Impact Factor
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Experimental Procedures
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(8):2282-2298. doi:10.7150/thno.30621 This issue Cite
Research Paper
Inhibition of protein disulfide isomerase in glioblastoma causes marked downregulation of DNA repair and DNA damage response genes
Shili Xu1, Yajing Liu2, Kai Yang3,7, Hanxiao Wang2, Andrea Shergalis1, Anahita Kyani1, Armand Bankhead III4,5, Shuzo Tamura1, Suhui Yang1, Xi Wang3,7, Chih-chen Wang3,7, Alnawaz Rehemtulla2, Mats Ljungman2,6, Nouri Neamati1 ![]()
1. Department of Medicinal Chemistry, College of Pharmacy, Rogel Cancer Center, University of Michigan, Ann Arbor, MI 48109, USA
2. Radiation Oncology, Rogel Cancer Center, Center for RNA, University of Michigan, Ann Arbor, MI 48109, USA
3. National Laboratory of Biomacromolecules, CAS Center for Excellence in Biomacromolecules, Institute of Biophysics, Chinese Academy of Sciences, Beijing 100101, China
4. Department of Biostatistics, University of Michigan, Ann Arbor, MI 48109, USA
5. Department of Computational Medicine and Bioinformatics, University of Michigan, Ann Arbor, MI 48109, USA
6. Environmental Health Sciences, Rogel Cancer Center, University of Michigan, Ann Arbor, MI 48109, USA
7. College of Life Sciences, University of Chinese Academy of Sciences, Beijing 10049, China
Received 2018-10-12; Accepted 2019-2-13; Published 2019-4-12
Abstract

Aberrant overexpression of endoplasmic reticulum (ER)-resident oxidoreductase protein disulfide isomerase (PDI) plays an important role in cancer progression. In this study, we demonstrate that PDI promotes glioblastoma (GBM) cell growth and describe a class of allosteric PDI inhibitors that are selective for PDI over other PDI family members.
Methods: We performed a phenotypic screening triage campaign of over 20,000 diverse compounds to identify PDI inhibitors cytotoxic to cancer cells. From this screen, BAP2 emerged as a lead compound, and we assessed BAP2-PDI interactions with gel filtration, thiol-competition assays, and site-directed mutagenesis studies. To assess selectivity, we compared BAP2 activity across several PDI family members in the PDI reductase assay. Finally, we performed in vivo studies with a mouse xenograft model of GBM combining BAP2 and the standard of care (temozolomide and radiation), and identified affected gene pathways with nascent RNA sequencing (Bru-seq).
Results: BAP2 and related analogs are novel PDI inhibitors that selectively inhibit PDIA1 and PDIp. Though BAP2 contains a weak Michael acceptor, interaction with PDI relies on Histidine 256 in the b' domain of PDI, suggesting allosteric binding. Furthermore, both in vitro and in vivo, BAP2 reduces cell and tumor growth. BAP2 alters the transcription of genes involved in the unfolded protein response, ER stress, apoptosis and DNA repair response.
Conclusion: These results indicate that BAP2 has anti-tumor activity and the suppressive effect on DNA repair gene expression warrants combination with DNA damaging agents to treat GBM.
Keywords: Protein disulfide isomerase, drug discovery, allosteric inhibition, cancer, DNA repair
Introduction
Glioblastoma (GBM) is the most common and most aggressive type of primary human brain tumor [1]. Surgery followed by radiotherapy in combination with temozolomide (TMZ) is the standard of care for GBM; however, only 5.5 % of patients survive more than 5 years after diagnosis [2]. As an alkylating agent that induces DNA damage, TMZ is toxic and exhibits limited prolongation of survival [3, 4]. No successful drug treatment for GBM has been approved for over three decades. Additionally, GBM tumor resistance may arise from therapy-resistant cancer stem cells (CSC). CSCs are a subpopulation of tumor cells that have self-renewal properties, are self-sustaining, and can differentiate into nontumorigenic cancer cell types [5]. CSCs can arise from neural stem cells and allow repopulation of the tumor mass after treatment. Transcription factors including Sox2 and OLIG2 maintain the stem cell-like properties of the cells [6, 7]. Thus, a critical unmet need exists for new, targeted chemotherapeutics with improved efficacy and safety for GBM patients.
Overexpression of protein disulfide isomerase (PDI) promotes cancer growth, suggesting that it represents a potential cancer therapeutic target [8-10]. PDI is a dithiol-disulfide oxidoreductase with molecular chaperone functions [11, 12]. Cysteines (Cys) in the catalytic sites of the homologous a- and a'-domains are essential for enzymatic function and can transition between oxidized (disulfide) and reduced (dithiol) states [13, 14]. Mainly located in the endoplasmic reticulum (ER), PDI mediates folding of specific polypeptides and is upregulated by the unfolded protein response (UPR) as a cancer survival mechanism [15]. Concordantly, PDI inhibition results in the accumulation of unfolded and misfolded proteins, ER stress, and cell death [16]. Several inhibitors of PDI have been identified, including PACMA31 for the treatment of ovarian cancer [17], 16F16 and LOC14 for the prevention of neurodegeneration [18, 19], CCF642 for the treatment of multiple myeloma [20], juniferdin for the inhibition of HIV-1 infection [21], bepristats and quercetin-3-rutinoside for the suppression of thrombus formation [22-24], and KSC-34 and analogs as a domain-selective PDI probes [25]. PDI inhibition results in synergistic cell killing in combination with TMZ [26] and sorafenib [27]. However, no PDI inhibitors have been approved for clinical use.
We previously validated PDI as a therapeutic target wherein PACMA31 was demonstrated to have anti-tumor activity [17]. PACMA31 has been demonstrated by our lab in this report and others to be non-specific towards PDI (PDIA1), and can inhibit other PDI family members, such as ERp57 [22]. Furthermore, we identified a potent PDI inhibitor, 35G8, that was toxic in a 2D cancer model [28]. However, 35G8, as a known redox-cycling molecule, does not possess drug-like properties. This prompted us to pursue a PDI inhibitor with a novel scaffold and more appropriate drug-likeness. In this study, we investigate chalcone-containing derivatives as PDI inhibitors and demonstrate that PDI promotes GBM cell growth. Chalcones (benzylideneacetophenones) are simple privileged molecules, and, although various chalcones have anti-cancer activities, some of their molecular targets have not been fully validated [29]. Therefore, an improved understanding of their mechanisms of cytotoxicity is critical for further development. Though the discovered chalcone compounds contain a Michael-acceptor moiety, a weak electrophile, our lead chalcone-containing compound BAP2 binds to an allosteric site on PDI, selectively inhibits PDIA1 and PDIp activity, and suppresses cell growth in a model with GBM patient-derived cells. To address the PDI binding nature of the BAP2 scaffold, we synthesized an additional 67 analogs and published our extensive findings on the structure-activity relationship in a separate study [30]. We further discovered that PDI knockdown and inhibition abrogate the stem-like phenotype of GBM cells. Bromouridine labeling and sequencing (Bru-seq) of nascent RNA demonstrated that PDI inhibition modulates transcriptional pathways associated with ER stress and the UPR. More significantly, PDI inhibition caused a global downregulation of DNA damage response (DDR) genes. These findings warrant further development of these compounds as a novel targeted approach for the treatment of GBM and in combination with DNA-damaging chemotherapy.
Experimental Procedures
Reagents. Control and PDI siRNAs were purchased from OriGene Technologies (Rockville, MD). Opti-MEM medium, Lipofectamine RNAiMAX transfection reagent, propidium iodide, and AlamarBlue Cell Viability Reagent were purchased from Life Technologies (Grand Island, NY). PDI (1:4000, #3501), E2F1 (1:500, #3742), RAD51 (1:500, #8875S), Sox2 (1:1000, #3579), phospho-histone H2A.X (1:500, #9718), PARP (1:1000, #5625), and cleaved caspase 3 (1:1000, #9664) antibodies were purchased from Cell Signaling Technology (Danvers, MA). Actin (1:3000, sc-47778), BRCA2 (1:1000, sc-135731), ATR (1:1000, sc-515173), ATM (1:1000, sc-135663), WRN (1:1000, sc-135807), and HSPA6 (1:1000, sc-376193) antibodies were purchased from Santa Cruz Biotechnology (Dallas, TX). CD133/1 (AC133)-APC antibody was purchased from Milteny Biotec (Auburn, CA). Secondary antibodies were purchased from Cell Signaling (anti-rabbit, 1:7500, #35568 and anti-mouse, 1:5000, #35518).
Cell culture. GBM cell lines U87MG, NU04, and U118MG were kindly provided by Dr. Alan L. Epstein (University of Southern California, Los Angeles, CA) and were maintained in RPMI-1640 (Life Technologies) supplemented with 10 % FBS (Fisher Scientific, Pittsburgh, PA). A172 cells were obtained from the American Type Culture Collection (ATCC). All cell lines were authenticated with STR DNA profiling (University of Michigan) and matched to reference profiles from the ATCC database. Cells were grown as monolayers at 37 °C in a humidified atmosphere of 5 % CO2. Four patient-derived primary cell lines (HF2303, HF2587, HF2927, and HF3016 cells) were provided by Dr. Tom Mikkelsen and Dr. Ana C. deCarvalho (Henry Ford Hospital, Detroit, MI). Establishment of primary tumor cell culture was described previously [31]. Primary GBM cell lines were maintained in neurosphere medium composed of DMEM/F-12 supplemented with N-2 (Gibco), 0.5 mg/ml BSA (Sigma), 25 µg/ml gentamicin (Gibco), 0.5 % antibiotic/antimycotic (Invitrogen), 20 ng/ml bFGF, and 20 ng/ml EGF (Peprotech). Cells were maintained in culture up to 20 passages. Cells were checked for Mycoplasma contamination with PlasmoTest kit (InvivoGen).
Bioinformatics analysis. Rembrandt gene expression data was sourced from Array Express and processed using the robust multiarray analysis algorithm. Rembrandt patient metadata was sourced from the Georgetown Database of Cancer [32]. Gravendeel normalized gene expression data was sourced from GEO and original paper supplemental data [33]. GBM and LGG TCGA data were downloaded from the Genome Data Analysis Center (GDAC) Firehose [34]. Kruskal-Wallis and survival analysis statistics were calculated using the R statistical programming language [35]. GSEAv2.2.3 was used with v6 gene sets sourced from MSigDB. To quantify enrichment significance, 10,000 gene set permutations were performed using weighted mode scoring and Pearson metric.63
PDI reductase assay. PDI activity was measured as described previously [36]. Briefly, purified recombinant PDI was incubated with indicated compounds at 37 °C for 1 h in buffer (100 mM sodium phosphate, 2 mM EDTA, 8 μM DTT, pH 7.0), followed by addition of DTT (100 μM) and bovine insulin (130 μM). The a and a'c domains in the PDI assay were incubated for 45 minutes at room temperature before addition of insulin. Resulting aggregation of reduced insulin B chain was measured at 650 nm.
Denatured and reduced RNase A (drRNase A) assay. drRNase assay was performed as described [37]. Briefly, oxidative renaturation of denatured and reduced RNase A was measured spectrophotometrically in the presence of cCMP as an RNase A substrate. Tris-acetate buffer (pH 8.0), 4.5 mM cCMP, GSH and GSSG (at concentrations to yield a redox buffer of chosen composition), and 1.4 µM PDI were equilibrated at 25 °C. The assay was initiated by addition of reduced, denatured RNase A. The hydrolysis of cCMP resulting from the gain in ribonuclease activity was recorded as an increase in absorbance at 296 nm.
PDI knockdown. PDI knockdown was modified from a previous approach [36]. Control or PDI siRNA (100 nM) with Lipofectamine RNAiMAX transfection reagent was added to cells in Opi-MEM medium. After 10 h, 1 mL of RPMI-1640 + 20 % FBS was added to each well. 24 h after siRNA addition, cell culture medium was replaced with RPMI-1640 + 10 % FBS.
MTT assay. MTT growth inhibition assay was conducted as described [38]. Cells were seeded and allowed to attach overnight. Cells were treated with compounds and, after 72 h, were incubated with 0.3 mg/mL 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (Amresco, Solon, OH) for an additional 3 h at 37 °C. After removal of the supernatant, DMSO was added, and absorbance was read at 570 nm. Percent cell growth inhibition was expressed as (1 - A/C) × 100 % (A and C are absorbance values from experimental and control cells, respectively). Standard error of the mean (SEM) was calculated based on IC50 values obtained from at least three independent experiments.
Alamar Blue viability assay. Neurospheres were dissociated and seeded at 5,000 cells/well in 96-well plates. Cells were treated after 24 h. Alamar Blue reagent was added to cells per manufacturer protocol after 72 h. Fluorescence signals were determined by EnVision Xcite Multilabel Reader (PerkinElmer, Waltham, MA).
Colony formation assay. Briefly, 600 (NU04 and U87MG) or 300 (HCT116) cells were seeded per well in 24-well (NU04 and U87MG) or 96-well (HCT116) plates and allowed to attach. After 24 h, serial dilutions of compounds were added to the culture medium and incubated for 24 h. Cells were cultured until colonies formed (12 days (NU04 and U87MG) or 7 days (HCT116)), stained with crystal violet solution (0.25 % crystal violet, 10 % formaldehyde (37 % v/v), 80 % methanol) for 1 h, and thoroughly washed with water before imaging.
Sphere formation assay. Sphere formation assay was adapted from previous studies [39]. Spheres were dissociated and 50 cells/μl were seeded in 96-well plates. After 24 h, cells were treated with indicated compounds and incubated for 7 days in neurosphere medium. The number of floating spheres in nine individual fields per sample was counted.
Thermal shift assay. The thermal shift of PDI (0.3 mg/ml in 100 mM Na2PO4, pH 7.0) in the presence or absence of compound was determined with the Protein Thermal Shift kit (Applied Biosystems), following the kit protocol. PDI was mixed with buffer, compound at indicated concentrations, and 1× ROX dye to a total volume of 20 μL in a 384-well plate. The plate was heated from 25 to 99 °C at a rate of 1 °C/min in the ViiA7 Real-Time PCR instrument (Thermo Fisher Scientific). Melting curves and temperatures were calculated using the Boltzmann method in the Protein Thermal Shift software (Thermo Fisher Scientific).
Cellular thermal shift assay. For the cellular thermal shift assay [40, 41], U87MG or A172 cells were seeded at 2 × 106 cells/100 mm dish and allowed to attach overnight. Cells were treated with indicated concentrations of compounds, or DMSO as the negative control, for 2 h at 37 °C, 5 % CO2. After treatment, cells were trypsinized, washed with DPBS, and suspended in 600 μL DPBS. Cells were split into 100 μL aliquots, heated at indicated temperatures for 3 min in the Veriti Thermal Cycler (Applied Biosystems), and incubated for 3 min at room temperature. The cells were flash-frozen twice and spun at 14,000 × g for 20 min at 4 °C. Supernatants were collected and loaded onto a 10 % polyacrylamide gel at a volume of 16 μL, with 4 μL 5× SDS loading dye. Subsequently, Coomassie staining using standard procedure or Western blotting was performed.
Western blot. Western blot was modified from previously described protocols [42]. Briefly, cells were washed with PBS and lysed in 20 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, 1 % Triton X-100, 1 % SDS, pH 7.5 with 1× protease inhibitor (Sigma-Aldrich, St. Louis, MO) and phosphatase inhibitor (Alfa Aesar, Ward Hill, MA) cocktails. Protein concentration was determined by the BCA assay (Thermo Scientific, Rockford, IL). Proteins were resolved on 10 % acrylamide gels via SDS-PAGE and electrotransferred to immobilon-FL PVDF membranes (Millipore, Billerica, MA). After incubation with StartingBlock (TBS) blocking buffer (Thermo Scientific), membranes were probed with primary antibodies in TBST with 5 % FBS, subsequently with DyLight 800 conjugated anti-rabbit IgG (Thermo Scientific) in TBST with 5 % milk and 0.01 % SDS and scanned using the ODYSSEY CLx infrared imaging system (LI-COR, Lincoln, NE).
Competition assay. Full-length recombinant PDI (15 μM) was incubated with BAP2, BAP30, or PACMA31 at indicated concentrations in PBS for 1 h at 37 °C followed by 1 h incubation with 20 μM PACMA57. Solutions were then mixed with 5× SDS sample buffer for SDS/PAGE. Gel fluorescence was analyzed using FluorChem M system (ProteinSimple, San Jose, CA).
Gel filtration. Full-length PDI-BAP2 complex was prepared by overnight incubation of 5 μM PDI at room temperature with 200 μM BAP2, before being subjected to a Superdex 200 HR 10/60 column (GE Healthcare) and chromatographed at 1 ml/min using 50 mM Tris/HCl buffer (pH 7.6) containing 150 mM NaCl and 2 mM EDTA. The main peak was collected, and aliquots were stored at -80 °C for the PDI reductase assay.
AMS assay. 20 μM isolated a or a'c domains from PDI were incubated with or without 40 μM BAP2 or PACMA31 for 1 h in PBS at 25 °C. Free thiols were blocked with 2 mM AMS (Invitrogen). Samples were analyzed by SDS-PAGE and stained with Coomassie blue.
Site-directed mutagenesis. Forward and reverse primers for H256A PDI were ordered from Integrated DNA Technologies (Forward Primer: GGTGAAATCAAGACTGCCATCCTGCTGTTC; Reverse Primer: GAACAGCAGGATGGCAGTCTTGATTTCACC). Site-directed mutagenesis was performed with the QuikChange II XL Site-Directed Mutagenesis Kit (Agilent Technologies) following manufacturer protocol with some modification. Briefly, reactions containing 10 ng DNA template, 100 ng primers, dNTP mix, reaction buffer, QuikSolution reagent, and 2.5 U of PfuUltra HF DNA polymerase were cycled at 95 °C for 1 min, 18 cycles of 95 °C for 50 s, 56 °C for 50 s, 68 °C for 7 min, and 68 °C for 6 min. Individual colonies were grown in 5 ml LB containing 200 μg/ml ampicillin for 8-16 h at 37 °C. DNA was purified (QIAprep Spin Miniprep Kit, Qiagen) and mutations were validated with Sanger sequencing conducted by the University of Michigan DNA Sequencing Core (Figure S1). Plasmids were transformed into BL21(DE3) competent cells (Promega) following the QuikChange II XL Site-Directed Mutagenesis protocol.
Protein purification. Wild-type and mutant PDI expression and purification were performed as previously described [17] with slight modifications. The a and a'c domains of PDI in PQE30 vectors were transformed into Express Iq Competent E. coli (New England BioLabs, Inc.). PDIp in a pET23b vector and ERp57 in a PQE30 vector were transformed into Rosetta BL21(DE3) pLysS (Novagen) and Express Iq Competent E. coli (New England BioLabs, Inc.), respectively. Bacteria were grown in LB medium with 200 μg/ml ampicillin (EMD Biosciences, La Jolla, CA) at 37 °C and incubated at an OD600 of 0.5 for 4 h with 1 mM isopropyl β-D-1-thiogalactopyranoside (GoldBio) or OD600 of 0.8 for 16 hours at 16 °C for PDIp and ERp57. Cells were harvested by centrifugation and re-suspended in Buffer A (20 mM sodium phosphate, pH 7.3). Cells were lysed by sonication and the cell debris was removed by centrifugation. The supernatant was applied to Ni-nitrilotriacetic acid beads (Qiagen, Hilden, Germany), equilibrated with 10 ml of Buffer A and incubated at 4 °C, overnight. After incubation, the nickel beads were added to a column, washed in Buffer A and then in Buffer B (20 mM sodium phosphate, 0.5 M sodium chloride and 50 mM imidazole, pH 7.3). The his-tagged proteins were eluted using Buffer C (20 mM sodium phosphate and 250 mM EDTA, pH 7.3) and eluent was dialyzed in 100 mM sodium phosphate buffer (pH 7.0) with 2 mM EDTA.
Stemness marker analysis. 1 × 106 cells/25 cm2 flask were seeded and treated with indicated compounds at 10 μM after 24 h. Cells were incubated for 72 h before collection and dissociation. CD133 staining was carried out per manufacturer protocol. Propidium iodide (PI) was included as a cell viability marker. Percentage of CD133-positive/PI negative cells was determined and recorded with the BD FACSAria (BD Biosciences).
Bromouridine (Bru)-RNA sequencing. Bru-RNA sequencing (Bru-seq) was performed as previously described, with some modification [43]. U87MG cells were treated with BAP2 at 3 µM for 4 h, and Bru was added for the last 30 min of treatment to a final concentration of 2 mM and incubated with the drug. Cells were collected, and total RNA was isolated with TRIzol reagent. Bru-labeled RNA was captured from total RNA by incubation with anti-BrdU antibodies (BD Biosciences) conjugated to magnetic beads (Dynabeads, Goat anti-Mouse IgG; Invitrogen) under gentle agitation at room temperature for 1 h. Bru-containing RNA population was isolated and sequenced. Sequencing reads were mapped to the HG19 reference genome. Pre-ranked gene lists were generated for each treatment by ranking genes by fold change in transcription compared to control. Sequencing results were filtered using cutoff values of gene size >300 base pairs and mean reads per kilobase per million > 0.5.
The dataset was interrogated using the Database for Annotation, Visualization and Integrated Discovery (DAVID), IPA, and GSEA. Genes with over 8-fold change in transcription (142 upregulated and 95 downregulated) after BAP2 treatment were submitted to DAVID (david.abcc.ncifcrf.gov) for functional annotation [44, 45]. Upregulated and downregulated genes were categorized into 235 and 81 functional gene clusters, respectively. For IPA, a dataset of ~2,500 genes (cutoff value: 2-fold change) was used. The identified pathways were ordered by p-value and maximum number of genes in the pathway. A pre-ranked gene list of 9380 genes was also analyzed for gene enrichment using GSEA gene sets based on the Kolmogorov-Smirnov statistic. For each gene set, an enrichment score (ES) was normalized to account for the difference in gene set size, and a false discovery rate (FDR) was calculated based on the normalized enrichment score (NES) values.
Xenograft implantation and treatment. 250,000 U87MG or D54 cells were implanted subcutaneously into each flank of four to six-week old Athymic Nude-Foxn1nu mice in 100 µL RPMI:BD Matrigel Basement Membrane Matrix (1:1) suspension. Caliper measurement was conducted twice weekly and tumor volume was determined as V = (length/2) X (width2). Mice were randomized into four groups when tumors reached 100 mm3 and treatments: vehicle control, TMZ alone (5mg/kg in Ora-Plus (Perrigo) via oral gavage five days/week) (LKT Laboratories), BAP2 alone (20 mg/kg via intraperitoneal injection in 5 % DMSO / 65 % 1,2-propanediol / 30 % saline five days/week), or local radiation (Kimtron Medical, IC-320) alone (2 Gy × 5 doses/week) was given for two weeks. Western blot was performed on mouse tumor tissue. Briefly, cells and snap-frozen tissues were washed with PBS and lysed in 20 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, 1 % Triton X-100, 1 % SDS, pH 7.5 with 1× protease inhibitor (Sigma-Aldrich) and phosphatase inhibitor (Alfa Aesar) cocktails. Protein concentration was determined by the BCA assay (Thermo Scientific). Proteins were resolved on 10 % or 4-12% acrylamide gels (Invitrogen) via SDS-PAGE and electrotransferred to immobilon-FL PVDF membrane (Millipore, Billerica, MA). After incubation with StartingBlock (TBS) blocking buffer (Thermo Scientific) or 5 % bovine serum albumin (BSA) (Thermo Scientific), membranes were probed with primary antibodies in TBST with 5 % FBS or BSA, subsequently with DyLight 800 conjugated anti-rabbit IgG (Thermo Scientific) in TBST with 5 % milk and 0.01 % SDS or 2.5% BSA and scanned using the ODYSSEY CLx infrared imaging system (LI-COR) or ChemiDocTM imager (Bio-Rad). Alternatively, Horseradish peroxidase-conjugated anti-rabbit or mouse IgG (H+L) (Jackson ImmunoResearch) were used as secondary antibodies and Pierce™ ECL (Thermal Scientific) or ECL™ prime (GE Healthcare) were used as substrate.
Statistical analysis. Significance was evaluated with one-way ANOVA or student t-test using Prism (version 6.01, La Jolla, CA). Differences were statistically significant at p < 0.05.
Results
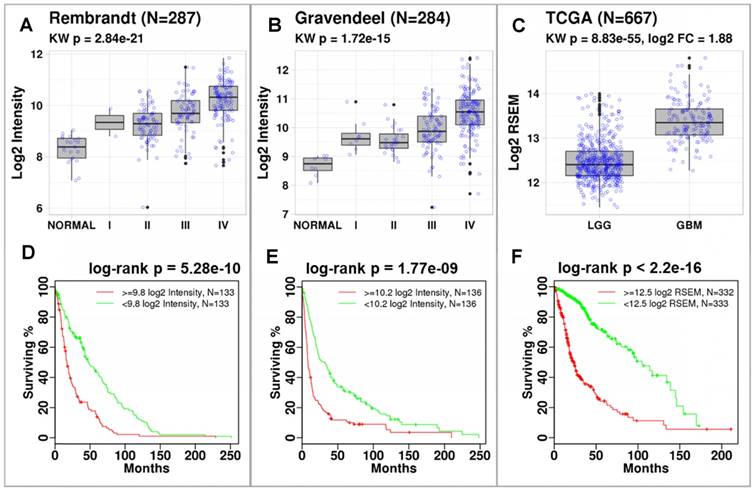
P4HB expression correlates with tumor grade and is associated with reduced overall survival. Using data published by Rembrandt (n = 287) and Gravendeel (n = 284), we discovered that P4HB (gene name for PDIA1) was significantly overexpressed in brain tumors compared to normal brain tissues (Figure 1). Differences in expression were quantified using a Kruskal-Wallis test across glioma grade and normal tissues (Rembrandt: p = 2.84E-21 (Figure 1A) and Gravendeel: p = 1.72E-15 (Figure 1B)). In both datasets, P4HB expression was associated with progression. In TCGA dataset (n = 667), P4HB expression was higher in patients with GBM compared to patients with lower grade glioma (LGG) (p = 8.83E-55, Figure 1C). Patients with high P4HB expression (regardless of grade) exhibit reduced overall survival in Rembrandt (p = 5.28E-10, Figure 1D) [32], Gravendeel (p = 1.77E-9, Figure 1E) [33], and TCGA (p = 2.2E-16, Figure 1F) datasets. Compared to other PDI family members, mRNA expression of P4HB is significantly upregulated in GBM patient samples, which further supports P4HB as a critical GBM tumor growth gene (Figure S2). These findings suggest that overexpression of PDI may be linked to brain tumor progression and aggressiveness.
P4HB expression increases with cancer grade and is associated with reduced survival. Significant differences in P4HB expression were observed using Kruskal-Wallis (KW) statistics across glioma grade and normal tissue. P4HB had significantly increased expression in patients with GBM compared to patients with LGG in the Rembrandt (a), Gravendeel (b), and TCGA (c) datasets. Patients with increased P4HB expression (regardless of grade) exhibited reduced survivability in three glioma datasets (d-f). Populations were stratified using median P4HB expression per dataset.
To investigate the role of PDI in cell proliferation, we silenced PDI expression in two GBM cell lines (Figure S3A). PDI knockdown significantly decreased cell proliferation (Figure S3B) and viability (Figure S3C), suggesting that PDI promotes GBM cell growth and survival. Furthermore, the shRNA data in Project Achilles (http://portals.broadinstitute.org/achilles) show that PDI knockdown results in negative shRNA scores (Figure S3D). shRNA score is the log2-based change in shRNA compared with pooled shRNA in several cancer cell lines and a negative shRNA score is indicative of decreased cell proliferation. Our data show that PDI knockdown reduced cell viability in over 95 % of cancer cell lines transfected, further supporting PDI as a drug target in cancer.
Chalcone-containing compounds inhibit PDI. We performed a two-step screening of a subset of our in-house drug-like small molecule libraries (Figure 2A) [28]. From an initial screen for cytotoxicity, 443 compounds with high potency were identified and tested for PDI inhibition. The screening campaign provided us with two lead compounds that contained the chalcone or benzylideneacetophenone scaffold (BAPs): BAP1 (IC50 = 0.83 ± 0.20 µM) and BAP2 (IC50 = 0.93 ± 0.09 µM) (Figure 2B, Table S1).
BAP2 derivatives are potent PDI inhibitors. (a) 20,000 drug-like compounds were screened in the MTT assay. Compounds that inhibited 40 % of cancer cell growth in the MTT assay were screened in the PDI reductase assay. Numbers indicate compounds identified after each screening filter. (b) Inhibitory effects of BAP1 and BAP2 in the PDI reductase assay. (c) Inhibition profile of BAP analogs 3-6 in the PDI reductase assay. (d) PDI-BAP2 complex preparation by gel filtration. (e) The reductase and isomerase activity of PDI-BAP2 complex.
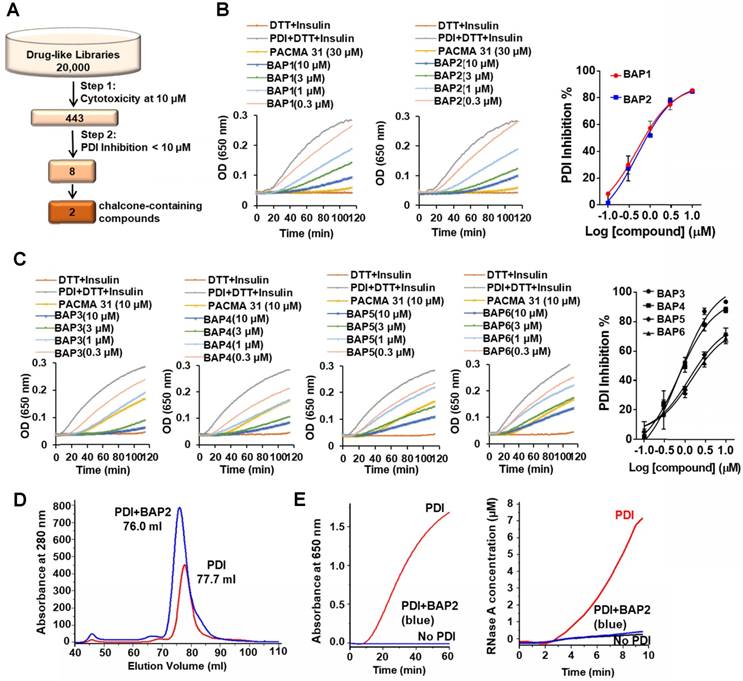
We next identified 41 analogs of BAP1 and BAP2 with drug-like properties and characterized their PDI inhibitory potential (Figure S4, Table S1). Four of 41 additional BAP2 derivatives inhibited PDI with an IC50 value < 5 µM: BAP3 - 6 (Figure 2C, Table S1). Thus, modest structural changes around the chalcone scaffold had marked effects on PDI inhibition. Gel filtration analysis demonstrated that BAP2 forms a stable complex with PDI, and binding induces a conformational change (Figure 2D). PDI incubated with BAP2 was eluted with a smaller volume. After removing the excess BAP2 via gel filtration, the 76 mL eluted sample lost the isomerase and reductase activity, which may indicate that PDI and BAP2 form a stable complex (Figure 2E). However, it is unclear whether BAP2 and PDI form intermolecular covalent bond(s) and whether the binding of BAP2 to PDI is irreversible.
To further validate that BAP2 targets PDI, we tested the ability of BAP2 to stabilize PDI using the thermal shift assay. BAP2 stabilized PDI to thermal unfolding, similar to endogenous ligand estradiol [46], in both the thermal shift assay and cellular thermal shift assay (CETSA) [40] (Figure S5). The CETSA measures the ability of intracellular proteins to withstand thermal degradation in the presence of inhibitors. In this study, BAP2 stabilizes PDI to thermal degradation by about +1-2 °C (Figure S5C). These results demonstrate intracellular target engagement of PDI by BAP2. To further determine the selectivity of BAP2 binding to the proteome, we performed CETSA and examined the total lysate on a Coomassie-stained gel (Figure S5D). We did not observe any visible stabilization of any other proteins in the lysate by BAP2; we did, however, observe stabilization of a protein around 70 kDa by estradiol, likely estrogen receptor alpha, the main target of estradiol. We did not observe stabilization of PDI with the total cell lysate likely because many proteins share a similar molecular weight (~57 kDa).
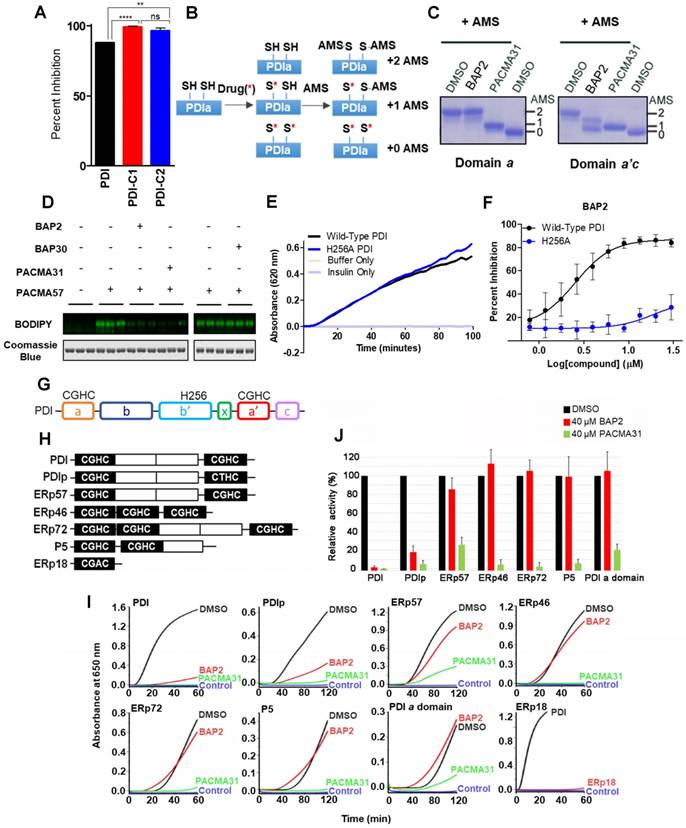
BAP2 is a selective allosteric PDI inhibitor. To determine how BAP2 interacts with PDI to inhibit its activity, we first tested two PDI mutants with one active catalytic site in each mutant: PDI-C1 (SGHS in a domain, CGHC in a' domain) and PDI-C2 (CGHC in a domain, SGHS in a' domain) [47]. BAP2 inhibited PDI-C1 and PDI-C2 more potently than PDI (Figure 3A), but there was no difference in the potency of BAP2 between the two mutants. To examine active site cysteine accessibility, we used 4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid (AMS), which reacts with cysteine thiols and adds ~ 500 Da for each modification, to modify isolated BAP2-treated a and a'c domains (Figure 3B). Incubation with BAP2 blocked AMS binding to the a'c domain, but not the a domain (Figure 3C). In contrast, PACMA31 blocked AMS binding to cysteines in both a and a'c domains. In addition, BAP2 prevented covalent binding of PACMA57 (fluorescent analog of PACMA31) to PDI active-site cysteines, whereas BAP30 (an inactive derivative) had no significant effect (Figure 3D).
BAP2 targets an allosteric site in PDI selectively. (a) Comparison of inhibitory effects of BAP2 on active sites in the a and a' domains. PDI (CGHC in a, CGHC in a'), PDI-C1 (SGHS in a, CGHC in a') and PDI-C2 (CGHC in a, SGHS in a') at 0.5 μM were incubated with or without 10 μM BAP2 at 37 °C for 1 h. Statistical significance determined via unpaired t test. ** p < 0.01; **** p < 0.0001. ns: not significant. (b) Schematic of AMS reactivity with active-site thiols on PDI. (c) Thiol reactivity of the active sites of a and a'c domains of PDI with AMS before and after BAP2 treatment. (d) BAP2 inhibited binding of PACMA57, a fluorescent analog of PACMA31, to PDI. (e) Activity of wild-type and H256A PDI in the PDI reductase assay. (f) BAP2 treatment in the PDI reductase assay with wild-type and H256A PDI. (g) Domain architecture of PDI. Active site CGHC residues and b' domain His256 are shown. (h) Domain architecture of PDI family members with the CXXC active site tested. (i) Inhibition of PDI family members (1 μM) by BAP2 (40 μM) and PACMA31 (40 μM) in the PDI reductase assay. (j) Reductase activity calculated by the maximal slope of the curve relative to lag time. Activity of protein incubated with DMSO was used as 100 %. Data expressed as mean ± S.D. (n ≥ 3).
To further explore BAP2 binding to the active site cysteines, we purified the a and a'c domains of PDI, which have isolated reductase activity [48]. PACMA31 (100 μM) and 35G8 (1 μM) inhibit reductase activity of the isolated domains, whereas inhibitors estradiol, bepristat 1a, and BAP2 did not inhibit the activity of the isolated domains (Figure S6). These results indicate that BAP2 may not bind directly to the a or a'c domains. To explore the possibility that BAP2 did not bind in the active site, we tested the activity of a saturated analog of BAP2 (Figure S6B). Without the Michael acceptor, the analog retained modest potency, which suggests that inhibition of PDI is not solely dependent on the alpha-beta unsaturated ketone scaffold.
His256 is an important residue for substrate recognition in the b' domain of PDI family members [49, 50]. Several PDI inhibitors, including quercetin 3-rutinoside [48] and bepristat 1a [22], bind in the b' domain; thus, we sought to determine whether BAP2 may also bind in the hydrophobic substrate-binding pocket. BAP2 and analogs contain a key hydroxyl group required for activity, similar to estradiol, an endogenous PDI ligand that binds in the b' domain of PDIp [50]. Reductase capabilities of PDI were not hindered by the histidine-to-alanine mutation H256A (Figure 3E); however, BAP2 was much less active against the H256A mutant than the wild-type protein (Figure 3F). These results indicate that BAP2 interaction with His256 is important for activity. BAP2 binding in the b' domain may induce a conformational change in PDI that blocks the a' active site (Figure 3G).
We further evaluated BAP2 activity against a panel of PDI family members containing the CXXC active site (PDI, PDIp, ERp57, ERp46, ERp72, P5, and ERp18) (Figure 3H), and a single catalytic domain a of PDI. We found that BAP2 selectively inhibits PDI and PDIp, whereas PACMA31 inhibits all tested PDI family enzymes (Figure 3I, S7, S8). PDIp shares the highest degree of similarity with PDI among PDI family proteins in evolutionary divergence [51]. A conserved histidine residue responsible for substrate recognition is found in both PDI and PDIp, providing a potential explanation for activity against both family members. The b'xa' domain found in PDI and PDIp is not present in ERp46 or P5, which could explain why BAP2 is not active against those PDI family members. Furthermore, ERp57 and ERp72 lack the conserved histidine residue that BAP2 requires for activity. ERp18 displayed low disulfide reductase activity making it difficult to detect a significant effect of BAP2. Additionally, BAP2 did not inhibit the isolated PDI a domain, confirming that BAP2 inhibits catalytic activity by binding to an allosteric site located in the b'xa'c domain (Figure 3J).
BAP compounds reduce GBM cell growth and synergize with TMZ. Among the BAP derivatives generated, BAP2 was the most potent (IC50 value: 5.3 ± 1.8 µM in NU04) (Table S2). Derivatives inhibited colony formation in a 2D cell model of GBM (Figure 4A), and cytotoxicity correlated with PDI inhibitory activity (Figure 4B). Furthermore, BAP2 was active in patient-derived primary GBM cells (HF2303, HF2598, HF2927, and HF3016), whereas inactive analog BAP30 did not show significant cytotoxicity (Figure 4C). BAP2 also sensitized cancer cells to TMZ treatment in the colony formation assay (Figure S9).
BAP2 analogs inhibit GBM cell growth and prevent maintenance of stemness properties. (a) Treatment with BAP1-6 inhibited colony formation. (b) A significant correlation exists between PDI inhibition and compound cytotoxicity. (c) BAP2 induced cell death in patient-derived GBM cell lines. (d) PDI knockdown inhibited U87MG sphere formation. Five representative images were taken for each condition. (e) BAP analogs inhibited U87MG sphere formation. Five representative images are shown for each condition. (f) BAP2 impaired sphere formation in four patient-derived GBM cell lines, whereas BAP30 showed no significant inhibition. (g) BAP2 (10 μM), but not BAP30 (10 μM), reduced CD133 population in human GBM cells. Cells were treated with BAP2 or BAP30 for 72 h. (h) BAP2 downregulated Sox2 expression, as assessed by Western blot analysis. Means ± SEM were calculated from three experiments. Scale bars represent 200 μm. * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001.
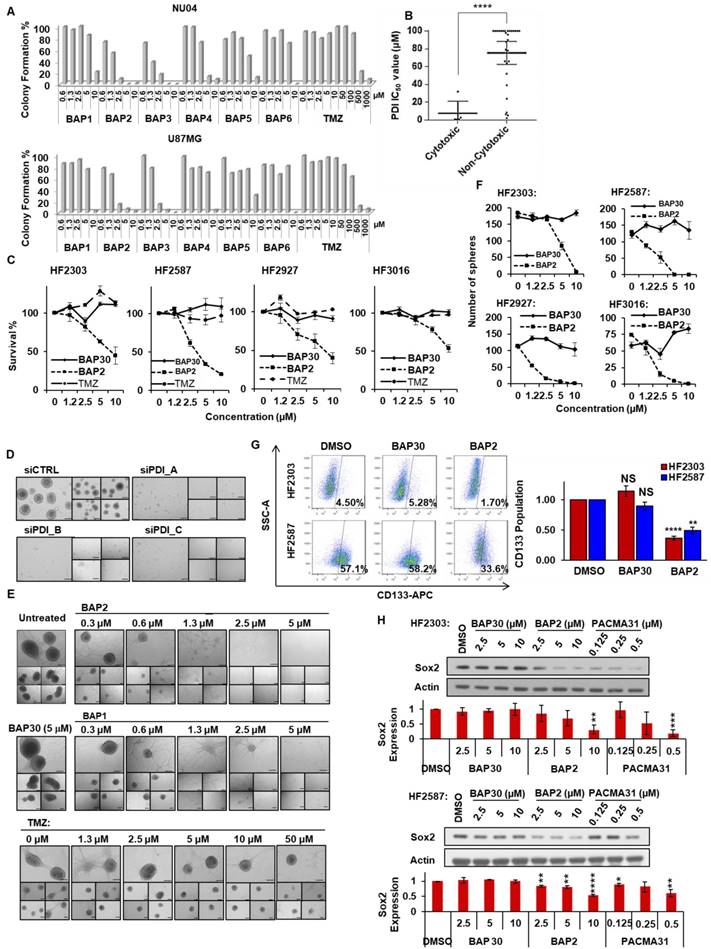
PDI supports maintenance of stemness properties in GBM. PDI knockdown impaired sphere formation (Figure 4D), a phenotype of CSCs [52], indicating that PDI may be necessary for CSC maintenance. Consistently, BAP2 analogs at non-toxic doses inhibited sphere formation, whereas BAP30 showed no significant inhibition (Figure 4E, S10A). Additionally, BAP2 dose-dependently inhibited sphere formation in patient-derived GBM cells, whereas BAP30 had no effect (Figure 4F, S10B). Furthermore, BAP2 treatment at a non-toxic concentration reduced the CD133-positive cell population in patient-derived GBM cells, whereas BAP30 did not (Figure 4G). In addition, Sox2 protein expression, as an orthogonal stemness marker, was dose-dependently abrogated by BAP2 and PACMA31, but not by BAP30, and confirms the reduction in GBM cell stemness induced by BAP2 (Figure 4H). Our findings of the role of PDI in supporting stemness properties in GBM is consistent with a recent report showing that inhibition of PDI induced differentiation of acute myeloid leukemia cells through the activation of CCAAT enhancer-binding protein α levels [53]. Taken together, these results suggest that our PDI inhibitors may target the GBM CSC population. Follow-up studies are needed to further investigate the mechanisms of PDI inhibition in suppressing the stemness properties in GBMs.
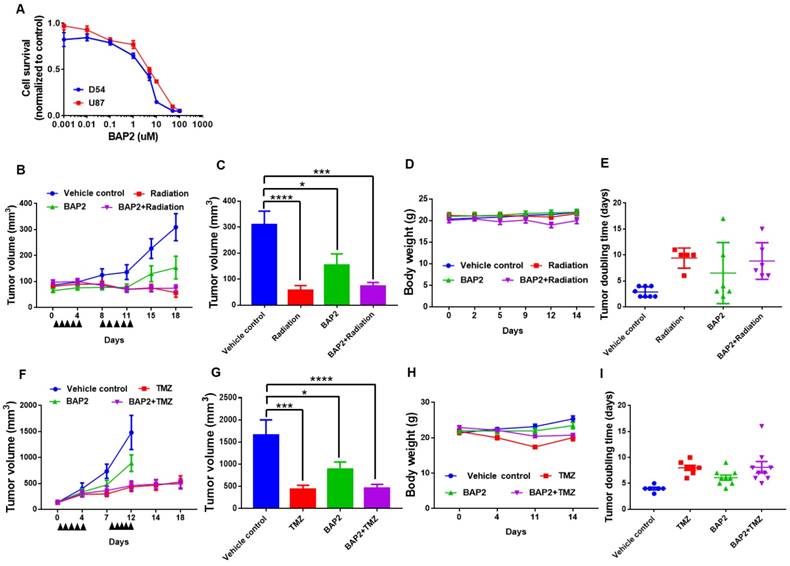
BAP2 suppresses tumor growth in a mouse model of GBM. An initial viability assay confirmed that BAP2 treatment significantly lowered viability at concentrations above 1 μM (Figure 5A). Mouse flank xenografts were established, and, when tumors were approximately 100 mm3, treatment was initiated using radiation (Figure 5B, 5E), TMZ (Figure 5F, 5I) or BAP2, each as a single agent, or BAP2+TMZ or radiation for five days/week for two weeks. BAP2 treatment caused tumor growth delay (p = 0.003 and 0.0345 compared to vehicle control for D54 and U87MG xenografts, respectively). Radiation and TMZ also caused tumor growth delay (p < 0.0001 (radiation) and p = 0.0012 (TMZ)) (Figure 5C, 5G). TMZ was more efficacious than BAP2 compared to vehicle control (p = 0.0347) (Figure 5G); however, mice treated with TMZ had a 15 % decrease in body weight, while mice treated with BAP2 exhibited minimal loss in body weight (Figure 5D, 5H). These results demonstrate that BAP2 may be as effective as TMZ, without the toxicity associated with DNA alkylating chemotherapy.
PDI inhibition is toxic to D54 and U87MG cells and delays tumor progression. (a) D54 and U87MG cells were seeded overnight and BAP2 was added for 72 h. Graph is plotted from three independent experiments and presented as mean ± SEM. D54 or U87MG cells were subcutaneously implanted into Athymic Nude-Foxn1nu mice and vehicle control, TMZ (5mg/kg in Ora-Plus (Perrigo) via oral gavage), BAP2 (20mg/kg via intraperitoneal injection in 5 % DMSO / 65 % 1,2-Propanediol / 30 % saline) or ionizing radiation (2 Gy/day), and TMZ+BAP2 or radiation+BAP2 were given for two weeks (all treatment administered daily for five days per week). Tumor measurement was performed twice weekly to determine volume that was plotted as mean ± SEM in (b) D54 and (f) U87MG xenografts, and tumor volume at Day 18 and Day 12 was used for statistical analysis in (c) D54 (g) and U87MG tumors. Body weight was also recorded twice weekly for mice bearing (d) D54 and (h) U87MG tumors. Tumor doubling time was calculated according to (http://www.chestx-ray.com/index.php/calculators/doubling-time) for (e) D54 and (i) U87MG xenografts. Study group sizes were: vehicle control - n = 6 (U87MG) or n = 8 (D54), TMZ - n = 6 (U87MG), radiation - n = 5 (D54), BAP2 - n = 9 (U87MG) or n = 6 (D54), TMZ+BAP2 - n = 9 (U87MG), and radiation+BAP2 - n = 6 (D54). * p < 0.05; *** p < 0.001; **** p < 0.0001 compared to control group.
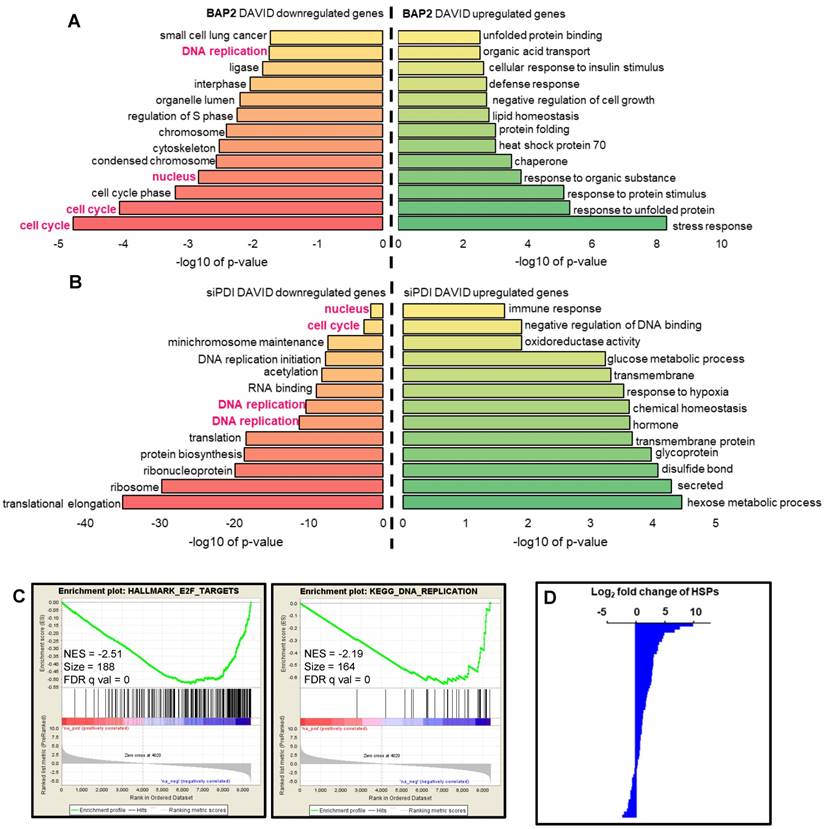
BAP2 affects transcription of genes involved in UPR, ER stress, and apoptosis. Nascent RNA Bru-seq analysis of BAP2-treated U87MG cells identified 237 differentially transcribed genes (≥ 8-fold difference). BAP2 treatment for four hours resulted in increased transcription of genes involved in the UPR, ER stress, and apoptosis (Figure 6A, Table S3). Furthermore, BAP2 decreased transcription of genes responsible for DNA replication and cell cycle function (Figure 6A, Table S4). Ingenuity Pathway Analysis (IPA) identified that 63 genes affected by BAP2 treatment belonged to the protein ubiquitination pathway, which is activated for protein degradation when unfolded and misfolded proteins accumulate during ER stress and UPR (Table S5). Bru-seq nascent RNA sequencing of cells treated with siPDI demonstrated that similar sets of genes decreased, including DNA replication, cell cycle, and nucleus (Figure 6B). GSEA was performed with genes preranked by log2FC values with cutoff values of gene size >300 base pairs and mean reads per kilobase per million >0.5. HALLMARK_E2F_TARGETS (NES = -2.51) and KEGG_DNA_REPLICATION (NES = -2.19) were the most significant gene sets downregulated following BAP2 treatment (Figure 6C). BAP2 had a pronounced effect on a larger family of heat shock proteins (HSP), further supporting the role of BAP2 in inducing ER stress (Figure 6D). Moreover, the BAP2 dataset matched gene set enrichment of other ER stress and UPR-inducing compounds (e.g. phospholipids [54], arsenite [55], and epoxomicin [56]) (Table S6,S7).
BAP2 affects DNA replication and other signaling pathways as assessed by Bru-seq. Functional terms represented by genes upregulated and downregulated at least eight-fold (log2(fold change) ≥ 3) by BAP2 (a) or siPDI (log2(fold change) ≥ 1) (b). Pathways highlighted in pink represent gene sets that were identified over both BAP2 and siPDI treatment. (c) BAP2 treatment enriched for downregulation of E2F targets and DNA replication gene sets. NES: normalized enrichment score. FDR q val: false discovery rate q value. Criteria for GSEA was false discovery rate < 0.05 %. (d) Transcription of 47 of 63 heat shock proteins increased after BAP2 treatment.
In particular, transcription of heat shock proteins, HSPA6 (876-fold), HSPH1 (99-fold), and DNAJB4 (28-fold), was upregulated by BAP2 (Figure S11A-C). Highly downregulated genes include mitochondrial methyltransferase-like gene METTL12 (125-fold), GTPase-activating ARHGAP22 (37-fold), and syntaxin 1A (27-fold), a gene that encodes for a membrane-trafficking protein (Figure S11D-F). Taken together, the nascent RNA transcriptome of BAP2-treated cells indicates PDI is inhibited, causing ER stress and eventual cell death.
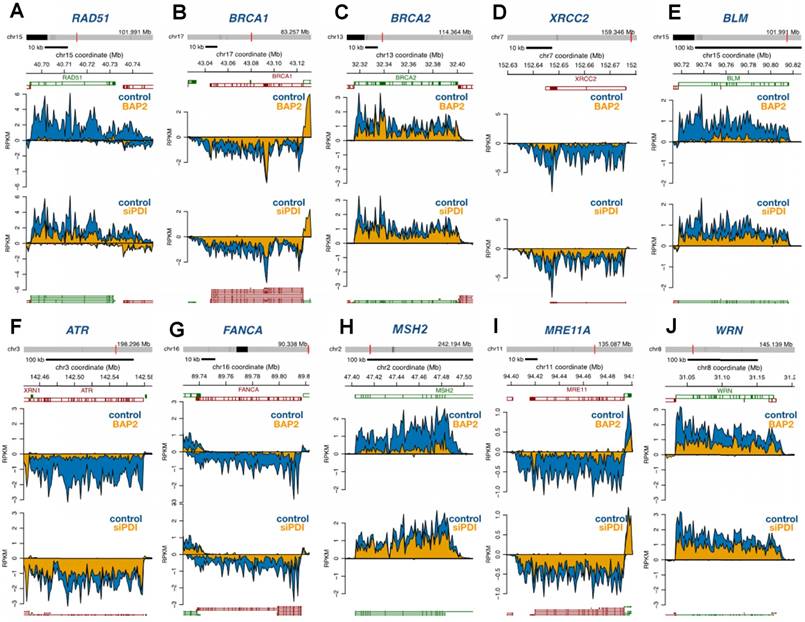
PDI inhibition downregulates DNA repair and DDR genes. Multiple genes involved in homologous recombination, Fanconi anemia, non-homologous end-joining, base excision repair, nucleotide excision repair, and mismatch repair pathways were significantly downregulated upon PDI knockdown or BAP2 treatment (Figure 7). From a more complete list of DNA repair and DDR genes, we observed a trend of downregulation not only by BAP2 and PDI-targeting siRNA but also by PACMA31 (Figure S12A, Table S8). PDI knockdown, BAP2, and PACMA31 all downregulated E2F genes (Gene Set: Hallmark E2F Targets), further supporting this hypothesis and demonstrating the similarities between BAP2 treatment and PDI knockdown (Figure S12B, Table S9). In the E2F targets gene set, BAP2 downregulated 65 % of the genes, PACMA31 downregulated 72 % of the genes, and siPDI downregulated 38 % of the genes. These observations link PDI activity to transcription of DNA repair and DDR genes. Additional similarities between BAP2, PACMA31, and PDI siRNA treatment were identified in genes related to hypoxia (Figure S12C). Furthermore, PACMA31 and BAP2 dose-dependently lower E2F1 and RAD51 protein expression in U87MG and D54 cells (Figure S13A). Decrease in protein expression of BRCA2, ATR, ATM, WRM, E2F1, and RAD51 upon BAP2 treatment was confirmed with Western blotting (Figure S13B). In vivo downregulation of DDR proteins was confirmed for BRCA2, ATR, RAD51, and E2F1 in response to BAP2+TMZ or radiation (Figure S13C, S13D). Although the mechanistic detail of such a link is beyond the scope of this current study, we hypothesize that PDI inhibition downregulates DNA repair genes such as RAD51 through decreased expression of E2F transcription factors [57].
BAP2 treatment and PDI siRNA knockdown cause global downregulation of DNA repair genes as assessed by Bru-seq. Four-hour treatment with BAP2 and siRNA-mediated PDI knockdown slows transcription of representative DNA repair genes including RAD51 (a), BRCA1 (b), BRCA2 (c), XRCC2 (d), BLM (e), ATR (f), FANCA (g), MSH2 (h), MRE11A (i), and WRN (j).
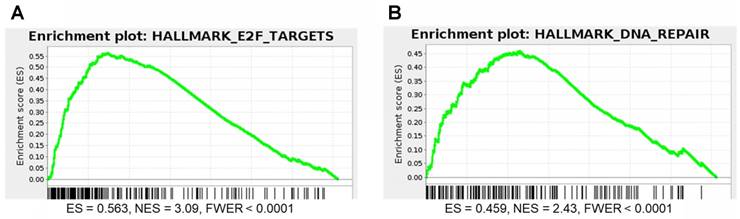
DNA repair and E2F gene sets are significantly enriched for genes that correlate with P4HB expression in brain cancer patient samples. We performed GSEA to identify genes that may have functional association with P4HB overexpression, using Pearson statistics in the combined TCGA LGG and GBM patient dataset (N = 676). E2F Targets (NES = 3.09 and FWER <0.0001) and DNA Repair (NES = 2.43 and FWER <0.0001) MSigDB Hallmark gene sets were significantly enriched for genes that correlate with P4HB mRNA expression (Figure 8A, 8B). E2F Targets gene set co-expression enrichment is consistent with in vitro reduced expression observed in response to PDI inhibition. Taken together, expression of P4HB correlates with expression of E2F and DNA repair genes suggesting a potential mechanism by which targeting PDI in cancer cells may sensitize them to radiation and chemotherapy.
E2F and DNA repair gene sets are significantly enriched for genes that correlate with P4HB expression. GSEA was performed to evaluate the functional enrichment of P4HB co-expressed genes using a Pearson statistic in the combined TCGA Low Grade Glioma and Glioblastoma patient dataset (N = 676). E2F (a) and DNA Repair (b) MSigDB Hallmark gene sets correlate with P4HB expression. ES: enrichment score. NES: normalized enrichment score. FWER: family-wise error rate.
Discussion
Glioblastomas are characterized by intratumoral histopathological heterogeneity and molecular heterogeneity [1]. These features contribute to therapeutic resistance and high recurrence rates leading to poor survival [58]. The molecular heterogeneity of GBM has hampered clinical translation of therapies targeting oncogenic pathways, hence inhibition of signaling pathways that are required for maintenance of a transformed phenotype, irrespective of oncogenic driver, is gaining traction.
GBM tumors may arise from neural stem cells, or hijack pathways used by stem cells for survival, and neural stem cell markers, such as Sox2 and OLIG2 are necessary for glioma growth [7, 59]. Thus, therapy-resistant CSCs within the tumor may contribute to GBM recurrence by repopulating the tumor mass following treatment [60-62]. Furthermore, ER stress can promote the transition of intestinal cells from epithelial stem cells to differentiated cells, suggesting a possible role for protein folding in regulating stemness [63]. Our report provides preliminary evidence that PDI may be critical for a stem cell phenotype. Here we describe a novel class of PDI inhibitors that inhibit the stem-like phenotype in patient-derived spheroid cultures.
Cell cultures derived from GBM patients preserve the complex genetic profiles of parental tumors and recapitulate aggressive growth characteristics when implanted in immunodeficient mice [64]. Therefore, we used a panel of patient-derived GBM cell lines to mimic tumor heterogeneity. In patient-derived neurosphere lines, BAP2 showed significant efficacy compared to TMZ, suggesting that BAP2 may overcome TMZ resistance in GBMs. Several studies have shown that PDI family members are associated with tumor progression and TMZ resistance in gliomas [26, 65]. PDI, encoded by P4HB, a gene overexpressed in GBM, may therefore be an impactful therapeutic target in GBM.
Posttranslational modifications in the active sites of PDI, including S-glutathionylation, S-nitrosylation, and succination, suppress its enzymatic activity resulting in accumulation of unfolded proteins and ER stress [66-68]. Most PDI inhibitors, including PACMA31, 16F16, and DTNB, also modify active-site cysteines through covalent binding [8]. Due to this binding characteristic, they lack selectivity among PDI family enzymes, wherein thioredoxin and other proteins that share the CGHC active site are also inhibited. The presence of a Michael acceptor moiety on BAP2 suggests the compound has weak electrophilic activity and binds to the active site cysteines of PDI. However, mutation of His256 blocks BAP2 activity even at high concentrations (100 μM) [77]. Furthermore, the para hydroxyl group that is critical for BAP2 activity is not forming a hydrogen bond to stabilize the carbonyl group, which is the key interaction for inducing Phase II enzymes such as glutathione transferases [69, 70]. Unlike active-site-targeting PDI inhibitors, BAP2 and analogs bind to an allosteric site located in the b'xa'c domain, resulting in a conformational change in PDI that hinders active-site reactivity. Additionally, BAP2 is active against PDI and PDIp, PDI family members that contain the histidine residue in the b' domain. Though ERp57 contains active site cysteines, BAP2 does not inhibit it, and ERp57 is one of the isoforms that does not contain the key histidine in the b' domain. More extensive analysis of the binding position of BAP analogs should be performed to optimize the scaffold further. Ongoing structural determination will provide a clearer visualization and understanding of this unique binding mode.
Resistance to DNA alkylating agent TMZ often develops because GBM cells upregulate O6-methylguanine-DNA methyltransferase (MGMT). Though MGMT depletion has not been an effective therapy for GBM, DNA repair is nonetheless an important mediator in the development of resistance to treatment and is an important aspect of the resistant nature of CSCs [71]. PDI has been linked to the process of DNA-nuclear matrix anchoring, a critical step to promote DNA repair [72]. In this study, we demonstrate that PDI inhibition by siRNA, PACMA31, and BAP2 decreases global transcription of DNA repair genes (Figure S12). These results suggest that PDI inhibition may be beneficial in combination with DNA alkylating agents such as TMZ to sensitize the tumor. Since BAP2 selectively inhibits PDI and PDIp, these may be the major PDI family members implicated in regulating transcription of DNA repair genes; however, it remains to be determined whether other PDI family members play a role in DNA repair and GBM resistance mechanisms. Furthermore, with evidence that PDI inhibition lowers E2F1 mRNA and protein expression, a potential mechanism linking PDI to the DNA damage response emerges. E2F1 expression may decrease in response to an ER stress signal triggered by PDI inhibition, which causes RAD51 downregulation. Further investigation into this mechanism is warranted to confirm the role of PDI in DNA repair and the DNA damage response.
The GBM tumor microenvironment is generally hypoxic, selecting for tumor cells and CSCs with an increasingly more invasive and aggressive phenotype. Tumor hypoxia is responsible for decreased drug penetration, increased metastases, and decreased overall survival. Tumor cells respond to hypoxia by activating the UPR and upregulating PDI [73-76]. Our Bru-seq studies show that both PDI knockdown and inhibition of its enzymatic activity highly correlate with downregulation of multiple hypoxic response gene sets, further supporting BAP2 analogs as a potential treatment for hypoxic tumors.
While the effects of BAP2 correlate with siPDI treatment, variations in individual genes may be observed because of the different modes of PDI inhibition. BAP2 is a small-molecule inhibitor of PDI and inhibits PDI in an acute manner, whereas siPDI inhibits PDI in a relatively slower manner. The cells may respond differently to the two different mechanisms of PDI inhibition; thus, comparing the transcriptomic consequences of the two perturbations should be done carefully. For example, PDI interacts with other proteins under various biological contexts. Such protein-protein interactions are interrupted by siPDI, while they could be maintained in the presence of BAP2.
In conclusion, we have discovered a selective small molecule inhibitor of PDI that binds in a novel allosteric binding site likely spanning the b'x and a' domains to block active site thiol activity. Of interest is the novel binding mode of BAP2 and how allosteric binding affects the conformation of the a' domain. An extensive SAR campaign supported the relevance of the b' domain for BAP2 analog activity, including the necessity of the hydroxyl group for potency [30]. Furthermore, BAP2 is toxic in a panel of primary GBM cell lines and sensitizes cells to TMZ. Finally, we have demonstrated that PDI inhibition downregulates DNA repair pathways and genes in the E2F pathway. Our findings provide further evidence that PDI inhibition may synergize with DNA alkylating agents and other therapies in which resistance is defined by an upregulation of DNA repair mechanisms. Furthermore, our findings support the exciting possibility of a druggable allosteric site in PDI for future development of selective PDI inhibitors for treating GBM.
Abbreviations
GBM: glioblastoma; PDI: protein disulfide isomerase; ER: endoplasmic reticulum; ATCC: American Type Culture Collection; GSEA: gene set enrichment analysis; DDR: DNA damage response; TCGA: The Cancer Genome Atlas; LGG: low grade glioma; TMZ: temozolomide; UPR: unfolded protein response; KW: Kruskal-Wallis; MTT: 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; AMS: 4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid; CSC: cancer stem cell; MGMT: O6-methylguanine-DNA methyltransferase; DTNB: 5,5'-dithiobis-(2-nitrobenzoic acid); HSP: heat shock protein.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work was funded by a grant from the NIH (CA193690) and National Key R&D Program of China (2017YFA0504000). The authors would like to thank Ding Xue for his gracious assistance with the study.
Author contributions
Conceptualization, N.N, A.R., C-C.W., and M.L.; Investigation & Formal Analysis, S.X., Y.L., K.Y., H.W., A.S., A.K., A.B., S.T., S.Y., and X.W.; Writing - Original Draft, S.X., N.N., and A.S.; Writing - Review & Editing, S.X., Y.L., K.Y., H.W., A.S., A.K., A.B., S.T., S.Y., X.W., C-C.W., A.R., M.L., and N.N.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Shergalis A, Bankhead A 3rd, Luesakul U, Muangsin N, Neamati N. Current challenges and opportunities in treating glioblastoma. Pharmacol Rev. 2018;70:412-45
2. Ostrom QT, Gittleman H, Liao P, Vecchione-Koval T, Wolinsky Y, Kruchko C. et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2010-2014. Neuro Oncol. 2017;19:V1-V88
3. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ. et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987-96
4. Sengupta S, Marrinan J, Frishman C, Sampath P. Impact of temozolomide on immune response during malignant glioma chemotherapy. Clin Dev Immunol. 2012 doi: 10.1155/2012/831090
5. Batlle E, Clevers H. Cancer stem cells revisited. Nat Med. 2017;23:1124-34
6. Gangemi RM, Griffero F, Marubbi D, Perera M, Capra MC, Malatesta P. et al. SOX2 silencing in glioblastoma tumor-initiating cells causes stop of proliferation and loss of tumorigenicity. Stem Cells. 2009;27:40-8
7. Ligon KL, Huillard E, Mehta S, Kesari S, Liu HY, Alberta JA. et al. Olig2-regulated lineage-restricted pathway controls replication competence in neural stem cells and malignant glioma. Neuron. 2007;53:503-17
8. Xu S, Sankar S, Neamati N. Protein disulfide isomerase: a promising target for cancer therapy. Drug Discov Today. 2014;19:222-40
9. Lee E, Lee DH. Emerging roles of protein disulfide isomerase in cancer. BMB Rep. 2017;50:401-10
10. Parakh S, Atkin JD. Novel roles for protein disulphide isomerase in disease states: a double edged sword? Front Cell Dev Biol. 2015;3:30
11. Hatahet F, Ruddock LW. Protein disulfide isomerase: a critical evaluation of its function in disulfide bond formation. Antioxid Redox Signal. 2009;11:2807-50
12. Shergalis A, Neamati N. Protein disulfide isomerase. In. Choi S, Ed. Encyclopedia of Signaling Molecules. New York: Springer. 2016:1-12
13. Kozlov G, Maattanen P, Thomas DY, Gehring K. A structural overview of the PDI family of proteins. FEBS J. 2010;277:3924-36
14. Wang C, Li W, Ren J, Fang J, Ke H, Gong W. et al. Structural insights into the redox-regulated dynamic conformations of human protein disulfide isomerase. Antioxid Redox Signal. 2013;19:36-45
15. Obacz J, Avril T, Le Reste PJ, Urra H, Quillien V, Hetz C. et al. Endoplasmic reticulum proteostasis in glioblastoma-From molecular mechanisms to therapeutic perspectives. Sci Signal. 2017:10 doi: 10.1126/scisignal.aal2323
16. Muller C, Bandemer J, Vindis C, Camare C, Mucher E, Gueraud F. et al. Protein disulfide isomerase modification and inhibition contribute to ER stress and apoptosis induced by oxidized low density lipoproteins. Antioxid Redox Signal. 2013;18:731-42
17. Xu S, Butkevich AN, Yamada R, Zhou Y, Debnath B, Duncan R. et al. Discovery of an orally active small-molecule irreversible inhibitor of protein disulfide isomerase for ovarian cancer treatment. Proc Natl Acad Sci U S A. 2012;109:16348-53
18. Hoffstrom BG, Kaplan A, Letso R, Schmid RS, Turmel GJ, Lo DC. et al. Inhibitors of protein disulfide isomerase suppress apoptosis induced by misfolded proteins. Nat Chem Biol. 2010;6:900-6
19. Kaplan A, Gaschler MM, Dunn DE, Colligan R, Brown LM, Palmer AG 3rd. et al. Small molecule-induced oxidation of protein disulfide isomerase is neuroprotective. Proc Natl Acad Sci U S A. 2015;112:E2245-52
20. Vatolin S, Phillips JG, Jha BK, Govindgari S, Hu J, Grabowski D. et al. Novel protein disulfide isomerase inhibitor with anticancer activity in multiple myeloma. Cancer Res. 2016;76:3340-50
21. Khan MM, Simizu S, Lai NS, Kawatani M, Shimizu T, Osada H. Discovery of a small molecule PDI inhibitor that inhibits reduction of HIV-1 envelope glycoprotein gp120. ACS Chem Biol. 2011;6:245-51
22. Bekendam RH, Bendapudi PK, Lin L, Nag PP, Pu J, Kennedy DR. et al. A substrate-driven allosteric switch that enhances PDI catalytic activity. Nat Commun. 2016;7:12579
23. Jasuja R, Passam FH, Kennedy DR, Kim SH, van Hessem L, Lin L. et al. Protein disulfide isomerase inhibitors constitute a new class of antithrombotic agents. J Clin Invest. 2012;122:2104-13
24. Zwicker JI, Schlechter BL, Stopa JD, Liebman H, Aggarwal A, Puligandla M. et al. Targeting protein disulfide isomerase with the flavonoid isoquercetin to improve hypercoagulability in advanced cancer. JCI Insight. 2019 doi: 10.1172/jci.insight.125851
25. Cole KS, Grandjean JMD, Chen K, Witt CH, O'Day J, Shoulders MD. et al. Characterization of an A-site selective protein disulfide isomerase A1 inhibitor. Biochemistry. 2018;57:2035-43
26. Sun S, Lee D, Ho AS, Pu JK, Zhang XQ, Lee NP. et al. Inhibition of prolyl 4-hydroxylase, beta polypeptide (P4HB) attenuates temozolomide resistance in malignant glioma via the endoplasmic reticulum stress response (ERSR) pathways. Neuro Oncol. 2013;15:562-77
27. Won JK, Yu SJ, Hwang CY, Cho SH, Park SM, Kim K. et al. Protein disulfide isomerase inhibition synergistically enhances the efficacy of sorafenib for hepatocellular carcinoma. Hepatology. 2017;66:855-68
28. Kyani A, Tamura S, Yang S, Shergalis A, Somanta S, Kuang Y. et al. Discovery and mechanistic elucidation of a class of PDI inhibitors for the treatment of glioblastoma. ChemMedChem. 2017;12:164-77
29. Mahapatra DM, Bharti SK, Asati V. Anti-cancer chalcones: structural and molecular target perspectives. Eur J Med Chem. 2015;98:69-114
30. Yang S, Shergalis A, Lu D, Kyani A, Lu Z, Ljungman M. et al. Design, synthesis, and biological evaluation of novel allosteric protein disulfide isomerase inhibitors. J Med Chem. 2019 doi: 10.1021/acs.jmedchem.8b01951
31. deCarvalho AC, Nelson K, Lemke N, Lehman NL, Arbab AS, Kalkanis S. et al. Gliosarcoma stem cells undergo glial and mesenchymal differentiation in vivo. Stem Cells. 2010;28:181-90
32. Madhavan S, Zenklusen JC, Kotliarov Y, Sahni H, Fine HA, Buetow K. Rembrandt: helping personalized medicine become a reality through integrative translational research. Mol Cancer Res. 2009;7:157-67
33. Gravendeel LA, Kouwenhoven MC, Gevaert O, de Rooi JJ, Stubbs AP, Duijm JE. et al. Intrinsic gene expression profiles of gliomas are a better predictor of survival than histology. Cancer Res. 2009;69:9065-72
34. Broad Institute TCGA Genome Data Analysis Center. Firehose stddata__2016_01_28 run. Broad Institute of MIT Harvard. 2016 doi:10.7908/C11G0KM9
35. R Core Team. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. 2016 https://www.R-project.org/
36. Xu S, Grande F, Garofalo A, Neamati N. Discovery of a novel orally active small-molecule gp130 inhibitor for the treatment of ovarian cancer. Mol Cancer Ther. 2013;12:937-49
37. Lyles MM, Gilbert HF. Catalysis of the oxidative folding of ribonuclease A by protein disulfide isomerase: pre-steady-state kinetics and the utilization of the oxidizing equivalents of the isomerase. Biochemistry. 1991;30:619-25
38. Carmichael J, DeGraff WG, Gazdar AF, Minna JD, Mitchell JB. Evaluation of a tetrazolium-based semiautomated colorimetric assay: assessment of chemosensitivity testing. Cancer Res. 1987;47:936-42
39. Ikushima H, Todo T, Ino Y, Takahashi M, Miyazawa K, Miyazono K. Autocrine TGF-beta signaling maintains tumorigenicity of glioma-initiating cells through Sry-related HMG-box factors. Cell Stem Cell. 2009;5:504-14
40. Molina DM, Jafari R, Ignatushchenko M, Seki T, Larsson EA, Dan C. et al. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science. 2013;341:84-7
41. Jafari R, Almqvist H, Axelsson H, Ignatushchenko M, Lundback T, Nordlund P. et al. The cellular thermal shift assay for evaluating drug target interactions in cells. Nat Protoc. 2014;9:2100-22
42. Xu S, Oshima T, Imada T, Masuda M, Debnath B, Grande F. et al. Stabilization of MDA-7/IL-24 for colon cancer therapy. Cancer Lett. 2013;335:421-30
43. Paulsen MT, Veloso A, Prasad J, Bedi K, Ljungman EA, Tsan YC. et al. Coordinated regulation of synthesis and stability of RNA during the acute TNF-induced proinflammatory response. Proc Natl Acad Sci U S A. 2013;110:2240-5
44. Dennis G J, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 2003;4:P3
45. Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44-57
46. Primm TP, Gilbert HF. Hormone binding by protein disulfide isomerase, a high capacity hormone reservoir of the endoplasmic reticulum. J Biol Chem. 2001;276:281-6
47. Wang L, Li SJ, Sidhu A, Zhu L, Liang Y, Freedman RB. et al. Reconstitution of human Ero1-L alpha/protein-disulfide isomerase oxidative folding pathway in vitro. Position-dependent differences in role between the a and a ' domains of protein-disulfide isomerase. J Biol Chem. 2009;284:199-206
48. Lin L, Gopal S, Sharda A, Passam F, Bowley SR, Stopa J. et al. Quercetin-3-rutinoside inhibits protein disulfide isomerase by binding to its b'x domain. J Biol Chem. 2015;290:23543-52
49. Yagi-Utsumi M, Satoh T, Kato K. Structural basis of redox-dependent substrate binding of protein disulfide isomerase. Sci Rep. 2015;5:13909
50. Fu XM, Wang P, Zhu BT. Characterization of the estradiol-binding site structure of human pancreas-specific protein disulfide isomerase: indispensable role of the hydrogen bond between His278 and the estradiol 3-hydroxyl group. Biochemistry. 2011;50:106-15
51. Galligan JJ, Petersen DR. The human protein disulfide isomerase gene family. Hum Genomics. 2012;6:6
52. Laks DR, Masterman-Smith M, Visnyei K, Angenieux B, Orozco NM, Foran I. et al. Neurosphere formation is an independent predictor of clinical outcome in malignant glioma. Stem Cells. 2009;27:980-7
53. Chlebowska-Tuz J, Sokolowska O, Gaj P, Lazniewski M, Firczuk M, Borowiec K. et al. Inhibition of protein disulfide isomerase induces differentiation of acute myeloid leukemia cells. Haematologica. 2018;103:1843-52
54. Oskolkova OV, Afonyushkin T, Leitner A, von Schlieffen E, Gargalovic PS, Lusis AJ. et al. ATF4-dependent transcription is a key mechanism in VEGF up-regulation by oxidized phospholipids: critical role of oxidized sn-2 residues in activation of unfolded protein response. Blood. 2008;112:330-9
55. Oh RS, Pan WC, Yalcin A, Zhang H, Guilarte TR, Hotamisligil GS. et al. Functional RNA interference (RNAi) screen identifies system A neutral amino acid transporter 2 (SNAT2) as a mediator of arsenic-induced endoplasmic reticulum stress. J Biol Chem. 2012;287:6025-34
56. Obeng EA, Carlson LM, Gutman DM, Harrington WJ Jr, Lee KP, Boise LH. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood. 2006;107:4907-16
57. Ren B, Cam H, Takahashi Y, Volkert T, Terragni J, Young RA. et al. E2F integrates cell cycle progression with DNA repair, replication, and G(2)/M checkpoints. Genes Dev. 2002;16:245-56
58. Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD. et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98-110
59. Annovazzi L, Mellai M, Caldera V, Valente G, Schiffer D. SOX2 expression and amplification in gliomas and glioma cell lines. Cancer Genomics Proteomics. 2011;8:139-47
60. Eramo A, Ricci-Vitiani L, Zeuner A, Pallini R, Lotti F, Sette G. et al. Chemotherapy resistance of glioblastoma stem cells. Cell Death Differ. 2006;13:1238-41
61. Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB. et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756-60
62. Murat A, Migliavacca E, Gorlia T, Lambiv WL, Shay T, Hamou MF. et al. Stem cell-related "Self-Renewal" signature and high epidermal growth factor receptor expression associated with resistance to concomitant chemoradiotherapy in glioblastoma. J Clin Oncol. 2008;26:3015-24
63. Heijmans J, van Lidth de Jeude JF, Koo BK, Rosekrans SL, Wielenga MC, van de Wetering M. et al. ER stress causes rapid loss of intestinal epithelial stemness through activation of the unfolded protein response. Cell Rep. 2013;3:1128-39
64. Lee J, Kotliarova S, Kotliarov Y, Li A, Su Q, Donin NM. et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell. 2006;9:391-403
65. Zou H, Wen C, Peng Z, Shao Y-Y, Hu L, Li S. et al. P4HB and PDIA3 are associated with tumor progression and therapeutic outcome of diffuse gliomas. Oncol Rep. 2018;39:501-10
66. Corazzari M, Gagliardi M, Fimia GM, Piacentini M. Endoplasmic reticulum stress, unfolded protein response, and cancer cell fate. Front Oncol. 2017;7:78
67. Townsend DM, Manevich Y, He L, Xiong Y, Bowers RR Jr, Hutchens S. et al. Nitrosative stress-induced s-glutathionylation of protein disulfide isomerase leads to activation of the unfolded protein response. Cancer Res. 2009;69:7626-34
68. Uehara T, Nakamura T, Yao D, Shi ZQ, Gu Z, Ma Y. et al. S-nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature. 2006;441:513-7
69. Dinkova-Kostova AT, Abeygunawardana C, Talalay P. Chemoprotective properties of phenylpropenoids, bis(benzylidene)cycloalkanones, and related Michael reaction acceptors: correlation of potencies as phase 2 enzyme inducers and radical scavengers. J Med Chem. 1998;41:5287-96
70. Amslinger S, Al-Rifai N, Winter K, Wormann K, Scholz R, Baumeister P. et al. Reactivity assessment of chalcones by a kinetic thiol assay. Org Biomol Chem. 2013;11:549-54
71. Annovazzi L, Mellai M, Schiffer D. Chemotherapeutic drugs: DNA damage and repair in glioblastoma. Cancers. 2017;9:E57
72. VanderWaal RP, Spitz DR, Griffith CL, Higashikubo R, Roti Roti JL. Evidence that protein disulfide isomerase (PDI) is involved in DNA-nuclear matrix anchoring. J Cell Biochem. 2002;85:689-702
73. Peñaranda Fajardo NM, Meijer C, Kruyt FA. The endoplasmic reticulum stress/unfolded protein response in gliomagenesis, tumor progression and as a therapeutic target in glioblastoma. Biochem Pharmacol. 2016;118:1-8
74. Graven KK, Molvar C, Roncarati JS, Klahn BD, Lowrey S, Farber HW. Identification of protein disulfide isomerase as an endothelial hypoxic stress protein. Am J Physiol Lung Cell Mol Physiol. 2002;282:L996-1003
75. Galmiche A, Sauzay C, Chevet E, Pluquet O. Role of the unfolded protein response in tumor cell characteristics and cancer outcome. Curr Opin Oncol. 2017;29:41-7
76. Manuel AM, Walla MD, Faccenda A, Martin SL, Tanis RM, Piroli GG. et al. Succination of protein disulfide isomerase links mitochondrial stress and endoplasmic reticulum stress in the adipocyte during diabetes. Antioxid Redox Signal. 2017;27:1281-96
77. Yang S, Shergalis A, Lu D, Kyani A, Liu Z, Ljungman M, Neamati N. Design, synthesis, and biological evaluation of novel allosteric protein disulfide isomerase inhibitors. J Med Chem. 2019;62:3447-74
Author contact
![]() Corresponding author: N.N. E-mail: neamatiedu; Phone: 734-647-2732
Corresponding author: N.N. E-mail: neamatiedu; Phone: 734-647-2732