Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(8):2361-2379. doi:10.7150/thno.29628 This issue Cite
Research Paper
Induction of nuclear protein-1 by thyroid hormone enhances platelet-derived growth factor A mediated angiogenesis in liver cancer
Ching-Ying Chen1,2, Sheng-Ming Wu3,4, Yang-Hsiang Lin5, Hsiang-Cheng Chi6, Syuan-Ling Lin1, Chau-Ting Yeh5, Wen-Yu Chuang7, Kwang-Huei Lin1,5,8 ![]()
1. Department of Biochemistry, College of Medicine, Chang-Gung University, Taoyuan, Taiwan
2. Department of Biomedical Sciences, College of Medicine, Chang-Gung University, Taoyuan, Taiwan
3. Division of Pulmonary Medicine, Department of Internal Medicine, Shuang Ho Hospital, Taipei Medical University, New Taipei City, Taiwan
4. Division of Pulmonary Medicine, Department of Internal Medicine, School of Medicine, College of Medicine, Taipei Medical University, Taipei, Taiwan
5. Liver Research Center, Chang Gung Memorial Hospital, Linkou, Taoyuan, Taiwan
6. Radiation Biology Research Center, Institute for Radiological Research, Chang Gung University / Chang Gung Memorial Hospital, Linkou, Taoyuan, Taiwan
7. Department of Pathology, Chang Gung Memorial Hospital and Chang Gung University, Taoyuan, Taiwan
8. Research Center for Chinese Herbal Medicine, College of Human Ecology, Chang Gung University of Science and Technology, Taoyuan, Taiwan
Received 2018-8-31; Accepted 2019-2-24; Published 2019-4-13
Citation:
Chen CY, Wu SM, Lin YH, Chi HC, Lin SL, Yeh CT, Chuang WY, Lin KH. Induction of nuclear protein-1 by thyroid hormone enhances platelet-derived growth factor A mediated angiogenesis in liver cancer. Theranostics 2019; 9(8):2361-2379. doi:10.7150/thno.29628. https://www.thno.org/v09p2361.htm
Other stylesAbstract

Background & Aims: Hepatocellular carcinoma (HCC) is among the leading causes of cancer deaths worldwide. Many studies indicate that disruption of cellular thyroid hormone signaling promotes HCC progression. However, the mechanisms underlying the regulation of genes downstream of thyroid hormone actions in HCC have remained elusive. In the current study, we identified NUPR1 (nuclear protein-1), a stress-induced protein that overexpresses in various neoplasia, is upregulated by triiodothyronine/thyroid hormone receptor (T3/TR) signaling and aimed to elucidate its role in angiogenesis in cancer progression.
Methods: Quantitative reverse transcription-PCR, luciferase promoter and chromatin immunoprecipitation assays were performed to identify the NUPR1 regulatory mechanism by T3/TR. In vitro and In vivo vascular formations were performed to detect the angiogenic function of NUPR1. Human angiogenesis arrays were performed to identify the downstream angiogenic pathway. The sorafenib resistant ability of TR/NUPR1 was further examined in vitro and in vivo. Clinical relevance of TR, NUPR1 and platelet-derived growth factor A (PDGFA) were investigate in HCC samples using qRT-PCR and western blot.
Results: Our experiments disclosed positive regulation of NUPR1 expression by T3/TR through direct binding to the -2066 to -1910 region of the NUPR1 promoter. Elevated NUPR1 and TR expression link to poor survival in clinical HCC specimens. An analysis of clinicopathological parameters showed that expression of NUPR1 is associated with vascular invasion and pathology stage. Functional studies revealed that NUPR1 induced endothelial cell angiogenesis in vitro and in vivo. Using a human angiogenesis array, we identified PDGFA as a target of NUPR1 in the downstream angiogenic pathway. NUPR1 induced transcription of PDGFA through direct binding to the corresponding promoter region, and inhibition of the PDGFA signaling pathway impaired angiogenesis in human umbilical vein endothelial cells (HUVECs). Notably, the angiogenic effects of NUPR1/PDGFA were mediated by the MEK/ERK signaling pathway. TR/NUPR1 expression increased cell viability and resistance to sorafenib treatment. Moreover NUPR1 expression was positively correlated with TRα, TRβ, and PDGFA expression.
Conclusions: We propose that the T3/TR/NUPR1/PDGFA/MEK/ERK axis has a vital role in hepatocarcinogenesis and suggest NUPR1 as a potential therapeutic target in HCC.
Keywords: Thyroid hormone receptor, NUPR1, Hepatocellular carcinoma, PDGFA, sorafenib
Introduction
Hepatocellular carcinoma (HCC) accounts for 80%-90% of primary liver cancers and is among the leading causes of cancer deaths worldwide [1]. The major risk factors for HCC include hepatitis B and hepatitis C virus infection, alcohol consumption, diabetes, and aflatoxins [2, 3]. HCC is usually diagnosed at an advanced stage, with patients typically presenting with upper-abdominal pain and weight loss. Treatment of HCC, which is stage-guided and varies according to the cause of the disease, includes surgery, liver transplantation, and targeted therapy [4]. Although diagnostic and treatment methods have improved, the 5-year survival rate has remained below 12%. Thus, a new therapeutic strategy is essential for improving the prognosis and treatment of HCC.
Thyroid hormone (3,3'-5-triiodo-L-thyronine; T3) is a potent mediator of a variety of physiological processes, including embryonic development, cellular differentiation, metabolism, and regulation of cell proliferation [5]. T3 binds to specific high-affinity thyroid hormone receptors (TRs), which are ligand-dependent transcription factors belonging to the nuclear receptor superfamily [6]. TRs regulate gene expression by binding to thyroid hormone response elements (TREs) in the promoter region of target genes. Human TRs are encoded by the TRα (THRA) and TRβ (THRB) genes, located on human chromosomes 17 and 3, respectively, and yield several additional isoforms through alternative splicing and differential promoter usage. These receptors are comprised of functional domains, including a ligand-binding domain, DNA-binding domain, and dimerization and transactivation domains [7]. Considerable evidence has accumulated to show that aberrant expression of TRs is associated with human cancers [8-10]. In one study, Cristofanilli et al. reported that hypothyroid patients have a reduced incidence of primary breast carcinoma and a reduced risk of developing the invasive disease [11]. Treatment with the antithyroid drug, propylthiouracil, in combination with tamoxifen increases survival in patients with glioblastoma multiforme [12]. Moreover, epidemiological studies have demonstrated that hyperthyroidism associates with an increased risk of ovarian cancer [13]. In our previous studies, genes and microRNA regulated by T3/TR signaling were shown to be involved in tumor cell invasion and migration in HCC [14-17]. However, these studies did not adequately elucidate the detailed molecular mechanism by which T3/TR signaling leads to hepatocarcinogenesis.
In the current study, we used oligonucleotide microarrays to screen for genes regulated by T3 in HepG2 cells stably expressing TRα (HepG2-TRα). Among the T3-regulated genes identified, nuclear protein-1 (NUPR1) was the most notably upregulated by T3 treatment. NUPR1, which shares biochemical homology with high mobility group (HMG)-like proteins [18], was first identified in the rat pancreas during acute pancreatitis [19, 20]. NUPR1 has been implicated in diverse functions, and its expression is crucial for tumor development. Previous studies have reported NUPR1 overexpression in primary human HCC samples, and shown that it influences cell proliferation, migration, invasion and sorafenib resistance through induction of the downstream genes, RELB, IER3, and RUNX2 [21]. NUPR1 was also shown to be involved in mitochondrial defect-derived glycolytic activation in liver cancer [22]. Here, we report that T3/TR induced NUPR1 expression through direct binding to thyroid hormone response element (TRE) in the NUPR1 promoter region. Functional studies revealed that NUPR1 induced endothelial cell angiogenesis in vitro and in vivo. Using a human angiogenesis array, we identified the gene encoding platelet-derived growth factor A (PDGFA) as a target of NUPR1 in the activation of the downstream angiogenic pathway. We further found that the angiogenic effects of NUPR1/PDGFA are mediated by the MEK/ERK signaling pathway. Also, NUPR1 mediated transcriptional induction of PDGFA through direct binding to the corresponding promoter region. The viability of tumor cell following the addition of sorafenib was significantly increased in TR/NUPR1 expression cell. Clinicopathological analyses revealed that elevated expression of NUPR1 and TRs in clinical HCC specimens were linked to poor survival; moreover, NUPR1 expression was positively correlated with TRα, TRβ, and PDGFA expression. Thus, this study demonstrates a mechanism that implicates the T3/TR/NUPR1/PDGFA axis in HCC, providing novel therapeutic targets for tumorigenesis.
Materials and Methods
Cell cultures
The human hepatoma cell lines, HepG2, Huh7, Mahlavu and J7 were routinely grown in Dulbecco Modified Eagle medium (DMEM) supplemented with 10% (v/v) fetal bovine serum (FBS). The human umbilical vein endothelial cells (HUVECs) line was growth in endothelial cell basal medium-2 (EBM-2; Lonza, CC-3156) supplemented with EBM-2 SingleQuots (Lonza, CC-4176). Stably transfected HepG2-TRα, HepG2-TRβ, and HepG2-neo cells were cultured in DMEM containing 10% (v/v) FBS with G418 [23]. NUPR1-overexpressing (ovNUPR1) and luciferase control Mahlavu cells were cultured in DMEM containing 10% (v/v) FBS with blasticidin. NUPR1-knockdown (shNUPR1) and luciferase control (shLuc) Huh7 cells were cultured in DMEM containing 10% (v/v) FBS with puromycin. T3 was purchased from Sigma-Aldrich (T2752). Serum was depleted of T3 (0 nM T3) using an AG 1-X8 resin (Bio-Rad, 40-1451). Cells were cultured at 37 °C in a humidified atmosphere of 95% air and 5% CO2.
Quantitative reverse transcription-polymerase chain reaction (qRT-PCR) analyses
Total RNA was extracted from cells using TRIzol reagent (Invitrogen). cDNA was synthesized from total RNA using a Superscript II kit (Invitrogen, 18064-014) according to the manufacturer's protocols. qRT-PCR was conducted in a 15-μl reaction mixture containing 25 nM forward and reverse primers, 1×SYBR Green reaction mix (Applied Biosystems), and varying quantities of the template. SYBR Green fluorescence was measured with an ABI PRISM 7500 sequence detection system (Applied Biosystems). Primers used to amplify NUPR1 were 5'-AAG CTG AGG GAG TGG AGA GG-3' (forward) and 5'-TAT TGT TGC TGC CAC CCT GG-3' (reverse).
Immunoblot analysis
Total cell lysates, nuclear extracts, and conditioned medium (CM) were isolated and fractionated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) on 8-12% gels. Separated proteins were transferred to a PVDF (polyvinylidene difluoride) membrane and blocked by incubating in 5% (w/v) nonfat milk. PVDF membranes were incubated with appropriate primary and horseradish peroxidase (HRP)-conjugated secondary antibodies, and immunoreactive proteins were subsequently detected by chemiluminescence using an ECL detection kit (Amersham Inc., RPN2232) and visualized with X-ray film. The intensities of immunoreactive bands were quantified using Image Gauge software (Fuji Film, Tokyo, Japan). The indicated antibodies against the following proteins were used: NUPR1 (made in-house), lamin A/C (Santa Cruz, sc-20681), GAPDH (Millipore, MAB374), β-actin (Chemicon, MAB1501R), PDGFA (Abcam, ab135881), fascin (Santa Cruz, sc-21743), phospho-PDGFRα (Santa Cruz, sc-12911), PDGFRα (R&D Systems, AF-307-NA), phospho-MEK1/2 (Cell Signaling, #9154), MEK1/2 (Cell Signaling, #4694), phospho-ERK1/2 (Cell Signaling, #4370), ERK1/2 (Cell Signaling, #9211), and TR (Santa Cruz, sc-739).
Luciferase reporter assay
Serial deletion mutants of the NUPR1 promoter (-2944/+96) were prepared by PCR amplification and inserted into a pGL2 plasmid and extracted using plasmid extraction kit (Tools#DPT-BA17). The effects of T3 on the transcriptional activity of the resulting NUPR1 promoter fragments were determined by co-transfecting HepG2-TRα cells with the corresponding NUPR1 promoter-reporter plasmids and LacZ/β-galactosidase expression vector (included to normalize transfection efficiency) and then treating with 0-100 nM T3 for 24 h. Cells were lysed and luciferase activity was measured. A mutant form of the NUPR1 promoter construct was created by changing the TRE sequence, aggtcaCCTGaggtca to ggggggCCTGgggggg, using site-direct mutagenesis.
The PDGFA promoter region (-3090/+16) and two additional deletion constructs, -2128/+16 and -1242/+16, were prepared and inserted upstream of a minimal thymidine kinase promoter (pA3-TK). PDGFA 5'-flanking DNA reporter construct and a pcDNA3-NUPR1 expression plasmid were co-transfected into 293TN cells for 24 h. Mutant forms of PDGFA promoters were created by changing the mutant 1 sequence tatttttaata to tctgtttactg and mutant 2 sequence ccaataca to ccgccgca using site-direct mutagenesis.
Chromatin immunoprecipitation (ChIP) assay
HepG2-TRα cells were treated with or without 100 nM T3 for 48 h. After harvesting cells, DNA and proteins were crosslinked by incubating with 1% formaldehyde for 10 min, and complexes were sonicated to obtain DNA fragments in the 200-500-bp range. DNA fragments were immunoprecipitated with nonspecific mouse immunoglobulin G or specific antibodies against TR. Immunoprecipitated DNA fragments were amplified using specific primers targeting the binding region. FURIN and GAPDH promoter regions were used as positive and negative controls, respectively. For PDGFA promoter ChIP assays, NUPR1-overexpressing Mahlavu cells were harvested and crosslinked. Lysates were sonicated and immunoprecipitated with nonspecific rabbit immunoglobulin G or specific antibodies against NUPR1, and amplified using specific primers targeting the binding region.
HUVECs in vitro tube formation assay
For tube formation assays, 48-well culture plates were coated with 70% matrigel (BD Matrigel Basement Membrane Matrix) in EBM-2 media. After 24 h, 3 × 105 HUVECs were mix with 30 μg CM and seeded onto matrigel-coated culture plates. After 16 h, HUVECs tube formation was observed under a microscope.
Chick chorioallantoic membrane assay
Day-0 fertilized chicken eggs were incubated for 7 days at 37 °C. After that, a 3 × 3 cm hole was opened in eggs, and a 1:1 mix of CM and matrigel mixture was implanted in the embryo; the hole was then sealed with tape. After a 4-day incubation, 6 mL of 20% cream, used to visualize vessels, was injected into chick chorioallantoic membrane.
In vivo matrigel plug angiogenesis assay and immunohistochemical staining
NUPR1-overexpressing, -knockdown, or control cells (2 × 106) in 100 μl PBS were mix with 150 μl matrigel and injected subcutaneously into the flanks of nude mice (BALB/cAnN.Cg-Foxn1nu/CrlNarl) (n > 3). Mice were sacrificed 2 weeks later, and tumor weight and size were measured, and hemoglobin content was determined using Drabkin's reagent (Sigma-Aldrich). Histological sections from xenografts were immunostained for the angiogenesis marker CD31 (Abcam, ab28364). All experimental protocol was approved by Chang-Gung Institutional Animal Care and Use Committee (IACUC Approval No. CGU16-112).
Human angiogenesis array
Human angiogenesis array analyses were performed according to the manufacturer's protocol (B&D System). Briefly, 250 μl of CM from NUPR1-overexpressing or control Mahlavu cells was diluted and mixed with a cocktail of biotinylated detection antibodies. The resulting sample/antibody complexes were bound on membranes by their conjugate immobilized capture antibody. After washing to remove unbound material, streptavidin-HRP and chemiluminescence detection reagents were added sequentially. The light was produced at each spot in proportion to the amount of analyte bound. Mean pixel densities were quantified using Image J.
Measurement of PDGF-AA by enzyme-linked immunosorbent assay (ELISA)
The levels of PDGF-AA proteins in NUPR1-overexpressing or -knockdown cells were quantified using commercially available ELISA kits (R&D Systems) according to the manufacturer's instructions.
Cell viability assay
Cells were seeded at 6-12 × 103 cells/well in 96-well plates with or without 100 nM T3 and incubated overnight at 37 °C. Cells were treated with 0-10 μM sorafenib (Santa Cruze, sc-20125A) for 24 h. The medium was replaced by 100 μl of fresh medium containing 0.5 mg/ml MTT (Sigma, M5655) per well. After incubation at 37 °C for 4 h, the medium was removed, then 100 μl of isopropanol was added to each well. Pipette up and down several times to make sure the converted dye dissolved completely. The absorbance of each well was measured at 570 nm/650 nm.
Sorafenib treatment for Xenograft models of tumor progression
To investigate the antitumor effects of sorafenib on thyroid hormone receptor overexpressing tumors, J7-neo and J7-TRα cells (1 x 106) were injected subcutaneously into the flanks of nude mice (BALB/cAnN.Cg-Foxn1nu/CrlNarl) (n > 6). After the tumors reached the size of approximately 100 mm3, mice were orally administered 30 mg/kg sorafenib twice a week for 20 days. Control mice received the only vehicle. Tumor growth was measured twice a week. Animals were sacrificed at the end of experiment. All experimental protocol followed the United States National Institutes of Health guidelines and the Guide for the Care and Use of Laboratory Animals issued by the Chang-Gung Institutional Animal Care and Use Committee.
Human HCC specimens
Paired human HCC specimens (n = 158) were obtained from the Taiwan Liver Cancer Network (TLCN). Total RNA was reverse transcript to cDNA, and mRNA expression level was analyzed using qRT-PCR. Expression of NUPR1 and TR protein was determined by western blot analysis. The protocol was approved by the Medical Ethics and Human Clinical Trial Committee at Chang-Gung Memorial Hospital (IRB:103-4866B).
Statistical analysis
Data are presented as means ± SD of at least three independent experiments. Statistical analyses were performed using Student t-test and one-way analysis of variance (ANOVA). P-values < 0.05 were considered significant.
Results
NUPR1 is upregulated by T3/TR
To identify genes regulated by T3 in hepatoma cells, we performed oligonucleotide microarrays in HepG2-TRα cells [24]. Among the T3-regulated genes identified, NUPR1 was the most notably upregulated by T3 treatment (8-fold with 100 nM T3 for 48 h), and the result was verified by qRT-PCR (Figure 1A). Incubation of HepG2-TRα cells with 100 nM T3 for 48 h increased NUPR1 mRNA levels by 40-53-fold. Similar results were also observed in HepG2-TRβ cells, where treatment with 100 nM T3 for 48 h increased NUPR1 expression levels by 35-48-fold. NUPR1 mRNA levels trended higher in T3-treated HepG2-neo control cells, but this difference did not reach statistical significance.
The effect of T3/TR signaling on NUPR1 protein expression in HepG2 isogenic cell lines was assessed at different time points and different concentrations of T3 (Figure 1B). T3 induced a time- and concentration-dependent increase in NUPR1 protein levels in HepG2-TRα cells. Similar results were obtained in HepG2-TRβ cells. In contrast, exposure of control HepG2-neo cells to 100 nM T3 for 72 h did not significantly affect NUPR1 protein expression. The same phenomena were also observed in other hepatoma cell lines. J7-overexpression TRα and TRβ cells have shown increased NUPR1 expression with T3 treatment (Figure 1C). Notably, activated NUPR1 were also obtained in Huh7 cells, another HCC cell line that expresses detectable endogenous TR proteins (Figure S1) [25]. These results indicate that the effect of T3 on NUPR1 expression in TR-overexpressing cells depends on the level of TR proteins in these cells.
T3 induces expression of NUPR1 at the transcriptional level
To determine whether the regulation of NUPR1 expression by T3 occurred at the transcriptional level, we conducted promoter activity assays. To this end, the -2344 to +96-bp (-2344/+96) upstream region of the NUPR1 gene (transcription start site, +1), was inserted into the pGL2 plasmid to create the luciferase-based reporter plasmid, NUPR1-pGL2-luc. A bioinformatics search revealed putative TREs in the NUPR1 promoter, including a palindromic (Pal) TRE, a direct repeat 4 (DR4) TRE, and an inverted palindrome (F2) TRE. A series of pGL2-luc reporter constructs were transfected in HepG2-TRα cells, followed by treatment with 0-100 nM T3 for 24 h (Figure 1D). T3 at a concentration of 100 nM stimulated approximately a 3-fold increase in the activity of the full-length -2344/+96 NUPR1 promoter but had no effect on deletion mutant constructs lacking the most distal -2344/-1373 region, suggesting that this region contains a potential positive TRE. To confirm this and further refine the location of the TRE, we transfected HepG2-TRα cells with a luciferase-based reporter construct containing a -2201/-1798 fragment inserted upstream of a minimal thymidine kinase promoter. Subsequent transcription analyses showed that T3 increased the reporter activity, demonstrating that a possible TRE in the -2201/-1798 upstream region of NUPR1 is responsible for T3-induced transcription of the human NUPR1 gene. Notably, T3/TR-stimulated NUPR1 promoter activity was lost following mutation of the TRE sequence in the NUPR1 promoter. Collectively, these findings identify the -2201/-1798 region as the location of the TRE site in the NUPR1 promoter.
To further determine whether TR proteins directly target the TRE region of the NUPR1 promoter, we performed ChIP assays (Figure 1E). Immunoprecipitation with specific anti-TR antibodies followed by PCR amplification of precipitated DNA fragments clearly demonstrated that TR was recruited to the TRE binding site at -2066/-1910, whereas immunoprecipitation with control IgG yielded only background signals. The TRE-containing human FURIN gene was used as a positive control, whereas human GAPDH gene was used as the negative control. Collectively, these ChIP assay results reveal that the TR binds to the endogenous NUPR1 promoter in intact cells.
NUPR1 overexpression is linked to poor survival in clinical HCC specimens
To gain insight into the clinicopathological significance of NUPR1, NUPR1 expression in HCC was investigated. Expression of NUPR1 in 158 paired samples of human HCC tumor and their adjacent normal tissue were determined using qRT-PCR. NUPR1 was significantly overexpressed in HCC specimens compared with adjacent normal tissue (Figure 2A, left panel). An analysis of clinicopathological parameters showed that expression of NUPR1 was associated with vascular invasion and pathology stage (Figure 2A, right panel). Moreover, elevated TRα and TRβ levels were expressed in human HCC tumor specimens and were all correlated with vascular invasion (Figure S2A-B). Immunohistochemical study for angiogenesis marker CD31 and NUPR1 expression in clinical specimens showed that NUPR1 was highly expressed in tumors with vascular invasion (Figure S2C).
Figure 1
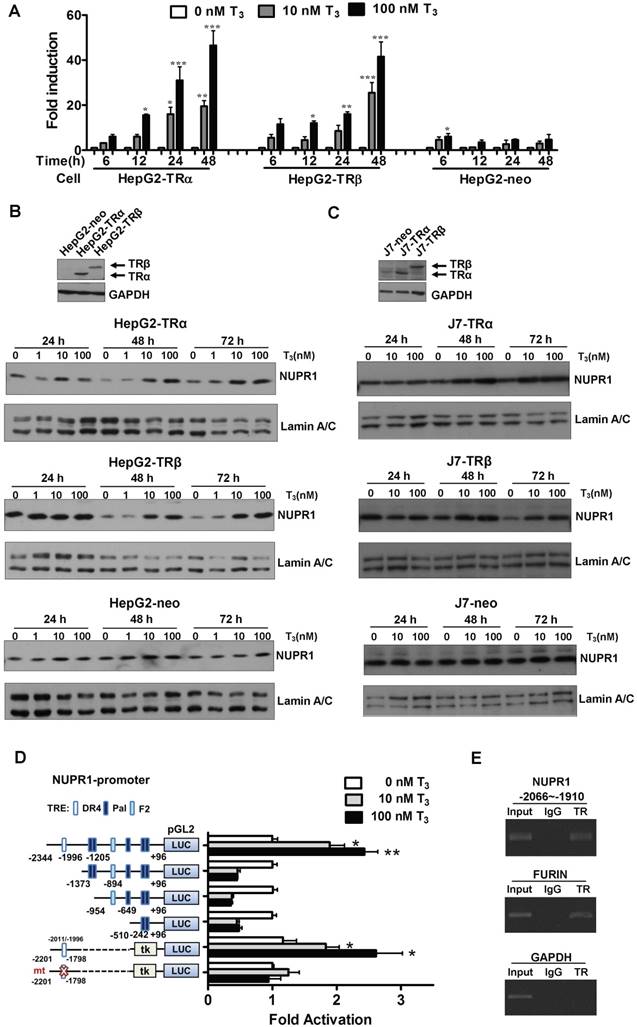
T3/TR signaling induces an increase in NUPR1 mRNA and protein expression. (A) HepG2-TRα, HepG2-TRβ, and HepG2-neo cells were treated with T3 (0-100 nM) for 6-48 h, and NUPR1 expression levels were measured using qRT-PCR. Expression of NUPR1 mRNA was normalized to that of 18s rRNA. (B-C) NUPR1 protein expression levels were determined in TR overexpression and control HepG2 and J7 cell lines treated for different times with different concentrations of T3. Lamin A/C was used as an internal control. (D) Schematic representation of the NUPR1 promoter, indicating potential TREs (squares). HepG2-TRα cells were transfected with serially deleted NUPR1 5'-flanking DNA pGL2-luc reporter constructs, and treated with 0-100 nM T3. After 24 h, cells were lysed and promoter activity was determined. (E) HepG2-TRα cell lysates were immunoprecipitated with nonspecific mouse IgG or antibodies against TR. FURIN and GAPDH promoter regions were used as positive and negative controls, respectively. Differences were analyzed using a one-way ANOVA (*p < 0.05, **p < 0.01, ***p < 0.001).
Figure 2
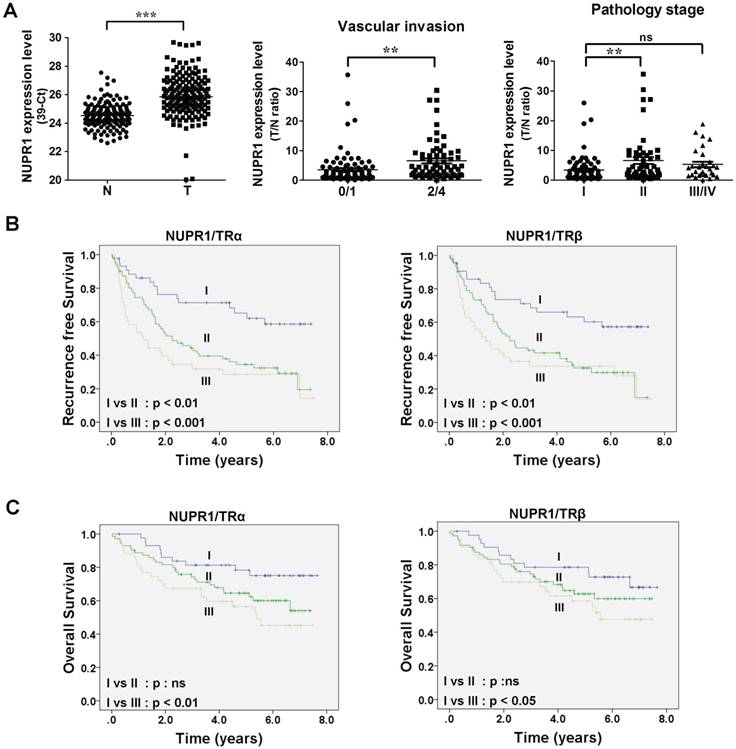
High NUPR1 expression is linked to poor prognosis. (A) Expression of NUPR1 in 158 paired human HCC (T) and adjacent normal (N) tissues were determined by qRT-PCR; expression of NUPR1 mRNA was normalized to that of 18s rRNA (left panel). NUPR1 expression levels were presented as 39-Ct values. Statistical significance was calculated using paired Student's t test. NUPR1 mRNA expression level were presented as the T/N ratio (right two panels). NUPR1 expression levels were highly correlated with vascular invasion (0/1 grade, n = 93; 2/4 grade, n = 65) and pathology stage (stage I, n = 76; stage II, n = 52; stage III/IV, n = 30). Differences were analyzed using one-way ANOVA (*p < 0.05, **p < 0.01, ***p < 0.001). (B-C) Recurrence-free survival and overall survival were analyzed using the Kaplan-Meier method. Median expression levels of NUPR1 and TRs were used as a cutoff. NUPR1 and TR expression levels were separated to three groups: group I, low levels of NUPR1 and TRs (TRα, n = 44; TRβ n = 43); group II, high levels of NUPR1 and low levels of TRs, or low levels of NUPR1 and high levels of TRs (TRα, n = 71; TRβ, n = 72); group III, high levels of NUPR1 and TRs (TRα, n = 43; TRβ, n = 43).
Tumor samples were then dichotomized into NUPR1 low- and high-expression groups, using the median T/N ratio of HCC specimens as a cutoff. Patients in the high NUPR1 expression group displayed worse recurrence-free survival (p = 0.009), but the overall survival was not significantly different. We then separated NUPR1 and TR expression levels into three groups: group I, low expression of NUPR1 and TRs; group II, high expression of NUPR1 and low expression of TRs, or low expression of NUPR1 and high expression of TRs; and group III, high expression of NUPR1 and TRs. Patients with high expression of NUPR1 and TRs showed significantly poorer recurrence-free survival (Figure 2B) and overall survival (Figure 2C) than those with low expression of NUPR1 and TRs. However, NUPR1, TRα, and TRβ were not an independent prognostic factor associated with survival (Table S1).
NUPR1 promotes angiogenesis in vitro
Clinical data showed an association of high expression of NUPR1 with vascular invasion. Previous reports have indicated that thyroid hormone sustains angiogenesis [26-28]. Accordingly, we analyzed the effect of conditioned medium (CM) derived from NUPR1-knockdown or control HepG2-TRα cells, incubated in the presence of 100 nM T3 or T3-depleted serum (0 nM T3), on tube formation by HUVECs. As shown in Figure 3A, CM from T3-stimulated HepG2-TRα cells promoted a prominent and significant increase in tube formation compared with T3-depleted cells.
Figure 3
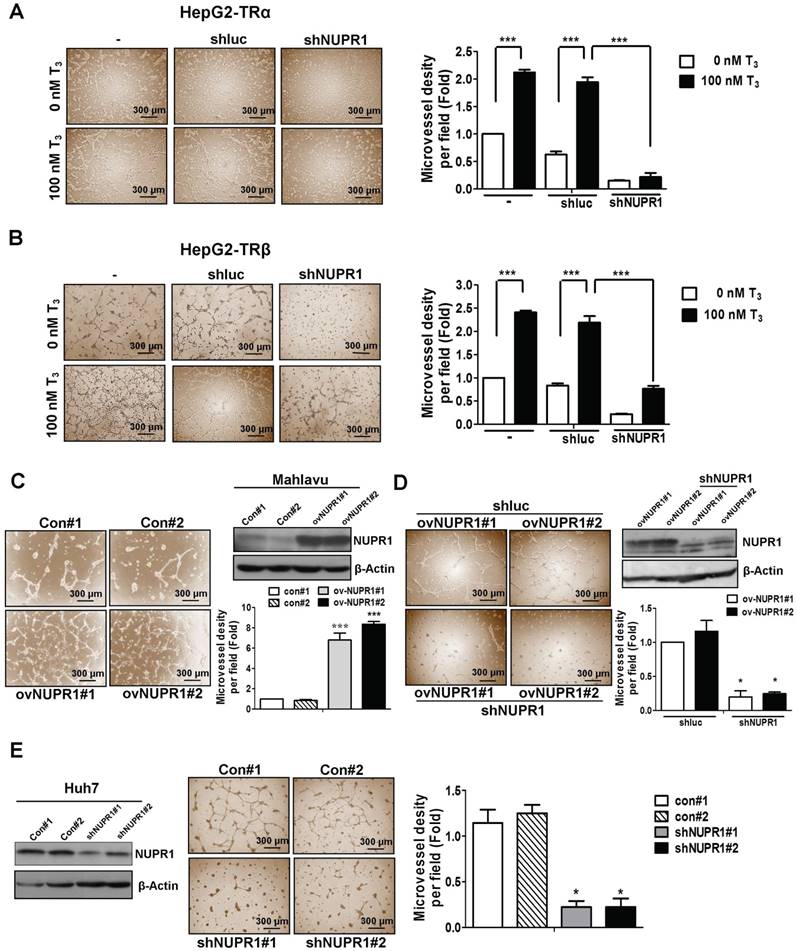
NUPR1 promotes cells angiogenesis in vitro. (A, B) CM was harvested from HepG2-TRα and HepG2-TRβ cells, transfected with or without luciferase or shNUPR1 plasmids, and treated with 0 nM or 100 nM T3 for 24 h. HUVECs were seeded onto matrigel-coated dishes and incubated with CM for 16 h. Representative images are shown. (C) HUVECs were mixed with CM from NUPR1-overexpressing or control Mahlavu cells for 16 h. Representative images are shown. (D) HUVECs were treated with CM from NUPR1-knockdown or control Mahlavu-NUPR1 cells for 16 h. The number of branching points were quantified. (E) HUVECs were seeded on matrigel and treated with CM from NUPR1-knockdown or control Huh7 cells for 16 h. Representative images are shown. Bar plot represents means ± SD (*p < 0.05, **p < 0.01, ***p < 0.001).
Notably, HUVECs tube formation was significantly decreased in T3-treated NUPR1-knockdown HepG2-TRα cells compared that induced by control cells. Similar results were observed in HepG2-TRβ cells (Figure 3B), J7-TRα cells and J7-TRβ cells (Figure S3A). To determine the angiogenesis function of NUPR1 in HCC cell lines, we stably expressed NUPR1 in the Mahlavu HCC cell line, which expresses relatively low endogenous levels of NUPR1 (Figure S3B). We then treated HUVECs with CM from NUPR1-overexpressing or control Mahlavu cells. These experiments showed that NUPR1-overexpressing cells significantly induced HUVECs tube formation compared with that induced by control cells (Figure 3C). Conversely, NUPR1-knockdown Mahlavu-NUPR1 cells inhibited HUVECs tube formation compared with control Mahlavu-NUPR1 cells (Figure 3D). To determine the consequences of NUPR1 depletion in hepatoma cells, we established NUPR1-knockdown and control Huh7 cells, and assessed the effect of CM from these cells on angiogenesis. In contrast to the angiogenic effect of NUPR1-overexpressing cells, NUPR1-knockdown Huh7 cells significantly decreased HUVECs tube formation (Figure 3E). These results indicate that T3/TR-upregulated NUPR1 promotes angiogenesis in vitro.
Expressed NUPR1 in hepatoma cells promotes angiogenesis in vivo
We next assessed whether NUPR1 promotes angiogenesis in vivo, we performed chick chorioallantoic membrane (CAM) assays. Vessel growth on chick chorioallantoic membranes of eggs treated with NUPR1-overexpressing CM was higher than control cells (Figure 4A). Conversely, CM derived from NUPR1-knockdown Huh7 cells significantly suppressed vessels formation on membranes of eggs (Figure 4B). To further validate the role of NUPR1 in vivo, matrigel plug angiogenesis assay was performed. Weights and volumes of tumors formed from NUPR1-overexpressing cells were not significantly different from those of tumors formed from control cells. However, the hemoglobin content in xenograft tumors from NUPR1-overexpressing cells was 8-11-fold higher than that in control xenograft tumors (Figure 4C). Histological analysis of sections from NUPR1 overexpressing xenograft tumors immunostained for the angiogenesis marker CD31 revealed that CD31-positive microvessel density areas were higher in NUPR1-overexpressing xenograft tumors than control tumors (Figure 4D). We further examine the effect of NUPR1-knockdown on matrigel plug angiogenesis assay. As expected, Huh7-shNUPR1-derived tumors displayed a significant decrease in blood vessels and hemoglobin content compared with control groups (Figure 4E-F). These data demonstrate that NUPR1 play an essential role in angiogenesis in hepatocellular carcinoma.
NUPR1-dependent angiogenesis is mediated by induction of PDGFA expression and secretion
To investigate the mechanism underlying NUPR1-induced angiogenesis, we profiled the expression of 55 angiogenesis related proteins in CM from NUPR1-overexpressing or control Mahlavu cells. These screens showed that expression levels of the angiogenesis-related proteins, PDGF-AA (platelet-derived growth factor AA), TGF-β1 (tumor-derived growth factor-β1), CCL3 (C-C motif chemokine ligand 3) and VEGF-C (vascular endothelial growth factor-C) were higher in NUPR1-overexpressing cells compared with control cells (Figure 5A). Of these, the gene encoding PDGFA, which is known to be critical for HCC progression [29, 30], was most notably upregulated with NUPR1 overexpression. Thus, we selected PDGF-AA for further study. To validate the PDGF-AA expression level in NUPR1-overexpressing and knockdown cells, we performed ELISA and western blotting. ELISAs showed that secreted PDGFA levels were increased in CM from NUPR1-overexpressing cells and decreased in CM from NUPR1-knockdown cells compared with that from control cells (Figure 5B). A similar phenomenon was observed in western blot data (Figure 5C), confirming that NUPR1 enhances PDGFA expression/secretion.
To validate the involvement of PDGFA in promoting angiogenesis, we knocked down PDGFA expression in NUPR1-overexpressing Mahlavu cells and assessed HUVECs tube formation following treatment with Mahlavu cell-derived CM. HUVECs tube formation induced by CM from PDGFA-knockdown NUPR1-overexpressing cells was reduced compared with control cells (Figure 5D). Furthermore, pretreatment of HUVECs with a neutralizing antibody against human PDGFRα significantly decreased tube formation by CM from NUPR1-overexpressing cells (Figure 5E). Conversely, we found that treatment of HUVECs with recombinant human PDGFA protein in the presence of NUPR1-knockdown CM significantly increased tube formation (Figure 5F). Collectively, these results demonstrate that the pro-angiogenic activity of NUPR1 is mediated by PDGFA.
Figure 4
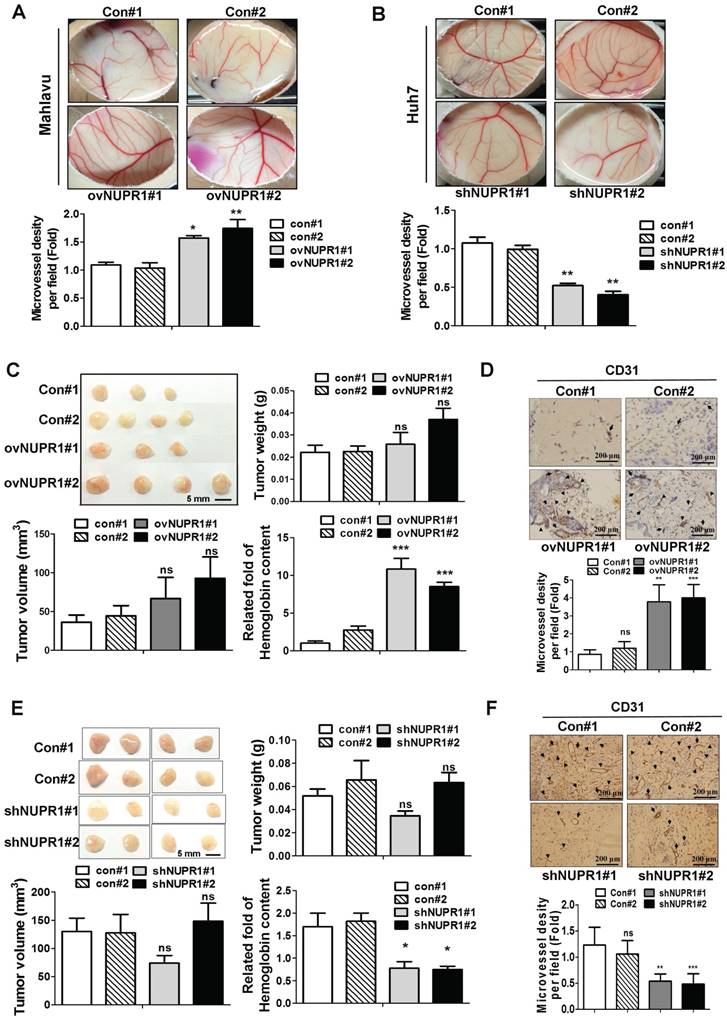
NUPR1 promotes angiogenesis in vivo. (A, B) A 1:1 mixture of matrigel and CM from NUPR1-overexpressing Mahlavu cells, or NUPR1-knockdown Huh7 cells, or their respective controls, were implanted into the embryo. After a 4-day incubation, vessels on the chick chorioallantoic membrane were counted. (C, E) NUPR1-overexpressing Mahlavu cells, or NUPR1-knockdown Huh7 cells, or their respective controls, were mixed with matrigel and injected subcutaneously into the flanks of nude mice (n > 3/group). Two weeks later, mice were sacrificed and tumor weight, tumor size, and hemoglobin content were measured. (D, F) Histological sections from C, E xenografts were immunostained for the angiogenesis marker CD31. CD31-positive blood vessels are indicated by arrows. Bar plot represents means ± SD (*p < 0.05, **p < 0.01, ***p < 0.001).
Figure 5
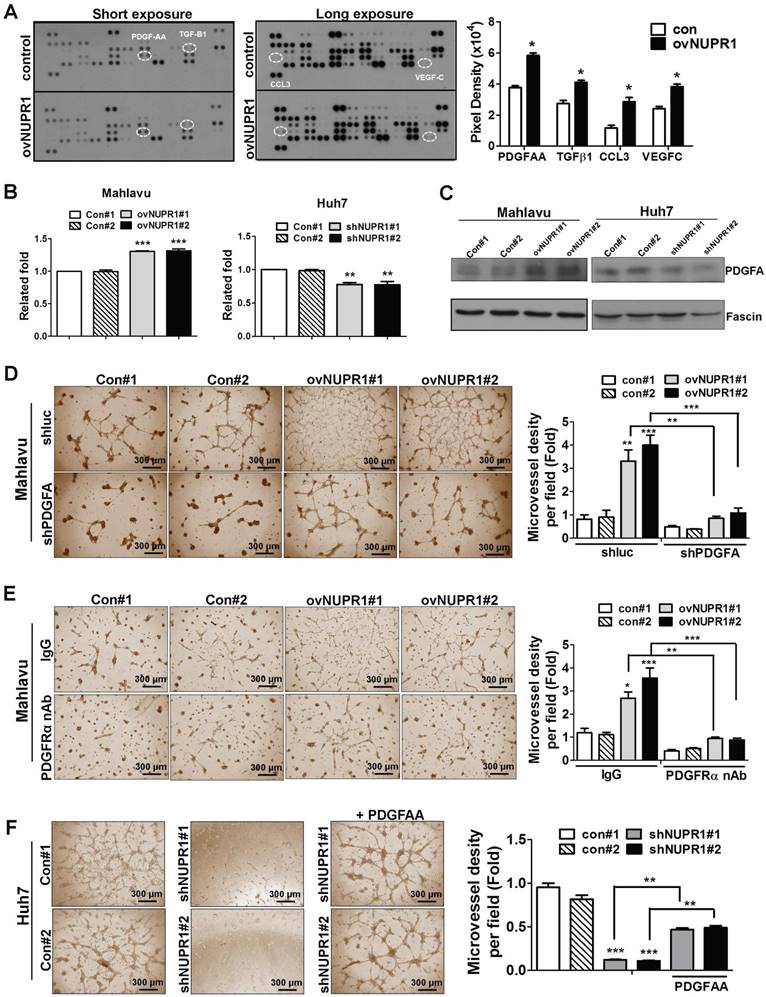
NUPR1 acts through induction of PDGFA expression to promote angiogenesis. (A) Human-specific angiogenesis antibody array membranes were incubated with CM from NUPR1-overexpressing or control Mahlavu cells. Quantification of mean pixel densities were shown. (B, C) Relative levels of PDGF-AA secreted by NUPR1-overexpressing Mahlavu cells, or NUPR1-knockdown Huh7 cells, or their respective controls, were analyzed by ELISA and western blotting. Results were shown as the fold-change in PDGF-AA secretion compared with control cell lines. Fascin was used as a loading control. (D) HUVECs were seeded on matrigel and treated with CM from NUPR1-overexpressing or control Mahlavu cells transfected with control shLuc or shPDGFA plasmid for 16 h. (E) HUVECs were pretreated with 10 μg/ml nonspecific rabbit IgG or PDGFRα neutralizing antibodies for 1 h, then seeded onto matrigel together with CM from NUPR1-overexpressing or control Mahlavu cells for 16 h. (F) HUVECs were seeded onto matrigel and treated with CM from NUPR1-knockdown or control Huh7 cells. The third panel shows HUVECs treated with CM from NUPR1-knockdown Huh7 cells together with 20 ng/ml PDGFA ligand. Bar plot represents means ± SD (*p < 0.05, **p < 0.01, ***p < 0.001; one-way ANOVA).
NUPR1 promotes angiogenesis through a PDGFA/MEK/ERK signaling cascade
To investigate the signaling pathway downstream of NUPR1/PDGFA that is involved in inducing angiogenesis, we treated HUVECs with CM from NUPR1-overexpressing Mahlavu cells or NUPR1-knockdown Huh7 cells, or their respective controls, and performed western blotting. These analyses showed that expression levels of the phosphorylated forms of PDGFRα, MEK1/2, and ERK1/2 proteins were increased in HUVECs cultured with CM from NUPR1-overexpressing cells compared with control cells (Figure 6A). Conversely, the phosphorylated forms of these proteins in HUVECs were decreased after treatment with CM from NUPR1-knockdown cells (Figure 6B). To further clarify the signaling pathway involved in angiogenesis in vitro, we treated HUVECs with CM from NUPR1-overexpressing or control cells containing the ERK inhibitor U0126 or dimethylsulfoxide (DMSO). As shown in Figure 6C, treatment with ERK inhibitor-containing CM significantly disrupted NUPR1-mediated tube formation. Furthermore, the blood vessels in chick embryo chorioallantoic membranes were decreased in the presence of the ERK inhibitor (Figure 6D). These results suggest that NUPR1 acts through induction of PDGFA expression to promote endothelial tube formation, an effect that requires activation of the MEK/ERK signaling pathway.
NUPR1 drives transcriptional activation of PDGFA through direct binding to the promoter region
To further explore the mechanisms by which NUPR1 regulates PDGFA transcription, we conducted promoter activity assays. Although the consensus sequence of the NUPR1 binding site has not been demonstrated, NUPR1 has been reported to share biochemical homology with HMG-like proteins [18], which bind to an A/T-rich sequence. To confirm this, we created a luciferase-based reporter plasmid by inserting an upstream promoter region (-3090/+16) of the PDGFA gene into a minimal thymidine kinase promoter. In addition to the full-length promoter region, two deletion constructs, -2128/+16 and -1242/+16, were prepared. Predicated potential NUPR1 binding sites are indicated by squares. The -3090/+16 region of PDGFA promoter increased luciferase transcriptional activity by approximately 2-fold, whereas other PDGFA deletion reporter constructs had no effect (Figure 6E, left panel). These data indicate that the potential NUPR1 binding site in the PDGFA promoter is located within the -3090/-2128 region. To confirm this and further refine the NUPR1 binding site, we generated PDGFA promoter -3090/+16 with first binding site mutant (mutant 1) or two binding sites mutant (mutant 2) and the results showed that promoter activity was significantly decreased in PDGFA promoter mutant 2 compared with wild type promoter, demonstrating that a possible NUPR1 binding site in the -2231/-2027 upstream region of PDGFA (Figure 6E, right panel). To further determine whether NUPR1 proteins directly target the PDGFA promoter, we performed ChIP assays (Figure 6F). The results clearly demonstrated that NUPR1 was recruited to the PDGFA promoter at -2231/-2027 region, whereas control IgG yielded only background signals. Collectively, these ChIP assay results reveal that the NUPR1 binds to the endogenous PDGFA promoter in intact cells.
TR-NUPR1 increase hepatoma cells chemoresistance
Chemotherapy resistance is a major cause of treatment failure in a malignant tumor. Up to now, sorafenib is the only approved drug for treatment of advanced HCC to inhibit angiogenesis and growth of tumor cell. However, it is increasingly reported that drug resistance often develops and the mechanism remains unclear [31, 32]. Our previous studies indicated that T3/TR signaling promoted chemoresistance in hepatoma cells [14, 33]. Elevated thyroid hormone was associated with poor outcomes in advanced HCC patients receiving sorafenib treatment [34]. NUPR1 has also been reported that it participated in sorafenib resistance [21]. To investigate whether TR-NUPR1 is involved in sorafenib resistance, TR overexpression HepG2 and J7 cells were treated with sorafenib and cell viability was evaluated. Cell viability following the addition of sorafenib was significantly increased in T3-treated HepG2-TRα and HepG2-TRβ cells. In contrast, exposure of control HepG2-neo cells to T3 did not significantly affect cell viability (Figure 7A). Similar results were obtained in J7-TRα and J7-TRβ cells (Figure 7B). Moreover, rescue experiments have been performed by depletion of NUPR1 expression after T3 treatment. Knockdown of NUPR1 suppressed cell viability in T3-treated TR overexpressing HepG2 and J7 cell lines following the addition of sorafenib (Figure 7C-D). These results suggest that TR increases hepatoma cells chemoresistance through upregulating NUPR1.
Figure 6
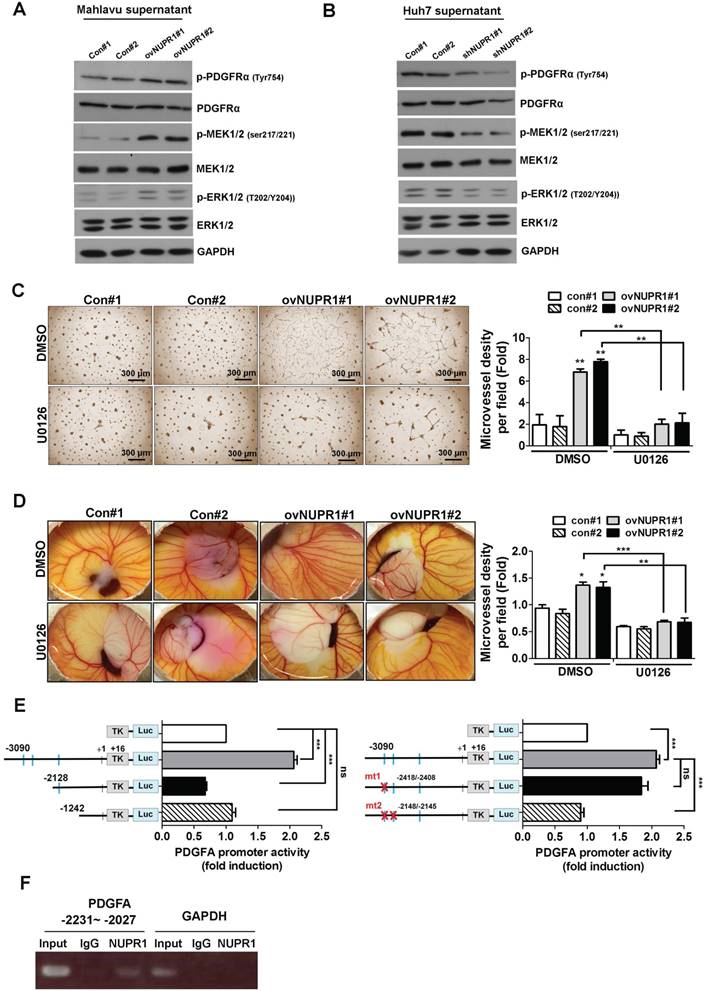
NUPR1 promotes angiogenesis through a PDGFA/MEK/ERK signaling cascade. (A, B) The expression levels of total and phosphorylated forms of PDGFRα, MEK1/2, and ERK1/2 proteins in HUVECs cultured with CM from NUPR1-overexpressing, -knockdown, or control Mahlavu and Huh7 cells for 48 h were analyzed by western blotting. GAPDH was used as a loading control. (C) HUVECs were pretreated with DMSO (vehicle control) or the ERK inhibitor U0126 (10 μM) for 1 h, then seeded on matrigel together with CM from NUPR1-overexpressing and control Mahlavu cells for 16 h. (D) Chick embryo were treated with DMSO or U0126 for 1 h, after which a mixture of matrigel and CM from NUPR1-overexpressing or control Mahlavu cells was implanted on the chick embryo. After a 4-day incubation, the number of vessel branching points were quantified by image analysis. (E) Serially deleted pA3TK-PDGFA promoter-report constructs and pcDNA3-NUPR1 expression plasmid were co-transfected in 293TN cells, after 24 h, cells were lysed and promoter activity was determined (left panel). Wild type and mutant forms of PDGFA promoter constructs were co-transfected with pcDNA3-NUPR1 plasmid in 293TN cells, after 24 h, cells were lysed and promoter activity was determined (right panel). (F) NUPR1-overexpression Mahlavu cell lysates were immunoprecipitated with nonspecific rabbit IgG or antibodies against NUPR1. GAPDH promoter region was used as negative control, respectively. Bar plot represents means ± SD (*p < 0.05, **p < 0.01, ***p < 0.001; one-way ANOVA).
Figure 7
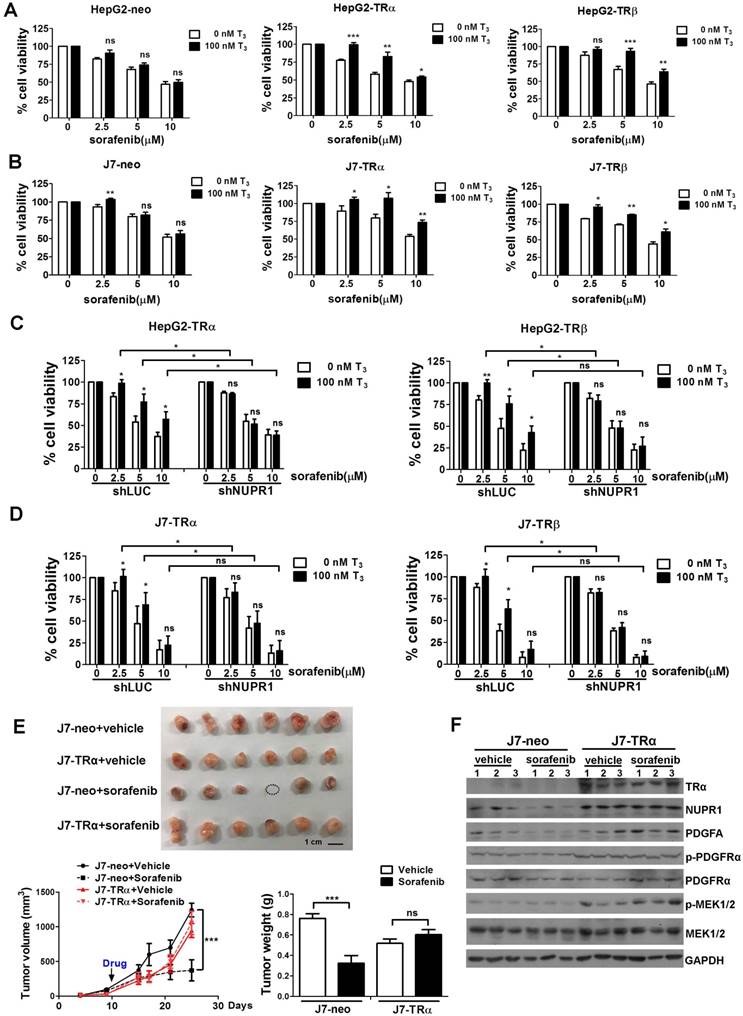
TR-NUPR1 induces sorafenib resistance in hepatoma cells in vitro and in vivo. (A-B) Cells were seeded at 6-12 × 103 cells/well in 96-well plates with or without 100 nM T3 and incubated overnight at 37 °C. After that, cells were treated with 0-10 μM sorafenib for 24 h. The MTT assay absorbance of each well was measured at 570 nm/650 nm. (C-D) NUPR1 was knockdown in TR overexpression HepG2 and J7 cell lines and cells were treated with T3 and sorafenib. The MTT assay absorbance of each well was measured at 570 nm/650 nm. (E) 1 x 106 of J7-neo and J7-TRα cells were injected subcutaneously into the flanks of nude mice (n > 6). After the tumors reached the size of approximately 100 mm3, mice were administered 30 mg/kg sorafenib by oral gavage twice a week for 20 days. Control mice received only the vehicle (DMSO). Tumor growth was measured twice a week. Mice were sacrificed and the tumors were removed, weighed, and processed for western blot analysis. (F) TRα, NUPR1, PDGFA, PDGFRα and MEK1/2 expression level were determined by western blot from J7-neo or J7-TRα xenografts. GAPDH was used as a loading control. Data represents means ± SD (*p < 0.05, **p < 0.01, ***p < 0.001).
Figure 8
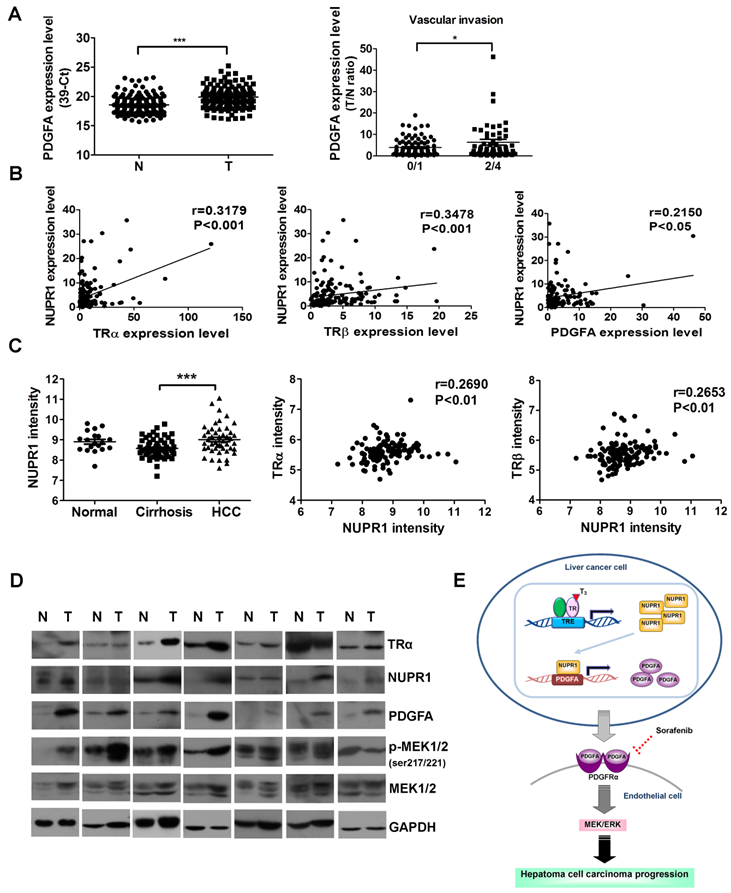
NUPR1 expression is positively correlated with TR and PDGFA expression in hepatoma specimens. (A) Expression of PDGFA in 158 paired human HCC (T) and adjacent normal (N) tissues were determined by qRT-PCR; expression of PDGFA mRNA was normalized to that of 18s rRNA (left panel). PDGFA expression levels were highly correlated with vascular invasion. Differences were analyzed using one-way ANOVA (*p < 0.05, **p < 0.01, ***p < 0.001). (B) Spearman correlations of TRα, TRβ, and PDGFA with NUPR1 in 158 paired human specimens were analyzed and shown. (C) NUPR1 expression levels were analyzed in the public Oncomine database. Left panel: The Mas liver database showed that NUPR1 expression levels in HCC tissues were higher than those in cirrhotic tissue (normal, n = 19; cirrhosis, n = 58; HCC, n = 47). Spearman analyses showed that TRα and TRβ expression were positively correlated with NUPR1 expression. Expression levels of these genes are presented as microarray intensities. (D) Expression of angiogenic related proteins, PDGFA, p-MEK1/2, NUPR1 and TRα protein expression levels in paired HCC tissues were determined by western blotting. GAPDH was used as a loading control. (E) Schematic model of the regulation of angiogenesis in hepatoma cell carcinoma progression by the T3/TR/NUPR1/PDGFA/MEK/ERK pathway.
To explore the effect of sorafenib resistance mediated by T3/TR-induced NUPR1 expression in vivo, J7-neo and J7-TRα cells were injected subcutaneously into the flanks of nude mice. After the tumors reached the size of approximately 100 mm3, mice were administered sorafenib by oral gavage. Concordant with in vitro results, TR overexpression conferred sorafenib resistance. In contrast, exposure of control J7-neo xenografts to sorafenib significantly affect tumor growth and tumor weight (Figure 7E). In addition, western blot analyses showed that NUPR1, PDGFA, p-PDGFRα and p-MEK1/2 expression levels were higher in J7-TRα xenografts compared with J7-neo xenografts (Figure 7F). Moreover, these genes were suppressed in sorafenib-treated groups compared with vehicle-treated groups in J7-neo xenografts, but its expression was not affected or only slightly decreased in sorafenib-treated groups in J7-TRα xenografts. Collectively, these findings confirm that T3/TR mediated sorafenib resistance via NUPR1 and its downstream targets.
NUPR1 is positively correlated with TRα, TRβ, and PDGFA expression in clinical HCC specimens
The clinicopathological significance of PDGFA expression in HCC was also investigated. PDGFA was significantly overexpressed in 158 paired HCC specimens compared with adjacent normal tissue (Figure 8A, left panel). An analysis of clinicopathological parameters showed that expression of PDGFA was associated with vascular invasion (Figure 8A, right panel). A linear regression analysis further revealed a significant positive correlation between NUPR1 and TRα, TRβ, PDGFA levels, based on the T/N ratio (Figure 8B). Furthermore, an analysis of the public Oncomine database containing 124 human hepatoma samples (Mas liver) revealed that NUPR1 expression levels were higher in HCC samples than in cirrhosis samples (Figure 8C, left panel). A Spearman analysis showed that TRα and TRβ expression were positively correlated with NUPR1 expression (Figure 8C, right panel). In addition, western blot analyses showed that angiogenic-related genes- PDGFA, p-MEK1/2, NUPR1 and TRα expression levels were increased in HCC tissues in 7 representatives paired specimens (Figure 8D). Clinical investigations reveal associations among TR, NUPR1, and PDGFA, supporting an important role of NUPR1 in HCC progression.
Discussion
Liver cancer is one of the leading causes of cancer deaths worldwide [1-3], at least in part because of the high angiogenic and metastatic potential of liver tumor cells. High levels of angiogenesis-related factors are significantly associated with rapid recurrence and poor survival [35], but the underlying mechanisms are not fully understood. Hence, identification of novel therapeutic targets is crucial for the prognosis of HCC. Here, our findings support an oncogenic role of NUPR1 in HCC, suggesting that the PDGFA/MEK/ERK signaling pathway is involved in TR/NUPR1-induced angiogenesis and sorafenib chemoresistance in HCC (Figure 8E).
Accumulating evidence has implicated aberrant expression of TRs in human cancers [5, 8, 9, 36]. Cristofanilli et al. reported that hypothyroid patients have a reduced incidence of primary breast carcinoma in association with a lower risk of developing the invasive disease [11]. Moreover, treatment with the antithyroid drug, propylthiouracil, in combination with tamoxifen increased survival in patients with glioblastoma [12]. Consistent with this, epidemiological studies have demonstrated that hyperthyroidism is associated with an increased risk of ovarian cancer [13]. Interestingly, treatment of hypothyroidism with levothyroxine (T4) was shown to be associated with a significantly reduced risk of colorectal cancer [37], and a previous case-control study revealed that hypothyroidism was associated with a significantly elevated risk of HCC in women [38]. Our previous studies also indicated that numerous oncogenes and suppressor genes regulated by T3/TR could promote or suppress HCC progression [14-17, 25, 39, 40]. Collectively, these observations demonstrated that T3/TR signaling could play dual roles in carcinogenesis, possibly reflecting the various TR isoforms, functions of T3/TR in different tissues, and different stages of tumor development. Thus, how T3/TR switches between an oncogenic role and tumor suppressor role will require further investigation.
Several reports have demonstrated that NUPR1 is involved in the progression of various cancers. Elevated NUPR1 level has been found in pancreatic cancer patients [20], and NUPR1 expression has been shown to promote pancreatic cancer cell metastasis and invasion. Jung et al. reported that expression of NUPR1 in early-stage breast cancers is associated with poorer prognosis [41]. Moreover, knockdown of NUPR1 was shown to inhibit tumor growth and deregulate autophagic flux and impairs autolysosomal clearance in human non-small-cell lung cancer [42, 43], and inhibit anti-apoptotic and tumor cell promoter activity in colorectal cancer [44]. In the current study, we provide the first demonstration that T3/TR-induced increases in NUPR1 upregulate expression of PDGFA, which promotes angiogenic effects in HCC through activation of the MEK/ERK signaling pathway. A clinicopathological analysis revealed that patients with high expression of NUPR1 and TRs displayed significantly poorer overall survival than those with low expression of NUPR1 and TRs. Consistent with our data, Emma et al. found that NUPR1 expression was significantly higher in primary human HCC tissues, and further showed that NUPR1 increased cell growth, migration, and invasion [21]. Lee et al. also identified NUPR1 as a key modulator of a mitochondrial respiratory defect during liver cancer progression [22]. In addition, Bak et al. reported that NUPR1 is activated by hepatitis B virus X protein, the primary risk factor in HCC, through a Smad4 pathway, and modulates cell growth and survival in liver cancer [45]. These reports support the conclusion that NUPR1 plays an important role in hepatocarcinogenesis.
Angiogenesis, a hallmark of cancer, is induced early during the multistage development of invasive cancers [46]. Here, we provide the first evidence that NUPR1 acts through transactivation of PDGFA to promote endothelial tube formation in HCC. PDGFA is a member of the PDGF family, which consists of five isoforms that regulate angiogenesis and participate in cancer progression [47]. Each isoform binds to a dimeric form of one of two different receptors—PDGFRα or PDGFRβ—to activate downstream signal pathways, with PDGFA binding to PDGFRα [48]. Several studies have reported that PDGFA is involved in tumor development. PDGFA expression is elevated in oral cancer, pancreatic cancer, and mantle cell lymphoma, and its expression was shown to promote tumor cell migration, invasion, and angiogenesis [49-51]. In addition, it has been reported that higher expression of PDGFA is associated with hepatic fibrogenesis in chronic hepatitis C [30]. PDGFA also contributes to HCC progression by enhancing NRF2 expression [29]. In the current study, we found that the angiogenic effects of NUPR1 were disrupted by a PDGFRα neutralizing antibody and an ERK inhibitor, demonstrating that these actions of NUPR1 are mediated by activation of PDGFA/MEK/ERK signaling pathway.
Sorafenib, a multikinase inhibitor with activity against Raf kinase, VEGF receptors, PDGFRs and other tyrosine kinases, has been approved for the treatment of advanced HCC [52]. Sorafenib improves survival in HCC patients, reduces tumor cell proliferation and angiogenesis, and increases apoptosis [31]. However, the benefits of sorafenib are modest [32], and its mechanism of action is incompletely understood. Recently, Emma et al. reported that NUPR1 is involved in sorafenib resistance in HCC, showing that NUPR1 knockdown decreased cell growth and increased tumor cell sensitivity to sorafenib treatment [21]. Several studies have also demonstrated that sorafenib impairs tumor cell proliferation and viability through the MEK/ERK pathway. Our previous studies revealed that T3/TR upregulated TRAIL and Bcl-xL, suppressed FoxO1 and Bim to promote hepatoma cells metastasis and chemotherapies resistance [14, 33]. Higher value of thyroid-stimulating hormone (TSH) and free T4 (FT4) were associated with unfavorable tumor progression and overall survival in advanced HCC patients received sorafenib treatment. An index of thyroid function can significantly predicted opposite clinical outcomes in advanced HCC patients receiving sorafenib or chemotherapy treatment [34]. Consistent to our results, we confirmed the sorafenib resistance effect of T3/TR and this ability was mediated through NUPR1 by directly regulating the levels of PDGFA expression and secretion. PDGFA expression may account for the increased angiogenesis of tumor cells that inhibited by sorafenib. Our findings thus suggest potential molecular mechanisms for sorafenib chemoresistance in HCC tumorigenesis.
In conclusion, we have identified NUPR1-mediated regulation of angiogenesis via the PDGFA/MEK/ERK cascade as a novel pathway of thyroid hormone receptor-dependent HCC progresssion. Modulation of this angiogenesis pathway may provide potential therapeutic targets for HCC.
Abbreviations
HCC: hepatocellular carcinoma; NUPR1: nuclear protein-1; T3: 3,3'-5-triiodo-L-thyronine; TR: thyroid hormone receptor; PDGFA: platelet-derived growth factor A; HUVECs: human umbilical vein endothelial cells; TREs: thyroid hormone response elements; HMG: high mobility group; qRT-PCR: quantitative reverse transcription-polymerase chain reaction; ChIP: chromatin immunoprecipitation; CM: conditioned medium; CAM: chick chorioallantoic membrane; TGF-β1: tumor-derived growth factor-β1; CCL3: C-C motif chemokine ligand 3; VEGF-C: vascular endothelial growth factor-C; ELISA: enzyme-linked immunosorbent assay; PDGFRα: platelet-derived growth factor receptor α.
Supplementary Material
Supplementary figures and table.
Acknowledgements
This work was supported by grants from Chang Gung Memorial Hospital, Taoyuan, Taiwan (CMRPD1G0421, CMRPD1G0422, CRRPD1F0011, CRRPD1F0012, CRRPD1F0013, BMRP130, NMRPD1D1021, NMRPD1D1022 and NMRPD1D1023 to K. H. Lin) and from the Ministry of Science and Technology of the Republic of China (MOST 103-2320-B-182-018-MY3 to KHLin). We would like to thank the Taiwan Liver Cancer Network (TLCN) for providing the hepatoma tissue samples and related clinical data (all anonymous).
Author contributions
CYC performed most experiments and wrote the manuscript. SMW performed promoter assay and ChIP assay experiments. YHL, HCC, SLL, CTY, WYC contributed experience on methods. KHL supervised and reviewed the manuscripts.
Competing Interests
The authors have declared that no competing interest exists.
References
1. McGlynn KA, London WT. The global epidemiology of hepatocellular carcinoma: present and future. Clin Liver Dis. 2011;15:223-43 vii-x
2. Porru S, Placidi D, Carta A, Gelatti U, Ribero ML, Tagger A. et al. Primary liver cancer and occupation in men: a case-control study in a high-incidence area in Northern Italy. Int J Cancer. 2001;94:878-83
3. El-Serag HB. CURRENT CONCEPTS Hepatocellular Carcinoma. New Engl J Med. 2011;365:1118-27
4. Galun D, Srdic-Rajic T, Bogdanovic A, Loncar Z, Zuvela M. Targeted therapy and personalized medicine in hepatocellular carcinoma: drug resistance, mechanisms, and treatment strategies. J Hepatocell Carcinoma. 2017;4:93-103
5. Wu SM, Cheng WL, Lin CD, Lin KH. Thyroid hormone actions in liver cancer. Cell Mol Life Sci. 2013;70:1915-36
6. Cheng SY. Multiple mechanisms for regulation of the transcriptional activity of thyroid hormone receptors. Rev Endocr Metab Disord. 2000;1:9-18
7. Cheng SY, Leonard JL, Davis PJ. Molecular aspects of thyroid hormone actions. Endocr Rev. 2010;31:139-70
8. Chi HC, Chen CY, Tsai MM, Tsai CY, Lin KH. Molecular functions of thyroid hormones and their clinical significance in liver-related diseases. Biomed Res Int. 2013;2013:601361
9. Aranda A, Martinez-Iglesias O, Ruiz-Llorente L, Garcia-Carpizo V, Zambrano A. Thyroid receptor: roles in cancer. Trends Endocrinol Metab. 2009;20:318-24
10. Gonzalez-Sancho JM, Garcia V, Bonilla F, Munoz A. Thyroid hormone receptors/THR genes in human cancer. Cancer Lett. 2003;192:121-32
11. Cristofanilli M, Yamamura Y, Kau SW, Bevers T, Strom S, Patangan M. et al. Thyroid hormone and breast carcinoma. Primary hypothyroidism is associated with a reduced incidence of primary breast carcinoma. Cancer. 2005;103:1122-8
12. Hercbergs AA, Goyal LK, Suh JH, Lee S, Reddy CA, Cohen BH. et al. Propylthiouracil-induced chemical hypothyroidism with high-dose tamoxifen prolongs survival in recurrent high grade glioma: a phase I/II study. Anticancer Res. 2003;23:617-26
13. Ness RB, Grisso JA, Cottreau C, Klapper J, Vergona R, Wheeler JE. et al. Factors related to inflammation of the ovarian epithelium and risk of ovarian cancer. Epidemiology. 2000;11:111-7
14. Chi HC, Chen SL, Liao CJ, Liao CH, Tsai MM, Lin YH. et al. Thyroid hormone receptors promote metastasis of human hepatoma cells via regulation of TRAIL. Cell Death Differ. 2012;19:1802-14
15. Chung IH, Chen CY, Lin YH, Chi HC, Huang YH, Tai PJ. et al. Thyroid hormone-mediated regulation of lipocalin 2 through the Met/FAK pathway in liver cancer. Oncotarget. 2015;6:15050-64
16. Lin YH, Liao CJ, Huang YH, Wu MH, Chi HC, Wu SM. et al. Thyroid hormone receptor represses miR-17 expression to enhance tumor metastasis in human hepatoma cells. Oncogene. 2013;32:4509-18
17. Wu SM, Huang YH, Yeh CT, Tsai MM, Liao CH, Cheng WL. et al. Cathepsin H regulated by the thyroid hormone receptors associate with tumor invasion in human hepatoma cells. Oncogene. 2011;30:2057-69
18. Encinar JA, Mallo GV, Mizyrycki C, Giono L, Gonzalez-Ros JM, Rico M. et al. Human p8 is a HMG-I/Y-like protein with DNA binding activity enhanced by phosphorylation. J Biol Chem. 2001;276:2742-51
19. Mallo GV, Fiedler F, Calvo EL, Ortiz EM, Vasseur S, Keim V. et al. Cloning and expression of the rat p8 cDNA, a new gene activated in pancreas during the acute phase of pancreatitis, pancreatic development, and regeneration, and which promotes cellular growth. J Biol Chem. 1997;272:32360-9
20. Sandi MJ, Hamidi T, Malicet C, Cano C, Loncle C, Pierres A. et al. p8 expression controls pancreatic cancer cell migration, invasion, adhesion, and tumorigenesis. J Cell Physiol. 2011;226:3442-51
21. Emma MR, Iovanna JL, Bachvarov D, Puleio R, Loria GR, Augello G. et al. NUPR1, a new target in liver cancer: implication in controlling cell growth, migration, invasion and sorafenib resistance. Cell Death Dis. 2016;7:e2269
22. Lee YK, Jee BA, Kwon SM, Yoon YS, Xu WG, Wang HJ. et al. Identification of a mitochondrial defect gene signature reveals NUPR1 as a key regulator of liver cancer progression. Hepatology. 2015;62:1174-89
23. Lin KH, Shieh HY, Hsu HC. Negative regulation of the antimetastatic gene Nm23-H1 by thyroid hormone receptors. Endocrinology. 2000;141:2540-7
24. Shih CH, Chen SL, Yen CC, Huang YH, Chen CD, Lee YS. et al. Thyroid hormone receptor-dependent transcriptional regulation of fibrinogen and coagulation proteins. Endocrinology. 2004;145:2804-14
25. Lin YH, Wu MH, Liao CJ, Huang YH, Chi HC, Wu SM. et al. Repression of microRNA-130b by thyroid hormone enhances cell motility. J Hepatol. 2015;62:1328-40
26. Liu X, Zheng N, Shi YN, Yuan J, Li L. Thyroid hormone induced angiogenesis through the integrin alphavbeta3/protein kinase D/histone deacetylase 5 signaling pathway. J Mol Endocrinol. 2014;52:245-54
27. Mousa SA, Lin HY, Tang HY, Hercbergs A, Luidens MK, Davis PJ. Modulation of angiogenesis by thyroid hormone and hormone analogues: implications for cancer management. Angiogenesis. 2014;17:463-9
28. Pinto M, Soares P, Ribatti D. Thyroid hormone as a regulator of tumor induced angiogenesis. Cancer Lett. 2011;301:119-26
29. Liu D, Zhang Y, Wei Y, Liu G, Liu Y, Gao Q. et al. Activation of AKT pathway by Nrf2/PDGFA feedback loop contributes to HCC progression. Oncotarget. 2016;7:65389-402
30. Tanikawa AA, Grotto RM, Silva GF, Ferrasi AC, Sarnighausen VC, Pardini MI. Platelet-derived growth factor A mRNA in platelets is associated with the degree of hepatic fibrosis in chronic hepatitis C. Rev Soc Bras Med Trop. 2017;50:113-6
31. Ziogas IA, Tsoulfas G. Evolving role of Sorafenib in the management of hepatocellular carcinoma. World J Clin Oncol. 2017;8:203-13
32. Zhu YJ, Zheng B, Wang HY, Chen L. New knowledge of the mechanisms of sorafenib resistance in liver cancer. Acta Pharmacol Sin. 2017;38:614-22
33. Chi HC, Chen SL, Cheng YH, Lin TK, Tsai CY, Tsai MM. et al. Chemotherapy resistance and metastasis-promoting effects of thyroid hormone in hepatocarcinoma cells are mediated by suppression of FoxO1 and Bim pathway. Cell Death Dis. 2016;7:e2324
34. Chu YD, Lin KH, Huang YH, Lin CC, Hung CF, Yeh TS. et al. A novel thyroid function index associated with opposite therapeutic outcomes in advanced hepatocellular carcinoma patients receiving chemotherapy or sorafenib. Asia Pac J Clin Oncol. 2018
35. Fernandez M, Semela D, Bruix J, Colle I, Pinzani M, Bosch J. Angiogenesis in liver disease. J Hepatol. 2009;50:604-20
36. Kim WG, Cheng SY. Thyroid hormone receptors and cancer. Biochim Biophys Acta. 2013;1830:3928-36
37. Rennert G, Rennert HS, Pinchev M, Gruber SB. A case-control study of levothyroxine and the risk of colorectal cancer. J Natl Cancer Inst. 2010;102:568-72
38. Hassan MM, Kaseb A, Li D, Patt YZ, Vauthey JN, Thomas MB. et al. Association between hypothyroidism and hepatocellular carcinoma: a case-control study in the United States. Hepatology. 2009;49:1563-70
39. Chen RN, Huang YH, Yeh CT, Liao CH, Lin KH. Thyroid hormone receptors suppress pituitary tumor transforming gene 1 activity in hepatoma. Cancer Res. 2008;68:1697-706
40. Liao CH, Yeh CT, Huang YH, Wu SM, Chi HC, Tsai MM. et al. Dickkopf 4 positively regulated by the thyroid hormone receptor suppresses cell invasion in human hepatoma cells. Hepatology. 2012;55:910-20
41. Jung SH, Lee A, Yim SH, Hu HJ, Choe C, Chung YJ. Simultaneous copy number gains of NUPR1 and ERBB2 predicting poor prognosis in early-stage breast cancer. Bmc Cancer. 2012;12:382
42. Guo X, Wang W, Hu J, Feng K, Pan Y, Zhang L. et al. Lentivirus-mediated RNAi knockdown of NUPR1 inhibits human nonsmall cell lung cancer growth in vitro and in vivo. Anat Rec (Hoboken). 2012;295:2114-21
43. Mu Y, Yan X, Li D, Zhao D, Wang L, Wang X. et al. NUPR1 maintains autolysosomal efflux by activating SNAP25 transcription in cancer cells. Autophagy. 2018;14:654-70
44. Li X, Martin TA, Jiang WG. COM-1/p8 acts as a tumour growth enhancer in colorectal cancer cell lines. Anticancer Res. 2012;32:1229-37
45. Bak Y, Shin HJ, Bak I, Yoon DY, Yu DY. Hepatitis B virus X promotes hepatocellular carcinoma development via nuclear protein 1 pathway. Biochem Biophys Res Commun. 2015;466:676-81
46. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74
47. Andrae J, Gallini R, Betsholtz C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008;22:1276-312
48. Chen PH, Chen X, He X. Platelet-derived growth factors and their receptors: structural and functional perspectives. Biochim Biophys Acta. 2013;1834:2176-86
49. Ong HS, Gokavarapu S, Tian Z, Li J, Xu Q, Cao W. et al. PDGFRA mRNA is overexpressed in oral cancer patients as compared to normal subjects with a significant trend of overexpression among tobacco users. J Oral Pathol Med. 2017;46:591-7
50. Sahraei M, Roy LD, Curry JM, Teresa TL, Nath S, Besmer D. et al. MUC1 regulates PDGFA expression during pancreatic cancer progression. Oncogene. 2012;31:4935-45
51. Palomero J, Vegliante MC, Rodriguez ML, Eguileor A, Castellano G, Planas-Rigol E. et al. SOX11 promotes tumor angiogenesis through transcriptional regulation of PDGFA in mantle cell lymphoma. Blood. 2014;124:2235-47
52. Keating GM, Santoro A. Sorafenib: a review of its use in advanced hepatocellular carcinoma. Drugs. 2009;69:223-40
Author contact
![]() Corresponding author: Kwang-Huei Lin, PhD., Department of Biochemistry, College of Medicine, Chang-Gung University, 259 Wen-Hwa 1 Road, Taoyuan, Taiwan, Republic of China. Tel./Fax: +886-3-2118263. E-mail: khlincgu.edu.tw
Corresponding author: Kwang-Huei Lin, PhD., Department of Biochemistry, College of Medicine, Chang-Gung University, 259 Wen-Hwa 1 Road, Taoyuan, Taiwan, Republic of China. Tel./Fax: +886-3-2118263. E-mail: khlincgu.edu.tw
Citation styles
APA
Chen, C.Y., Wu, S.M., Lin, Y.H., Chi, H.C., Lin, S.L., Yeh, C.T., Chuang, W.Y., Lin, K.H. (2019). Induction of nuclear protein-1 by thyroid hormone enhances platelet-derived growth factor A mediated angiogenesis in liver cancer. Theranostics, 9(8), 2361-2379. https://doi.org/10.7150/thno.29628.
ACS
Chen, C.Y.; Wu, S.M.; Lin, Y.H.; Chi, H.C.; Lin, S.L.; Yeh, C.T.; Chuang, W.Y.; Lin, K.H. Induction of nuclear protein-1 by thyroid hormone enhances platelet-derived growth factor A mediated angiogenesis in liver cancer. Theranostics 2019, 9 (8), 2361-2379. DOI: 10.7150/thno.29628.
NLM
Chen CY, Wu SM, Lin YH, Chi HC, Lin SL, Yeh CT, Chuang WY, Lin KH. Induction of nuclear protein-1 by thyroid hormone enhances platelet-derived growth factor A mediated angiogenesis in liver cancer. Theranostics 2019; 9(8):2361-2379. doi:10.7150/thno.29628. https://www.thno.org/v09p2361.htm
CSE
Chen CY, Wu SM, Lin YH, Chi HC, Lin SL, Yeh CT, Chuang WY, Lin KH. 2019. Induction of nuclear protein-1 by thyroid hormone enhances platelet-derived growth factor A mediated angiogenesis in liver cancer. Theranostics. 9(8):2361-2379.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.