Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(8):2252-2267. doi:10.7150/thno.30658 This issue Cite
Research Paper
Progerin accumulation in nucleus pulposus cells impairs mitochondrial function and induces intervertebral disc degeneration and therapeutic effects of sulforaphane
Xiaolong Xu1#, Di Wang1#, Chao Zheng1#, Bo Gao1#, Jing Fan1, Pengzhen Cheng1, Baohua Liu2, Liu Yang1,3 ![]() , Zhuojing Luo1,3
, Zhuojing Luo1,3 ![]()
1. Institute of Orthopedic Surgery, Xijing Hospital, Fourth Military Medical University, Xi'an 710032, People's Republic of China
2. Department of Biochemistry and Molecular Biology, Shenzhen University Health Science Center, Shenzhen 518060, People's Republic of China
3. Medical Research Institute, Northwestern Polytechnical University, Xi'an 710072, People's Republic of China
#These authors contributed equally to this work.
Received 2018-10-14; Accepted 2019-2-10; Published 2019-4-12
Citation:
Xu X, Wang D, Zheng C, Gao B, Fan J, Cheng P, Liu B, Yang L, Luo Z. Progerin accumulation in nucleus pulposus cells impairs mitochondrial function and induces intervertebral disc degeneration and therapeutic effects of sulforaphane. Theranostics 2019; 9(8):2252-2267. doi:10.7150/thno.30658. https://www.thno.org/v09p2252.htm
Other stylesAbstract

Progerin, a truncated unprocessed lamin A protein, causes tissue aging and degeneration. In this study we explored the role of progerin in the pathogenesis of intervertebral disc degeneration (IDD). We also examined the effect of sulforaphane (SFN) on progerin accumulation and mitochondrial dysfunction in IDD.
Methods: The role of progerin in IDD was explored using human nucleus pulposus (NP) tissues, rat NP cells, and Lmna G609G knock-in mice. Immunostaining, X-ray imaging, and Western blotting were performed to assess the phenotypes of intervertebral discs. Alterations in senescence and apoptosis were evaluated by SA-β-galactosidase, immunofluorescence, flow cytometry, and TUNEL assays. Mitochondrial function was investigated by JC-1 staining, transmission electron microscopy, and determination of the level of ATP and the activities of mitochondrial enzymes.
Results: The progerin level was elevated in degenerated human NP tissues. Lmna G609G/G609G mice displayed IDD, as evidenced by increased matrix metalloproteinase-13 expression and decreased collagen II and aggrecan expression and disc height. Furthermore, progerin overexpression in rat NP cells induced mitochondrial dysfunction (decreased ATP synthesis, mitochondrial membrane potential, and activities of mitochondrial complex enzymes), morphologic abnormalities, and disrupted mitochondrial dynamic (abnormal expression of proteins involved in fission and fusion), resulting in apoptosis and senescence. SFN ameliorated the progerin-induced aging defects and mitochondrial dysfunction in NP cells and IDD in Lmna G609G/G609G mice.
Conclusions: Progerin is involved in the pathogenesis of IDD. Also, SFN alleviates progerin‑induced IDD, which is associated with amelioration of aging defects and mitochondrial dysfunction. Thus, SFN shows promise for the treatment of IDD.
Keywords: Intervertebral disc degeneration, progerin, mitochondria, sulforaphane
Introduction
Low-back pain is a ubiquitous musculoskeletal disorder that affects up to 80% of individuals during their lifetime, imposing a considerable socioeconomic and healthcare burden on society [1-3]. Intervertebral disc degeneration (IDD) contributes to lower‑back pain. Intervertebral discs (IVDs) are composed of the inner nucleus pulposus (NP) surrounded by the annulus fibrosus. NP cells, critical components of the IVD, maintain homeostasis of the extracellular matrix, thus regulating disc functions and deformation. The pathogenesis of IDD is complex, being influenced by several risk factors, including genetics, immune system status, excessive loading and aging [4-7]. These factors ultimately trigger NP cell senescence and apoptosis, causing IDD. Elucidation of the mechanisms underlying NP dysfunction would enhance our understanding of IDD pathogenesis.
Progerin, a truncated, permanently farnesylated prelamin A, causes the premature aging disorder termed Hutchinson-Gilford Progeria Syndrome (HGPS) [8-10]. The accumulation of progerin causes a variety of defects in HGPS cells, such as compromised nuclear architecture integrity [11, 12], increased DNA damage [13], impaired redox homeostasis [14], and mitochondrial dysfunction [15]. Remarkably, there are striking commonalities between several premature aging diseases and aging-associated diseases, and progerin expression in the general population [16] is related to physiological aging [14]. Moreover, progerin is involved in the pathogenesis of a range of diseases, such as osteoarthritis and dilated cardiomyopathy, as well as skin aging, all of which may be linked to apoptosis and oxidative stress [17-20]. However, the role of progerin in human IDD is unclear. Investigation of the link between increased progerin expression and IDD progression would aid in the identification of novel therapeutic targets and pharmacological regimens preventing or alleviating IDD.
Sulforaphane (SFN) is a natural isothiocyanate compound present in cruciferous vegetables, especially broccoli. The therapeutic efficacy of SFN is being evaluated in preclinical and clinical trials for a wide range of disorders, including dysregulated type 2 diabetes [21], memory impairment [22], autism spectrum disorder [23] and Huntington's disease [24]. The protective effects of SFN are associated with activation of the NF-E2-related factor2 (Nrf2) antioxidant network and induction of autophagy [13]. In addition, SFN exerts a protective effect on the mitochondria of pancreatic β-cells [25]. However, the effects of SFN on IDD, as well as the underlying mechanisms, are unknown. In this study, using human NP tissues, rat NP cells, and Lmna G609G knock-in mice, we characterized the role of progerin in IDD and provide the first evidence of a link between progerin accumulation in animal and human NP tissue/cells to IVD degeneration. We also examined the protective effect of SFN against progerin-induced aging defects, with a focus on mitochondrial function.
Methods
Patient samples
NP specimens were obtained from 20 patients (10 males and 10 females; mean age = 36.4 ± 17.4 years) with degenerative disc disease or scoliosis. The degree of IDD was assessed according to modified Pfirrmann grading system [26] by magnetic resonance imaging (MRI). Grade II (n = 5) and III (n = 5) samples were combined into a Grade II/III group, and Grade IV (n = 4) and V (n = 6) samples into a Grade IV/V group (Table S1). Ethics approval was obtained from the Institutional Review Board of Xijing Hospital of the Fourth Military Medical University, and informed consent was obtained from each donor.
Animals
Lmna G609G/G609G mice, generated as described previously [10], were kindly provided by Prof. Baohua Liu (Shenzhen University, China). All animals were maintained under pathogen-free conditions at the Experimental Animal Centre of the Fourth Military Medical University. The G609G/G609G mice were genotyped using the following primers: forward 5′-CTATTGCATGCTTCTCCTCAG-3′, and reverse 5′-TGAGCGCAGGTTGTACTCAG-3′ (Figure S1B). All experimental procedures were approved by the Fourth Military Medical University Animal Use and Care Committee. Starting at 4 weeks of age, G609G/G609G mice were administered SFN at 10 mg/kg in phosphate-buffered saline (PBS) intraperitoneally (ip) three times per week or vehicle, for 12 weeks [21]. Treatment with vehicle alone did not induce apparent damage or stress in wild‑type (WT) mice. The body weight of the mice was monitored weekly. The survival rate was analyzed by the Kaplan-Meier method, and statistical analysis was performed by log-rank (Mantel-Cox) test.
Reagents and antibodies
SFN was purchased from ApexBio Technology (Houston, TX, USA). The following antibodies were used for Western blotting: Lamin A/C mouse monoclonal (Cell Signaling Technology, CST, Beverly, MA, USA, #4777), progerin mouse monoclonal (Santa Cruz Biotechnology, Santa Cruz, CA, USA, #sc-81611), OPA1 mouse monoclonal (Santa Cruz Biotechnology, #sc-393296), Drp1 mouse monoclonal (Santa Cruz Biotechnology, #sc-101270), Mfn1 mouse monoclonal (Santa Cruz Biotechnology, #sc-166644), Mfn2 mouse monoclonal (Santa Cruz Biotechnology, #sc-100560), PGC1α rabbit polyclonal (Abnova, Taiwan, #PAB12601), AMPKα (CST, #5832), Phospho-AMPK (Thr172)α (CST, #2535), collagen II rabbit polyclonal (Proteintech, China, 15943-1-AP), aggrecan rabbit polyclonal (Proteintech, #13880-1-AP), MMP-13 rabbit polyclonal (Abcam, Cambridge, MA, USA, ab39012), GAPDH (Proteintech, #60004-1-lg), and β-actin mouse monoclonal (Sangon Biotech, China, #D190606). For immunofluorescence and immunohistochemical analyses, the following antibodies were used: Anti-aggrecan (rabbit polyclonal; Proteintech, #13880-1-AP), P16INK4a mouse monoclonal (Abcam, #ab54210), LAP2 rabbit polyclonal (Proteintech, #14651-1-AP), γH2AX (Proteintech, #10856-1-AP), H3K27me3 rabbit monoclonal (CST, #9733), lamin B1 rabbit polyclonal (Proteintech, #12987-1-AP), progerin mouse monoclonal (Santa Cruz, #sc-81611), and collagen II rabbit polyclonal (Abcam, #ab34712).
Cell culture and treatment
Rat NP cells were kindly generated and donated by Prof. Di Chen (Rush University) [27], and were maintained in Dulbecco's modified Eagle's medium (DMEM)/F-12 (1:1) (Gibco, Grand Island, NY, USA) containing 10% fetal bovine serum (FBS) (Invitrogen, Carlsbad, CA, USA) and 1% penicillin/streptomycin (Gibco) at 37°C under 5% CO2 and 20% O2. The culture medium was replaced every 3 days. The cells were used at passages 5-8. The pLVX-mCherry-progerin-puro and pLVX-mCherry-puro vectors were provided by Prof. Baohua Liu (Shenzhen University, China). Briefly, rat NP cells were seeded in 35 mm dishes (2 × 105 cells/dish) at 24 h before infection. The cells were incubated overnight with the transfection mixture, washed with PBS, and incubated with fresh medium for 48 h. Next, antibiotic-resistant (5 μg/mL puromycin) cells were selected and expanded as mass populations. The medium was replaced twice weekly, and the cultures were passaged at 90% confluency. The following experimental groups were analyzed: (1) Vector con, vector-transfected NP cells; (2) progerin, progerin-expressing NP cells. To assess the effects of SFN, the following experimental groups were used: (1) Vector con+vehicle, only vector-transfected NP cells treated with vehicle; (2) progerin+vehicle, progerin-expressing cells treated with vehicle; and (3) progerin+SFN, progerin-expressing cells treated with SFN (dissolved in dimethyl sulphoxide and added to growth medium at a final concentration of 5 μM [14]).
RNA extraction and quantitative real-time reverse transcription-PCR
Human NP tissues were immediately snap‑frozen in liquid nitrogen and stored at -80 °C until mRNA extraction. Total RNA was isolated from human NP tissue or NP cells using the Total RNA Kit (Omega Biotek, Norcross, GA, USA) according to the manufacturer's instructions. RNA concentrations were estimated spectrophotometrically using the ratio of the absorbance at 260 nm to that at 280 nm, and equal amounts of RNA were converted to cDNA using PrimeScript™ RT Master Mix (TaKaRa, Tokyo, Japan). cDNA was subjected to amplification by quantitative real-time reverse transcription-polymerase chain reaction (qRT-PCR) on a 7500 Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) with SYBR Premix Ex Taq (TaKaRa). The expression levels of target genes were normalized to that of GAPDH, and fold changes in expression were calculated by the comparative threshold cycle (Ct) method using the formula 2-(ΔΔct). The primers used are listed in Table S2.
Protein preparation and Western blotting
Human NP tissues and rat NP cells were lysed in RIPA buffer (Beyotime Biotech, Nantong, China) containing 1 mM phenylmethylsulfonyl fluoride. The lysate was centrifuged at 13,000 rpm for 10 min at 4 °C, and the supernatants were used for analysis. Equal amounts of protein (30 μg) were separated in sodium dodecyl sulphate-polyacrylamide gels and transferred to polyvinylidene fluoride membranes (Millipore, Billerica, MA, USA). After blocking with 5% skim milk in Tris-buffered saline supplemented with 0.1% Tween 20 for 1 h at room temperature, the membranes were incubated at 4 °C overnight with a primary antibody against lamin A/C (diluted 1:1,000), progerin (diluted 1:200), Mfn1 (diluted 1:200), Mfn2 (diluted 1:200), Drp1 (diluted 1:200), OPA1 (diluted 1:200), AMPK (diluted 1:1,000), phospho-AMPK (Thr172)α (diluted 1:1,000), collagen II (diluted 1:500), or MMP13 (diluted 1:2,000). The membranes were next incubated with the appropriate horseradish peroxidase-conjugated secondary antibodies (Affinity Biosciences, Cincinnati, OH, USA) at a 1:2,000 dilution for 40 min at room temperature. GAPDH (diluted 1:2,000) or β-actin (diluted 1:1,000) was used as the internal loading control. The bands were visualized with ECL-Plus Reagent (Millipore, Billerica, MA, USA), and the band density was quantified using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Histology and immunofluorescence assays
Human NP tissues were fixed in 4% paraformaldehyde for 48 h, dehydrated, and embedded in optimal cutting temperature compound (OCT). The spine specimens of mice were fixed in paraformaldehyde for 48 h, decalcified in 10% ethylenediaminetetraacetic acid (EDTA; pH 7.4) for 14 days, dehydrated, and embedded in OCT. Coronal-oriented sections (5 μm) of the L2-L3 or L4-L5 spine were processed for histological or histochemical staining. Hematoxylin and eosin (HE) and Safranin O (SO) staining of the mid-coronal sections was performed according to standard protocols and the resulting images examined for the presence of disc degeneration. Histological scores were evaluated using the modified Thompson grading scale as described previously [1, 28].
Immunostaining was performed using a standard protocol. Briefly, human NP tissue and mouse spinal sections were incubated in proteinase K or pepsin (0.2 mg/mL) for 10 min at room temperature for antigen retrieval, and subsequently blocked in blocking solution (QuickBlock™; Beyotime Biotech) for 1 h at room temperature. Rat NP cells were fixed in 4% paraformaldehyde for 15 min at room temperature and permeabilized in PBS supplemented with 0.1% Triton X-100. Nonspecific binding was reduced by incubation in blocking solution for 1 h at room temperature. Slides or cells were then incubated overnight at 4 °C with a primary antibody against aggrecan (diluted 1:100), P16INK4a (diluted 1:100), progerin (diluted 1:100), collagen II (diluted 1:100), LAP2 (diluted 1:50), γH2AX (diluted 1:50), H3K27me3 (diluted 1:50), or lamin B1 (diluted 1:100). Next, the cells were washed three times with PBS for 5 min each. For immunohistochemical staining, a horseradish peroxidase-streptavidin detection system (ZSGB-BIO, Beijing, China) was used. For immunofluorescence assays, the slides were incubated with fluorophore-conjugated secondary antibodies and 5 μg/mL 4′,6-diamidino-2-phenylindole (DAPI) at room temperature for 1 h in the dark. Immunostaining specificity was evaluated by omission of the primary antibody. Images were acquired using a confocal laser microscope (Nikon C2, Nikon, Tokyo, Japan) under identical imaging conditions using identical acquisition parameters, and fluorescence signals were quantified with ImageJ software.
Disc height measurement and image analysis
X-ray imaging of lumbar discs of Lmna G609G/G609G and WT male mice was performed using cabinet X-ray imaging and irradiation systems (Faxitron Bioptic, LLC, Wheeling, IL, USA). Digital images were obtained at 45 kVp under identical imaging conditions using identical acquisition parameters. The relative heights of the lumbar IVDs were measured as reported previously [7]. Percentage disc height was calculated as the average of three measurements per disc. The percentage disc height of each group was calculated as the average height of three discs (L2-L5) from three mice.
Transmission electron microscopy
Rat NP cells were harvested using trypsin and fixed in 2.5% glutaraldehyde in 0.1 M phosphate buffer for 2 h at 4 °C. The cells were post-fixed in 1% osmium tetroxide in 0.1 M phosphate buffer. Mouse IVDs were fixed in 2.5% glutaraldehyde in 0.1 M phosphate buffer (pH 7.2-7.4) overnight at 4 °C and decalcified in 10% EDTA (pH 7.4) for 7 days. Subsequently, NP cells and IVDs were dehydrated in a graded ethanol series, and infiltrated with propylene oxide to embedding medium (Epon 812). Ultrathin sections were cut using an LKB-V ultramicrotome, post-stained with uranyl acetate and lead citrate, and visualized using a transmission electron microscope (TEM, H-7650; Hitachi, Tokyo, Japan)[29].
Senescence-associated β-galactosidase activity assay
Senescence-associated β-galactosidase (SA-β-Gal) activity was assayed as described previously [30]. Briefly, rat NP cells were washed in PBS, fixed in fixative solution (2% formaldehyde and 2% glutaraldehyde) for 15 min, and stained with β-galactosidase staining solution at pH 6.0 overnight at 37 °C. Images were acquired using a Zeiss AX10 microscope.
Apoptosis assay
Apoptosis was assayed by flow cytometry with Annexin V-FITC/PI (BD Biosciences, San Diego, CA, USA) according to the manufacturer's instructions. Briefly, NP cells were harvested and resuspended in binding buffer. Annexin V-FITC and PI were added to the cells, followed by incubation for 15 min at room temperature. The cell-cycle distribution of the cells was determined by flow cytometry (FACSAria; BD Biosciences).
TUNEL assay
TUNEL assays of rat NP cells and disc tissue were performed using the One Step TUNEL Apoptosis Assay Kit (Beyotime Biotech) and the In Situ Cell Death Detection Kit (Roche, Mannheim, Germany), respectively. Briefly, cells or sections were permeabilized with freshly prepared 0.1% Triton X-100 and 0.1% sodium citrate for 15 min at room temperature, and rinsed with 50 μL of TUNEL reaction mixture for 60 min at 37 °C in the dark. Next, the cells or sections were incubated with DAPI for 10 min. Images were captured using a fluorescence microscope (Axio Imager A1; Carl Zeiss, Oberkochen, Germany).
Reactive oxygen species detection
Intracellular reactive oxygen species (ROS) were detected by staining with the oxidation-sensitive fluorescent dye, 2',7'-dichlorofluorescein diacetate (DCFDA) (Beyotime Biotech). Cells were washed twice with PBS and stained with 10 μM DCFDA in serum-free medium for 30 min in the dark. Images were acquired using a fluorescence microscope (Axio Imager A1; Carl Zeiss).
Tissue mitochondrial fractions preparation
Mitochondria were isolated using a Tissue Mitochondria Isolation Kit (Beyotime Biotech) according to the manufacturer's instructions. Briefly, freshly isolated disc tissues were mixed with the mitochondrial extraction reagent and homogenized, with the suspension centrifuged at 600 g for 5 min at 4°C; the supernatant was then centrifuged at 11,000 g for 10 min at 4°C; the pellet contained the mitochondria.
Measurement of intracellular ATP content
Intracellular ATP content was measured using the ATP Bioluminescence Assay Kit (Beyotime Biotech) according to the manufacturer's instructions.
Measurement of mitochondrial membrane potential
Mitochondrial membrane potential was assessed using the JC-1 Assay Kit (Beyotime Biotech) according to the manufacturer's instructions. Briefly, rat NP cells cultured in 24-well plates were stained with JC-1 staining solution for 20 min at 37 °C in the dark and washed twice with the buffer provided with the kit. Fluorescence intensity was measured using a fluorescence microscope (Axio Imager A1; Carl Zeiss) with single excitation (argon-ion 488 nm) and dual emission (shift from green [530 nm] to red [590 nm]) [31]. The red:green fluorescence ratio reflects changes in the mitochondrial membrane potential.
Mitochondrial complex activity assay
Mitochondria were isolated from NP cells as described previously [32]. Briefly, rat NP cells were collected and resuspended in 1.0 mL of hypotonic buffer (10 mM NaCl, 2.5 mM MgCl2, 10 mM Tris base; pH 7.5) and homogenized on ice using a glass homogenizer (Fisher Scientific, Pittsburgh, PA, USA). The homogenates were centrifuged at 1,300 g for 5 min at 4 °C. The supernatant was collected and centrifuged at 17,000 g for 15 min at 4 °C, and the pellet (containing mitochondria) was resuspended in 100 μL of isotonic buffer (210 mM mannitol, 5 mM Tris base, 70 mM sucrose, 1 mM EDTA·2Na; pH 7.5). The activities of reduced nicotinamide adenine dinucleotide (NADH)-ubiquinone oxidoreductase (complex I), succinate-CoQ oxidoreductase (complex II), ubiquinol cytochrome c reductase (complex III), and Mg2+-ATPase (complex V) were determined as described previously [33].
Statistical analysis
Data are expressed as means ± standard errors of the mean (SEM). Differences between two or among multiple groups were analyzed by Student's t-test or one-way analysis of variance followed by Tukey's multiple-comparison post hoc test, respectively. All statistical analyses were performed using Prism software (ver. 6.0; GraphPad Inc., La Jolla, CA, USA) and SPSS software (ver. 19.0; IBM Corp., Armonk, NY, USA). A P-value <0.05 was considered to reflect statistical significance.
Results
Progerin is upregulated in degenerated human NP tissue
A total of 20 degenerated discs from 20 patients were evaluated in terms of progerin expression. IDD was graded using the Pfirrmann system based on MRI data (Figure S1A). Immunofluorescence (IF) and immunohistochemical staining of NP tissue sections revealed more pronounced degeneration in the Grade IV/V group compared to the Grade II/III group, including decreased aggrecan expression and increased numbers of TUNEL- and p16INK4a-positive cells (Figure S1B). Also, the IF assay detected progerin in sections from the Grade IV/V and II/III groups (Figure 1A). The proportion of progerin‑positive cells was considerably higher in the Grade IV/V compared to the Grade II/III group (Figure 1B). qRT-PCR revealed a significantly higher level of progerin-encoding mRNA in the Grade IV/V compared to the Grade II/III group (Figure 1C); this was confirmed at the protein level by Western blotting of NP tissues from IDD patients (Figure 1D). Furthermore, a significant linear correlation was evident between the progerin level and the Pfirrmann grade (Figure S1C). Thus, progerin upregulation correlated with IDD progression.
IDD is accelerated in LMNA G609G/G609G mice
To explore the link between excessive progerin expression and IDD progression, LmnaG609G knock-in mice, which exhibit excessive progerin accumulation, were generated as described previously following same strategy of Carlos group [10]. The genotypes of WT, heterozygous (G609G/+), and homozygous (G609G/G609G) mice were confirmed by genomic analysis of Lmna exon 11. Western blotting was used to assess lamin A/C and progerin levels in tissue samples from WT, G609G/+, and G609G/G609G mice (Figures S2A, S2B). Homozygous mice exhibited a lower growth rate, higher rate of premature deaths, and a lower body weight, than WT or heterozygous animals (Figures S2A, S2C, S2D). X-ray imaging showed that the disc height of G609G/G609G mice was dramatically lower than that of WT mice at 12 weeks of age (Figure 2A), indicative of disc degeneration and aging. To further confirm a role for progerin in IDD progression, the lumbar discs of 16-week-old male G609G/G609G and WT mice were subjected to histological analysis. HE and SO staining of NP sections from G609G/G609G mice revealed condensed NP and elevated histological scores (Figure 2B); NP tissues accumulated large amounts of progerin. As expected, this decreased the collagen II and aggrecan expression levels, and increased the number of TUNEL‑positive cells (Figure 2C).
Figure 1
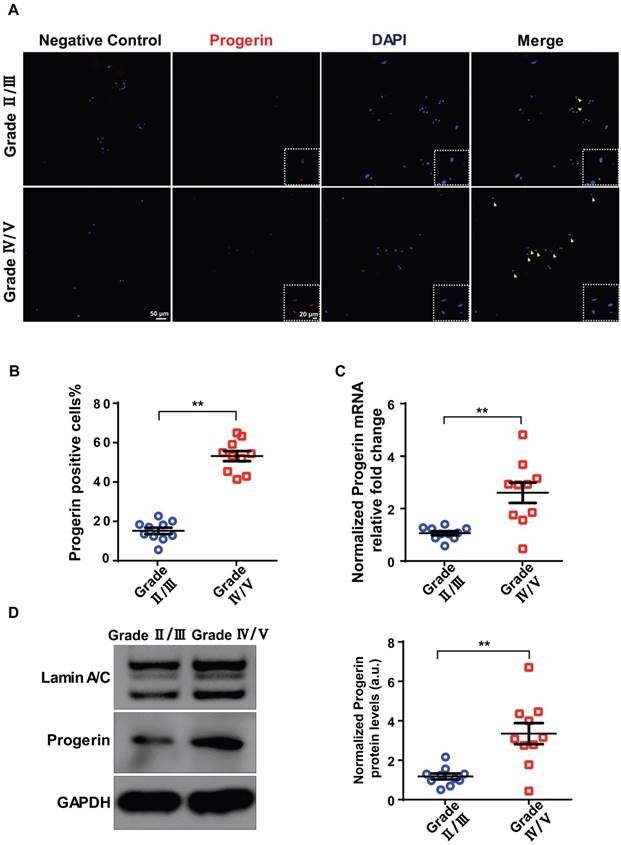
Progerin expression in human NP tissues. (A) IF staining of progerin in human NP tissues from the Grade II/III and Grade IV/V groups. Nuclei were stained with DAPI. Arrows indicate progerin-positive cells. (B) Progerin-positive cells in human NP tissues from the Grade II/III and Grade IV/V groups; We selected 10 random fields in 3 sections per individual for quantitative analysis. A total of 235-600 cells were analyzed from each individual; n = 10. **P < 0.01. (C) qRT-PCR analysis of progerin mRNA levels in NP tissues from the Grade II/III and Grade IV/V groups; n = 10; **P < 0.01. (D) Western blotting analysis of progerin in NP tissues from the Grade II/III and Grade IV/V groups; n = 10; **P < 0.01. Data represent mean ± SEM.
Figure 2
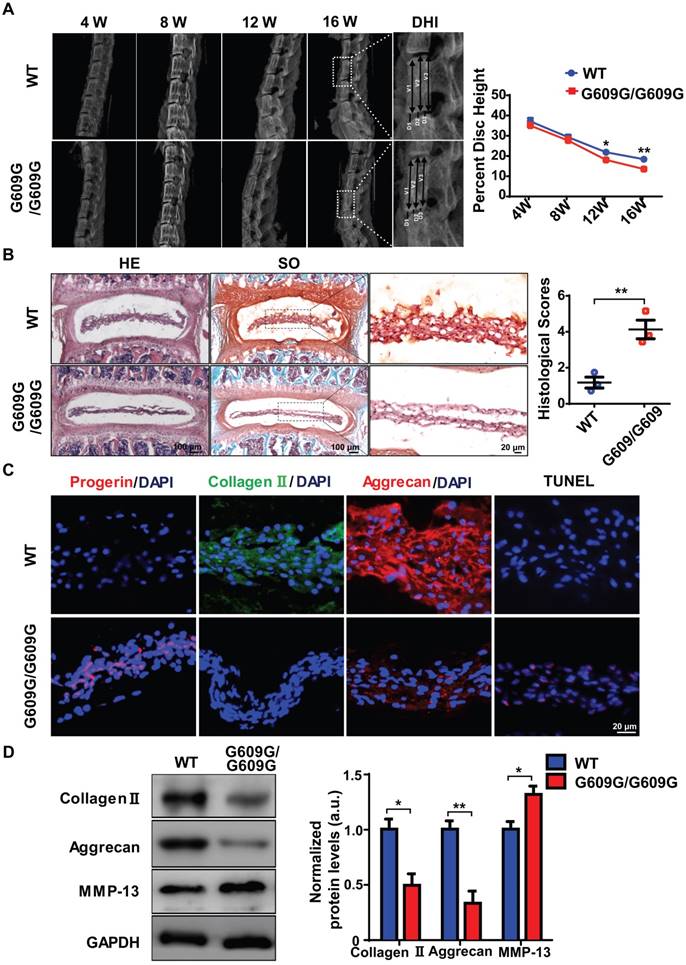
LMNA G609G/G609G mice show accelerated IDD. (A) Representative X-ray images and disc height indices (DHI) of the IVD of WT and G609G/G609G mice; n = 3; *P < 0.05, **P < 0.01. DHI% was calculated as: DHI% = (D1+D2+D3) × 100% / (V1+V2+V3+D1+D2+D3), where D indicates disc height and V indicates vertebra length. (B) HE and safranin O staining and histological scores of the IVDs of WT and G609G/G609G mice; n = 3; **P < 0.01. (C) Representative IF images of progerin, collagen II, and aggrecan expression and TUNEL staining of IVDs in WT and G609G/G609G mice. Nuclei were stained with DAPI. (D) Representative Western blots of collagen II, aggrecan, and MMP-13 in the IVDs of WT and G609G/G609G mice; n = 3; *P < 0.05, **P < 0.01. Data represent mean ± SEM. TUNEL, terminal deoxynucleotidyl transferase-mediated dUTP nick‑end labelling; MMP, matrix metalloproteinase.
Western blotting showed that the collagen II and aggrecan levels decreased, and that of matrix metalloproteinase 13 (MMP-13) increased, suggestive of accelerated extracellular matrix (ECM) degradation and IVD degeneration in G609G/G609G mice (Figure 2D). HGPS cells exhibit elevated levels of oxidative stress and mitochondrial dysfunction [14, 15, 34, 35]. To evaluate the effect of progerin on NP cells, we measured mitochondrial function via analysis of mitochondrial membrane potential and the ATP level. The mitochondrial membrane potential and ATP content were markedly decreased in LMNA G609G/G609G mice (Figures S2F, S2G). Furthermore, TEM showed that the mitochondrial morphology of LMNA G609G/G609G mice was disrupted (Figure S2E). Thus, excessive progerin expression induced NP degeneration and mitochondrial dysfunction.
Progerin causes aging-related defects and impairs mitochondrial function in NP cells
We next infected rat NP cells with mCherry-progerin-expressing or vector-only-expressing lentiviruses (Figure S3A). After infection, the nuclei of progerin-expressing cells became abnormally shaped (attributable to nuclear envelope blebbing) but those of vector-only-expressing cells remained normal (Figure S3B). An abnormal nuclear morphology is a hallmark of primary HGPS cells [15]. Moreover, progerin expression decreased the levels of the nuclear‑architecture-associated proteins lamin B1 and lamina-associated polypeptide2 (LAP2), and the extent of heterochromatin-associated trimethylation of lysine 27 on histone 3 (H3K27me3); but increased the expression of serine-139 phosphorylated H2AX (γH2AX) and the proportion of SA-β-gal positive cells (Figures 3A and S3C). Consistent with the findings in G609G/G609G mice, progerin overexpression also increased the numbers of TUNEL‑positive and apoptotic cells compared to those of the vector control (Figures 3B and 3C). In addition, the levels of mRNAs encoding Acan and Col2a1 were significantly lower, and that of the mRNA encoding MMP-13 was significantly higher, in progerin-overexpressing cells. Thus, such cells exhibited features characteristic of IVD degeneration (Figure S3D).
Consistent with the findings in G609G/G609G mice, the progerin group evidenced a higher ROS level (Figure 3D), disruption of mitochondrial membrane potential (Figure 3E), decreased ATP production (Figure 3F), and reduced activities of mitochondrial complex enzymes (Figure 3G) compared to the vector control group. Overall, the data clearly show that upregulation of progerin expression in NP cells induces severe oxidative stress and mitochondrial dysfunction, triggering aging-related defects and apoptosis in vitro.
Progerin disrupts mitochondrial morphology
We used TEM to examine the effect of progerin on mitochondrial morphology. Most NP cell mitochondria in the vector control group exhibited integrated structures, excessive progerin expression was associated with swollen and fragmented mitochondria (Figure 4A). MitoTracker staining was performed to visualize the mitochondria of NP cells. Compared to the vector control group, NP cells of the progerin-overexpressing group exhibited a significantly greater proportion of swollen mitochondria (Figure 4B), consistent with the observation that excessive progerin expression induces changes in mitochondrial structure and morphology in NP cells of LMNA G609G/G609G mice. We next measured the levels of the mitochondrial morphology-related proteins dynamin-related peptide1 (Drp1, regulating fission), optic atrophy 1 (OPA1, controlling mitochondrial inner‑membrane fusion), and mitofusin1/2 (Mfn1/2, modulating outer‑membrane fusion). Compared to the vector control group, the Drp1 expression level was increased and those of Mfn1/2 decreased, in the progerin group, indicating less mitochondrial fusion and more fission (Figure 4C). Moreover, the level of mRNA encoding Drp1 was significantly increased, and the levels of mRNAs encoding Mfn1/2 and Tfam dramatically decreased, perhaps associated with the observed mitochondrial morphological and functional defects (Figure S4). AMP-activated protein kinase (AMPK) and the peroxisome proliferator-activated receptor-γ coactivator1α (PGC1α) are key regulators of mitochondrial biogenesis and dynamics [15, 31]. Western blotting showed that the PGC1α protein level and the extent of AMPK phosphorylation were lower in the progerin group (Figure 4D). Together, the results indicate that progerin accumulation in NP cells disrupts mitochondrial morphology.
SFN ameliorates progerin-induced aging defects and mitochondrial dysfunction
SFN protects against cellular senescence [13, 14] and represses matrix degradation in animal models of osteoarthritis [36, 37]. Moreover, SFN prepared from a concentrated broccoli sprout extract reduced the fasting blood glucose level in obese patients with dysregulated type 2 diabetes, indicating that SFN is a both clinically potent and safe [21]. We explored whether SFN could rescue progerin-induced aging defects in NP cells. Progerin-expressing cells were treated with vehicle or SFN at 5 μM for 72 h. SFN significantly decreased the proportion of SA-β-Gal‑positive NP cells (Figure S5A). Also, endogenous DNA damage was attenuated in the SFN-treated progerin group, as indicated by increased expression of LAP2, H3K27me3, and lamin B1; and decreased expression of γH2AX (Figure 5A). SFN also markedly reduced the proportions of TUNEL‑positive and apoptotic cells (Figures S5B, S5C).
Figure 3
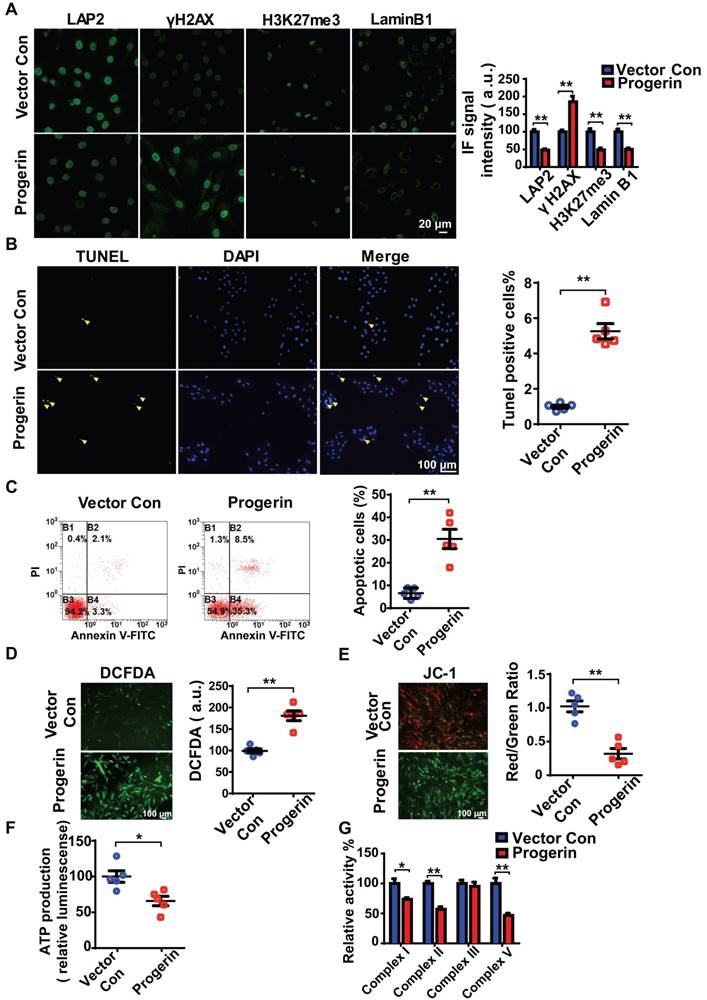
Progerin induces aging-related defects and mitochondrial dysfunction in NP cells. (A) Representative IF images and quantification of LAP2, γH2AX, H3K27me3, and lamin B1 expression in the vector con and progerin groups; n = 5; **P < 0.01. (B) TUNEL staining of NP cells in the vector con and progerin groups. Nuclei were stained with DAPI. The number of TUNEL-positive cells was significantly greater in the progerin group than in the vector con group; n = 5; **P < 0.01. (C) Representative flow cytometry dot plots of apoptosis after Annexin V-FITC/PI dual staining. The relative number of apoptotic cells was greater in the progerin group compared to the vector con group; n = 5; **P < 0.01. (D) Representative images and quantification of DCFDA-based ROS levels in the vector con and progerin groups; n = 5; **P < 0.01. (E) JC-1 staining. The red: green fluorescence ratio reflects changes in the mitochondrial membrane potential of NP cells in the vector con and progerin groups; n = 5; **P < 0.01. (F) ATP production in the vector con and progerin groups; n = 5; *P < 0.05. (G) Relative activities of mitochondrial complex enzymes in NP cells in the vector con and progerin groups; n = 4; *P < 0.05, **P < 0.01. Data represent mean ± SEM. LAP2, lamina-associated polypeptide2; γH2AX, serine-139 phosphorylated H2AX; H3K27me3, heterochromatin-associated tri-methylated lysine 27 on histone 3; SA-β-gal, senescence-associated β-galactosidase; DCFDA, 2',7'-dichloro-dihydrofluorescein-diacetate; JC-1, 5,5‑,6,6‑tetrachloro-1,1‑,3,3‑tetraethylbenzimidazole-carbocyanine iodide.
Figure 4
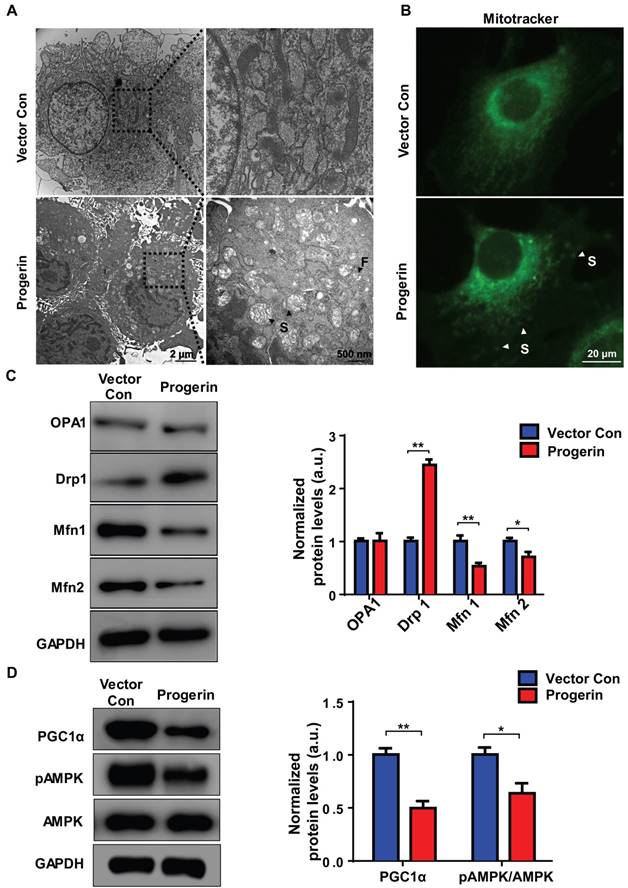
Progerin disrupts mitochondrial morphology and mitochondrial dynamics. (A) Representative TEM images of mitochondria in NP cells from the vector con and progerin groups; F, Fragmented mitochondria; S, swollen mitochondria. (B) Representative fluorescence images of mitochondria in NP cells; S, swollen mitochondria. (C) Representative Western blots of OPA1, Drp1, Mfn1, and Mfn2 in the vector con and progerin groups (n = 5). GAPDH was used as the control. *P < 0.05, **P < 0.01. (D) Representative Western blots of PGC1α, pAMPK, and AMPK in the vector con and progerin groups; n = 5; *P < 0.05, **P < 0.01. Data represent mean ± SEM. OPA1, optic atrophy1; Drp1, dynamin-related peptide1; Mfn1, mitofusin 1; Mfn2, mitofusin2; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; PGC1α, peroxisome proliferator-activated receptor-γ coactivator1α; AMPK, AMP-activated protein kinase.
As expected, SFN exerted a potent antioxidant effect, and protected mitochondrial function in vitro. The ROS level was significantly decreased, and the mitochondrial membrane potential and ATP production were increased, in SFN- compared to vehicle-treated cells (Figures 5B-5D). Interestingly, in NP cells, SFN partially reversed the effect of progerin on the levels of proteins controlling mitochondrial dynamics; specifically, the Drp1 level decreased and those of Mfn1/2 increased, suggesting a reversal of the shift between mitochondrial fusion and fission (Figure 5E). Furthermore, PGC1α expression and AMPK phosphorylation were markedly upregulated in the SFN-treated group (Figure 5F). Also, progerin-induced mitochondrial deformities and swelling were significantly attenuated in the SFN-treated group (Figures S5D, S5E).
Figure 5
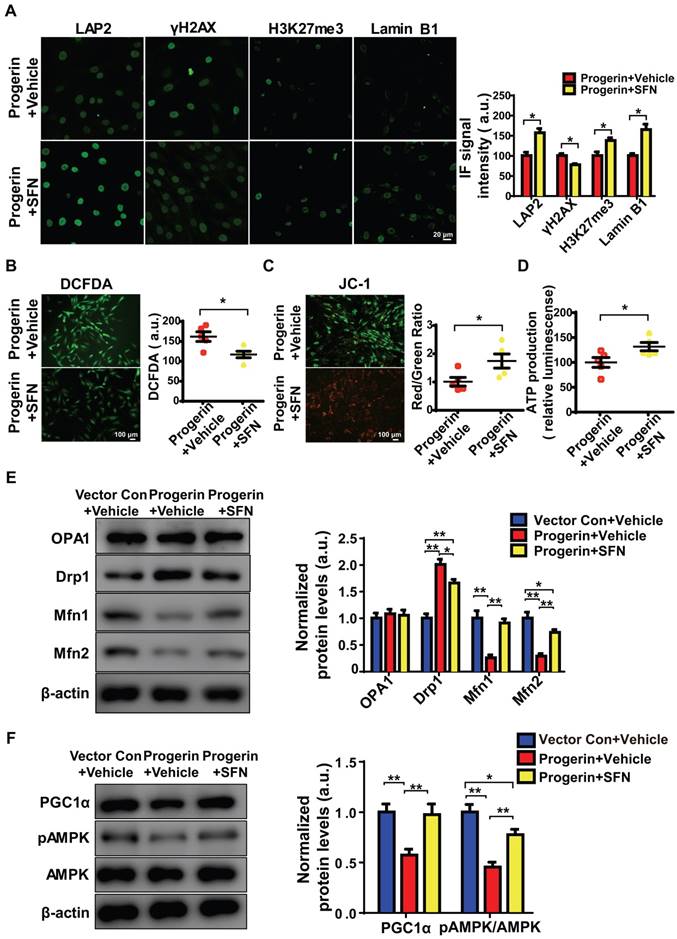
SFN ameliorates progerin-induced aging defects and mitochondrial dysfunction. (A) IF of LAP2, γH2AX, H3K27me3, and lamin B1 in the progerin+vehicle and progerin+SFN groups; n = 5; *P <0.05. (B) Representative images and quantification of ROS levels in the progerin+vehicle and progerin+SFN groups; n = 5; *P < 0.05. (C) JC-1 staining. The red: green fluorescence ratio reflects changes in the mitochondrial membrane potential of NP cells in the progerin+vehicle and progerin+SFN groups; n = 5; *P < 0.05. (D) ATP production in the progerin+vehicle and progerin+SFN groups; n = 5; *P < 0.05. (E) Western blotting analysis of OPA1, Drp1, Mfn1, and Mfn2 in NP cells from the vector+vehicle, progerin+vehicle, and progerin+SFN groups; n = 5; *P < 0.05, **P < 0.01. (F) Western blotting analysis of PGC1α, pAMPK, and AMPK in NP cells from the vector+vehicle, progerin+vehicle, and progerin+SFN groups; n = 5. β-actin was used as the control. *P < 0.05, **P < 0.01. Data represent mean ± SEM. SA-β-gal, senescence-associated β-galactosidase; DCFDA, 2',7'-dichloro-dihydrofluorescein-diacetate; JC-1, 5,5‑,6,6‑tetrachloro-1,1‑,3,3‑tetraethylbenzimidazole-carbocyanine iodide; OPA1, optic atrophy-1; Drp1, dynamin-related peptide1; Mfn1, mitofusin1; Mfn2, mitofusin2; PGC1α, peroxisome proliferator-activated receptor-γ coactivator1α; AMPK, AMP-activated protein kinase.
SFN attenuates progerin-induced IDD in LMNA G609G/G609G mice
We next explored whether SFN protected against IDD progression. Four-week old male G609G/G609G mice were treated with SFN (10 mg/kg, intraperitoneally [ip], three times per week), or vehicle for 12 weeks. At the end of treatment, the percentage disc height of SFN-treated G609G/G609G mice was significantly greater than that of vehicle-treated mice (Figure 6A). In line with the X-ray findings, safranin-O staining intensity increased and the histological scores decreased in NP sections from SFN-treated compared to vehicle-treated G609G/G609G mice (Figure 6B).
Figure 6
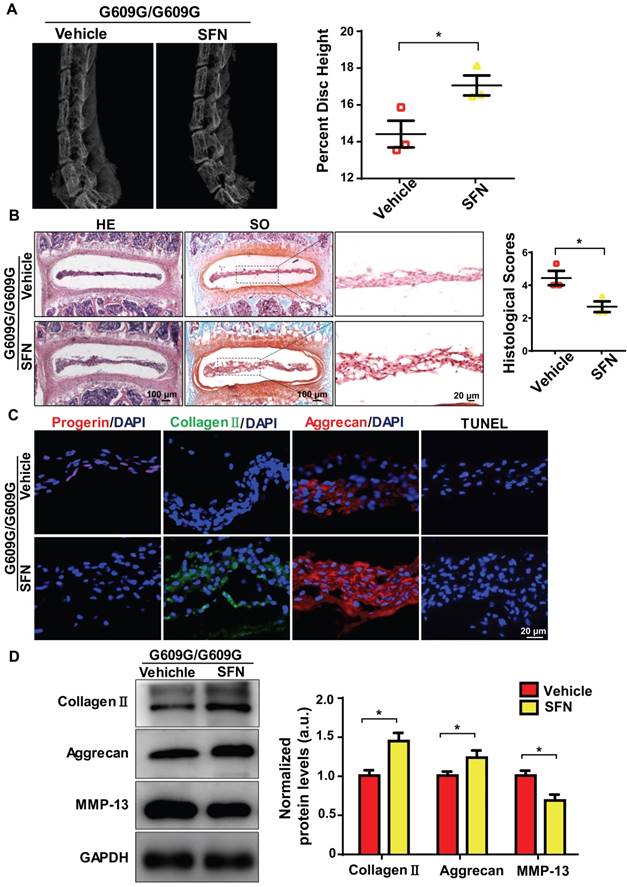
SFN attenuates progerin-induced IDD in LMNA G609G/G609G mice. (A) Representative X-ray images and disc height indices (DHI) of the IVDs of vehicle-treated and SFN-treated G609G/G609G mice; n = 3; *P < 0.05. (B) HE and safranin O staining and histological scores of the IVDs of vehicle-treated and SFN-treated G609G/G609G mice; n = 3; *P < 0.05. (C) Representative IF images of progerin, collagen II, and aggrecan expression and TUNEL staining of IVDs from vehicle-treated and SFN-treated G609G/G609G mice. Nuclei were stained with DAPI. (D) Western blotting analysis of collagen II, aggrecan, and MMP-13 in IVDs in vehicle-treated and SFN-treated G609G/G609G mice; n = 3; *P < 0.05. Data represent mean ± SEM. TUNEL, terminal deoxynucleotidyl transferase-mediated dUTP nick‑end labelling; MMP, matrix metalloproteinase.
Moreover, IF analysis demonstrated that progerin expression and the proportion of TUNEL‑positive cells decreased, and aggrecan and collagen II expression levels increased, in the SFN- versus vehicle-treated group (Figure 6C). Likewise, Western blotting revealed increased aggrecan and collagen II expression and decreased MMP-13 expression in the former group, indicating that ECM degradation and degeneration were attenuated in the SFN-treated group (Figure 6D). Together, the data suggest that SFN attenuates progerin-induced IDD in Lmna G609G/G609G mice.
Discussion
Progerin is expressed not only in HGPS but also during normal aging, which increases the importance of its accumulation in human cells and tissues [10, 38, 39]. Here, we provide the first experimental evidence that progerin is upregulated in degenerated human NP tissues. Lmna G609G knock-in mice (which exhibit progerin accumulation) displayed accelerated IVD degeneration, further indicating the essential role played by progerin in disc degeneration. Furthermore, progerin induced mitochondrial dysfunction and NP degeneration by disrupting mitochondrial dynamics. In both rat NP cells and G609G knock-in mice, SFN delayed the progression of progerin-induced IDD by ameliorating defects associated with aging and mitochondrial dysfunction. Collectively, the data suggest that progerin is involved in IDD pathogenesis and that SFN is a promising treatment for IDD.
Progerin expression is induced by mutation at position 1,824 (C3T) of the LMNA gene; this activates a cryptic splice site in exon 11 [9, 40]. The splice site is functional, and triggers progerin accumulation over time in cells from both young and old healthy individuals [41]. Cao et al. transfected a splicing reporter into multiple cell lines to evaluate activation of the cryptic splice site in LMNA, and found that such activation increased with increasing passage number, indicating that both spontaneous sporadic use of the cryptic splice site and progerin expression are relevant in terms of normal aging [42]. Several types of stress, including oxidative stress [43], telomere shortening, impaired DNA repair, and epigenetic defects [44], promote activation of the cryptic splice site and expression of both prelamin A [45] and progerin [42]. Most of these stress factors also induce disc degeneration [46-48]. A pathological relationship between progerin expression and chondrocyte and cartilage defects has been reported [18], but the cited study did not evaluate IVDs. The NP is the largest bodily avascular tissue and exhibits a very low turnover rate [49]. Thus, progerin may accumulate in the NP to induce cellular defects. Progerin accumulation has been reported to cause severe defects associated with aging, including laminar disturbance, disorganized heterochromatin, DNA damage accumulation, and telomere damage; eventually, this results in cellular senescence, apoptosis, and impairment of organ function [50, 51]. These data are consistent with our findings; excessive progerin expression triggered apoptosis of degenerated NP cells. Moreover, progerin overexpression in rat NP cells induced nuclear deformation and increased DNA damage, triggering cell senescence and apoptosis in line with the fact that DNA damage drives disc degeneration in humans. Indeed, DNA repair-deficient Ercc1-/- mice exhibit features indicative of disc degeneration, including loss of matrix proteoglycan, reduced disc height, and increased cellular senescence [7, 52].
Research on IVD diseases and low-back pain is aided by the use of small‑animal models that recapitulate the features of human disease. However, induction of disc degeneration in current small-animal models requires surgical or dietary manipulation. Lmna G609G knock-in mice closely mimic the genetics and pathophysiology of human HGPS [10], but the disc effects in terms of phenotype degeneration remain unclear. We found that Lmna G609G/G609G mice exhibited IDD acceleration, indicating that progerin accumulation is linked to IDD progression. Our findings suggest that the Lmna G609G/G609G mouse is a valuable model for investigation of the pathogenetic mechanisms of disc degeneration; the mouse recapitulates the loss of IVD height, ECM degradation, and enhanced apoptosis characteristic of human IDD.
Mitochondrial dysfunction is associated with both aging and disease development [53-55]. An elevated ROS level, impaired mitochondrial function, and decreased levels of mitochondrial proteins have been reported in fibroblasts from patients with HGPS [15, 56]. Our findings indicate that accumulation of progerin in rat NP cells disrupts mitochondrial structure and function, as evidenced by an increased ROS level, disruption of the mitochondrial membrane potential, reduced ATP production, and changes in the activities of the mitochondrial enzymic complex. These results support the notion that progerin-induced mitochondrial dysfunction is linked to susceptibility to degenerative IVD disease and low- back pain. Furthermore, this is the first report to show that progerin accumulation degrades mitochondrial function in NP cells. Mitochondria play essential roles in the pathogenesis of IDD and are therefore promising therapeutic targets [57, 58].
Mitochondria are highly dynamic organelles, with functionality dependent on the balance between fusion and division [59]. Changes in mitochondrial dynamics during IDD progression remain unclear. The PGC1α level and extent of AMPK phosphorylation decreased in progerin-expressing NP cells, perhaps reflecting changes in mitochondrial dynamics and function. A previous study found that lamin A directly interacted with SIRT1, activating the latter protein; progerin compromised the appropriate nuclear matrix localization of that protein and reduced its deacetylase activity [60], in turn reducing LKB1deacetylation and downregulating AMPK phosphorylation and activity [61, 62]. Moreover, lamin A serves as an endogenous activator of SIRT6, and this activity is impaired by progerin [63]. SIRT6 deficiency reduced AMPK phosphorylation and activation [64]. AMPK directly phosphorylates (activates) PGC1α [65], a transcriptional co-activator exhibiting self-enhancement of expression, to promote the transcription of nuclear-encoded mitochondrial genes. We thus hypothesize that the effect of progerin accumulation on AMPK and PGC1α signaling may compromise the function of SIRT1 or SIRT6. AMPK modulates mitochondrial dynamics by decreasing the levels of the mitochondrial fission proteins Drp1 and Fis1, and simulating those of the fusion proteins OPA1 and Mfn1/2 [66]. Also, AMPK activation prevented mitochondrial fission by decreasing the Drp1 and Fis1 levels in apoptotic endothelial cells in diabetes [67]. In PGC1α-over- and under-expressing cells, PGC1α regulated Mfn1, Mfn2, and Drp1 protein production, and phosphorylation, in turn modulating mitochondrial fusion/fission [68-70]. Therefore, the AMPK/PGC1α signaling pathway is a key regulator of mitochondrial dynamics, controlling both mitochondrial fusion and fission. Consistent with previous reports, we found that progerin accumulation in rat NP cells decreased the production of the fusion proteins Mfn1/2, and increased that of the fission protein Drp1, thus disturbing mitochondrial dynamics. Western blotting showed that the PGC1α protein level and extent of AMPK phosphorylation were lower in the progerin group. Thus, SFN modulates both mitochondrial dynamics and biogenesis, perhaps via the AMPK/PGC1α signaling pathway.
We explored whether SFN delayed IDD and/or reduced IVD degeneration in Lmna G609G/G609G mice. SFN protected against IVD degeneration, as indicated by an increased disc height and amelioration of matrix degradation and apoptosis. SNF enhanced protein degradation and induced progerin clearance in cultured HGPS fibroblasts [13]; these effects may in part explain why SFN ameliorates IVD degeneration. In addition, SFN protects mitochondria during adipogenesis, and in diabetes, by activating AMPK and PGC1α [71, 72]. We found that SFN ameliorated aging defects and mitochondrial dysfunction in rat NP cells. SFN exhibits a remarkable ability to modulate both the progerin level and mitochondrial homeostasis. Thus, fine-tuning of pathophysiological progerin accumulation/mitochondrial homeostasis may be of therapeutic utility. Our results suggest SFN to have potential for the treatment of IDD.
In conclusion, our findings provide insights into the mechanism of IVD degeneration, suggesting that clearance of progerin and improvement of mitochondrial function would delay IDD progression. The findings of this study increase our understanding of the therapeutic potential of SFN for IDD.
Abbreviations
AMPK: AMP-activated protein kinase; DAPI: 4′,6-diamidino-2-phenylindole; DCFDA: 2',7'-dichlorofluorescein diacetate; DMEM: Dulbecco's modified Eagle's medium; ECM: extracellular matrix; EDTA: ethylenediaminetetraacetic acid; FBS: foetal bovine serum; FOXO3: forkhead box O3; H3K27me3: heterochromatin-associated tri-methylated lysine 27 on histone 3; HE: Hematoxylin and eosin; HGPS: Hutchinson-Gilford Progeria Syndrome; IDD: intervertebral disc degeneration; IF: Immunofluorescence; ip: intraperitoneally; IVD: intervertebral disc; LAP2: lamina-associated polypeptide2; Mfn1/2: mitofusin1/2; MMP: matrix metalloproteinase; MRI: magnetic resonance imaging; NP: nucleus pulposus; Nrf2: NF-E2-related factor2; OCT: optimal cutting temperature compound; OPA1: optic atrophy1; PBS: phosphate-buffered saline; PGC1α: peroxisome proliferator-activated receptor-γ coactivator1α; ROS: reactive oxygen species; SA-β-Gal: Senescence-associated β-galactosidase; SFN: sulforaphane; SO: Safranin O; WT: wild‑type; γH2AX: serine-139 phosphorylated H2AX.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (81730065, 81772377, 81572192).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Choi H, Tessier S, Silagi ES, Kyada R, Yousefi F, Pleshko N. et al. A novel mouse model of intervertebral disc degeneration shows altered cell fate and matrix homeostasis. Matrix Biol. 2018;70:102-22
2. Cassidy JD. Saskatchewan health and back pain survey. Spine (Phila Pa 1976). 1998;23:1923
3. Vos T, Allen C, Arora M, Barber RM, Bhutta ZA, Brown A. et al. Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet. 2016;388:1545-602
4. Mayer JE, Iatridis JC, Chan D, Qureshi SA, Gottesman O, Hecht AC. Genetic polymorphisms associated with intervertebral disc degeneration. Spine J. 2013;13:299-317
5. Miller JD, Ganat YM, Kishinevsky S, Bowman RL, Liu B, Tu EY. et al. Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell. 2013;13:691-705
6. Bian Q, Ma L, Jain A, Crane JL, Kebaish K, Wan M. et al. Mechanosignaling activation of TGFbeta maintains intervertebral disc homeostasis. Bone Res. 2017;5:17008
7. Vo N, Seo HY, Robinson A, Sowa G, Bentley D, Taylor L. et al. Accelerated aging of intervertebral discs in a mouse model of progeria. J Orthop Res. 2010;28:1600-7
8. Harhouri K, Navarro C, Depetris D, Mattei MG, Nissan X, Cau P. et al. MG132-induced progerin clearance is mediated by autophagy activation and splicing regulation. EMBO Mol Med. 2017;9:1294-313
9. Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L. et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature. 2003;423:293-8
10. Osorio FG, Navarro CL, Cadinanos J, Lopez-Mejia IC, Quiros PM, Bartoli C. et al. Splicing-directed therapy in a new mouse model of human accelerated aging. Sci Transl Med. 2011;3:106ra7
11. Egesipe AL, Blondel S, Cicero AL, Jaskowiak AL, Navarro C, Sandre-Giovannoli A. et al. Metformin decreases progerin expression and alleviates pathological defects of Hutchinson-Gilford progeria syndrome cells. NPJ Aging Mech Dis. 2016;2:16026
12. Cao K, Capell BC, Erdos MR, Djabali K, Collins FS. A lamin A protein isoform overexpressed in Hutchinson-Gilford progeria syndrome interferes with mitosis in progeria and normal cells. Proc Natl Acad Sci U S A. 2007;104:4949-54
13. Gabriel D, Roedl D, Gordon LB, Djabali K. Sulforaphane enhances progerin clearance in Hutchinson-Gilford progeria fibroblasts. Aging Cell. 2015;14:78-91
14. Kubben N, Zhang W, Wang L, Voss TC, Yang J, Qu J. et al. Repression of the Antioxidant NRF2 Pathway in Premature Aging. Cell. 2016;165:1361-74
15. Xiong ZM, Choi JY, Wang K, Zhang H, Tariq Z, Wu D. et al. Methylene blue alleviates nuclear and mitochondrial abnormalities in progeria. Aging Cell. 2016;15:279-90
16. Pacheco LM, Gomez LA, Dias J, Ziebarth NM, Howard GA, Schiller PC. Progerin expression disrupts critical adult stem cell functions involved in tissue repair. Aging (Albany NY). 2014;6:1049-63
17. Attur M, Ben-Artzi A, Yang Q, Al-Mussawir HE, Worman HJ, Palmer G. et al. Perturbation of nuclear lamin A causes cell death in chondrocytes. Arthritis Rheum. 2012;64:1940-9
18. Mateos J, De la Fuente A, Lesende-Rodriguez I, Fernandez-Pernas P, Arufe MC, Blanco FJ. Lamin A deregulation in human mesenchymal stem cells promotes an impairment in their chondrogenic potential and imbalance in their response to oxidative stress. Stem Cell Res. 2013;11:1137-48
19. Messner M, Ghadge SK, Goetsch V, Wimmer A, Dorler J, Polzl G. et al. Upregulation of the aging related LMNA splice variant progerin in dilated cardiomyopathy. PLoS One. 2018;13:e0196739
20. McClintock D, Ratner D, Lokuge M, Owens DM, Gordon LB, Collins FS. et al. The mutant form of lamin A that causes Hutchinson-Gilford progeria is a biomarker of cellular aging in human skin. PLoS One. 2007;2:e1269
21. Axelsson AS, Tubbs E, Mecham B, Chacko S, Nenonen HA, Tang Y. et al. Sulforaphane reduces hepatic glucose production and improves glucose control in patients with type 2 diabetes. Sci Transl Med. 2017;9:eaah4477
22. Lee S, Kim J, Seo SG, Choi BR, Han JS, Lee KW. et al. Sulforaphane alleviates scopolamine-induced memory impairment in mice. Pharmacol Res. 2014;85:23-32
23. Singh K, Connors SL, Macklin EA, Smith KD, Fahey JW, Talalay P. et al. Sulforaphane treatment of autism spectrum disorder (ASD). Proc Natl Acad Sci U S A. 2014;111:15550-5
24. Liu Y, Hettinger CL, Zhang D, Rezvani K, Wang X, Wang H. Sulforaphane enhances proteasomal and autophagic activities in mice and is a potential therapeutic reagent for Huntington's disease. J Neurochem. 2014;129:539-47
25. Carrasco-Pozo C, Tan KN, Gotteland M, Borges K. Sulforaphane Protects against High Cholesterol-Induced Mitochondrial Bioenergetics Impairments, Inflammation, and Oxidative Stress and Preserves Pancreatic beta-Cells Function. Oxid Med Cell Longev. 2017;2017:3839756
26. Pfirrmann CW, Metzdorf A, Zanetti M, Hodler J, Boos N. Magnetic resonance classification of lumbar intervertebral disc degeneration. Spine (Phila Pa 1976). 2001;26:1873-8
27. Oh CD, Im HJ, Suh J, Chee A, An H, Chen D. Rho-Associated Kinase Inhibitor Immortalizes Rat Nucleus Pulposus and Annulus Fibrosus Cells: Establishment of Intervertebral Disc Cell Lines With Novel Approaches. Spine (Phila Pa 1976). 2016;41:e255-61
28. Norcross JP, Lester GE, Weinhold P, Dahners LE. An in vivo model of degenerative disc disease. J Orthop Res. 2003;21:183-8
29. He X, Bi XY, Lu XZ, Zhao M, Yu XJ, Sun L. et al. Reduction of Mitochondria-Endoplasmic Reticulum Interactions by Acetylcholine Protects Human Umbilical Vein Endothelial Cells From Hypoxia/Reoxygenation Injury. Arterioscler Thromb Vasc Biol. 2015;35:1623-34
30. Zhao Y, Jia Z, Huang S, Wu Y, Liu L, Lin L. et al. Age-Related Changes in Nucleus Pulposus Mesenchymal Stem Cells: An In Vitro Study in Rats. Stem Cells Int. 2017;2017:6761572
31. Sun L, Zhao M, Yu XJ, Wang H, He X, Liu JK. et al. Cardioprotection by acetylcholine: a novel mechanism via mitochondrial biogenesis and function involving the PGC-1alpha pathway. J Cell Physiol. 2013;228:1238-48
32. Gao J, Feng Z, Wang X, Zeng M, Liu J, Han S. et al. SIRT3/SOD2 maintains osteoblast differentiation and bone formation by regulating mitochondrial stress. Cell Death Differ. 2018;25:229-40
33. Feng Z, Zou X, Jia H, Li X, Zhu Z, Liu X. et al. Maternal docosahexaenoic acid feeding protects against impairment of learning and memory and oxidative stress in prenatally stressed rats: possible role of neuronal mitochondria metabolism. Antioxid Redox Signal. 2012;16:275-89
34. Viteri G, Chung YW, Stadtman ER. Effect of progerin on the accumulation of oxidized proteins in fibroblasts from Hutchinson Gilford progeria patients. Mech Ageing Dev. 2010;131:2-8
35. Rivera-Torres J, Acin-Perez R, Cabezas-Sanchez P, Osorio FG, Gonzalez-Gomez C, Megias D. et al. Identification of mitochondrial dysfunction in Hutchinson-Gilford progeria syndrome through use of stable isotope labeling with amino acids in cell culture. J Proteomics. 2013;91:466-77
36. Davidson RK, Jupp O, de Ferrars R, Kay CD, Culley KL, Norton R. et al. Sulforaphane represses matrix-degrading proteases and protects cartilage from destruction in vitro and in vivo. Arthritis Rheum. 2013;65:3130-40
37. Javaheri B, Poulet B, Aljazzar A, de Souza R, Piles M, Hopkinson M. et al. Stable sulforaphane protects against gait anomalies and modifies bone microarchitecture in the spontaneous STR/Ort model of osteoarthritis. Bone. 2017;103:308-17
38. Burtner CR, Kennedy BK. Progeria syndromes and ageing: what is the connection? Nat Rev Mol Cell Biol. 2010;11:567-78
39. Scaffidi P, Misteli T. Lamin A-dependent nuclear defects in human aging. Science. 2006;312:1059-63
40. De Sandre-Giovannoli A, Bernard R, Cau P, Navarro C, Amiel J, Boccaccio I. et al. Lamin a truncation in Hutchinson-Gilford progeria. Science. 2003;300:2055
41. Rodriguez S, Coppedè F, Sagelius H, Eriksson M. Increased expression of the Hutchinson-Gilford progeria syndrome truncated lamin A transcript during cell aging. European Journal of Human Genetics. 2009;17:928-37
42. Cao K, Blair CD, Faddah DA, Kieckhaefer JE, Olive M, Erdos MR. et al. Progerin and telomere dysfunction collaborate to trigger cellular senescence in normal human fibroblasts. Journal of Clinical Investigation. 2011;121:2833-44
43. Hamczyk MR, Del CL, Andres V. Aging in the Cardiovascular System: Lessons from Hutchinson-Gilford Progeria Syndrome. Annu Rev Physiol. 2018;80:27-48
44. Kubben N, Misteli T. Shared molecular and cellular mechanisms of premature ageing and ageing-associated diseases. Nat Rev Mol Cell Biol. 2017;18:595-609
45. Ragnauth CD, Warren DT, Liu Y, McNair R, Tajsic T, Figg N. et al. Prelamin A acts to accelerate smooth muscle cell senescence and is a novel biomarker of human vascular aging. Circulation. 2010;121:2200-10
46. Feng C, Yang M, Lan M, Liu C, Zhang Y, Huang B. et al. ROS: Crucial Intermediators in the Pathogenesis of Intervertebral Disc Degeneration. Oxidative Medicine and Cellular Longevity. 2017;2017:1-12
47. Jiang W, Zhang X, Hao J, Shen J, Fang J, Dong W. et al. SIRT1 protects against apoptosis by promoting autophagy in degenerative human disc nucleus pulposus cells. Scientific Reports. 2015:4
48. Kepler CK, Ponnappan RK, Tannoury CA, Risbud MV, Anderson DG. The molecular basis of intervertebral disc degeneration. Spine J. 2013;13:318-30
49. Choi H, Johnson ZI, Risbud MV. Understanding nucleus pulposus cell phenotype: a prerequisite for stem cell based therapies to treat intervertebral disc degeneration. Curr Stem Cell Res Ther. 2015;10:307-16
50. Vidak S, Foisner R. Molecular insights into the premature aging disease progeria. Histochem Cell Biol. 2016;145:401-17
51. Hilton BA, Liu J, Cartwright BM, Liu Y, Breitman M, Wang Y. et al. Progerin sequestration of PCNA promotes replication fork collapse and mislocalization of XPA in laminopathy-related progeroid syndromes. FASEB J. 2017;31:3882-93
52. Vo N, Niedernhofer LJ, Nasto LA, Jacobs L, Robbins PD, Kang J. et al. An overview of underlying causes and animal models for the study of age-related degenerative disorders of the spine and synovial joints. J Orthop Res. 2013;31:831-7
53. Zhang B, Xu L, Zhuo N, Shen J. Resveratrol protects against mitochondrial dysfunction through autophagy activation in human nucleus pulposus cells. Biochem Biophys Res Commun. 2017;493:373-81
54. Seo DY, Lee SR, Kim N, Ko KS, Rhee BD, Han J. Age-related changes in skeletal muscle mitochondria: the role of exercise. Integr Med Res. 2016;5:182-6
55. Cedikova M, Pitule P, Kripnerova M, Markova M, Kuncova J. Multiple roles of mitochondria in aging processes. Physiol Res. 2016;65:S519-31
56. Lattanzi G, Marmiroli S, Facchini A, Maraldi NM. Nuclear damages and oxidative stress: new perspectives for laminopathies. Eur J Histochem. 2012;56:e45
57. Zhang B, Xu L, Zhuo N, Shen J. Resveratrol protects against mitochondrial dysfunction through autophagy activation in human nucleus pulposus cells. Biochemical and Biophysical Research Communications. 2017;493:373-81
58. Feng C, Liu H, Yang M, Zhang Y, Huang B, Zhou Y. Disc cell senescence in intervertebral disc degeneration: Causes and molecular pathways. Cell Cycle. 2016;15:1674-84
59. Xue RQ, Sun L, Yu XJ, Li DL, Zang WJ. Vagal nerve stimulation improves mitochondrial dynamics via an M3 receptor/CaMKKbeta/AMPK pathway in isoproterenol-induced myocardial ischaemia. J Cell Mol Med. 2017;21:58-71
60. Liu X, Liu B, Ghosh S, Yang X, Zheng H, Zheng B. et al. Resveratrol Rescues SIRT1-Dependent Adult Stem Cell Decline and Alleviates Progeroid Features in Laminopathy-Based Progeria. Cell Metabolism. 2012;16:738-50
61. Lan F, Cacicedo JM, Ruderman N, Ido Y. SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP-activated protein kinase activation. J Biol Chem. 2008;283:27628-35
62. Zheng Z, Chen H, Li J, Li T, Zheng B, Zheng Y. et al. Sirtuin 1-Mediated Cellular Metabolic Memory of High Glucose Via the LKB1/AMPK/ROS Pathway and Therapeutic Effects of Metformin. Diabetes. 2011;61:217-28
63. Ghosh S, Liu B, Wang Y, Hao Q, Zhou Z. Lamin A Is an Endogenous SIRT6 Activator and Promotes SIRT6-Mediated DNA Repair. Cell Reports. 2015;13:1396-406
64. Cui X, Yao L, Yang X, Gao Y, Fang F, Zhang J. et al. SIRT6 regulates metabolic homeostasis in skeletal muscle through activation of AMPK. American Journal of Physiology-Endocrinology and Metabolism. 2017;313:e493-e505
65. Ruderman NB, Xu XJ, Nelson L, Cacicedo JM, Saha AK, Lan F. et al. AMPK and SIRT1: a long-standing partnership? Am J Physiol Endocrinol Metab. 2010;298:e751-60
66. Xue R-Q, Sun L, Yu X-J, Li D-L, Zang W-J. Vagal nerve stimulation improves mitochondrial dynamicsvia an M3 receptor/CaMKKβ/AMPK pathway in isoproterenol-induced myocardial ischaemia. Journal of Cellular and Molecular Medicine. 2017;21:58-71
67. Bhatt MP, Lim YC, Kim YM, Ha KS. C-peptide activates AMPKalpha and prevents ROS-mediated mitochondrial fission and endothelial apoptosis in diabetes. Diabetes. 2013;62:3851-62
68. Cannavino J, Brocca L, Sandri M, Grassi B, Bottinelli R, Pellegrino MA. The role of alterations in mitochondrial dynamics and PGC-1α over-expression in fast muscle atrophy following hindlimb unloading. The Journal of Physiology. 2015;593:1981-95
69. Soriano FX, Liesa M, Bach D, Chan DC, Palacin M, Zorzano A. Evidence for a mitochondrial regulatory pathway defined by peroxisome proliferator-activated receptor-gamma coactivator-1 alpha, estrogen-related receptor-alpha, and mitofusin 2. Diabetes. 2006;55:1783-91
70. Peng K, Yang L, Wang J, Ye F, Dan G, Zhao Y. et al. The Interaction of Mitochondrial Biogenesis and Fission/Fusion Mediated by PGC-1α Regulates Rotenone-Induced Dopaminergic Neurotoxicity. Molecular Neurobiology. 2017;54:3783-97
71. Choi KM, Lee YS, Kim W, Kim SJ, Shin KO, Yu JY. et al. Sulforaphane attenuates obesity by inhibiting adipogenesis and activating the AMPK pathway in obese mice. J Nutr Biochem. 2014;25:201-7
72. Fernandes RO, Bonetto JH, Baregzay B, de Castro AL, Puukila S, Forsyth H. et al. Modulation of apoptosis by sulforaphane is associated with PGC-1alpha stimulation and decreased oxidative stress in cardiac myoblasts. Mol Cell Biochem. 2015;401:61-70
Author contact
![]() Corresponding authors: Liu Yang, Ph.D., Institute of Orthopedic Surgery, Xijing Hospital, Fourth Military Medical University, Xi'an, People's Republic of China. E-mail: yangliuedu.cn. Tel: 86-29-84775291. Zhuojing Luo, Ph.D., Institute of Orthopedic Surgery, Fourth Military Medical University, Xi'an, People's Republic of China. E-mail: zjluoedu.cn. Tel:86-13809188665.
Corresponding authors: Liu Yang, Ph.D., Institute of Orthopedic Surgery, Xijing Hospital, Fourth Military Medical University, Xi'an, People's Republic of China. E-mail: yangliuedu.cn. Tel: 86-29-84775291. Zhuojing Luo, Ph.D., Institute of Orthopedic Surgery, Fourth Military Medical University, Xi'an, People's Republic of China. E-mail: zjluoedu.cn. Tel:86-13809188665.
Citation styles
APA
Xu, X., Wang, D., Zheng, C., Gao, B., Fan, J., Cheng, P., Liu, B., Yang, L., Luo, Z. (2019). Progerin accumulation in nucleus pulposus cells impairs mitochondrial function and induces intervertebral disc degeneration and therapeutic effects of sulforaphane. Theranostics, 9(8), 2252-2267. https://doi.org/10.7150/thno.30658.
ACS
Xu, X.; Wang, D.; Zheng, C.; Gao, B.; Fan, J.; Cheng, P.; Liu, B.; Yang, L.; Luo, Z. Progerin accumulation in nucleus pulposus cells impairs mitochondrial function and induces intervertebral disc degeneration and therapeutic effects of sulforaphane. Theranostics 2019, 9 (8), 2252-2267. DOI: 10.7150/thno.30658.
NLM
Xu X, Wang D, Zheng C, Gao B, Fan J, Cheng P, Liu B, Yang L, Luo Z. Progerin accumulation in nucleus pulposus cells impairs mitochondrial function and induces intervertebral disc degeneration and therapeutic effects of sulforaphane. Theranostics 2019; 9(8):2252-2267. doi:10.7150/thno.30658. https://www.thno.org/v09p2252.htm
CSE
Xu X, Wang D, Zheng C, Gao B, Fan J, Cheng P, Liu B, Yang L, Luo Z. 2019. Progerin accumulation in nucleus pulposus cells impairs mitochondrial function and induces intervertebral disc degeneration and therapeutic effects of sulforaphane. Theranostics. 9(8):2252-2267.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.