Impact Factor
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(5):1510-1522. doi:10.7150/thno.29620 This issue Cite
Research Paper
p66Shc Contributes to Liver Fibrosis through the Regulation of Mitochondrial Reactive Oxygen Species
Yan Zhao1*, Zhecheng Wang1*, Dongcheng Feng2, Huanyu Zhao1, Musen Lin3, Yan Hu3, Ning Zhang3, Li Lv1, Zhenming Gao2, Xiaohan Zhai4, Xiaofeng Tian2, Jihong Yao1 ![]()
1. Department of Pharmacology, Dalian Medical University, Dalian 116044, China.
2. Department of Surgery, The Second Affiliated Hospital of Dalian Medical University, Dalian 116023, China.
3. Department of Pharmacy, The Second Affiliated Hospital of Dalian Medical University, Dalian 116023, China.
4. Department of Pharmacy, The First Affiliated Hospital of Dalian Medical University, Dalian 116011, China.
*These authors contributed equally to this work.
Received 2018-8-31; Accepted 2019-1-15; Published 2019-2-20
Abstract

Background: p66Shc is a redox enzyme that mediates mitochondrial reactive oxygen species (ROS) generation. p66Shc inhibition confers protection against liver injury, however, its functional contribution to liver fibrosis remains unclear. The aim of this study is to explore the involvement of p66Shc in liver fibrosis and underlying mechanism of p66Shc by focusing on mitochondrial ROS.
Methods: p66Shc-silenced mice were injected with carbon tetrachloride (CCl4). Primary hepatic stellate cells (HSCs) were performed with p66Shc silencing or overexpression prior to TGF-β1 stimulation.
Results: p66Shc expression was progressively elevated in mice with CCl4-induced liver fibrosis, and p66Shc silencing in vivo significantly attenuated fibrosis development, reducing liver damage, oxidative stress and HSC activation, indicated by the decreased α-SMA, CTGF and TIMP1 levels. Furthermore, in primary HSCs, p66Shc-mediated mitochondrial ROS production played a vital role in mitochondrial morphology and cellular metabolism. Knockdown of p66Shc significantly inhibited mitochondrial ROS production and NOD-like receptor protein 3 (NLRP3) inflammasome activation, which were closely associated with HSC activation, indicated by the decreased α-SMA, CTGF and TIMP1 levels. However, p66Shc overexpression exerted the opposite effects, which were suppressed by a specific mitochondrial ROS scavenger (mito-TEMPO). More importantly, p66Shc expression was significantly increased in human with liver fibrosis, accompanied by NLRP3 inflammasome activation.
Conclusions: p66Shc is a key regulator of liver fibrosis by mediating mitochondrial ROS production, which triggers NLRP3 inflammasome activation.
Keywords: p66Shc, mitochondrial reactive oxygen species, liver fibrosis, hepatic stellate cell, NLRP3 inflammasome
Introduction
Hepatic stellate cells (HSCs) activation is responsible for liver fibrosis resulting from various chronic liver injury, as activated HSCs are implicated in the predominant source of extracellular matrix (ECM) accumulation. [1]. Physiologically, quiescent HSCs are required for retinoid homeostasis and ECM remodeling. Once activated, quiescent HSCs turn into myofibroblasts, accompanied with ECM overexpression [2]. However, no medicinal candidates specifically target HSCs, which limits the current antifibrotic therapy [3].
Oxidative stress is considered as a pivotal driver of liver fibrosis.[4]. Reactive oxygen species (ROS) accumulation promotes HSCs activation and transdifferentiation into myofibroblast, which are characterized by excessive ECM deposition, resulting in subsequent scar formation. Oxidative stress was detected both in human and mouse with liver fibrosis [5]. Furthermore, antioxidant treatment or antioxidant gene overexpression can prevent HSC activation in liver fibrosis [6, 7].
Oxidative stress is associated with numerous downstream molecules that contribute to liver fibrosis. In light of the recent study, NOD-like receptor protein 3 (NLRP3) inflammasome, is identified as a novel contributor of liver fibrosis [8]. NLRP3 inflammasome activation triggers the release of interleukin (IL)-1β and IL-18, contributing to HSC activation [9, 10]. Although intracellular ROS provokes NLRP3 inflammasome activation [11-13], the source and regulation of ROS remain to be clarified.
The mitochondrial ROS accounts for the majority of intracellular ROS [14]. The 66 kDa isoform of Shc (p66Shc), is vital for mitochondria-dependent oxidative balance [15]. Under cellular stress conditions, p66Shc catalyzes electron transfer from Cytochrome c to oxygen, which gives rise to the enhancement of hydrogen peroxide (H2O2) [16]. In response to obesity, ischemic myocardial injury, and Alzheimer's disease, circulating and tissue ROS levels and ROS-mediated damage are decreased in p66Shc knockout mice [17-19]. More recently, we identified p66Shc as a crucial mediator of hepatocyte oxidative stress. The protective effects of p66Shc downregulation partly attributes to a reduction of cellular ROS in hepatocytes [20-22]. However, the potential role of p66Shc in liver fibrosis needs to be further clarified. Additionally, increased expression of p66Shc mRNA in humans is significantly correlated with the degree of liver fibrosis [23], suggesting that p66Shc may contribute to liver fibrosis.
In this study, we explored whether p66Shc is involved in liver fibrosis. First, p66Shc expression in human and mouse fibrotic liver tissues was detected. Second, p66Shc knockdown was performed both in vivo and in vitro. Third, we elucidated the underlying mechanisms by which p66Shc participates in liver fibrosis through NLRP3 inflammasome activation.
Methods
Mice and liver injury
C57BL/6 mice (male) were injected with lentivirus-p66Shc-shRNA or lentivirus-scramble (1× 109 viral particles/mouse) via the tail vein. After 7 days, carbon tetrachloride (CCl4) was employed by intraperitoneal injections eight times during four weeks. Each injection contained a CCl4 dose of 1, 2, and 4 ml/kg diluted in corn oil. Mice were euthanized 48 h after the last injection. Animal experiments were applied according to the Guidelines for the Care and Use of Laboratory Animals, and were approved by the Institutional Ethics Committee of Dalian Medical University (Dalian, China).
Human liver samples
Twelve normal and twelve liver fibrosis tissue samples were obtained from the Second Affiliated Hospital of Dalian Medical University (Dalian, China). The informed consent was signed by all patients. Liver samples were handled by the approval of the Ethics Committee of Dalian Medical University.
Histologic examination and immunohistochemical staining
Liver tissue sections were collected and fixed in 4% paraformaldehyde at room temperature at least overnight, and were applied to Hematoxylin-eosin (H&E) and Masson staining. Fibrosis stage was assessed according to the Ishak score [24].
Immunohistochemistry (IHC) of α-SMA, or p66Shc was performed on paraffin sections (4 μm). Section were incubated with p66Shc or α-SMA (Proteintech) antibody overnight, and were visualized with DAB and hematoxylin. The number of positive cells was quantified with ImageJ software.
Biochemical assays
Alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were assessed with assay kits, respectively (Jiancheng Corp., Nanjing, China).
Hydrogen peroxide (H2O2) content and superoxide dismutase (SOD) activity were assessed with assay kits (Jiancheng Corp.).
HSC isolation and culture
Primary rat HSCs were isolated with proteinase/ collagenase digestive enzymes as previously described [25], and were maintained in DMEM (Gibco, Carlsbad, CA, USA) at 37 ℃ with 5% CO2.
Cell transfection
Primary HSCs were transfected with small interfering RNA (siRNA), pcDNA plasmid or negative control (GenePharma, Suzhou, China) for 48 h using Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA). The p66Shc siRNA sequence was sense 5'-GCA ACU UGA AGC UGG CCA ATT-3', antisense 5'-UUG GCC AGC UUC AAG UUG CTT-3'. The NLRP3 siRNA sequence was sense: 5'-GAU CCU AUU UGA AGA GUG U-3', antisense 5'-GAU CAA CCU CUC UAC CAG A-3'. Then cells were incubated with transforming growth factor-β1 (TGF-β1, 2 ng/ml for 24 h, Promega, USA) for different assays.
Western blotting
Proteins were separated via SDS-PAGE (10-12%). The relevant primary antibodies that were specific for p66Shc (BD Biosciences, USA); cleaved caspase-1 (Affinity Biosciences, USA); Col1a1 (Abcam Ltd., Cambridge, UK);IL-18 (ABclonal Biotechnology, Wuhan, China); α-SMA, NLRP3, adaptor apoptosis- associated speck-like protein (ASC), superoxide dismutase 2 (SOD2), mitochondrial uncoupling protein 1 (UCP1), IL-1β, Cytochrome c, β-actin and voltage-dependent ion channel (VDAC, Proteintech Group, Wuhan, China), followed by secondary antibodies incubation.
RNA isolation and quantificational reverse transcription-polymerase chain reaction (qRT-PCR)
TRIzol (Invitrogen) was applied for total RNA extraction. Then cDNA synthesis and RNA abundance were performed with primeScriptTM RT reagent kit and SYBR Premix Ex TaqTM II (TaKaRa, Japan), respectively. Expression levels normalized to β-actin levels in each sample were determined by calculating ΔΔCt. The primer sequences were shown in Table 1.
Primer sequences
| Gene | Forward primer | Reverse primer |
|---|---|---|
| p66Shc mouse | GTCCGACTACCTGTGTTCCTT | CAGCAGGATTGGCCAGCTT |
| Col1a1 mouse | TGACTGGAAGAGCGGAGAGATCT | TTCGGGCTGATGTACCAGTTC |
| α-SMA mouse | TGCCGAGCGTGAGATTGTC | CGTTCGTTTCCAATGGTGATC |
| β-actin mouse | AGAGGGAAATCGTGCGTGAC | CAATAGTGATGACCTGGCCGT |
| CTGF rat | GCCTACCGACTGGAAGACACA | CAGCCTGCAGAAGGTATTGTCA |
| TIMP1 rat | TGGTTCCCTGGCATAATCTGA | GGATCTGATCTGTCCACAAGCA |
| β-actin rat | GGAAATCGTGCGTGACATTAAAG | CGGCAGTGGCCATCTCTT |
| p66Shc human | GAGGCCATCAGTCTGGTGTGT | GCTGGTGGAGACGGTGAGA |
| β-actin human | ACCCTGAAGTACCCCATCGAG | ACATGATCTGGGTCATCTTCTCG |
Dual immunofluorescence microscopy
After fixed in 4% paraformaldehyde, The cells and liver tissue sections were incubated with primary antibody at 4 ℃ overnight followed by secondary antibody incubation (Proteintech) at 37 ℃ for 1 h. Subsequently, DAPI (Beyotime Institute of Biotechnology, Hangzhou, China) was used for nuclear staining. Immunofluorescence images were collected under 80i Nikon confocal microscope.
Mitochondrial ROS level determination
Mitochondrial ROS level was determined by visualization with MitoSOX Red (Invitrogen) or MitoTracker Red CMXRos (Invitrogen). Briefly, primary rat HSCs were plated on coverslips and incubated with 5 μM MitoSOX Red or 200 nM MitoTracker at 37 ℃ for 10 min. The nuclei were identified by Hoechst 33342 staining (Beyotime Institute of Biotechnology) in the dark for 5 min. Immunofluorescence images were collected under 80i Nikon confocal microscope.
Cells were stimulated with 150 μg/ml monosodium urate (MSU), and mitochondrial respiratory complex inhibiter (10 μM rotenone; 10 μM thenoyltrifluoroacetone, TTFA; 40 μg/ml antimycin A) for 6 h, or 100 μM mito-TEMPO for 3 h. MSU, a well-known NLRP3 inflammasome activator, was used to experimentally induce inflammasome activation. The reagents above were from Sigma (Missouri, USA).
Mitochondrial membrane potential
JC-1 probe (Beyotime Institute of Biotechnology) was carried out for mitochondrial membrane potential detection. Cells were loaded with JC-1 probe according to the instruction. Then, nuclei were detected by DAPI staining (Beyotime Institute of Biotechnology) in the dark. The red and green JC-1 fluorescence ratio was calculated.
Transmission electron microscopy (TEM)
TEM was performed to observe the mitochondrial morphology. Primary HSCs were fixed and embedded. Then ultrathin sections (50 μm thick) were prepared. The sections were dehydrated in a gradient of ethanol, and embedded in epoxy resin. Lead citrate and uranyl acetate were used for staining. The images were obtained using a transmission electron microscope (JEOL, Peabody, MA).
Mitochondria isolation
Mammalian mitochondrial isolation kit (BioVision, Milpitas, CA, USA) was performed for mitochondria isolation. The samples were homogenized in Mitochondrial Isolation Buffer using a precooled glass homogenizer. The samples were stored on ice and then centrifuged at 4 ℃. The mitochondria were resuspended in Storage Buffer.
Intracellular adenosine triphosphate (ATP) level assay
Intracellular ATP levels were measured with assay kit in accordance with the assay instructions (Beyotime Institute of Biotechnology).
Statistical analysis
Student's unpaired t test (two-group comparisons) and one-way ANOVA test (multi-group comparisons) were performed using GraphPad Prism. Data are expressed as the means±standard deviation (SD). P<0.05 was taken significant.
Results
p66Shc expression is correlated with liver fibrosis in mice
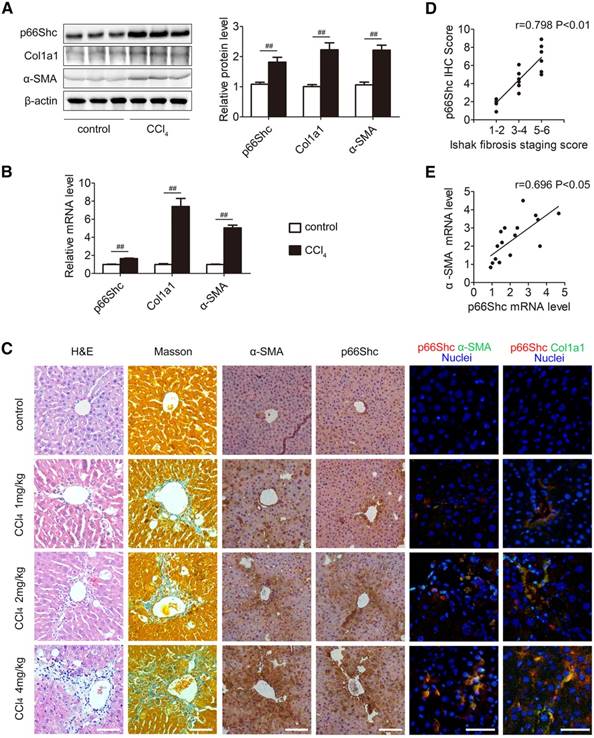
Western blotting and qRT-PCR analyses indicated that p66Shc, Col1a1 and α-SMA expression were strongly increased in fibrotic samples induced by CCl4 in comparison to the control (Figure 1A-B). H&E staining, Masson staining and IHC for α-SMA exhibited mild to severe fibrosis with different dosages of CCl4 treatment. As shown in Figure 1C-D, IHC analyses revealed that there was a positive correlation between the increased p66Shc expression and liver fibrosis progression. Notably, the p66Shc expression pattern coincided with that of α-SMA and Col1a1 expression in the fibrotic livers (Figure 1C), indicating enhanced p66Shc expression in activated HSCs. In addition, p66Shc mRNA was increased in the fibrotic livers, correlating with HSC activation, as observed by the induction of α-SMA mRNA (Figure 1E). Taken together, p66Shc expression is positively correlated with liver fibrosis progression.
p66Shc expression is correlated with liver fibrosis in mice. (A) p66Shc, Col1a1, α-SMA protein levels, n=3. (B) p66Shc, Col1a1, α-SMA mRNA levels, n=6. (C) H&E staining, Masson staining, IHC staining for α-SMA and p66Shc, and dual immunofluorescence staining for p66Shc and α-SMA, as well as for p66Shc and Col1a1. Scale bar, 200 μm. (D) Pearson's correlation analyses of p66Shc IHC scores with Ishak scores of Masson staining (r=0.798, P<0.01). (E) Pearson's correlation analyses of p66Shc mRNA with α-SMA mRNA (r=0.696, P<0.05). ##P<0.01.
p66Shc silencing attenuates liver fibrosis in mice
Then, we focused on the contribution of p66Shc to liver fibrosis. p66Shc silencing via lentiviral transduction was delivered into mice. Western blotting and qRT-PCR were carried out to assess the efficiency of p66Shc knockdown in vivo. As shown in Figure 2A-B, p66Shc expression was inhibited (approximately 70~80% reduction) by p66Shc silencing. As expected, p66Shc silencing markedly recovered SOD2 and UCP1 protein, SOD activity, as well as decreased H2O2 content and Cytochrome c release, indicating that intracellular ROS may contribute to the mechanism of p66Shc in liver fibrosis. Moreover, α-SMA and Col1a1, the most abundant ECM protein in the fibrotic livers, were notably abolished by concomitant p66Shc silencing (Figure 2B-E). Consistently, Masson staining assays revealed that p66Shc knockdown inhibited collagen accumulation (Figure 2F-G). p66Shc silencing also alleviated histological liver damage, evidenced by H&E staining (Figure 2F), and decreased serum ALT and AST concentrations (Figure 2H). The results demonstrated that p66Shc knockdown attenuates liver injury and decelerates liver fibrosis in vivo.
p66Shc silencing attenuates liver fibrosis in mice. p66Shc silencing was induced via lentivirus delivered to C57BL/6 mice exposed to CCl4 (2 ml/kg). (A) Liver p66Shc mRNA expression, n=6. (B) Liver p66Shc, SOD2, UCP1, Col1a1, α-SMA protein, n=3. (C) H2O2 content, n=8. (D) SOD activity, n=8. (E) Cytochrome c expression in the cytoplasm and mitochondria, n=3. (F) H&E and Masson staining. Scale bar, 200 μm. (G) Ishak score of Masson staining. (H) Serum ALT and AST levels, n=8. (I) Liver NLRP3 inflammasome protein expression, n=3. (J) Liver CTGF and TIMP1 mRNA levels, n=6. ##P<0.01, #P<0.05.
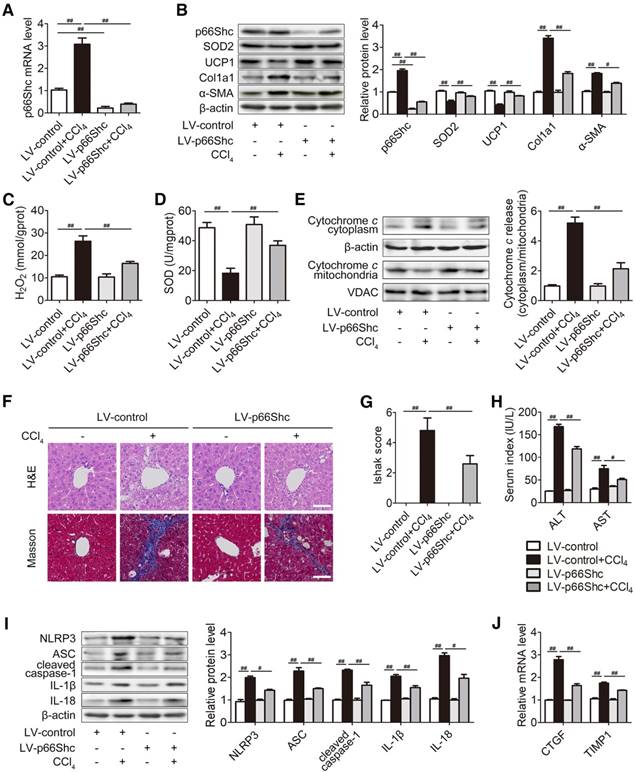
Since the NLRP3 inflammasome acts as a novel regulator of HSCs activation and ECM production, the contribution of p66Shc to NLRP3 inflammasome activation was determined. NLRP3 inflammasome complex (NLRP3, ASC, cleaved caspase-1, IL-1β and IL-18) protein were increased in CCl4-treated mice, and this increase was blocked by p66Shc silencing (Figure 2I). Furthermore, p66Shc knockdown significantly attenuated HSC activation, indicated by the decrease in CTGF and TIMP1 mRNA levels (Figure 2J). Taken together, these results suggest that p66Shc silencing inhibits HSC activation, which may be related to NLRP3 inflammasome activation.
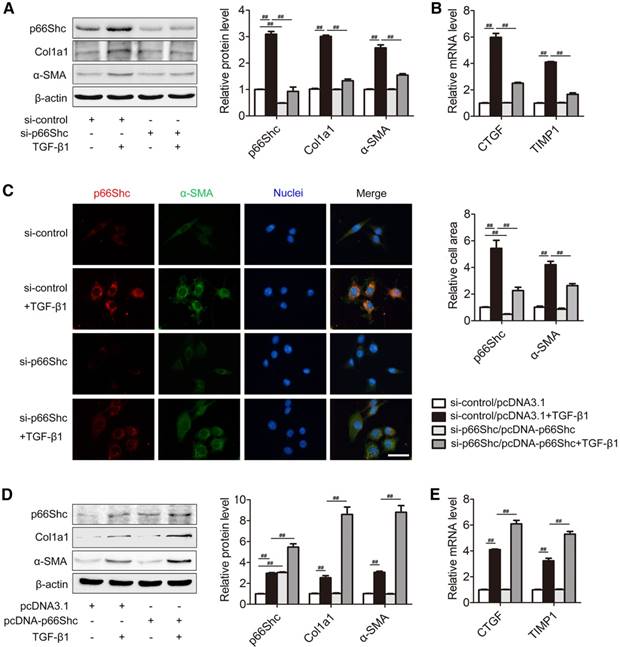
p66Shc contributes to HSC activation in vitro
Activated HSCs are responsible for ECM production and liver fibrosis [26], and TGF‐β1 is implicated as a key driver of HSC activation [27]. To assess whether p66Shc is involved in HSCs activation in vitro, primary HSCs were transfected with p66Shc siRNA (Figure 3A-C) or pcDNA-p66Shc (Figure 3D-E) following TGF-β1 challenge. We found that p66Shc knockdown by siRNA significantly blunted the Col1a1 and α-SMA protein, as well as CTGF and TIMP1 mRNA, two specific markers of HSC activation (Figure 3A-B). Double immunofluorescence staining showed that p66Shc localized with α-SMA- positive cells, and the increase in p66Shc and α-SMA induced by TGF-β1 was eliminated by transfection with p66Shc siRNA. Additionally, during the process of activation, primary HSCs acquired contractile and fibrogenic myofibroblast-like phenotype and enhanced HSC activation markers (α-SMA, CTGF and TIMP1), which was reversed by p66Shc siRNA (Figure 3C). In contrast, p66Shc overexpression amplified HSCs activation induced by TGF-β1 (Figure 3D-E). These results indicate that p66Shc contributes to HSC activation in vitro.
p66Shc contributes to HSC activation in vitro. Primary HSCs were transfected with p66Shc siRNA (A-C) or pcDNA-p66Shc (D, E) and then exposed to TGF-β1. (A) p66Shc, Col1a1, α-SMA protein, n=3. (B) CTGF and TIMP1 mRNA, n=6. (C) Dual immunofluorescence of p66Shc and α-SMA. Representative immunofluorescence images in the left panel. Scale bar, 200 μm. Representative cell area in the right panel. (D) p66Shc, Col1a1, α-SMA protein expression, n=3. (E) CTGF and TIMP1 mRNA expression, n=6. ##P<0.01.
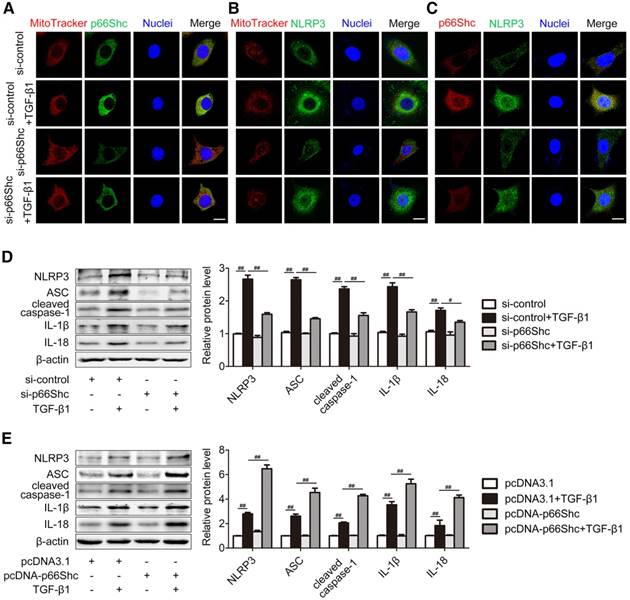
p66Shc promotes HSC activation via NLRP3 inflammasome activation
To study the underlying mechanism of p66Shc on liver fibrosis, primary HSCs were transfected with p66Shc siRNA (Figure 4A-D) or pcDNA vector (Figure 4E). During chronic liver injury, inflammation provokes HSC activation and NLRP3 inflammasome is associated with primary mouse HSCs or LX-2 HSCs activation [9, 28]. As shown in Figure 4A-B, both p66Shc (green) and NLRP3 (green) colocalized with mitochondria that were stained with MitoTracker (red). Furthermore, double immunofluorescence staining showed that p66Shc (red) and NLRP3 (green) were colocated in HSCs (Figure 4C). Therefore, p66Shc and NLRP3 were closely associated in mitochondria. Consistent with the results in vivo, p66Shc knockdown significantly impaired NLRP3 inflammasome activation in HSCs exposed to TGF-β1 stimulation, while p66Shc overexpression enhanced NLRP3 inflammasome activation (Figure 4D-E). Thus, p66Shc contributes to HSC activation through NLRP3 inflammasome activation.
p66Shc triggers NLRP3 inflammasome activation through the regulation of mitochondrial ROS
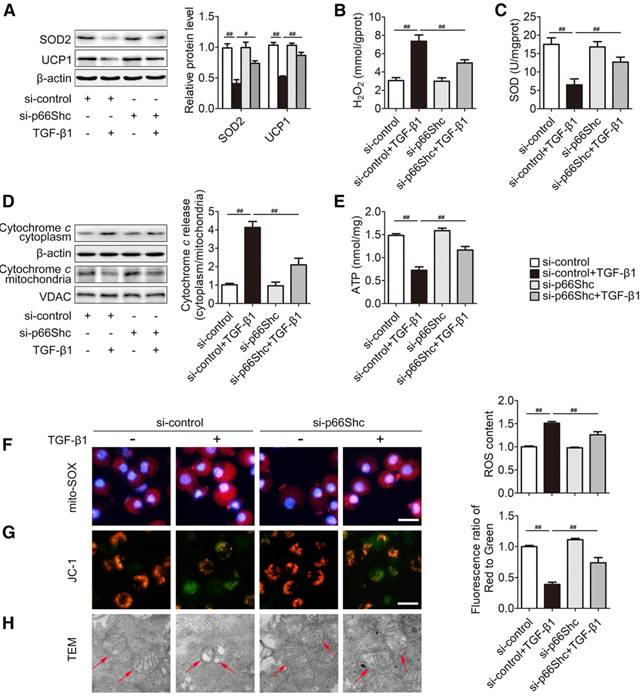
Mitochondrial ROS exerts a crucial role on NLRP3 inflammasome activation [29]. Then, we analyzed whether p66Shc-mediated mitochondrial ROS is involved in NLRP3 inflammasome activation. In primary HSCs, p66Shc knockdown triggered a notable increase in SOD2 and UCP1 protein, SOD activity and ATP content, as well as a dramatic decrease in H2O2 and Cytochrome c release to ameliorate oxidative stress in response to TGF-β1 (Figure 5A-E). Additionally, mitochondrial ROS was assessed by mitoSOX that served as a mitochondrial superoxide indicator. As shown in Figure 5F, mitochondrial ROS production was enhanced after exposure to TGF-β1 and was successfully reduced by p66Shc siRNA. Furthermore, the role of p66Shc in mitochondrial function was also characterized. The dysfunction of mitochondrial membrane potential was induced by TGF-β1 treatment, indicated by JC-1 monomers with green in the cytoplasm; however, p66Shc knockdown improved the normalization of mitochondrial membrane potential, shown by enhanced JC-1 monomers with red in the mitochondria (Figure 5G). In addition, p66Shc siRNA also substantially rescued the swollen mitochondria with disorganized and fragmented cristae induced by TGF-β1 (Figure 5H). As shown in Figure S1, p66Shc siRNA improved oxygen consumption rate (OCR) in response to TGF-β1. Collectively, these findings indicate that p66Shc knockdown attenuates mitochondrial ROS production and mitochondrial dysfunction in HSCs.
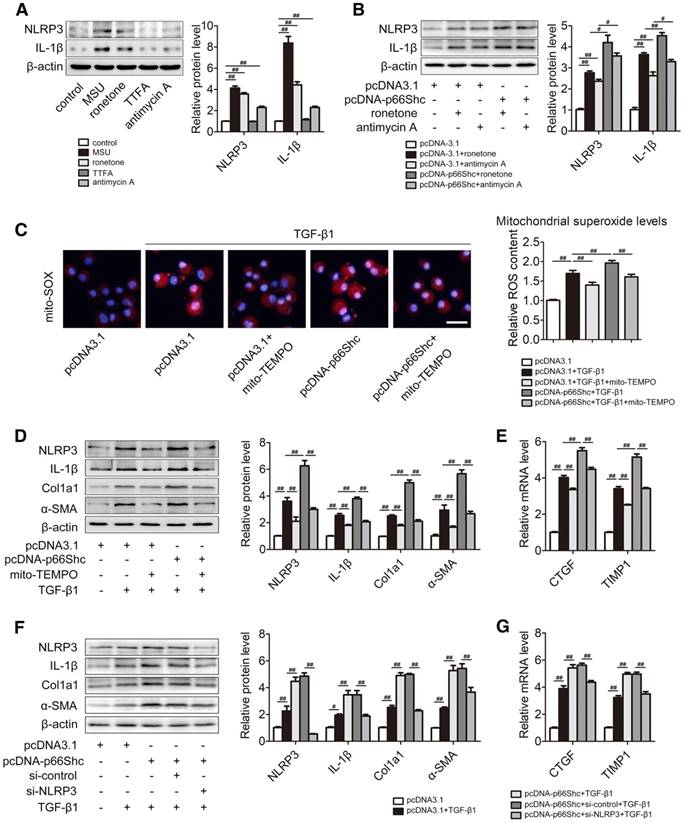
Next, we focused on the association between mitochondrial ROS and NLRP3 inflammasome activation in primary HSCs. Mitochondrial electron transport is the principal intracellular producer of ROS. Rather than complex II, respiratory complexes I and III are generally considered the main ROS producers in mitochondria [14]. Consistently, NLRP3 and IL-1β expression were increased as a result of mitochondrial ROS overproduction induced by rotenone (complex I inhibitor) and antimycin A (complex III inhibitor), respectively. Meanwhile, little or no effect was observed with the complex II inhibitor TTFA (Figure 6A). These data provide the evidence that mitochondrial ROS can trigger NLRP3 inflammasome activation. Furthermore, p66Shc overexpression amplified NLRP3 inflammasome activation in the presence of rotenone and antimycin A (Figure 6B). To further examine the effect of mitochondrial ROS production mediated by p66Shc on NLRP3 activation, primary HSCs were transfected with pcDNA-p66Shc in the absence or presence of mito-TEMPO, a specific mitochondrial ROS scavenger. Under TGF-β1 stimulation, p66Shc overexpression resulted in robust ROS production, which was blocked by mito-TEMPO, as detected by Mito-SOX (Figure 6C). Consequently, the high levels of NLRP3 and IL-1β induced by p66Shc overexpression were significantly abrogated by mito-TEMPO (Figure 6D). As expected, the reduction in mitochondrial ROS induced by mito-TEMPO substantially attenuated liver fibrosis (Col1a1 and α-SMA) as well as HSC activation (CTGF and TIMP1) in the presence of p66Shc overexpression (Figure 6D-E). Similar results were obtained with LX-2 cells. Moreover, p66Shc overexpression elevated the expression of Col1a1, α-SMA, CTGF and TIMP1, which was attenuated by NLRP3 siRNA (Figure 6F-G). Considering all the above findings, these data suggest that the underlying mechanism of the effect of p66Shc on NLRP3 activation in liver fibrosis is likely mediated through mitochondrial ROS.
p66Shc upregulation and NLRP3 inflammasome activation are involved in human liver fibrosis
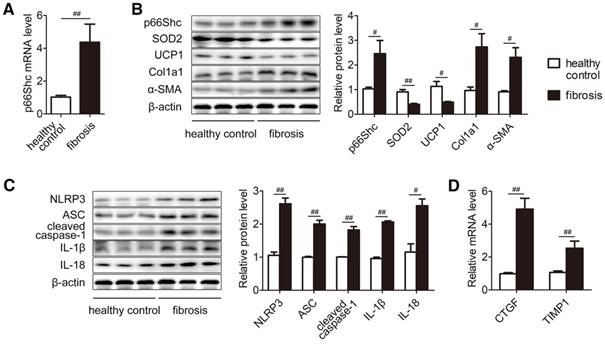
To dissect the biological relevance of p66Shc during liver fibrosis in clinical situations, the expression of p66Shc was compared between healthy control and fibrosis patients. Consistently, p66Shc mRNA and protein were elevated significantly, and dysregulation of antioxidant responses were detected as evidenced by decreased SOD2 and UCP1 levels. Moreover, the accumulation of collagen and α-SMA were observed in fibrotic livers (Figure 7A-B). Intriguingly, the NLRP3 inflammasome was activated (Figure 7C). Furthermore, CTGF and TIMP1 mRNA levels were dramatically higher in fibrotic livers than in healthy control livers (Figure 7D). Taken together, p66Shc upregulation and NLRP3 inflammasome activation are involved in human liver fibrosis.
Discussion
Our study firstly addressed the crucial role of p66Shc in liver fibrosis. First, p66Shc expression was significantly elevated in patients with fibrosis and in murine fibrotic models, and positively correlated with liver fibrosis stage in mice. Second, p66Shc silencing alleviated liver fibrosis in vivo. Third, p66Shc functioned as a contributor of HSC activation by activating the NLRP3 inflammasome. Therefore, p66Shc is a novel regulator of HSC activation and therapeutic target in liver fibrosis.
HSC activation represents the dominant event in liver fibrosis [26]. When exposed to pathophysiological cues, HSCs become activated and adopt a myofibroblast-like phenotype. These cells have been identified as the main producers of collagens and other ECM components, making them as the main concern for antifibrotic therapies [3]. In this study, enhanced p66Shc expression was related to liver fibrosis, especially in fibrotic patients. In addition, p66Shc expression was enhanced in activated HSCs (α-SMA-positive HSCs) and correlated with fibrosis progression. More importantly, p66Shc silencing blocked HSC activation induced by CCl4 administration. In vitro, we further found that p66Shc expression was remarkably elevated by TGF-β1 in primary HSCs. Knockdown of p66Shc apparently abolished the increase of CTGF and TIMP1 levels, two markers of HSC activation, whereas p66Shc overexpression exacerbated CTGF and TIMP1 abundance in response to TGF-β1 stimulation in primary HSCs. Additionally, we found that TGF-β1-induced primary HSCs was less responsive to apoptosis after inhibition and overexpression of p66Shc (data not shown). Therefore, p66Shc is an important mediator of HSC activation in liver fibrosis.
p66Shc is essential for HSC activation via NLRP3 inflammasome activation. Primary HSCs were transfected with p66Shc siRNA (A-D) or pcDNA-p66Shc vector (E) and further exposed to TGF-β1. (A, B) The colocalization of p66Shc and mitochondria (MitoTracker), as well as NLRP3 and mitochondria (MitoTracker). Scale bar, 12.5 μm. (C) Dual immunofluorescence of p66Shc and NLRP3. Scale bar, 12.5 μm. (D, E) NLRP3 inflammasome protein expression, n=3. ##P<0.01, #P<0.05.
p66Shc knockdown attenuates mitochondrial ROS production and mitochondrial dysfunction in primary HSCs. p66Shc knockdown was carried out by p66Shc siRNA in the presence of TGF-β1. (A) SOD2 and UCP1 protein levels, n=3. (B) H2O2 content, n=8. (C) SOD activity, n=8. (D) Cytochrome c expression in the cytoplasm and mitochondria; n=3. (E) ATP content, n=8. Representative fluorescence images of MitoSOX (F)- and JC-1 (G)-stained cells. Scale bar, 200 μm. (H) Mitochondrial morphology was determined via TEM (1500×, magnification, red arrow). ##P<0.01, #P<0.05.
Although, liver fibrosis has much in common with fibrosis in other organs, such as the lungs and kidneys [2], our findings are not applicable to all fibrosis processes. In renal fibrosis, p66Shc overexpression occurs paralleled with mitochondrial ROS overproduction and the fibrosis process [30]. However, p66Shc knockout ob/ob mice exhibited higher subcutaneous adipose tissue (SAT) collagen production and no protection against visceral adipose tissue (VAT) fibrosis [18]. Additionally, p66Shc knockout mice exhibit interstitial and alveolar fibrosis after cigarette smoke exposure [31]. Unlike in HSCs, the prohibitive apoptosis by p66Shc silencing interferes with the macrophage “clearance” from alveolar spaces and increases M2 cytokines and enzymes levels in macrophages, which can promote TGF-β expression, collagen deposition, and fibrosis in the surrounding areas. The different effects of p66Shc activation and silencing in different cells, tissues and organs may result from tissue specificity and the different mechanisms underlying fibrosis progression [26, 32].
Adaptor protein p66Shc is an attractive isoform of ShcA due to its additional CH2 domain, which is responsible for its redox property [16]. p66Shc is expressed in many tissues, where it contributes to organ dysfunction by promoting mitochondrial ROS production [33]. p66Shc-/- mice exhibit decreased intracellular ROS levels, and display increased resistance to oxidative stress [34]. Mitochondrial ROS represents the majority of intracellular ROS, particularly in persistent dysfunction including liver fibrosis [35]. p66Shc is identified as a central molecular of the mitochondrial oxidative stress [15]. p66Shc is partially located in mitochondria, where it promotes Cytochrome c release and functions as an oxidoreductase, resulting in H2O2 formation, the main ROS production [16]. In this study, we found that p66Shc silencing attenuated CCl4/TGF-β1-induced ECM and mitochondrial ROS accumulation both in vivo and in vitro. Mitochondria-localized p66Shc was involved in mitochondrial membrane potential, mitochondrial morphology and cellular metabolism by mitochondrial ROS accumulation. Additionally, in primary HSCs, we found that the release of Cytochrome c induced by TGF-β1 was restrained by p66Shc knockdown, along with a reduction in H2O2 content. Similar results were obtained in vivo. These findings indicated that p66Shc contributes to liver fibrosis via mitochondrial ROS.
p66Shc triggers NLRP3 inflammasome activation through regulation of mitochondrial ROS production. (A) Primary HSCs were stimulated with MSU and mitochondrial complex inhibitor (rotenone, TTFA and antimycin A). NLRP3 and IL-1β protein expression, n=3. (B) p66Shc overexpression was carried out in primary HSCs by pcDNA-p66Shc vector and then rotenone or antimycin A was stimulated. NLRP3 and IL-1β protein expression, n=3. (C-E) p66Shc overexpression was applied in primary HSCs by pcDNA-p66Shc vector transfection and then mito-TEMPO was stimulated under TGF-β1 treatment. (C) Representative fluorescence images of MitoSOX. (D) NLRP3, IL-1β, Col1a1, α-SMA protein levels, n=3. (E) CTGF and TIMP1 mRNA expression, n=6. (F-G) Primary HSCs were co-transfected with pcDNA-p66Shc and NLRP3 siRNA following TGF-β1 challenge. (F) NLRP3, IL-1β, Col1a1, α-SMA protein levels, n=3. (G) CTGF and TIMP1 mRNA expression, n=6. ##P<0.01, #P<0.05.
p66Shc upregulation and NLRP3 inflammasome activation are involved in human liver fibrosis. Relative protein expression and mRNA were measured in patients with liver fibrosis. (A) p66Shc mRNA levels. (B) p66Shc, SOD2, UCP1, Col1a1, α-SMA protein levels. (C) NLRP3 inflammasome protein levels. (D) CTGF and TIMP1 mRNA levels. n=12. ##P<0.01, #P<0.05.
NLRP3 inflammasome activation is a novel contributor in liver fibrosis [9]. Mitochondrial ROS plays a crucial role in NLRP3 inflammasome activation. Evidence is emerging to support that ROS generated by the mitochondrial respiratory chain exerts a pivotal effect on NLRP3 inflammasome activation [29]. Robust mitochondrial ROS production was driven by mitochondrial complex I inhibitor rotenone and complex III inhibitor antimycin A [36]. In primary HSCs, we found that rotenone and antimycin A stimulation caused NLRP3 inflammasome activation, and furthermore, mitochondrial ROS production regulated by p66Shc was involved in NLRP3 inflammasome activation. Confocal laser scanning microscopy showed that NLRP3 and p66Shc colocalized with mitochondria in HSCs. Importantly, knockdown of p66Shc significantly impaired NLRP3 inflammasome activation, while p66Shc overexpression abolished the results described above. In addition, treatment of primary HSCs and LX-2 cells with mito-TEMPO, a specific mitochondrial ROS scavenger, abrogated mitochondrial ROS release and blocked the NLRP3 inflammasome activation induced by p66Shc overexpression. As a result, mito-TEMPO suppressed the increase in Col1a1 and α-SMA enhanced by p66Shc overexpression in activated HSCs. Moreover, the mitochondrial ROS production blocked by p66Shc silencing attenuated liver fibrosis, based on Masson staining and Col1a1 expression in vivo. Thus, p66Shc-triggered NLRP3 inflammasome activation is mainly dependent on mitochondrial ROS, which promotes HSC activation and liver fibrosis.
Accumulating evidence reveals that ROS is crucial for HSC activation in liver fibrosis. In light of the important role played by ROS in liver fibrosis, strategies aimed at blocking ROS production are currently being researched [37]. Indeed, antioxidants were found to be beneficial against liver fibrosis in mice [38]. Nevertheless, the clinical benefits of antioxidant agents are not promising [39]. The main factor considered to account for this failure is that exogenous antioxidant agents may function differently from endogenous agents [40]. p66Shc is crucially involved in the intracellular redox balance and oxidative stress levels. The reported protection achieved by p66Shc deletion is primarily due to the reduction in intracellular free radicals [17, 19, 41]. In this study, our data suggest that p66Shc silencing protected mice from liver fibrosis by blunting the production of free radicals and that the ROS mediator p66Shc is a crucial trigger for liver fibrosis. Therefore, the development of treatment strategies controlling p66Shc function may represent a more effective alternative to combat liver fibrosis, and may be developed into more specific and powerful drugs against liver fibrosis. Furthermore, analysis of a large set of clinical samples is required to validate the clinical correlation between p66Shc expression and the stage of liver fibrosis.
Collectively, for the first time, our study demonstrated that p66Shc is essential for HSC activation and liver fibrosis through NLRP3 inflammasome activation. These findings identify p66Shc as a critical regulator of liver fibrosis and provide mechanistic insights into liver fibrosis progression that can be exploited for pharmacological intervention.
Abbreviations
ASC: adaptor apoptosis-associated speck-like protein; CTGF: connective tissue growth factor; FCCP: phenylhydrazone; H2O2: hydrogen peroxide; HSC(s): hepatic stellate cell(s); IL: interleukin; MSU: monosodium urate; NLRP3: NOD-like receptor protein 3; OCR: oxygen consumption rate; p66Shc: 66 kDa isoform of the growth factor adapter Shc; ROS: reactive oxygen species; SOD(2): superoxide dismutase (2); TIMP1: tissue inhibitor of metalloproteinase-1; TTFA: thenoyltrifluoroacetone; UCP1: mitochondrial uncoupling protein 1.
Supplementary Material
Supplementary figures.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (Nos. 81773799, 81473266, and 81603369).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Mallat A, Lotersztajn S. Cellular mechanisms of tissue fibrosis. 5. Novel insights into liver fibrosis. Am J Physiol Cell Physiol. 2013;305:C789-799
2. Schuppan D, Kim YO. Evolving therapies for liver fibrosis. J Clin Invest. 2013;123:1887-1901
3. Poelstra K. Liver fibrosis in 2015: Crucial steps towards an effective treatment. Nat Rev Gastroenterol Hepatol. 2016;13:67-68
4. Samarakoon R, Dobberfuhl AD, Cooley C, Overstreet JM, Patel S, Goldschmeding R. et al. Induction of renal fibrotic genes by TGF-beta1 requires EGFR activation, p53 and reactive oxygen species. Cell Signal. 2013;25:2198-2209
5. Yadav D, Hertan HI, Schweitzer P, Norkus EP, Pitchumoni CS. Serum and liver micronutrient antioxidants and serum oxidative stress in patients with chronic hepatitis C. Am J Gastroenterol. 2002;97:2634-2639
6. Li X, Wang X, Han C, Wang X, Xing G, Zhou L. et al. Astragaloside IV suppresses collagen production of activated hepatic stellate cells via oxidative stress-mediated p38 MAPK pathway. Free Radic Biol Med. 2013;60:168-176
7. Zhang F, Ni C, Kong D, Zhang X, Zhu X, Chen L. et al. Ligustrazine attenuates oxidative stress-induced activation of hepatic stellate cells by interrupting platelet-derived growth factor-beta receptor-mediated ERK and p38 pathways. Toxicol Appl Pharmacol. 2012;265:51-60
8. Wree A, Eguchi A, McGeough MD, Pena CA, Johnson CD, Canbay A. et al. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology. 2014;59:898-910
9. Szabo G, Csak T. Inflammasomes in liver diseases. J Hepatol. 2012;57:642-654
10. Jiang S, Zhang Y, Zheng JH, Li X, Yao YL, Wu YL. et al. Potentiation of hepatic stellate cell activation by extracellular ATP is dependent on P2X7R-mediated NLRP3 inflammasome activation. Pharmacol Res. 2017;117:82-93
11. Kim SR, Kim DI, Kim SH, Lee H, Lee KS, Cho SH. et al. NLRP3 inflammasome activation by mitochondrial ROS in bronchial epithelial cells is required for allergic inflammation. Cell Death Dis. 2014;5:e1498
12. Minutoli L, Puzzolo D, Rinaldi M, Irrera N, Marini H, Arcoraci V. et al. ROS-Mediated NLRP3 Inflammasome Activation in Brain, Heart, Kidney, and Testis Ischemia/Reperfusion Injury. Oxid Med Cell Longev. 2016;2016:2183026
13. Cai SM, Yang RQ, Li Y, Ning ZW, Zhang LL, Zhou GS. et al. Angiotensin-(1-7) Improves Liver Fibrosis by Regulating the NLRP3 Inflammasome via Redox Balance Modulation. Antioxid Redox Signal. 2016;24:795-812
14. Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483-495
15. Pesaresi MG, Amori I, Giorgi C, Ferri A, Fiorenzo P, Gabanella F. et al. Mitochondrial redox signalling by p66Shc mediates ALS-like disease through Rac1 inactivation. Hum Mol Genet. 2011;20:4196-4208
16. Giorgio M, Migliaccio E, Orsini F, Paolucci D, Moroni M, Contursi C. et al. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell. 2005;122:221-233
17. Derungs R, Camici GG, Spescha RD, Welt T, Tackenberg C, Spani C. et al. Genetic ablation of the p66(Shc) adaptor protein reverses cognitive deficits and improves mitochondrial function in an APP transgenic mouse model of Alzheimer's disease. Mol Psychiatry. 2017;22:605-614
18. Ciciliot S, Albiero M, Menegazzo L, Poncina N, Scattolini V, Danesi A. et al. p66Shc deletion or deficiency protects from obesity but not metabolic dysfunction in mice and humans. Diabetologia. 2015;58:2352-2360
19. Akhmedov A, Montecucco F, Braunersreuther V, Camici GG, Jakob P, Reiner MF. et al. Genetic deletion of the adaptor protein p66Shc increases susceptibility to short-term ischaemic myocardial injury via intracellular salvage pathways. Eur Heart J. 2015;36:516-526a
20. Yan H, Jihong Y, Feng Z, Xiaomei X, Xiaohan Z, Guangzhi W. et al. Sirtuin 1-mediated inhibition of p66shc expression alleviates liver ischemia/reperfusion injury. Crit Care Med. 2014;42:e373-381
21. Shan W, Gao L, Zeng W, Hu Y, Wang G, Li M. et al. Activation of the SIRT1/p66shc antiapoptosis pathway via carnosic acid-induced inhibition of miR-34a protects rats against nonalcoholic fatty liver disease. Cell Death Dis. 2015;6:e1833
22. Gao L, Shan W, Zeng W, Hu Y, Wang G, Tian X. et al. Carnosic acid alleviates chronic alcoholic liver injury by regulating the SIRT1/ChREBP and SIRT1/p66shc pathways in rats. Mol Nutr Food Res. 2016;60:1902-1911
23. Tomita K, Teratani T, Suzuki T, Oshikawa T, Yokoyama H, Shimamura K. et al. p53/p66Shc-mediated signaling contributes to the progression of non-alcoholic steatohepatitis in humans and mice. J Hepatol. 2012;57:837-843
24. Ishak K, Baptista A, Bianchi L, Callea F, De Groote J, Gudat F. et al. Histological grading and staging of chronic hepatitis. J Hepatol. 1995;22:696-699
25. Maschmeyer P, Flach M, Winau F. Seven steps to stellate cells. J Vis Exp. 2011
26. Hernandez-Gea V, Friedman SL. Pathogenesis of liver fibrosis. Annu Rev Pathol. 2011;6:425-456
27. Hellerbrand C, Stefanovic B, Giordano F, Burchardt ER, Brenner DA. The role of TGFbeta1 in initiating hepatic stellate cell activation in vivo. J Hepatol. 1999;30:77-87
28. Watanabe A, Sohail MA, Gomes DA, Hashmi A, Nagata J, Sutterwala FS. et al. Inflammasome-mediated regulation of hepatic stellate cells. Am J Physiol Gastrointest Liver Physiol. 2009;296:G1248-1257
29. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221-225
30. Yuan Y, Chen Y, Zhang P, Huang S, Zhu C, Ding G. et al. Mitochondrial dysfunction accounts for aldosterone-induced epithelial-to-mesenchymal transition of renal proximal tubular epithelial cells. Free Radic Biol Med. 2012;53:30-43
31. Lunghi B, De Cunto G, Cavarra E, Fineschi S, Bartalesi B, Lungarella G. et al. Smoking p66Shc knocked out mice develop respiratory bronchiolitis with fibrosis but not emphysema. PLoS One. 2015;10:e0119797
32. Wolters PJ, Collard HR, Jones KD. Pathogenesis of idiopathic pulmonary fibrosis. Annu Rev Pathol. 2014;9:157-179
33. Kumar S, Kim YR, Vikram A, Naqvi A, Li Q, Kassan M. et al. Sirtuin1-regulated lysine acetylation of p66Shc governs diabetes-induced vascular oxidative stress and endothelial dysfunction. Proc Natl Acad Sci U S A. 2017;114:1714-1719
34. Migliaccio E, Giorgio M, Mele S, Pelicci G, Reboldi P, Pandolfi PP. et al. The p66shc adaptor protein controls oxidative stress response and life span in mammals. Nature. 1999;402:309-313
35. Kang JW, Hong JM, Lee SM. Melatonin enhances mitophagy and mitochondrial biogenesis in rats with carbon tetrachloride-induced liver fibrosis. J Pineal Res. 2016;60:383-393
36. Teixeira J, Basit F, Swarts HG, Forkink M, Oliveira PJ, Willems P. et al. Extracellular acidification induces ROS- and mPTP-mediated death in HEK293 cells. Redox Biol. 2018;15:394-404
37. Friedman SL, Sheppard D, Duffield JS, Violette S. Therapy for fibrotic diseases: nearing the starting line. Sci Transl Med. 2013;5:167sr161
38. Das N, Mandala A, Naaz S, Giri S, Jain M, Bandyopadhyay D. et al. Melatonin protects against lipid-induced mitochondrial dysfunction in hepatocytes and inhibits stellate cell activation during hepatic fibrosis in mice. 2017; 62.
39. Bjelakovic G, Gluud LL, Nikolova D, Bjelakovic M, Nagorni A, Gluud C. Antioxidant supplements for liver diseases. Cochrane Database Syst Rev. 2011 Cd007749
40. Spescha RD, Shi Y, Wegener S, Keller S, Weber B, Wyss MM. et al. Deletion of the ageing gene p66(Shc) reduces early stroke size following ischaemia/reperfusion brain injury. Eur Heart J. 2013;34:96-103
41. Giorgio M, Stendardo M, Migliaccio E, Pelicci PG. P66SHC deletion improves fertility and progeric phenotype of late-generation TERC-deficient mice but not their short lifespan. Aging Cell. 2016;15:446-454
Author contact
![]() Corresponding author: Jihong Yao. Address: Department of Pharmacology, Dalian Medical University, Dalian, 116044, China. Tel.: 86-0411-86110410; Fax: 86-0411-86110408; E-mail: yaojihong65edu.cn
Corresponding author: Jihong Yao. Address: Department of Pharmacology, Dalian Medical University, Dalian, 116044, China. Tel.: 86-0411-86110410; Fax: 86-0411-86110408; E-mail: yaojihong65edu.cn