Technologies (Shenzhen, China). Luciferase reporter constructs containing mutated versions of D4 sites were obtained from Biowit Technologies (Shenzhen, China). siE4bp4 (siRNA targeting E4bp4), siRev-erbα (siRNA targeting Rev-erbα) and siBmal1/siBMAL1 (siRNA targeting Bmal1/BMAL1) were obtained from TranSheep Bio-Tech (Shanghai, China).
Mice
The intestinal-specific Bmal1 knockout (Bmal1iKO) mice on a C57BL/6 background were generated by crossing mice carrying a Bmal1 exon 8 floxed allele (Bmal1fl/fl, The Jackson laboratory) with Villin-Cre mice [B6SJL-Tg(Vil-Cre)997Gum, The Jackson Laboratory]. E4bp4-/- mice were obtained from Dr. Masato Kubo at RIKEN Institute in Japan. Rev-erbα-/- and Clock-/- mice were obtained from Cyagen Biosciences Inc. (Guangzhou, China). All mice were bred and maintained in the SPF barrier at the Institute of Laboratory Animal Science, Jinan University. All mice were housed under a 12 hour light/12 hour dark cycle and fed ad libitum. 8-12 weeks old mice were used for experiments. All experiments were performed using protocols approved by the Institutional Animal Care and Use Committees of Jinan University.
Pharmacokinetic experiments
Methotrexate (50 mg/kg) was administered to Bmal1fl/fl and Bmal1iKO mice (male) by oral gavage. Blood samples were collected by retro-orbital bleeding at 0.083, 0.25, 0.5, 0.75, 1, 2, 4, and 6 h (n = 5 per time point). Small intestine (jejunum), liver, and kidney samples were collected at 0.25 and 0.75 h. The plasma and tissue samples were prepared as previously described [46]. MTX concentrations in plasma and tissues were determined by LC-MS/MS analysis.
Toxicity experiments
Two sets of toxicity experiments were performed. In the first set of experiments, Bmal1fl/fl and Bmal1iKO mice (male) received a single dose of MTX (1000 mg/kg) by oral gavage. Hepatic and renal toxicities were assessed by measuring ALT (alanine aminotransferase), AST (aspartate aminotransferase), BUN (blood urea nitrogen) and Scr (serum creatinine) levels 24 h post drug administration. ALT, AST, BUN and Scr were quantified using their respective biochemical assay kits (Nanjing Jiancheng Bioengineering Institute, Nanjing, China) according to the manufacturer's instructions. For the second set of experiments, Bmal1fl/fl and Bmal1iKO mice (male) received a single dose of MTX (1000 mg/kg) at ZT2 or ZT14 by oral gavage. In the following fifteen days, the numbers of mouse deaths were recorded daily and the survival rates were calculated.
Hematoxylin & Eosin (H&E) staining
Small intestine tissues were washed and fixed in 4% buffered formalin and embedded in paraffin for staining with H&E. Images were captured using a Zeiss Axio Imager M1 microscope. An image analyzer (Kontron Elektronik GmbH) coupled to a binocular microscope (Carl Zeiss) was used to quantify acquired images and to determine the villus height, crypt and enterocyte numbers.
Everted intestinal sac experiments
Mucosal-to-serosal transport of MTX in everted sacs from mouse intestines was determined as described previously [47]. In brief, Bmal1fl/fl and Bmal1iKO mice (male, n = 3 per group) were anesthetized with ether and sacrificed by exsanguination from the femoral artery and vein. Immediately after sacrifice, 10 cm segments from proximal jejunum were collected. Each intestinal segment was everted and both ends were tied after filling the sac with 400 μl Krebs-ringer buffer (KRB, pH 6.4). The everted sacs were incubated in KRB buffer for 10 min, and transferred to KRB buffer containing 20 μM MTX. After incubation, the serosal solution was collected. Intestinal sacs were weighed and homogenized in twice the mass of ice-cold saline. MTX concentrations in serosal and homogenate samples were determined by LC-MS/MS analysis.
In situ single-pass intestinal perfusion experiment
In situ perfusion experiments was performed with mice and a perfusion pump (PHD22/2000, Harvard Apparatus) as previously described [48]. In brief, Bmal1fl/fl and Bmal1iKO mice (male, n = 5) were fasted overnight before experimentation. Mice were anaesthetized with pentobarbital sodium (60 mg/kg) by intraperitoneal injection. A midline longitudinal incision was made to expose the abdomen, and then a segment of jejunum (10 cm) was separated. The intestinal segment was cannulated with a silicone tube at the proximal and distal end of the segment. After removal of the intestinal content and preincubation of the segment with transport medium (HBSS, pH 7.4) containing 20 μM MTX for 30 min, the effluents were collected every 30 min for 120 min. At the end of experiment, the lengths of intestinal segments were measured. MTX concentrations in perfusate samples were determined by LC-MS/MS analysis.
Cell culture
Caco-2, CT26, NIH3T3 and HEK293T cells were purchased from the Cell Bank of Chinese Academy of Sciences (Shanghai, China). The cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) at 37℃ in a 5% CO2 humidified atmosphere.
Cell transfection
The cells were seeded to 6-well-plates. Once reaching a 70% confluence, cells were transfected with overexpression plasmids or siRNA (sequences summarized in Supplementary Table 1) using JetPRIME (Polyplus Transfection, Ill kirch, France) according to the instruction manual. After 48 h, the cells were collected for RNA and protein analyses.
RNA isolation and analysis
Tissue samples (small intestine, liver, kidney and/or brain) from male mice (n = 6 per group) were harvested at different circadian time points. All samples were flash frozen in liquid nitrogen, and stored at -80°C until use. Total RNA was extracted from tissue or cell samples using TRIzol reagent (Invitrogen). 500 ng of total RNA was reverse transcribed to cDNA using Superscript II reverse transcriptase (Invitrogen). The mRNA expression was determined by real-time PCR using GoTaqR qPCR Master Mix (Vazyme). qPCR primers were designed using Primer 5 and the sequences are summarized in the Supplementary Table 1. qPCR reactions were performed following our published procedures [49]. A melting curve analysis was performed to verify the amplification specificity with a temperature gradient of 0.1 °C/s from 65 to 95 °C. Mouse Hmbs or human GAPDH gene were used as internal controls.
Cellular accumulation and efflux experiments
CT26 cells were plated and treated with siBmal1 (or siNC as the control) for 48 h. The culture medium was removed and replaced with HBSS buffer containing MTX (at a series of concentrations). At each time point (30, 60 and 90 min), the incubation solution was aspirated. After washing with ice-cold PBS twice, the cells were solubilized with 0.4 ml 50% methanol. MTX concentrations in cell samples were determined by LC-MS/MS analysis.
Cell viability assay
CT26 cells were seeded and treated with siBmal1 (or siNC as the control) for 48 h. The culture medium was replaced with fresh medium containing MTX (at a series of concentrations, n = 6), and allowed for incubation for another 2 days. Then, the cells were incubated with MTT (1 mg/ml, dissolved in serum-free medium) for 4 h. Cell residues were dissolved with 200 μl DMSO. The absorbance at 490 nm was determined using Synergy LX Multi-Mode Microplate Reader (BioTek, Winooski, VT, USA).
Western blotting
Western blotting was performed as described previously [49]. In brief, mouse tissues or cells were lysed in RIPA buffer (with 1 mM PMSF). Protein concentrations were determined using a BCA assay kit (Beyotime Biotechnology, China). The lysate was subjected to sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis. The proteins within the gel were transferred to polyvinylidene fluoride (PVDF) membranes (Millipore, Bedford, MA). Blots were probed with the primary antibodies, followed by horseradish peroxidase-conjugated secondary antibody. Protein bands were detected by enhanced chemiluminescence.
Luciferase reporter assays
Various versions of Mrp2 promoter luciferase constructs (-2000/+32, -900/+32, -400/+32, -200/+32, and -50/+32) were synthesized and cloned into the pGL3.0 vector. Cells were co-transfected with firefly luciferase reporter (200 ng), renilla luciferase reporter (pRL-nTK, 10 ng, Promega) and overexpression plasmids using the JetPrime transfection reagent. Cells were lysed in passive lysis buffer 24 h after transfection. Firefly and renilla luciferase activity were measured using Dual-Luciferase Reporter Assay System (Promega). Firefly luciferase activity was normalized to renilla luciferase activity, and expressed as relative luciferase unit (RLU).
Electrophoretic mobility shift assay (EMSA)
EMSA assay was performed with a chemiluminescent EMSA kit as previously described [50]. Nuclear proteins from E4bp4 or Dbp overexpressing HEK293T cells were extracted using a cytoplasmic/nuclear protein extraction kit according to the manufacturer's instructions. Nuclear protein samples were mixed with labeled DNA probe. For competition experiments, a 25-fold molar excess unlabeled DNA probe was premixed with the proteins for 20 min prior to the addition of labeled DNA probe. The mixture was subjected to non-denaturing polyacrylamide gel electrophoresis, followed by the transfer to a positively charged nylon membrane. The signals were detected by Omega Lum G imaging system (Aplegen, CA). Oligonucleotide sequences are provided in Supplementary Table 1.
Chromatin immunoprecipitation (ChIP)
ChIP assays were performed using SimpleChip plus Enzymatic Chromatin IP kit (Cell Signaling Technology, Beverly, MA, USA) following the manufacturer's protocol. In brief, the small intestine was cross-linked with 1.5% formaldehyde and digested with micrococcal nuclease. The sheared chromatin was immunoprecipitated overnight at 4°C using anti-E4BP4, anti-DBP or anti-IgG (as a control) antibody. The purified DNAs were subjected to qPCR analyses with primers encompassing the E4BP4 and DBP-binding sites in Mrp2 promoter (Supplementary Table 1). The amount of immunoprecipitated DNA was normalized to input DNA.
LC-MS/MS analysis
MTX quantification was performed using a UHPLC-MS/MS system consisting of a Dionex ultimate 3000 UHPLC system and AB 3500 triple-quadrupole mass spectrometer (AB Sciex, Ontario, Canada). The mass spectrometer was operated at the positive ESI mode. Chromatographic separations were conducted on an ACE Ultra-Core 2.5 Super C18 column (Phenomenex, Torrance, CA) at a flow rate of 0.3 ml/min. A gradient elution program consisting of mobile phase A (0.1% formic acid in water) and mobile phase B (0.1% formic acid in acetonitrile) was applied: 10% B (0-2 min), 10-90% B (2-6 min), 10% B (6-8 min).
TUNEL assay
Apoptosis was analyzed by TUNEL assay using Click-iT ® Plus TUNEL Assay (Life Technologies, Carlsbad, CA) according to manufacturer's instruction. Integral optical density (IOD, an index of apoptosis level) was calculated using Image-pro plus 6.0 (Media Cybernetics, Rockville, MD).
Statistical analysis
Data are presented as means ± SD (standard deviation). Student's t test was used to test for statistical differences between two groups. One-way or two-way ANOVA followed by Bonferroni post hoc test was used for multiple group comparisons. The homogeneity of variance assumption for ANOVAs was verified by Levene's test. Statistical analyses for survival were performed with the logrank test. The level of significance was set at p<0.05 (*).
Abbreviations
ABC, ATP-binding cassette; ABCC, ABC subfamily C; ALT, alanine aminotransferase; AST, aspartate aminotransferase; AUC, area under the curve; Bmal1, brain and muscle ARNT-like 1; BUN, blood urea nitrogen; ChIP, chromatin immunoprecipitation; Clock, circadian locomotor output cycles kaput; Cry, cryptochrome; CYPs, cytochromes P450; D-box, D site of albumin promoter; Dbp, D-site binding protein; DHFR, dihydrofolate reductase; DMEM, Dulbecco's modified Eagle's medium; E4bp4, E4 promoter-binding protein 4; EMSA, electrophoretic mobility shift assay; E-box, enhancer box; FBS, fetal bovine serum; Hlf, hepatic leukemia factor; KRB, Krebs-ringer; Mrp2, multidrug resistance-associated protein 2; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide; MTX, methotrexate; Tef, thyrotroph embryonic factor; Per, period; PVDF, polyvinylidene fluoride; qPCR, quantitative polymerase chain reaction; Rev-erbα, reverse erythroblastosis virus α; RevRE, Rev-erbα response element; RLU, relative luciferase unit; Scr, serum creatinine; siRNA, short interfering RNA; XMEs, xenobiotic-metabolizing enzymes; ZT, zeitgeber time; ZO-1, zonula occludens-1.
Supplementary Material
Supplementary figures and table.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Nos. 81722049 and 81573488), the Local Innovative and Research Teams Project of Guangdong Pearl River Talents Program (No. 2017BT01Y036), and the Natural Science Foundation of Guangdong Province (No. 2017A03031387).
Author contributions
Participated in research design: Yu, Zhang and Wu. Conducted experiments: Yu, Zhou, Xu, Guo, Chen, Zhang. Performed data analysis: Yu, Zhang, and Wu. Wrote or contributed to the writing of the manuscript: Yu and Wu.
Yu and Zhang contributed equally to this work.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Partch CL, Green CB, Takahashi JS. Molecular architecture of the mammalian circadian clock. Trends Cell Biol. 2014;24:90-99
2. Hastings M, O'Neill JS, Maywood ES. Circadian clocks: Regulators of endocrine and metabolic rhythms. J Endocrinol. 2007;195:187-198
3. Takahashi JS. Molecular components of the circadian clock in mammals. Diabetes Obes Metab. 2015;17:6-11
4. Rey G, Cesbron F, Rougemont J. et al. Genome-Wide and Phase-Specific DNA-Binding Rhythms of BMAL1 Control Circadian Output Functions in Mouse Liver. PLoS Biol. 2011;9:e1000595
5. Buhr ED, Takahashi JS. Molecular components of the Mammalian circadian clock. Handb Exp Pharmacol. 2013;217:3-27
6. Kume K, Zylka MJ, Sriram S. et al. mCRY1 and mCRY2 are essential components of the negative limb of the circadian clock feedback loop. Cell. 1999;98:193-205
7. Preitner N, Damiola F, Lopez-Molina L. et al. The orphan nuclear receptor REV-ERBα controls circadian transcription within the positive limb of the mammalian circadian oscillator. Cell. 2002;110:251-260
8. Asher G, Schibler U. Crosstalk between components of circadian and metabolic cycles in mammals. Cell Metab. 2011;13:125-137
9. Zhang D, Tong X, Nelson BB. et al. The hepatic BMAL1/AKT/lipogenesis axis protects against alcoholic liver disease in mice via promoting PPARα pathway. Hepatology. 2018:13 [Epub ahead of print]
10. Nikolaeva S, Ansermet C, Centeno G. et al. Nephron-Specific Deletion of Circadian Clock Gene Bmal1 Alters the Plasma and Renal Metabolome and Impairs Drug Disposition. J Am Soc Nephrol. 2016;27:2997-3004
11. Early JO, Menon D, Wyse CA. et al. Circadian clock protein BMAL1 regulates IL-1β in macrophages via NRF2. Proc Natl Acad Sci U S A. 2018;115:8460-8468
12. Yang G, Chen L, Grant GR. et al. Timing of expression of the core clock gene Bmal1 influences its effects on aging and survival. Sci Transl Med. 2016;8:324ra16
13. Hoogerwerf WA, Hellmich HL, Cornélissen G. et al. Clock gene expression in the murine gastrointestinal tract: endogenous rhythmicity and effects of a feeding regimen. Gastroenterology. 2007;133:1250-1260
14. Sládek M, Rybová M, Jindráková Z. et al. Insight into the circadian clock within rat colonic epithelial cells. Gastroenterology. 2007;133:1240-1249
15. Matsu-Ura T, Dovzhenok A, Aihara E. et al. Intercellular Coupling of the Cell Cycle and Circadian Clock in Adult Stem Cell Culture. Mol Cell. 2016;64:900-912
16. Stokes K, Cooke A, Chang H. et al. The Circadian Clock Gene BMAL1 Coordinates Intestinal Regeneration. Cell Mol Gastroenterol Hepatol. 2017;4:95-114
17. Shimizu M. Modulation of intestinal functions by dietary substances: an effective approach to health promotion. J Tradit Complement Med. 2012;2:81-83
18. Malofeeva EV, Domanitskaya N, Gudima M. et al. Modulation of the ATPase and transport activities of broad-acting multidrug resistance factor ABCC10 (MRP7). Cancer Res. 2012;72:6457-6467
19. Chu XY, Strauss JR, Mariano MA. et al. Characterization of mice lacking the multidrug resistance protein MRP2 (ABCC2). J Pharmacol Exp Ther. 2006;317:579-589
20. Mottino AD, Hoffman T, Jennes L. et al. Expression of multidrug resistance-associated protein 2 in small intestine from pregnant and postpartum rats. Am J Physiol Gastrointest Liver Physiol. 2001;280:1261-1273
21. Oh JH, Lee JH, Han DH. et al. Circadian Clock Is Involved in Regulation of Hepatobiliary Transport Mediated by Multidrug Resistance-Associated Protein 2. J Pharm Sci. 2017;106:2491-2498
22. Johnson BP, Walisser JA, Liu Y. et al. Hepatocyte circadian clock controls acetaminophen bioactivation through NADPH-cytochrome P450 oxidoreductase. Proc Natl Acad Sci U S A. 2014;111:18757-18762
23. Zhang T, Yu F, Guo L. et al. Small Heterodimer Partner Regulates Circadian Cytochromes p450 and Drug-Induced Hepatotoxicity. Theranostics. 2018;8:5246-5258
24. Tang Q, Cheng B, Xie M. et al. Circadian Clock Gene Bmal1 Inhibits Tumorigenesis and Increases Paclitaxel Sensitivity in Tongue Squamous Cell Carcinoma. Cancer Res. 2017;77:532-544
25. Abolmaali K, Balakrishnan A, Stearns AT. et al. Circadian variation in intestinal dihydropyrimidine dehydrogenase (DPD) expression: a potential mechanism for benefits of 5FU chrono-chemotherapy. Surgery. 2009;146:269-273
26. Lévi F, Okyar A. Circadian clocks and drug delivery systems: impact and opportunities in chronotherapeutics. Expert Opin Drug Deliv. 2011;8:1535-1541
27. Murakami Y, Higashi Y, Matsunaga N. et al. Circadian clock-controlled intestinal expression of the multidrug-resistance gene mdr1a in mice. Gastroenterology. 2008;135:1636-1644
28. Gachon F, Olela FF, Schaad O. et al. The circadian PAR-domain basic leucine zipper transcription factors DBP, TEF, and HLF modulate basal and inducible xenobiotic detoxification. Cell Metab. 2006;4:425-436
29. Kitamura Y, Hirouchi M, Kusuhara H. et al. Increasing systemic exposure of methotrexate by active efflux mediated by multidrug resistance-associated protein 3 (mrp3/abcc3). J Pharmacol Exp Ther. 2008;327:465-473
30. J. Jolivet, K. H. Cowan, G. A. Curt, et al. The pharmacology and clinical use of methotrexate. N Engl J Med. 1983;18:1094-1104
31. Abdel-Daim MM, Khalifa HA, Abushouk AI. et al. Diosmin Attenuates Methotrexate-Induced Hepatic, Renal, and Cardiac Injury: A Biochemical and Histopathological Study in Mice. Oxid Med Cell Longev. 2017;2017:3281670
32. Song JG, Nakano S, Ohdo S. et al. Chronotoxicity and chronopharmacokinetics of methotrexate in mice: modification by feeding schedule. Jpn J Pharmacol. 1993;62:373-378
33. Masuda M, I'izuka Y, Yamazaki M. et al. Methotrexate is excreted into the bile by canalicular multispecific organic anion transporter in rats. Cancer Res. 1997;57:3506-3510
34. Mikkelsen TS, Thorn CF, Yang JJ. et al. PharmGKB summary: methotrexate pathway. Pharmacogenet Genomics. 2011;21:679-686
35. Stearns AT, Balakrishnan A, Rhoads DB. et al. Diurnal Rhythmicity in the Transcription of Jejunal Drug Transporters. J Pharmacol Sci. 2008;108:144-148
36. Shun Yamaguchi, Shigeru Mitsui, Lily Yan. et al. Role of DBP in the Circadian Oscillatory Mechanism. Mol Cell Biol. 2000;20:4773-4781
37. Mukherji A, Kobiita A, Ye T. et al. Homeostasis in intestinal epithelium is orchestrated by the circadian clock and microbiota cues transduced by TLRs. Cell. 2013;153:812-827
38. Duez H, van der Veen JN, Duhem C. et al. Regulation of bile acid synthesis by the nuclear receptor Rev-erbalpha. Gastroenterology. 2008;135:689-698
39. Chen Z, Shi T, Zhang L. et al. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family in multidrug resistance: A review of the past decade. Cancer Lett. 2016;370:153-164
40. Ripperger JA, Shearman LP, Reppert SM. et al. CLOCK, an essential pacemaker component, controls expression of the circadian transcription factor DBP. Genes Dev. 2000;14:679-689
41. Ando H, Yanagihara H, Sugimoto K. et al. Daily rhythms of P-glycoprotein expression in mice. Chronobiol Int. 2005;22:655-665
42. Oishi K, Miyazaki K, Kadota K. et al. Genome-wide expression analysis of mouse liver reveals CLOCK-regulated circadian output genes. J Biol Chem. 2003;278:41519-41527
43. Ohdo S, Inoue K, Yukawa E. et al. Chronotoxicity of methotrexate in mice and its relation to circadian rhythm of DNA synthesis and pharmacokinetics. Jpn J Pharmacol. 1997;75:283-290
44. Zeng H, Chen ZS, Belinsky MG. et al. Transport of methotrexate (MTX) and folates by multidrug resistance protein (MRP) 3 and MRP1: effect of polyglutamylation on MTX transport. Cancer Res. 2001;61:7225-7232
45. Gachon F, Firsov D. The role of circadian timing system on drug metabolism and detoxification. Expert Opin Drug Metab Toxicol. 2011;7:147-158
46. Zhang T, Zhao M, Lu D. et al. REV-ERBa regulates CYP7A1 through repression of liver receptor homolog-1. Drug Metab Dispos. 2018;46:248-258
47. Kitamura Y, Hirouchi M, Kusuhara H. et al. Increasing Systemic Exposure of Methotrexate by Active Efflux Mediated by Multidrug Resistance-Associated Protein 3 (Mrp3/Abcc3). J Pharmacol Exp Ther. 2008;327:465-473
48. Mols R, Brouwers J, Schinkel AH. et al. Intestinal perfusion with mesenteric blood sampling in wild-type and knockout mice: evaluation of a novel tool in biopharmaceutical drug profiling. Drug Metab Dispos. 2009;37:1334-1337
49. Yu F, Zhang T, Guo L. et al. Liver Receptor Homolog-1 Regulates Organic Anion Transporter 2 and Docetaxel Pharmacokinetics. Drug Metab Dispos. 2018;46:980-988
50. Hellman LM, Fried MG. Electrophoretic mobility shift assay (EMSA) for detecting protein-nucleic acid interactions. Nat Protoc. 2007;2:1849-1861
Author contact
![]() Corresponding author: Baojian Wu, Ph.D., College of Pharmacy, Jinan University. E-mail: bj.wucom or tbaojianwuedu.cn
Corresponding author: Baojian Wu, Ph.D., College of Pharmacy, Jinan University. E-mail: bj.wucom or tbaojianwuedu.cn
Citation styles
APA
Yu, F., Zhang, T., Zhou, C., Xu, H., Guo, L., Chen, M., Wu, B. (2019). The Circadian Clock Gene Bmal1 Controls Intestinal Exporter MRP2 and Drug Disposition. Theranostics, 9(10), 2754-2767. https://doi.org/10.7150/thno.33395.
ACS
Yu, F.; Zhang, T.; Zhou, C.; Xu, H.; Guo, L.; Chen, M.; Wu, B. The Circadian Clock Gene Bmal1 Controls Intestinal Exporter MRP2 and Drug Disposition. Theranostics 2019, 9 (10), 2754-2767. DOI: 10.7150/thno.33395.
NLM
Yu F, Zhang T, Zhou C, Xu H, Guo L, Chen M, Wu B. The Circadian Clock Gene Bmal1 Controls Intestinal Exporter MRP2 and Drug Disposition. Theranostics 2019; 9(10):2754-2767. doi:10.7150/thno.33395. https://www.thno.org/v09p2754.htm
CSE
Yu F, Zhang T, Zhou C, Xu H, Guo L, Chen M, Wu B. 2019. The Circadian Clock Gene Bmal1 Controls Intestinal Exporter MRP2 and Drug Disposition. Theranostics. 9(10):2754-2767.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.