Theranostics
13.3
Impact Factor
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Results and Discussion
Conclusions
Methods
Abbreviations
Supplementary Material
Acknowledgements
References
Introduction
Results and Discussion
Conclusions
Methods
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(5):1453-1473. doi:10.7150/thno.29987 This issue Cite
Research Paper
Oncogenic MSH6-CXCR4-TGFB1 Feedback Loop: A Novel Therapeutic Target of Photothermal Therapy in Glioblastoma Multiforme
Yaodong Chen1, Pengfei Liu2, Peng Sun2, Jian Jiang1, Yuanbo Zhu2, Tianxiu Dong1, Yingzhe Cui2, Yuan Tian2, Tingting An1, Jiuwei Zhang1, Zizhuo Li1 ![]() , Xiuhua Yang1
, Xiuhua Yang1 ![]()
1. Department of Abdominal Ultrasonography, The First Affiliated Hospital of Harbin Medical University, Harbin, 150001, China
2. Department of Magnetic Resonance, The First Affiliated Hospital of Harbin Medical University, Harbin, 150001, China
Received 2018-9-16; Accepted 2019-1-20; Published 2019-2-20
Citation:
Chen Y, Liu P, Sun P, Jiang J, Zhu Y, Dong T, Cui Y, Tian Y, An T, Zhang J, Li Z, Yang X. Oncogenic MSH6-CXCR4-TGFB1 Feedback Loop: A Novel Therapeutic Target of Photothermal Therapy in Glioblastoma Multiforme. Theranostics 2019; 9(5):1453-1473. doi:10.7150/thno.29987. https://www.thno.org/v09p1453.htm
Other stylesAbstract

Glioblastoma multiforme (GBM) has been considered the most aggressive glioma type. Temozolomide (TMZ) is the main first-line chemotherapeutic agent for GBM. Decreased mutS homolog 6 (MSH6) expression is clinically recognized as one of the principal reasons for GBM resistance to TMZ. However, the specific functions of MSH6 in GBM, in addition to its role in mismatch repair, remain unknown.
Methods: Bioinformatics were employed to analyze MSH6 mRNA and protein levels in GBM clinical samples and to predict the potential cancer-promoting functions and mechanisms of MSH6. MSH6 levels were silenced or overexpressed in GBM cells to assess its functional effects in vitro and in vivo. Western blot, qRT-PCR, and immunofluorescence assays were used to explore the relevant molecular mechanisms. Cu2(OH)PO4@PAA nanoparticles were fabricated through a hydrothermal method. Their MRI and photothermal effects as well as their effect on restraining the MSH6-CXCR4-TGFB1 feedback loop were investigated in vitro and in vivo.
Results: We demonstrated that MSH6 is an overexpressed oncogene in human GBM tissues. MSH6, CXCR4 and TGFB1 formed a triangular MSH6-CXCR4-TGFB1 feedback loop that accelerated gliomagenesis, proliferation (G1 phase), migration and invasion (epithelial-to-mesenchymal transition; EMT), stemness, angiogenesis and antiapoptotic effects by regulating the p-STAT3/Slug and p-Smad2/3/ZEB2 signaling pathways in GBM. In addition, the MSH6-CXCR4-TGFB1 feedback loop was a vital marker of GBM, making it a promising therapeutic target. Notably, photothermal therapy (PTT) mediated by Cu2(OH)PO4@PAA + near infrared (NIR) irradiation showed outstanding therapeutic effects, which might be associated with a repressed MSH6-CXCR4-TGFB1 feedback loop and its downstream factors in GBM. Simultaneously, the prominent MR imaging (T1WI) ability of Cu2(OH)PO4@PAA could provide visual guidance for PTT.
Conclusions: Our findings indicate that the oncogenic MSH6-CXCR4-TGFB1 feedback loop is a novel therapeutic target for GBM and that PTT is associated with the inhibition of the MSH6-CXCR4-TGFB1 loop.
Keywords: glioblastoma multiforme, MSH6-CXCR4-TGFB1 feedback loop, photothermal therapy, magnetic resonance imaging, Cu2(OH)PO4
Introduction
Glioblastoma multiforme (GBM) has been considered the most aggressive glioma of the central nervous system, with a median overall survival barely exceeding 16 months and a 5-year survival rate of less than 5% [1,2]. Despite the existence of multimodal therapies, typically consisting of maximal resection combined with adjuvant radiotherapy and chemotherapy, the clinical outcome of GBM patients remains miserable, with a high recurrence rate [3,4]. The primary reason is the singular feature of invasive growth, which impedes the ability of surgical ablation to remove the GBM completely [5]. Therefore, elucidating the key mechanisms responsible for GBM progression is critical for identifying effective therapeutic approaches.
The epithelial-to-mesenchymal transition (EMT) has been considered one of the major factors that account for the invasive property of GBM cells [6]. EMT is a complex biological process involving multiple biochemical steps that convert a polarized sheet of epithelial cells to a mesenchymal phenotype, which is characterized by weak cell adhesion and enhanced migratory and invasive capabilities [7]. In addition, several studies have shown that EMT is also related to morphological alterations, resistance to chemotherapy/radiotherapy, anoikis and stem-like properties [8]. Various cytokines, such as epidermal growth factor (EGF) and transforming growth factor (TGF), initially trigger EMT in cancer cells. In response to these specific ligands, receptor-mediated signaling pathways trigger the activation of multiple intracellular transcription factors, such as members of the SNAI, TWIST and ZEB families, which can regulate changes in gene expression patterns that orchestrate EMT [9]. These transcription factors were defined as EMT regulatory factors. Significantly, these EMT regulatory factors can affect proliferation, cell cycle, stemness, angiogenesis and antiapoptotic effects, in addition to EMT-related migration and invasion [10,11].
Standard therapy for GBM is generally acknowledged as the maximum safe open resection followed by adjuvant radio-/chemotherapy with temozolomide (TMZ), which is the most important first-line chemotherapeutic agent against this malignancy [12]. TMZ, as an alkylating agent, can induce approximately 13 DNA adducts, among which the minor adduct O6-methylguanine (O6-meG) possesses the strongest cytotoxicity. O6-meG mispairs with thymine (T) to form O6-meG/T mismatches, which can activate the mismatch repair system (MMR) to perform futile repair cycles [13]. Subsequently, secondary lesions (single-stranded DNA gaps) accumulate in this incorrect repair process, leading to DNA double- strand breaks (DSBs) that ultimately induce apoptosis or senescence in GBM cells [14]. Nevertheless, GBM inevitably recurs in a TMZ-resistant form after initial treatment. It is generally recognized by clinicians that the decreasing MSH6 (an important component for MMR) in GBM can weaken the treatment outcome of TMZ [15,16]. However, many studies have already verified that mutations in MSH6 frequently occur during TMZ treatment, which seriously impairs the effects of MSH6 [17]. Therefore, some investigators have intended to overexpress MSH6 to improve TMZ efficacy. However, such strategies have ignored an important question: what are the functions of MSH6 in addition to mismatch repair? This work is the first to confirm that MSH6 is an oncogene that can promote gliomagenesis, proliferation (G1 phase), migration and invasion (EMT), stemness, angiogenesis and antiapoptotic effects through the MSH6-CXCR4- TGFB1 feedback loop, which could impact EMT regulatory factors in GBM.
Recently, image-guided theranostics have been considered as an emerging strategy for high-accuracy tumor treatment via visualization of the targeted region [18]. In particular, the use of single-matter with both intrinsic imaging and therapeutic capabilities has attracted increasing interest. Such a “one-for-all” strategy represents a facile/time-saving synthesis and avoids the in vivo side effects caused by complicated compositions [19]. With respect to therapeutic techniques, phototherapy currently attracts considerable attention, typically photothermal therapy (PTT), which relies on a photoabsorptive nanomaterial to trigger hyperthermia and destroy cancer tissues [20]. To date, many imaging methods have been combined with cancer treatment to enhance accuracy. Among these medical imaging techniques, magnetic resonance imaging (MRI) is the main imaging modality for the clinical diagnosis of GBM because of its unique craniocerebral imaging ability [21]. Hence, this work aims to implement MRI-guided PTT for GBM by employing a single-matter-based theranostic agent.
Cu2(OH)PO4 appears to be a potential candidate for MRI-guided PTT. First, the strong photoabsorption region of Cu2(OH)PO4 locates in 800-1400 nm, satisfying the biological PTT window in the near infrared (NIR) region [22]. Second, Cu2+ is a paramagnetic ion with an unpaired 3d electron and thus many copper-containing compounds (such as CuO) could be used as MRI contrast agents in T1WI [23]. Nevertheless, little effort has been devoted to the synthesis of Cu2(OH)PO4-based nanomaterials for biomedical applications due to difficulties in controlling morphologies, dimensions, and surface modifications, all of which are required for in vivo administration. Fortunately, Guo et al. previously fabricated 4 nm polyacrylic acid (PAA)-coated Cu2(OH)PO4 nanoparticles (Cu2(OH)PO4@PAA) with a desirable PTT effect [24]. We noticed that the steady PTT temperature of Cu2(OH)PO4@PAA could reach up to 52 °C under 1064 nm irradiation, which is suitable for hyperthermic ablation of GBM. In addition, the dimensions of Cu2(OH)PO4@PAA have great potential for passing through the blood-brain barrier [25]. More importantly, the application of Cu2(OH)PO4 as an MRI contrast agent has not been investigated. However, if this development occurs, Cu2(OH)PO4 will doubtless facilitate craniocerebral imaging of GBM by MRI. Therefore, this work evaluated the availability of Cu2(OH)PO4@PAA for MRI-guided PTT in GBM.
Results and Discussion
MSH6 was highly expressed in human GBM
Bioinformatics, as an important tool for cancer research, can provide predictions and guidance for our study [26]. To obtain objective information, we used online analysis tools. After retrieving online databases, it was determined that the MSH6 protein was principally distributed in the nucleus (Figure 1A). In addition, GBM/normal differential expression analysis showed that MSH6 expression in GBM and lower grade glioma (LGG; WHO grades I-III) tissues was higher than that in normal tissues at the mRNA level, while GBM showed significantly higher protein levels (Figure 1B, C). Patient survival analysis revealed that high MSH6 expression predicted poor overall survival (OS) and disease-free survival (DFS) for all gliomas and LGG, rather than for GBM alone (Figure 1D). This phenomenon is likely due to the influence of TMZ, which can utilize the cell damage repair function of MSH6 to exert antitumor effects or induce mutations in MSH6 [13,17]. These results suggested that MSH6 might exert vital functions to promote the progression of GBM.
Figure 1
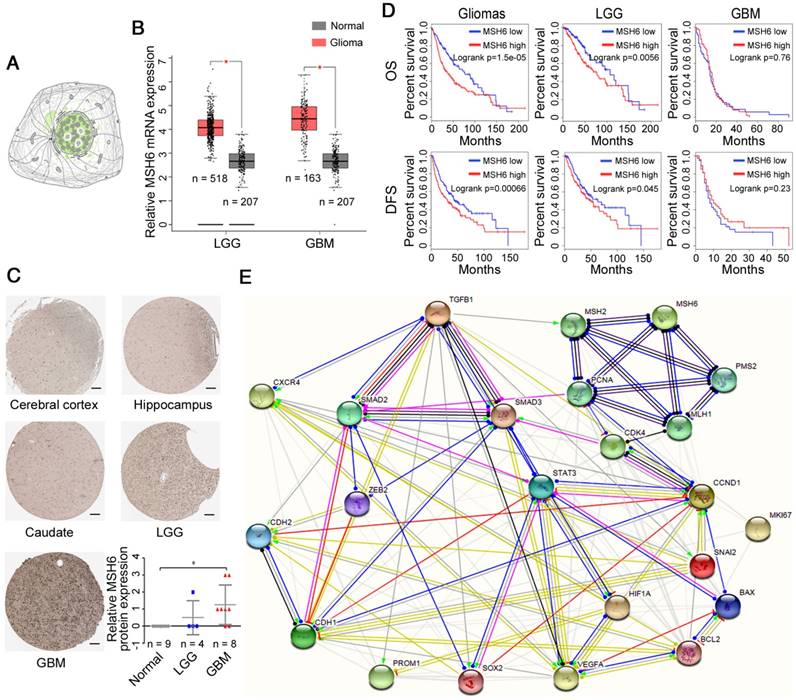
Prediction of MSH6 expression level and functions. (A) The distribution of MSH6 protein in GBM cells (Green, The Human Protein Atlas). (B) MSH6 mRNA levels in normal, LGG and GBM tissues (GEPIA). (C) MSH6 protein levels in normal, LGG and GBM tissues (The Human Protein Atlas) (scale bar represents 100 μm). (D) Patient survival analysis of gliomas, LGG and GBM (GEPIA). (E) Protein-protein interactions of MSH6 and relevant factors (STRING). Error bars represent the standard deviation. * P < 0.05.
Then, we investigated the relationships between MSH6 and some pivotal functional genes using Pearson correlation analysis. The results revealed that a group of genes exhibited positive correlations with the expression of MSH6 at the mRNA level, including the proliferation-associated genes Ki67, AURKA and AURKB; the cell cycle-associated genes Cyclin A2, Cyclin B1, Cyclin D1 and Cyclin E1; the antiapoptosis- associated gene Bcl-2; the EMT-associated genes Slug, ZEB1, ZEB2, E-cadherin and N-cadherin; the stem cell-associated genes CD133, SOX2, Nestin, HMMR, HOXA7 and HOXA10; the angiogenesis-associated gene HIF1A; and representative genes of several vital signaling pathways, including STAT3, ERK2, ERK1, JNK, p38, p65, Smad2, and Smad3 (Figure S1). To further verify the relationships between MSH6 and the proteins encoded by the aforementioned genes, we used cBioPortal to analyze the changes in these genes at the protein level before and after MSH6 alteration. The results showed that the expression of p-EGFR (Tyr1173) and p-STAT3 (Tyr705) significantly decreased, while that of E-cadherin, Cyclin E1 and Bcl-2 increased after MSH6 alteration (Table S1). All of these results indicated that MSH6 may be involved in regulating the biological behaviors of tumor cells, which are manipulated by the corresponding functional genes.
Furthermore, to clarify how MSH6 cooperates with these factors to promote the progression of GBM, we employed STRING to analyze the interactions of MSH6 with the proteins encoded by the aforementioned 40 genes and developed a relationship map (Figure S2). Then, we removed some irrelevant factors based on the previous mRNA and protein results to generate a clearer relationship map (Figure 1E). These predictive results were employed to guide the following experiments.
Knockdown of MSH6 significantly suppressed the growth, migration and invasion of GBM cells
To explore the influences of MSH6 suppression on the malignant biological behaviors of GBM, we employed siRNA-MSH6 (si-MSH6) to inhibit MSH6 in the U87MG, U251 and T98G cell lines. Cell growth assays revealed that si-MSH6 cells grew much more slowly than siRNA-negative control (si-NC) cells, and these significant differences started from the second day (Figure 2A and Figure S3A). Cell cycle analysis showed remarkable G1 arrest in GBM cells with silenced MSH6 (Figure 2B and Figure S3B). Apoptosis assays showed significant apoptosis after silencing MSH6 in GBM cells (Figure 2C and Figure S3C).
In many cases, genes related to antiapoptotic effects can impact the motility of tumor cells [27]. Thus, we examined the changes in motility after silencing MSH6 in GBM cells using Transwell assays, which revealed that MSH6 knockdown remarkably reduced the migration and invasion abilities of GBM cells (Figure 2D, E and Figure S3D, E).
Overexpression of MSH6 promoted gliomagenesis and the progression of GBM
The expression of MSH6 in U87MG, U251 and T98G cells was detected using western blot analysis, and the results revealed that the MSH6 protein levels in U87MG cells were higher than those in U251 and T98G cells (Figure S4). To further investigate the changes in the biological behaviors of GBM cells induced by MSH6 overexpression, stable cell lines were established by infecting GBM cells with a negative control lentivirus (referred to as U251-Con and T98G-Con) or an MSH6 lentivirus (referred to as U251-MSH6 and T98G-MSH6). After selection, stable clones were obtained, and the western blot results confirmed remarkable MSH6 overexpression (Figure S5). The growth curve assays indicated that GBM-MSH6 cell proliferation was faster than that of control cells, and an obvious significant difference emerged starting on the third day (Figure 3A). Accordingly, colonies yielded from GBM-MSH6 cells were more competitive in quantity and size compared to those from GBM-Con cells (Figure 3B and Figure S6). These phenomena may be due to the acceleration of the G1 phase of the cell cycle after elevating MSH6 (Figure 3C).
To investigate whether MSH6 could influence gliomagenesis in vivo, U251-Con, U251-MSH6, T98G- Con or T98G-MSH6 cells were injected subcutaneously in the flank of nude mice. Tumor growth was monitored. Earlier tumor formation was noticed in the U251-MSH6 group (5.32 ± 0.26 days) than in the U251-Con group (8.53 ± 0.78 days). As shown in Figure 3D and Figure S7, the average tumor weight and volume of the U251-MSH6 group were approximately 2.5-fold higher than those of the U251-Con group at 4 weeks postinoculation. Notably, T98G, which is not tumorigenic in nude mice (identified by the American Type Culture Collection), acquired a tumorigenic ability after overexpressing MSH6, while the cells of the T98G-Con group exhibited necrosis. Hematoxylin and eosin (H&E) staining is a pathological diagnostic method that is currently recognized as one of the most objective and direct methods for tumor diagnosis and analysis. Thus, H&E staining was used to determine tumor growth in all groups and incremental angiogenesis and microangiogenesis in the U251-MSH6 group (Figure 3E and Figure S8). In addition, clinical ultrasonic imaging (USI) and MRI were employed to monitor the evolution of the tumors and changes in certain tumor characteristics that can be detected by specific imaging techniques. B-mode ultrasonography showed that the tumors of the U251-MSH6 group grew faster than those of the U251-Con group, and the internal echogenicity of the U251-MSH6 group was slightly higher than that of the U251-Con group. Additionally, color Doppler flow imaging (CDFI) and color power angiography (CPA) revealed increased angiogenesis and microangiogenesis, respectively, in the U251-MSH6 group, which were consistent with the results of the H&E staining. Ultrasonic elastosonography (USE) results clearly showed a hard nature of both the U251-MSH6 group and the U251-Con group (Figure 3F and Figure S9). In the MRI observation, the average tumor size of the U251-MSH6 group was remarkably greater than that of the U251-Con group, which is consistent with the above USI results. In addition, tumors of the U251-Con group and the U251-MSH6 group all exhibited low signal intensity in T1WI, high signal intensity in T2WI/T2-SPIR, and low-intermediate signal intensity in T2-FLAIR. All of these observations were consistent with the conventional MRI performance of GBM (Figure 3G and Figure S10). No tumors formed in the T98G-Con group, while the tumors of the T98G-MSH6 group showed imaging features similar to those of the U251-MSH6 group. We found no obvious necrosis inside the tumors, even when the tumors reached a size up to 2 cm, in USI, MRI or H&E staining, which might be related to the enhanced angiogenesis and microangiogenesis induced by MSH6.
Figure 2
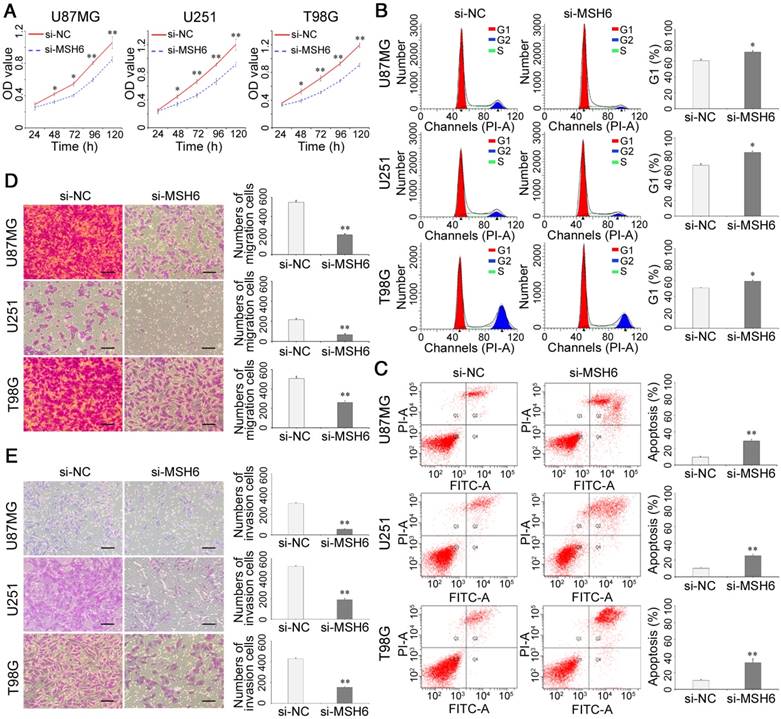
The growth, migration and invasion of GBM cells after silencing MSH6. (A) Cell viability assays, (B) cell cycle assays, (C) cell apoptosis assays, (D) cell migration assays (scale bar represents 200 μm), and (E) cell invasion assays (scale bar represents 200 μm) of U87MG, U251 and T98G cells after silencing MSH6. Error bars represent the standard deviation, n = 3. * P < 0.05, ** P < 0.01.
Figure 3
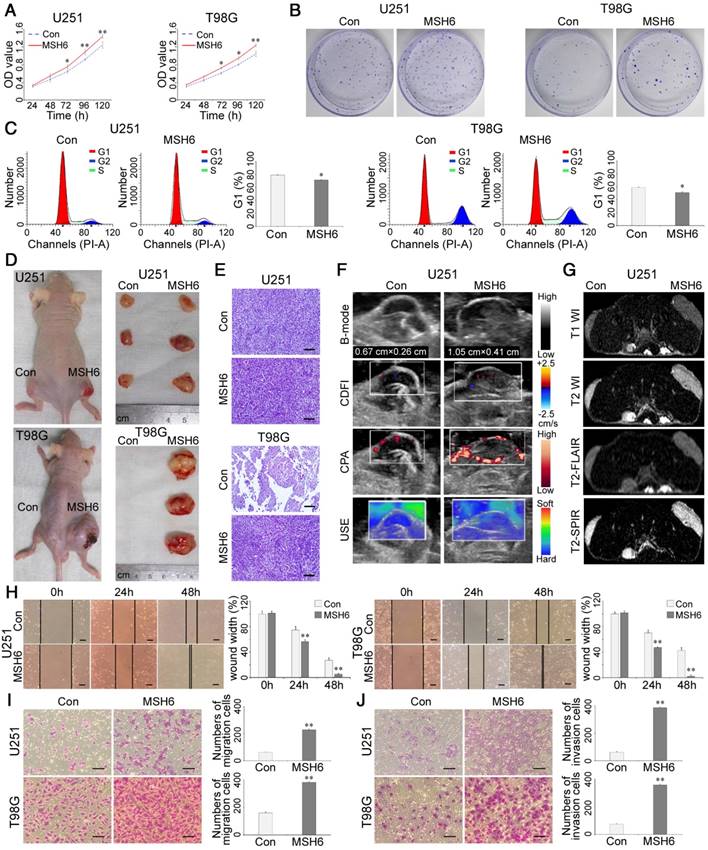
Gliomagenesis and the progression of GBM after overexpressing MSH6. (A) Cell viability assays, (B) colony-forming assays, and (C) cell cycle assays of U251 and T98G cells after overexpressing MSH6. (D) Representative photographs of U251-Con/MSH6 or T98G-Con/MSH6 tumor-bearing nude mice and corresponding tumors at 4 weeks postinoculation. (E) H&E staining of tumors from U251-Con/MSH6 or T98G-Con/MSH6 tumor-bearing nude mice (scale bar represents 100 μm). (F) USI and (G) MRI of tumors from U251-Con/MSH6 tumor-bearing nude mice. (H) Wound-healing assays (scale bar represents 200 μm), (I) cell migration assays (scale bar represents 200 μm), and (J) cell invasion assays (scale bar represents 200 μm) of U251 and T98G cells after overexpressing MSH6. Error bars represent the standard deviation, n = 3. * P < 0.05, ** P < 0.01.
MSH6 has been demonstrated to be associated with migration and invasion after knockdown of MSH6 in GBM cells (Figure 2D, E and Figure S3D, E). Therefore, we explored whether MSH6 overexpression could enhance the cell motility of GBM cells. Positive results were obtained by conducting wound- healing assays, in which GBM-MSH6 cells covered the scratched “wound” faster than GBM-Con cells (Figure 3H). Furthermore, Transwell assays confirmed that GBM-MSH6 cells possessed stronger migration and invasion abilities than GBM-Con cells (Figure 3I, J). Notably, the tumors in the U251-MSH6 group and the T98G-MSH6 group infiltrated and destroyed the surrounding skin, which could partly reflect their enhanced invasiveness (Figure 3D).
Figure 4
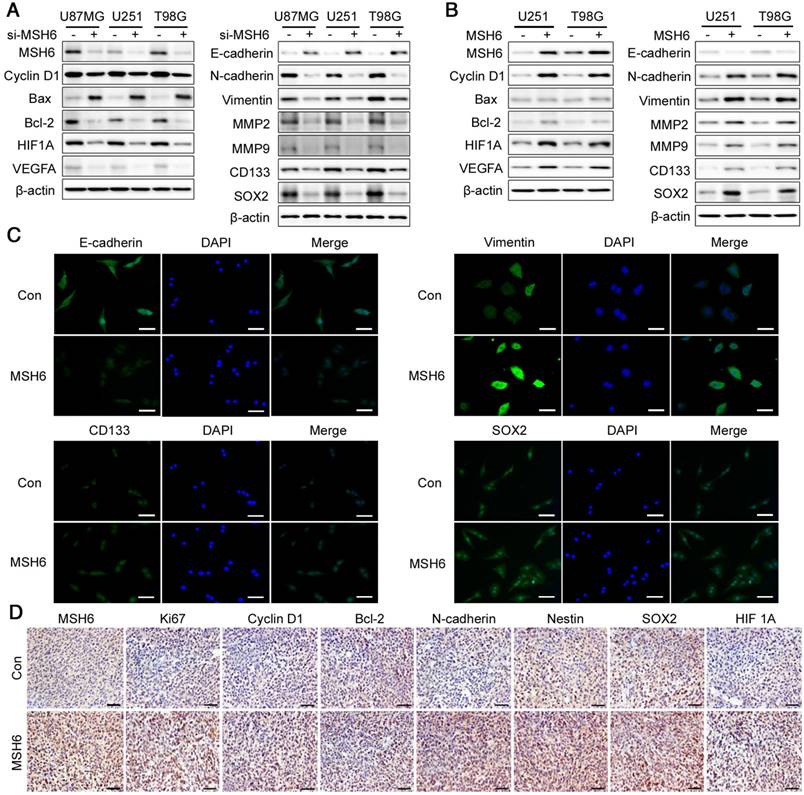
The functions of MSH6 at the molecular level. (A) The expression of some typical regulatory factors and markers was detected by western blot assays after silencing MSH6 or (B) overexpressing MSH6. (C) The protein expression and subcellular localization of E-cadherin, Vimentin, CD133 and SOX2 were detected using immunofluorescence assays in U251 cells (scale bar represents 100 μm). (D) The expression of some representative tumor markers was determined using immunohistochemistry assays in xenograft tumor tissues from U251-Con and U251-MSH6 cells (scale bar represents 50 μm).
MSH6 promoted the progression of GBM at the molecular level
To further elucidate the functions of MSH6 in GBM at the molecular level, we detected a series of typical regulatory factors and markers after silencing or elevating MSH6, including the following: the G1 phase regulatory factor Cyclin D1; the proapoptotic factor Bax; the antiapoptotic factor Bcl-2; the epithelial marker E-cadherin; the mesenchymal markers N- cadherin, Vimentin, MMP2, and MMP9; the stemness markers CD133 and SOX2; and the angiogenesis regulatory factors HIF1A and VEGFA. The levels of Cyclin D1, Bcl-2, N-cadherin, Vimentin, MMP2, MMP9, CD133, SOX2, HIF1A and VEGFA were decreased in MSH6-silenced GBM cells compared to those in the negative control, whereas the proapoptotic factor Bax and the epithelial marker E-cadherin remarkably increased (Figure 4A and Figure S11). Conversely, MSH6 overexpression exerted the opposite effects on the above factors and markers, except Bax, which remained constant, as there was no obvious apoptosis after oncogene overexpression (Figure 4B). Immunofluorescence assays further validated the increases in Vimentin, CD133 and SOX2 and the decreases in E-cadherin in U251-MSH6 and T98G-MSH6 cells (Figure 4C and Figure S12). The immunohistochemical assay of tumor tissues segmented from the U251-Con/MSH6 tumor-bearing nude mice showed that Cyclin D1, Bcl-2, N-cadherin, Nestin, SOX2, and HIF1A were increased in the U251-MSH6 group. Simultaneously, the well-known proliferation marker Ki67 increased after elevating MSH6 (Figure 4D). These results further indicate that MSH6 is an oncogene that promotes proliferation, the G1 phase of the cell cycle, EMT-related migration and invasion, stemness, angiogenesis and antiapoptotic effects by regulating the expression of corresponding regulatory factors and markers at the molecular level.
HIF1A is a well-known factor involved in the adaptive metabolic response to hypoxia [28]. However, we noticed an interesting phenomenon that MSH6 increased the accumulation of HIF1A protein when GBM cells were grown under normoxia. To our knowledge, protein accumulation is a common result of protein synthesis and degradation [29], and we believe that HIF1A is no exception. Previous researches have indicated that HIF1A is not directly accumulated by hypoxia but by the decreased intracellular pH value, which can be induced by hypoxic conditions [30]. Analogously, aerobic glycolysis (the Warburg effect), as one of the major patterns of energy supply in cancer cells, can also decrease the intracellular pH value [31]. Then, the low pH value promotes HIF1A protein synthesis by triggering specific signaling pathways or transcription factors, such as the STAT3 signaling pathway, its most common collaborator [32,33]. In addition, the low pH value can inhibit the degradation of HIF1A protein by decreasing its hydroxylation [34]. In this study, the rapid proliferation of GBM cells induced by MSH6 was undoubtedly accompanied by a high metabolism, which could keep the pH value constant or further reduced. Under this condition, the degradation of HIF1A protein was likely to remain constant or slow down. Simultaneously, MSH6 overexpression might promote the synthesis of HIF1A protein by activating the STAT3 signaling pathway. Ultimately, increased synthesis and constant or reduced degradation resulted in the incremental protein accumulation of HIF1A under normoxia.
MSH6 regulated the STAT3 and Smad2/3 signaling pathways
Given the remarkable effects of MSH6 on gliomagenesis and the progression of GBM cells, the analysis of signaling pathways related to gliomagenesis and tumor progression that may be activated by MSH6 was conducted through detecting the expression of the AKT, STAT3, ERK1/2, JNK, p38, p65, and Smad2/3 as well as their corresponding phosphorylated forms using western blot assays [35-37]. The results indicated that silencing MSH6 decreased the phosphorylation of STAT3 and Smad2/3 (Figure 5A, Figure S13 and Figure S14), whereas MSH6 overexpression increased the phosphorylation of these factors (Figure 5B and Figure S15). Immunofluorescence assays revealed increases in p-STAT3 and p-Smad2/3, especially for nuclear translocation, after elevating MSH6 in U251 and T98G cells (Figure 5C). These results indicated that MSH6 could promote gliomagenesis and the progression of GBM by regulating the STAT3 and Smad2/3 signaling pathways.
MSH6 regulated gliomagenesis and the progression of GBM through Slug and ZEB2
EMT is precisely orchestrated by EMT regulatory factors of the SNAI, TWIST and ZEB families, and the most important factors are Snail, Slug, Twist, ZEB1 and ZEB2 [38]. These transcription factors have been shown to regulate EMT, tumorigenesis, proliferation, the cell cycle, antiapoptotic effects, stemness and angiogenesis. Additionally, many studies have demonstrated that several signaling pathways can lead to the activation of these transcription factors [39,40]. Considering that p-STAT3 and p-Smad2/3 could be readily affected by MSH6, we hypothesized that MSH6 may regulate the aforementioned functions through some of these transcription factors, which were impacted by p-STAT3 and p-Smad2/3. Therefore, we first detected the transcription and translation of the transcription factors Snail, Slug, Twist, ZEB1 and ZEB2 after silencing or overexpressing MSH6. The expression of Slug and ZEB2 was obviously influenced by MSH6 at the mRNA and protein levels (Figure 6A, B, Figure S16 and Figure S17). In addition, immunofluorescence showed an increase in Slug and ZEB2 in the nuclei of U251-MSH6 and T98G-MSH6 cells (Figure 6C and Figure S18), suggesting that MSH6 could regulate the expression of Slug and ZEB2. Previously, Yang et al. and Qi et al. revealed that both Slug and ZEB2 could regulate proliferation, the cell cycle, apoptosis, migration and invasion in GBM cells, showing functions similar to MSH6 [41, 42]. On the basis of the above results, we hypothesized that MSH6 regulated gliomagenesis and the progression of GBM through Slug and ZEB2. To confirm this possibility, we then knocked down Slug or ZEB2 using siRNA in MSH6-overexpressing GBM cells. As expected, silencing Slug or ZEB2 abolished the proliferation, migration and invasion induced by MSH6 in U251-MSH6 and T98G-MSH6 cells (Figure 6D, E, F and Figure S19). In addition, at the molecular level, western blot results showed that the expression of representative markers of the cell cycle, antiapoptotic effects, EMT, stemness and angiogenesis induced by increased MSH6 was also reversed by silencing Slug or ZEB2 (Figure 6G). These results demonstrated that Slug and ZEB2 could mediate the regulating effects of MSH6 on proliferation, the cell cycle, antiapoptotic effects, migration, invasion, stemness and angiogenesis of GBM cells.
MSH6, CXCR4 and TGFB1 established a triangular feedback loop
TGFB1 has been demonstrated to be a vital regulator of EMT [43]. Hence, we next explored the relationship between MSH6 and TGFB1. Silencing MSH6 decreased the expression of TGFB1 (Figure 7A and Figure S20), whereas MSH6 overexpression increased the expression of this factor (Figure 7B). Immunofluorescence assays also verified the increase in TGFB1 in U251 and T98G cells after MSH6 overexpression (Figure 7C and Figure S21). Zhu et al. reported that CXCR4 could regulate the expression of TGFB1, and our study confirmed this finding (Figure 7D) [44]. Moreover, we discovered that CXCR4 and MSH6 could promote the expression of each other (Figure 7A-D, Figure S20 and Figure S21). Therefore, we hypothesized that MSH6 could impact TGFB1 by regulating CXCR4. This hypothesis was confirmed by the fact that the increase in TGFB1 could be reversed when CXCR4 was inhibited in GBM-MSH6 cells (Figure 7E).
Then, we investigated the effect of TGFB1 on the regulation of MSH6. It was found that 10 ng/mL TGFB1 could inhibit the expression of MSH6, p-STAT3, and Slug while increasing the expression of CXCR4, p-Smad2/3, and ZEB2. Furthermore, TGFB1 had no discernable effect on these factors at concentrations lower than 10 ng/mL (Figure 7F). To determine whether 10 ng/mL TGFB1 could restrain the expression of p-STAT3 and Slug by impeding MSH6, we introduced 10 ng/mL TGFB1 into U251-MSH6 and T98G-MSH6 cells. As expected, the overexpression of MSH6 reversed the inhibitory effect of TGFB1 on p-STAT3 and Slug (Figure 7G).
Figure 5
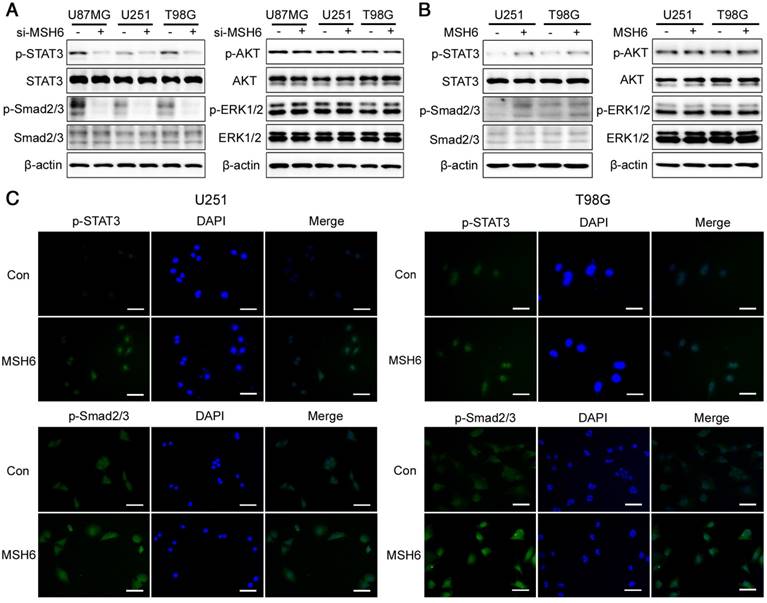
The STAT3 and Smad2/3 signaling pathways were regulated by MSH6. (A) The expression of some signaling pathway-related proteins was detected by western blot assays after silencing MSH6 or (B) overexpressing MSH6. (C) The protein expression and subcellular localization of p-STAT3 and p-Smad2/3 were detected using immunofluorescence assays in U251 and T98G cells (scale bar represents 100 μm).
Figure 6
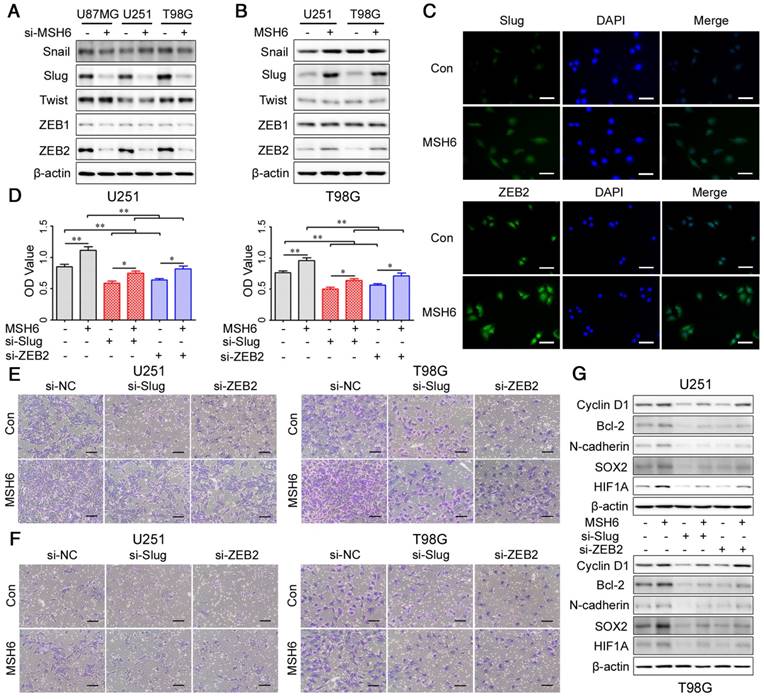
The EMT regulatory factors Slug and ZEB2 were regulated by MSH6. (A) The expression of five EMT regulatory factors was evaluated by western blot assays after silencing MSH6 or (B) overexpressing MSH6. (C) The protein expression and subcellular localization of Slug and ZEB2 were detected using immunofluorescence assays in U251 cells (scale bar represents 100 μm). (D) Cell viability assays, (E) cell migration assays (scale bar represents 200 μm) and (F) cell invasion assays (scale bar represents 200 μm) of U251 and T98G cells after silencing Slug or ZEB2 while simultaneously overexpressing MSH6. (G) The expression of some representative tumor markers was detected by western blot assays after silencing Slug or ZEB2 while simultaneously overexpressing MSH6 in U251 and T98G cells. Error bars represent the standard deviation, n = 3. * P < 0.05, ** P < 0.01.
On the basis of the above results, we propose a triangular relationship among MSH6, CXCR4 and TGFB1 as the MSH6-CXCR4-TGFB1 feedback loop. For tumor cells, the theory of “grow-or-go” illustrated that proliferation and invasion are mutually exclusive behaviors [45]. In the MSH6-CXCR4-TGFB1 feedback loop, TGFB1 is more inclined to promote GBM cell invasion, while MSH6 is more inclined to boost proliferation [46, 47]. TGFB1 suppressed MSH6 at high doses, possibly because of the “grow-or-go” behavior of GBM cells.
To detect the effects of Slug and ZEB2 on upstream factors, we inhibited Slug and ZEB2 using the corresponding si-Slug and si-ZEB2 in U87MG, U251 and T98G cells. As shown in Figure 7H, Slug could promote CXCR4, MSH6, TGFB1, p-STAT3, p-Smad2/3 and ZEB2 expression. Unlike Slug, ZEB2 could only regulate p-STAT3 and Slug (Figure 7I). Nonetheless, the increase in ZEB2 could not reverse the expression of p-STAT3 and Slug when 10 ng/mL TGFB1 was used to inhibit MSH6 expression, which suggested that the regulatory capability of ZEB2 for p-STAT3 is weaker than that of MSH6.
The MSH6-CXCR4-TGFB1 feedback loop could be a therapeutic target for GBM
Based on the aforementioned discoveries, this work elucidated a novel molecular mechanism in GBM. Specifically, the oncogenic MSH6-CXCR4- TGFB1 feedback loop can accelerate gliomagenesis, proliferation (G1 phase), migration and invasion (EMT), stemness, angiogenesis and antiapoptotic effects by regulating the p-STAT3/Slug and p- Smad2/3/ZEB2 signaling pathways in GBM (Figure 8A). By employing principal component analysis of GEPIA, the genes MSH6, CXCR4, TGFB1, Slug, ZEB2, Ki67, Cyclin D1, Bcl-2, N-cadherin, CD133, SOX2, and HIF1A were revealed to be effective biomarkers for GBM and could effectively distinguish the brain cortex, LGG and GBM (Figure 8B). Moreover, we demonstrated that the probability of alterations in MSH6, CXCR4 and TGFB1 was very low in GBM (Figure 8C). These discoveries suggested that the MSH6-CXCR4-TGFB1 feedback loop could be regarded as a potential therapeutic target for GBM.
Previous studies have shown that TMZ can treat GBM by utilizing the mismatch repair function of MSH6, while the deficiency of MSH6 decreases the sensitivity of GBM to TMZ [15,16]. Unfortunately, TMZ can induce mutations in MSH6 after long-term treatment, resulting in TMZ insensitivity [17]. This work confirmed that MSH6 is an oncogene. Based on these data, we proposed that TMZ could utilize or inactivate the oncogenic MSH6 to damage GBM. In other words, the oncogenic MSH6 is a vital target of TMZ. Thus, the aforementioned TMZ insensitivity can be attributed to a deficiency of target content.
Figure 7

The MSH6-CXCR4-TGFB1 feedback loop. (A) CXCR4 and TGFB1 expression was evaluated by western blot assays after silencing MSH6 or (B) overexpressing MSH6. (C) The protein expression and subcellular localization of CXCR4 and TGFB1 were detected using immunofluorescence assays in U251 cells (scale bar represents 100 μm). (D) The expression of MSH6-related regulatory factors was evaluated by western blot assays after silencing CXCR4 or (E) simultaneously overexpressing MSH6. (F) The expression of MSH6-related regulatory factors was evaluated by western blot assays after adding different doses of TGFB1. (G) The expression of MSH6, p-STAT3, STAT3 and Slug was evaluated by western blot assays after adding 10 ng/mL TGFB1 while simultaneously overexpressing MSH6. (H) The expression of MSH6-related regulatory factors was evaluated by western blot assays after silencing Slug or (I) ZEB2 in U87MG, U251, and T98G cells.
Figure 8
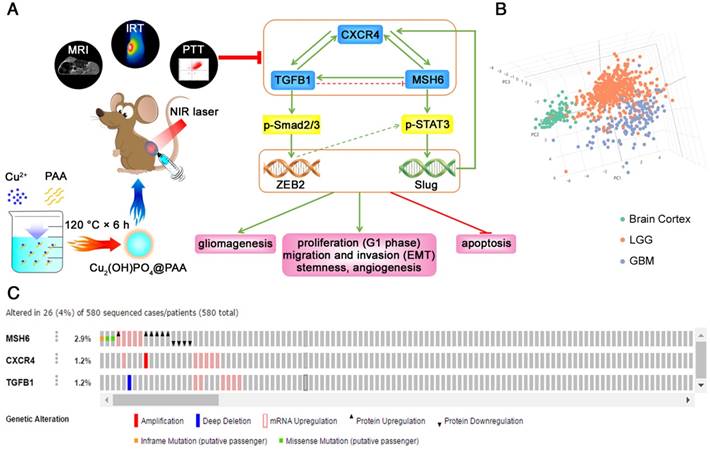
The MSH6-CXCR4-TGFB1 feedback loop as a therapeutic target for GBM. (A) Schematic illustration of the synthesis method and theranostic functions of Cu2(OH)PO4@PAA (left) and the molecular mechanism of MSH6-facilitated gliomagenesis and progression (right). (B) Principal component analysis of the MSH6-related genes set in the normal brain cortex, LGG and GBM (GEPIA). (C) The alterations of MSH6, CXCR4 and TGFB1 in GBM tissues (cBioPortal).
Developing a more effective multitarget treatment is a better strategy than overexpressing MSH6 to increase TMZ efficacy. Our previous research revealed that GBM can be eliminated by hyperthermia through inhibiting p-STAT3 [48]. Additionally, based on the results of a clinical trial of 30 patients with malignant glioma (WHO grades III-IV), Sun et al. indicated that radiofrequency hyperthermia could induce growth retardation, growth termination or even necrosis of malignant gliomas [49]. Moreover, Jin et al. discovered that hyperthermia depressed the proliferation and invasion of mouse malignant melanoma by inhibiting TGFB1 [50]. Considering that the MSH6-CXCR4-TGFB1 feedback loop was the upstream regulator of p-STAT3, we hypothesized that PTT (a new technology for hyperthermia) could restrain GBM by inhibiting the MSH6-CXCR4-TGFB1 feedback loop.
Preparation and characterization of the new MRI contrast agent Cu2(OH)PO4@PAA
To implement imaging-guided PTT for GBM, we need to fabricate a theranostic nanoparticle. Cu2(OH) PO4 is a “one-for-all” multifunctional theranostic agent that combines both tumor imaging and therapy. In addition, we aimed to treat GBM with Cu2(OH)PO4 because the temperature produced is suitable for GBM and mild for the brain [51]. Moreover, the potential MRI capability of Cu2(OH)PO4 could be expected because it contains paramagnetic ion Cu2+.
Herein, PAA-modified Cu2(OH)PO4 nanoparticles were synthesized according to Guo's report [24]. Figure 9A, B displays transmission electron microscopy (TEM) images of Cu2(OH)PO4@PAA nanoparticles 4 nm in size (Figure 9C). The zeta-potential value of Cu2(OH)PO4@PAA was -49.72 mV. The X-ray diffraction (XRD) pattern shown in Figure 9D indicated a well index to the standard Cu2(OH)PO4 (JCPDS file no. 360404). The success of surface decoration with PAA was proven by Fourier transformation infrared spectroscopy (FT-IR) analysis (Figure 9E). The peaks at 2940, 1713, 1584, and 1411 cm-1 are ascribed to the C-H stretching vibration mode, -COOH stretching, COO- asymmetric stretching and COO- symmetric stretching, respectively, all of which originate from the PAA ligand [52]. The broadened peak at 1055 cm-1 represents the stretching vibration mode of phosphate ions from Cu2(OH)PO4 [53]. Figure 9F presents the absorbance spectra of Cu2(OH)PO4@PAA powder. The typical photoabsorption band is located at 800-1300 nm, which includes the entire PTT biological treatment area. We next performed a photothermal conversion test, which revealed that Cu2(OH)PO4@ PAA aqueous dispersions had more obvious time- dependent and concentration-dependent temperature increments compared with those of the pure water control, indicating the photothermal role of Cu2(OH)PO4@PAA (Figure 9G, H).
MRI, a prevailing bioimaging technique, has been widely used for the clinical diagnosis of GBM. To obtain a clear diagnosis of GBM, MRI contrast agents are typically applied to enhance the visualization of GBM on MRI. To date, Gd3+/Mn2+-based T1-weighted and Fe3O4-based T2-weighted MRI contrast agents have been widely investigated [54]. Notably, Cu2+ is a paramagnetic ion that possesses an unpaired 3d electron. As such, copper compounds may also be used as MRI contrast agents. Perlman et al. reported that CuO NPs could shorten the T1 relaxation time, leading to bright contrast in T1WI [55]. Hence, the analog Cu2(OH)PO4@PAA holds potential as an MRI contrast agent. However, to the best of our knowledge, the exploration of Cu2(OH)PO4 as an MRI contrast agent has not yet been reported. In this work, we found that the longitudinal relaxivity (r1) of Cu2(OH)PO4@PAA was 0.62 mM-1s-1 (Figure 9I), which is superior to that of CuO NPs (0.38 mM-1s-1) [55]. Considering that materials that shorten the T1 of protons in water generally possess effective contrast-enhanced capability in T1WI, we detected this potential and found that the T1 signal intensity was enhanced by Cu2(OH)PO4@PAA and positively correlated with its concentration in T1WI (Figure 9J and Figure S22). This effect is a prerequisite for the in vivo application of MRI contrast. Therefore, we then assessed the MR imaging capability of Cu2(OH) PO4@ PAA on U251 tumor-bearing mice. MRI signals of the tumor region could be clearly identified starting from 3 h after intratumorally injecting the nanoparticles. The area of detectable MRI signals enlarged with time and spread to the whole tumor at 6 h. Then, the MRI signals retreated from the tumor at 12 h and completely vanished at 24 h (Figure 9K). Altogether, we have reasons to believe that Cu2(OH)PO4@PAA can serve as a potential MRI contrast agent.
Figure 9
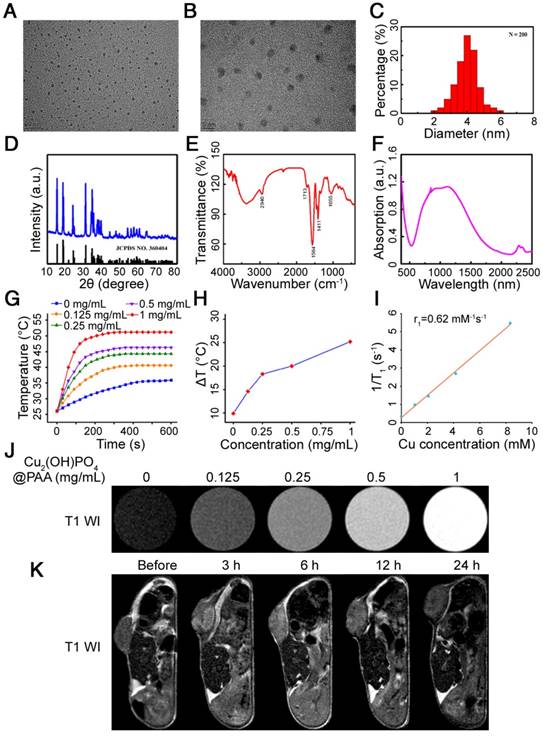
Characterization of Cu2(OH)PO4@PAA. (A, B) TEM analyses, (C) size distribution, (D) XRD pattern, (E) FT-IR spectra, and (F) powder absorbance of Cu2(OH)PO4@PAA. (G) Temperature variations in Cu2(OH)PO4@PAA dispersions (0-1mg/mL) upon laser irradiation. (H) Temperature increments induced by NIR irradiation (10 min) in Cu2(OH)PO4@PAA dispersions (0-1 mg/mL). (I) The r1 of Cu2(OH)PO4@PAA. (J) T1-weighted MRI images of Cu2(OH)PO4@PAA aqueous dispersions (0-1 mg/mL). (K) T1-weighted MRI images of U251 tumor-bearing nude mice before/after intratumoral injection with Cu2(OH)PO4@PAA dispersions.
PTT with Cu2(OH)PO4@PAA restrained the MSH6-CXCR4-TGFB1 feedback loop
It has been reported that tumors can be eliminated by the PTT effect within a short duration of several minutes when the on-site temperature is over a certain threshold [56]. However, an accurate critical temperature for GBM has not been reported. Hence, in this work, we explored the lowest therapeutic temperature for hyperthermia with 10 min that completely killed GBM cells, which was determined to be 50 °C (Figure 10A and Figure S23). Therefore, our subsequent experiments that refer to hyperthermia or PTT all used a therapeutic dose of 50 °C × 10 min.
Identifying the biosafety of Cu2(OH)PO4@PAA plays a critical role for appraising its utilizability in biomedical diagnosis and treatment, particularly its cytotoxicity to the major organs for human drug metabolism, such as the liver and kidney [57,58]. To this end, MTT assays were employed to detect the cytotoxic effects of Cu2(OH)PO4@PAA on human normal hepatic cells (LO2), human renal tubular epithelial cells (HK2), human umbilical vein endothelial cells (HUVECs) and GBM cells. No significant cytotoxicity was observed from the Cu2(OH)PO4@PAA nanoparticles (less than or equal to 1 mg/mL) (Figure S24). Then, the photothermal therapeutic effectiveness of Cu2(OH)PO4@PAA + NIR was evaluated in vitro. MTT assays showed that a water bath at 50 °C and Cu2(OH)PO4@PAA + NIR-mediated PTT could dramatically kill GBM cells, which was in sharp contrast to the intact cells of the other three groups (Figure 10B and Figure S25). To identify the type of cell death, we employed fluorescein isothiocyanate (FITC)-conjugated Annexin V and propidium iodide (PI) staining assays. A water bath at 50 °C and Cu2(OH)PO4@PAA + NIR-mediated PTT could result in the apoptosis of GBM cells (Figure 10C and Figure S26). Then, we investigated the relative molecular mechanisms of PTT for the treatment of GBM. The MSH6-CXCR4-TGFB1 feedback loop and its downstream factors were detected by western blot assays, which confirmed that the expression levels of the members of the MSH6- CXCR4-TGFB1 feedback loop and its downstream factors were all decreased after treatment of 50 °C heating or Cu2(OH)PO4@PAA + NIR-mediated PTT (Figure 10D and Figure S27).
Prior to PTT in vivo, we first investigated the photothermal conversion efficiency of Cu2(OH)PO4@ PAA on U251 tumor-bearing nude mice using infrared thermal imaging (IRT). As shown in Figure 10E, F, under NIR irradiation, the existence of Cu2(OH)PO4@PAA clearly promoted the temperature increment of the tumor region, and the temperature slightly exceeded 50 °C starting from 120 s after NIR irradiation, which was suitable to irreversibly eliminate the GBM tissue. Subsequently, we assessed the therapeutic efficacy of Cu2(OH)PO4@PAA + NIR-mediated PTT on four groups of mice (n = 5, each group). As shown in Figure 10G, the tumors gradually diminished or disappeared after Cu2(OH) PO4@PAA + NIR treatment. In contrast, the tumors in the other three groups grew obviously. These results were further confirmed by the quantified weights and volumes of the tumor specimens removed from the mice on the 14th day after the corresponding treatment (Figure 10H). In addition, effective surveillance after treatment often provides more information for the accurate evaluation of therapeutic efficacy. To reduce complications and the mortality of the mice during monitoring, clinical USI was selected because it is noninvasive, accurate, and demonstrated detection diversity. B-mode ultrasonography revealed that PTT mediated by Cu2(OH)PO4@PAA + NIR could obviously reduce the size of the tumors within 7 days after treatment and even ultimately achieve complete tumor ablation. In contrast, the length and width of the tumors in the other three groups grew rapidly and eventually doubled on the 14th day (Figure S28A). These results were consistent with the results of the macroscopic observations and final pathological specimen analysis (Figure 10G, H), but more detailed and accurate. Moreover, the variation tendencies of angiogenesis and microangiogenesis detected by CDFI and CPA, respectively, were in accordance with that of the tumor size (Figure 10I and Figure S28B). Notably, USE detection indicated tumor softening on the 3rd day after treatment with PTT mediated by Cu2(OH)PO4@PAA + NIR (Figure S28C), which might be attributed to the apoptosis and necrosis of tumors after treatment [59]. Pathological examination is considered the gold standard of therapeutic efficacy evaluation; thus, H&E staining was carried out in the tumor slices to further confirm the above findings. The results were as expected; PTT mediated by Cu2(OH)PO4@PAA + NIR induced the necrosis and apoptosis of numerous tumor cells. In contrast, the tumors of the other three groups grew well, with more angiogenesis and microangiogenesis (Figure 10J and Figure S29). Finally, we extracted proteins from the tumors and studied the antitumor mechanism of Cu2(OH)PO4@PAA + NIR using western blot assays, revealing that the expression levels of the members of the MSH6-CXCR4-TGFB1 feedback loop and its downstream factors were all decreased after treatment by Cu2(OH)PO4@PAA + NIR-mediated PTT (Figure 10K). Overall, in vitro and in vivo experiments demonstrated the prominent therapeutic effects of PTT mediated by Cu2(OH)PO4@ PAA + NIR in GBM, which might be associated with the inhibition of the MSH6-CXCR4-TGFB1 loop.
Of note, there was no obvious body weight loss for mice in the four groups throughout the entire process (Figure S30). Moreover, the H&E results of the major organs, including the heart, lung, liver, kidney and spleen, showed almost no detectable lesions (Figure S31). These results further confirmed the good biocompatibility of Cu2(OH)PO4@PAA nanoparticles and the high biological safety of PTT [60].
To date, surgical operation is still the primary choice for GBM patients. For residual tumor tissues that cannot be completely removed by means of resection due to aggressive growth of GBM, PTT can be considered to assist in eliminating residual tumor during the operation. Applications of PTT for GBM remain in the nascent stage of exploration, and potential limitations or challenges currently focus on the design and preparation of photothermal agents (PTAs) [61]. PTAs need to be elaborately engineered to obtain the following features: excellent biocompatibility, high photothermal conversion efficiency, outstanding real-time fluorescence or photoacoustic imaging ability, powerful chemotherapeutic drug- carrying capacity, excellent ability to penetrate the BBB, and prominent targeted delivery capability [62, 63]. The targeting of PTT to tumor tissues can be achieved through the following aspects: intratumoral injection of PTAs, selective NIR irradiation, and EPR effect and active targeting of PTAs [64]. Considering that the primary purpose of this study was to explore the impact of PTT on the MSH6-CXCR4-TGFB1 loop, we used intratumoral injection and selective NIR irradiation to easily achieve targeted PTT for GBM in ectopic flank tumor models. Going forward, to achieve targeted PTT in orthotopic tumor models or clinical trials of GBM, intravenous injection may be a better strategy to achieve more efficient delivery and improved PTA intratumoral distribution [65] and, hence, better therapeutic effects, although this method requires a more sophisticated PTA design. In addition, researches involving the physiology and pathology of the central nervous system and nanomaterial-tissue interactions are highly desirable to accelerate the clinical translation of PTT for GBM [66]. With the accumulation of these studies and knowledge, PTT or its combination with current therapies may provide promising therapeutic modalities for the treatment of GBM.
Conclusions
In summary, this work provided the first evidence that PTT could effectively treat GBM through restraining the oncogenic MSH6-CXCR4- TGFB1 feedback loop and its downstream factors. We revealed MSH6 overexpression in human GBM tissues and elucidated a novel mechanism of MSH6- mediated gliomagenesis, proliferation (G1 phase), migration and invasion (EMT), stemness, angiogenesis and antiapoptotic effects in GBM. Briefly, the MSH6-CXCR4-TGFB1 feedback loop could accelerate the gliomagenesis and progression of GBM through regulating the p-STAT3/Slug and p-Smad2/3/ZEB2 signaling pathways. The results of the principal component analysis and alteration analysis confirmed that MSH6, CXCR4 and TGFB1 were vital markers of GBM and had low alteration rates, making the MSH6-CXCR4-TGFB1 feedback loop a promising therapeutic target. For the phenomenon of decreasing MSH6-induced TMZ failure in GBM, we concluded that the oncogenic MSH6 is a principal target of TMZ; thus, deficiency in target content will undoubtedly lead to GBM insensitivity to TMZ. PTT as a multitarget treatment possesses great potential for treating GBM. To devise a more controllable and manipulable strategy for the thermal ablation of GBM, this work employed theranostic Cu2(OH)PO4@PAA nanoparticles for MRI-guided PTT. Both in vitro and in vivo experiments confirmed the excellent MRI (T1WI) and PTT functions of the nanoparticles. Significantly, the outstanding therapeutic effects of MRI-guided PTT mediated by Cu2(OH)PO4@PAA + NIR was demonstrated to be associated with the inhibition of the MSH6-CXCR4-TGFB1 loop.
Methods
Bioinformatics prediction
GEPIA (http://gepia.cancer-pku.cn/index.html) was applied to conduct tumor/normal differential expression analysis, patient survival analysis, Pearson correlation analysis and principal component analysis [67]. The Human Protein Atlas (http://www.protein atlas.org) was employed to determine the distribution of the MSH6 protein in cancer cells and to contrast the MSH6 protein levels detected by the MSH6 antibody (CAB009091) in normal brain, LGG and GBM tissues [68]. The cBioPortal for Cancer Genomics (http:// www.cbioportal.org) provided the alterations in MSH6, CXCR4 and TGFB1 in GBM tissues and the differences in protein expression between MSH6- altered and MSH6-unaltered GBM tissues [69]. The prediction of protein-protein interactions was carried out using STRING (https://string-db.org) [70].
Cell lines and animals
Human GBM cell lines (U87MG, U251 and T98G), HUVECs, LO2 cells, and HK2 cells were originally supplied by ATCC (Manassas, VA, USA). U87MG cells, U251 cells, T98G cells, HUVECs, and LO2 cells were maintained in a 37 °C incubator with humidified atmosphere containing 5% CO2 and cultured with Dulbecco's modified Eagle's medium, while RPMI 1640 medium was suitable for HK2 cells. All media were purchased from GE Healthcare (Chicago, IL, USA) and supplemented with 10% FBS. BALB/c nude mice (4-5 weeks old) were supplied by Charles River Japan (Beijing, China) and maintained in a pathogen-free environment. The animal experiment scheme was reviewed and approved by the Committee on the Use of Live Animals in Teaching and Research of Harbin Medical University (Harbin, Heilongjiang, China).
Figure 10
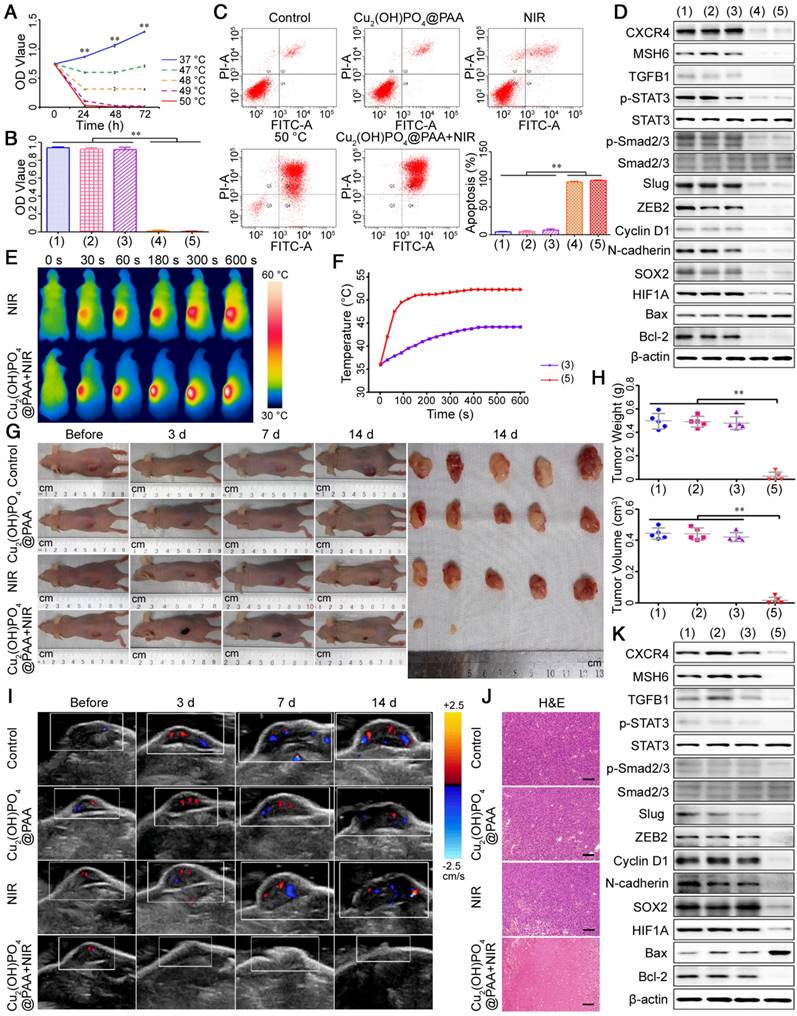
PTT with Cu2(OH)PO4@PAA. (A) Cell viability assays of U251 cells after hyperthermia at different temperatures (n = 3). (B) Cell viability assays (n = 3) and (C) cell apoptosis assays (n = 3) of U251 cells after different treatments. (D) The expression of MSH6-related regulatory factors and markers was evaluated by western blot assays after different treatments in U251 cells. (E) IRT images of U251 tumor-bearing mice and (F) corresponding temperature profiles. (G) Representative photographs of U251 tumor-bearing mice after different treatments and corresponding tumors on the 14th day. (H) The weights and volumes of tumors from U251 tumor-bearing mice after different treatments (n = 5). (I) CDFI images of U251 tumor-bearing mice before and 3 days, 7 days and 14 days after different treatments. (J) H&E staining of tumors from U251 tumor-bearing mice after different treatments (scale bar represents 100 μm). (K) The expression of MSH6-related regulatory factors and markers from U251 tumor-bearing mice evaluated by western blot assays after different treatments. Groups: (1) control; (2) Cu2(OH)PO4@PAA; (3) NIR; (4) 50 °C (water bath); and (5) Cu2(OH)PO4@PAA + NIR. Error bars represent the standard deviation. ** P < 0.01.
Transient transfection of siRNAs
Silencing of MSH6, CXCR4, Slug and ZEB2 was achieved by transfecting specific siRNAs (GenePharma, Shanghai, China), which required the use of the Lipofectamine 2000 reagent (Invitrogen, Carlsbad, California, USA). The sequences of the siRNAs are shown in Table S2. After transfection, the cells were cultured for 48 h before subsequent examination or treatment. In addition, we selected the best siRNA, which was used for the following experiments, for each gene using western blot, as follows: siRNA- MSH6-3, siRNA-CXCR4-3, siRNA-Slug-1 and siRNA- ZEB2-2 (Figure S32). In addition, to avoid potential off-target effects, we employed a second siRNA for MSH6 (siRNA-MSH6-1, referred to as si-MSH6-1) to repeat the experimental results of MSH6 silencing in U87MG cells.
Lentivirus-mediated overexpression of MSH6
To overexpress MSH6, MSH6 cDNA was cloned into the pLVX vector between the EcoRI and BamHI sites and confirmed by DNA sequencing. Viral packaging was performed using ViraPower Lentiviral Packaging Mix (Invitrogen, Carlsbad, California, USA) in 293T cells. The packed virus was concentrated and purified by a Lenti-X Concentrator (Takara, Beijing, China). The resulting construct was named the MSH6 lentivirus. The negative control lentivirus was an empty vector.
MTT assay
The viability of cells was tested by MTT (Sigma‑Aldrich, Merck KGaA, Darmstadt, Germany). In brief, cells were seeded in 96-well plates at a density of 8×103 cells per well. The number of viable cells was determined at 48 h after the indicated treatments unless otherwise indicated. Cells were incubated with medium containing MTT (0.05 mg/ mL, 200µL) for 4 h. Then, the intracellular formazan crystals were dissolved by dimethylsulfoxide (100 µL/well). The optical absorption value, which was read by a microplate reader (ELx808, BioTek Instruments, Winooski, VT, USA), represents the number of viable cells. The above experiments were repeated 3 times.
Plate colony-forming assay
Three thousand GBM cells were cultured in a culture dish with a 6-cm diameter. After incubation for 2 weeks, colonies were fixed with methanol, stained with 0.5% crystal violet for 15 min and then analyzed with a light microscope (IX51, Olympus, Tokyo, Japan).
Wound-healing assay
Cell mobility was estimated by a wound-healing assay. Briefly, a scratch was made in a confluent monolayer of GBM cells using a pipette tip. Then, images were collected with a microscope (IX51, Olympus, Tokyo, Japan) after 0, 24 and 48 h. The experiments were repeated 3 times.
Transwell assays
Transwell chambers (BD Biosciences, San Jose, CA, USA) were employed to evaluate the invasion and migration capabilities of GBM cells. The lower compartment of the chamber contained medium with 10% FBS, while the corresponding upper compartment was seeded with GBM cells suspended in FBS- free medium. Then, the GBM cells were incubated for 24-48 h at 37 °C with 5% CO2. In contrast to the migration assay, the invasion assay included 5 mg/mL of Matrigel (BD Biosciences, Franklin Lakes, NJ, USA) covering the upper surface of the polycarbonate membrane (8-μm pore size). After incubation, the cells that failed to pass through the polycarbonate membrane were removed, whereas the migratory or invasive cells were fixed and stained as described above and then analyzed with a light microscope (IX51, Olympus, Tokyo, Japan). The experiments were repeated 3 times.
Cell cycle analysis
For cell cycle analysis, the GBM cells were harvested, washed with cold PBS and fixed with prechilled 75% ethanol. Subsequently, the cells underwent RNase (10 mg/mL) digestion and PI (1 mg/mL) staining, after which a FACSCalibur flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA) was employed to detect and analyze the cell cycle of the stained cells. The experiments were repeated 3 times.
Analysis of apoptosis
Annexin V-FITC/PI Apoptosis Detection kit was purchased from 4A Biotech (Beijing, China). The GBM cells were harvested and then stained in the dark with Annexin V-FITC and PI for 15 min, after which a FACSCalibur flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA) was employed to detect and analyze the spontaneous apoptosis of the stained cells. The experiments were repeated 3 times.
Western blot analysis
The cells were harvested in radioimmunoprecipitation assay buffer and lysed on ice. From each sample, equal amounts of protein were employed for sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred to PVDF membranes. After being blocked with 5% nonfat milk/TBST, the membrane was successively incubated with the appropriate primary antibody and a secondary antibody. Every incubation was followed by 3 washes with TBST. Protein signals were determined using a chemiluminescent substrate (Thermo Fisher Scientific, Waltham, MA, USA). Primary antibodies: MSH6, N-cadherin, Vimentin, E-cadherin, p-AKT, p-STAT3, p-Smad2/3, p-ERK, p-p38, p-JNK, p-p65, STAT3, Smad2/3, ERK, p38, JNK and p65 antibodies were obtained from Cell Signaling Technology (Danvers, MA, USA); Ki67, Nestin, CD133, SOX2, Cyclin D1, Bax, Bcl-2, MMP2, MMP9, HIF1A, VEGFA, Snail, Slug, Twist, ZEB1, ZEB2, AKT, TGFB1 and β-actin antibodies were supplied by Proteintech (Wuhan, Hubei, China); CXCR4 antibody was obtained from Abcam (Shanghai, China). Secondary antibodies: horseradish peroxidase (HRP)-conjugated affinipure goat anti-mouse IgG and HRP- conjugated affinipure goat anti-rabbit IgG were purchased from Proteintech (Wuhan, Hubei, China).
Quantitative real-time polymerase chain reaction (qRT-PCR)
Total cellular RNA was isolated by applying a Total RNA Extraction Kit (CORNING, Suzhou, Jiangsu, China) and then transcribed into cDNA using reverse transcriptase (TOYOBO, Osaka, Japan). The primer sequences are shown in Table S3. qRT-PCR was performed by employing the SYBR Green Master Mix (TOYOBO, Osaka, Japan) on an ABIPRISM 7900HT instrument (Applied Biosystems, Grand Island, NY, USA).
Immunofluorescence
GBM cells cultured in 24-well plates were fixed with 4% paraformaldehyde and then incubated with goat serum containing 0.1% Triton-100. Subsequently, a sequential incubation process with the appropriate primary antibody and a FITC-conjugated secondary antibody (Proteintech, Wuhan, Hubei, China) was conducted in GBM cells. The nuclei of GBM cells were counterstained with DAPI before images were captured using a fluorescence microscope (BX53, Olympus, Tokyo, Japan).
Subcutaneous GBM experiments
After intraperitoneal injection of an anesthetic agent (sodium pentobarbital; 1%, 5 mL/kg), BALB/c nude mice were randomized into the U251 group and T98G group (n = 3, each group) for subcutaneous inoculation of corresponding GBM cells. For both groups, GBM-Con cells were inoculated in the left flank, while GBM-MSH6 cells were inoculated in the right flank. Observations of tumor formation and growth were continued for 4 weeks using USI, MRI or pathology. The tumor volume was calculated by the following formula: V = length × width2/2.
USI
In vivo evaluation of tumor growth, angiogenesis, microangiogenesis and hardness degree was conducted on an Ultrasound System (iU Elite, Philips Healthcare, Amsterdam, The Netherlands). The specific imaging modalities used were B-mode ultrasonography, CDFI, CPA and USE, respectively.
MRI
MRI detection was carried out on an MRI System (Achieva 3.0T TX, Philips Healthcare, Amsterdam, The Netherlands) with clinical craniocerebral imaging sequences, including T1WI, T2WI, T2-FLAIR and T2-SPIR. In addition, the imaging sequence of T1 mapping was used to measure the longitudinal relaxation time (T1). In vitro, the MRI signals were determined using EP tubes (1.5 mL) filled with Cu2(OH)PO4@PAA dispersions (0-1 mg/mL). In vivo, U251 or T98G tumor-bearing mice were used as the models for MRI detection. Moreover, the acquisition of MRI signals generated by Cu2(OH)PO4@PAA in the tumor region was performed before/after intratumoral injection of the nanoparticles (1 mg/mL, 100 μL).
Immunohistochemical staining
GBM tissue samples fixed with formaldehyde were embedded in paraffin. Subsequently, the GBM tissue samples were sectioned for immunostaining with an antibody specific for the targeted protein. Immunohistochemical staining was conducted by employing the streptavidin-peroxidase complex. Then, images were obtained using a light microscope (BX53, Olympus, Tokyo, Japan).
Preparation of Cu2(OH)PO4@PAA
Cu(NO3)2·3H2O and Na2HPO4·12H2O were purchased from Aladdin (Shanghai, China). PAA was supplied by Sigma-Aldrich (Merck KGaA, Darmstadt, Germany). A hydrothermal method was used to fabricate the Cu2(OH)PO4@PAA nanoparticles. Briefly, 10 mL of PAA (35 mg/mL) was added to 20 mL of Cu(NO3)2·3H2O (14.5 mg/mL) with magnetic stirring. Then, equal amount of Na2HPO4·12H2O solution (7.13 mg/mL) was slowly added to the above mixed solution with magnetic stirring. After adjusting the pH value of the final mixed solution to 7.0, the hydrothermal reaction was conducted using a 100 mL Teflon-lined autoclave in an electric oven (120 °C, 6 h). The cyan product was obtained by centrifugation (10000 rpm, 5 min), and then washed with ethanol and deionized water. Ultimately, the product was homogeneously dispersed in deionized water.
Characterization of Cu2(OH)PO4@PAA
TEM analysis of the sample was performed on a JEM-2010 microscope (JEOL, Akishima, Tokyo, Japan) with a 100 kV acceleration voltage. The zeta-potential of the sample was detected by DLS (Zeta PALS BI-90 Plus, Brookhaven Instruments, Holtsville, NY, USA). The phase nature of the sample was investigated by XRD (XD-D1, Shimadzu, Kyoto, Japan). FT-IR spectra were measured with an AVATAR360 spectrometer (Thermo Fisher Scientific, Waltham, MA, USA). The optical properties were determined by a U-4100 spectrophotometer (Hitachi, Tokyo, Japan).
Photothermal conversion of Cu2(OH)PO4@PAA
Cu2(OH)PO4@PAA aqueous suspensions at different concentrations were placed into quartz tubes (inner diameter of 0.5 cm and length of 2.5 cm). The 1064-nm NIR laser (0.8 W/cm2) was employed to irradiate every sample for 10 min at the position of 0.5 cm above liquid surface. Meanwhile, the changes of temperature were monitored by a thermal imager (E60, FLIR, Portland, OR, USA) and imaged every 30 seconds.
In vitro PTT with Cu2(OH)PO4@PAA
GBM cells (8×104/mL, 100 μL/well) were incubated in 96-well plates for 24 h, after which the initial medium was discarded. Subsequently, isometric medium containing Cu2(OH)PO4@PAA (1 mg/mL) was added to each well, which was replaced by fresh medium (100 μL/well) after a 6-h incubation. Then, the 1064-nm NIR laser (0.8 W/cm2) was employed to irradiate every well for 10 min at the position of 0.5 cm above liquid surface. Meanwhile, temperature changes were monitored by a thermal imager. Subsequent assays were performed after a 48-h incubation.
In vivo PTT with Cu2(OH)PO4@PAA
U251 tumor-bearing nude mice were randomized into four groups (n = 5, each group) when the tumor size reached approximately 200 mm3. The mice in group 1 served as the no treatment control group, while those in groups 2, 3 and 4 were intratumorally injected with Cu2(OH)PO4@PAA solution (1 mg/mL, 100 μL), irradiated with NIR, and treated with both interventions, respectively. The treatment was conducted at 3 h postinjection using a 1064-nm NIR laser at a power density of 0.8 W/cm2, after which USI was employed to evaluate the therapeutic effectiveness of each treatment.
Statistical analysis
SPSS 22.0 software (IBM SPSS, Armonk, NY, USA) was employed to analyze and identify the statistical differences among the groups. Data from at least three independent experiments are shown as the mean ± standard deviation. The existence of statistically significant difference was considered when the P value was less than 0.05.
Abbreviations
AURKA: aurora kinase A; AURKB: aurora kinase B; CDFI: color Doppler flow imaging; CPA: color power angiography; CXCR4: C-X-C motif chemokine receptor 4; EGFR: epidermal growth factor receptor; EMT: epithelial-to-mesenchymal transition; GBM: glioblastoma multiforme; HIF1A: hypoxia inducible factor 1 subunit alpha; HMMR: hyaluronan- mediated motility receptor; HOXA7: homeobox A7; HOXA10: homeobox A10; IRT: infrared thermal imaging; MRI: magnetic resonance imaging; MSH6: mutS homolog 6; NIR: near infrared; PAA: polyacrylic acid; PTT: photothermal therapy; r1: longitudinal relaxivity; Slug: snail family transcriptional repressor 2; Smad2/3: smad family member 2/3; Snail: snail family transcriptional repressor 1; SOX2: SRY-box 2; STAT3: signal transducer and activator of transcription 3; T1: longitudinal relaxation time; TGFB1: transforming growth factor beta 1; TMZ: temozolomide; Twist: twist family bHLH transcription factor 1; USE: ultrasonic elastosonography; VEGFA: vascular endothelial growth factor A; ZEB1: zinc finger E-box binding homeobox 1; ZEB2: zinc finger E-box binding homeobox 2.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This study was financially supported by the National Natural Science Foundation of China (grant no. 81771894) and the Fund of Scientific Research Innovation of The First Affiliated Hospital of Harbin Medical University (grant no. 2018B009).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Delgado-López PD, Corrales-García EM. Survival in glioblastoma: a review on the impact of treatment modalities. Clin Transl Oncol. 2016;18:1062-71
2. Ostrom QT, Gittleman H, Liao P, Rouse C, Chen Y, Dowling J. et al. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2007-2011. Neuro Oncol. 2014;16:iv1-63
3. Murakami R, Hirai T, Nakamura H, Furusawa M, Nakaguchi Y, Uetani H. et al. Recurrence patterns of glioblastoma treated with postoperative radiation therapy: relationship between extent of resection and progression-free interval. Jpn J Radiol. 2012;30:193-7
4. Shah AH, Graham R, Bregy A, Thambuswamy M, Komotar RJ. Recognizing and correcting failures in glioblastoma treatment. Cancer Invest. 2014;32:299-302
5. Sayegh ET, Kaur G, Bloch O, Parsa AT. Systematic review of protein biomarkers of invasive behavior in glioblastoma. Mol Neurobiol. 2014;49:1212-44
6. Kahlert UD, Nikkhah G, Maciaczyk J. Epithelial-to-mesenchymal(-like) transition as a relevant molecular event in malignant gliomas. Cancer Lett. 2013;331:131-8
7. Iser IC, Pereira MB, Lenz G, Wink MR. The epithelial-to-mesenchymal transition-like process in glioblastoma: an updated systematic review and in silico investigation. Med Res Rev. 2017;37:271-313
8. Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010;29:4741-51
9. Iwadate Y. Epithelial-mesenchymal transition in glioblastoma progression. Oncol Lett. 2016;11:1615-20
10. Savary K, Caglayan D, Caja L, Tzavlaki K, Bin Nayeem S, Bergstrom T. et al. Snail depletes the tumorigenic potential of glioblastoma. Oncogene. 2013;32:5409-20
11. Siebzehnrubl FA, Silver DJ, Tugertimur B, Deleyrolle LP, Siebzehnrubl D, Sarkisian MR. et al. The ZEB1 pathway links glioblastoma initiation, invasion and chemoresistance. EMBO Mol Med. 2013;5:1196-212
12. Ohba S, Hirose Y. Current and future drug treatments for glioblastomas. Curr Med Chem. 2016;23:4309-16
13. Zhang J, Stevens MF, Bradshaw TD. Temozolomide: mechanisms of action, repair and resistance. Curr Mol Pharmacol. 2012;5:102-14
14. Quiros S, Roos WP, Kaina B. Processing of O6-methylguanine into DNA double-strand breaks requires two rounds of replication whereas apoptosis is also induced in subsequent cell cycles. Cell Cycle. 2010;9:168-78
15. Naumann SC, Roos WP, Jöst E, Belohlavek C, Lennerz V, Schmidt CW. et al. Temozolomide- and fotemustine-induced apoptosis in human malignant melanoma cells: response related to MGMT, MMR, DSBs, and P53. Br J Cancer. 2009;100:322-33
16. Felsberg J, Thon N, Eigenbrod S, Hentschel B, Sabel MC, Westphal M. et al. Promoter methylation and expression of MGMT and the DNA mismatch repair genes MLH1, MSH2, MSH6 and PMS2 in paired primary and recurrent glioblastomas. Int J Cancer. 2011;129:659-70
17. Yip S, Miao J, Cahill DP, Iafrate AJ, Aldape K, Nutt CL. et al. MSH6 mutations arise in glioblastomas during temozolomide therapy and mediate temozolomide resistance. Clin Cancer Res. 2009;15:4622-9
18. Zhou H, Qian W, Uckun FM, Wang L, Wang YA, Chen H. et al. IGF1 receptor targeted theranostic nanoparticles for targeted and image-guided therapy of pancreatic cancer. ACS Nano. 2015;9:7976-91
19. Li Z, Liu J, Hu Y, Howard KA, Li Z, Fan X. et al. Multimodal imaging-guided antitumor photothermal therapy and drug delivery using bismuth selenide spherical sponge. ACS Nano. 2016;10:9646-58
20. Mou J, Lin T, Huang F, Chen H, Shi J. Black titania-based theranostic nanoplatform for single NIR laser induced dual-modal imaging-guided PTT/PDT. Biomaterials. 2016;84:13-24
21. Brower V. MRI study identifies three subtypes of glioblastoma. Lancet Oncol. 2015;16:e484
22. Wang G, Huang B, Ma X, Wang Z, Qin X, Zhang X. et al. Cu2(OH)PO4, a near-infrared-activated photocatalyst. Angew Chem Int Ed Engl. 2013;52:4810-3
23. Ge R, Lin M, Li X, Liu S, Wang W, Li S. et al. Cu2+-loaded polydopamine nanoparticles for magnetic resonance imaging-guided pH- and near-infrared-light-stimulated thermochemotherapy. ACS Appl Mater Interfaces. 2017;9:19706-16
24. Guo W, Qiu Z, Guo C, Ding D, Li T, Wang F. et al. Multifunctional theranostic agent of Cu2(OH)PO4 quantum dots for photoacoustic image-guided photothermal/photodynamic combination cancer therapy. ACS Appl Mater Interfaces. 2017;9:9348-58
25. Mc Carthy DJ, Malhotra M, O'Mahony AM, Cryan JF, O'Driscoll CM. Nanoparticles and the blood-brain barrier: advancing from in-vitro models towards therapeutic significance. Pharm Res. 2015;32:1161-85
26. Batzoglou S, Schwartz R. Computational biology and bioinformatics. Bioinformatics. 2014;30:i1-2
27. Wang J, Wang Q, Cui Y, Liu ZY, Zhao W, Wang CL. et al. Knockdown of Cyclin D1 inhibits proliferation, induces apoptosis, and attenuates the invasive capacity of human glioblastoma cells. J Neurooncol. 2012;106:473-84
28. Soni S, Padwad YS. HIF-1 in cancer therapy: two decade long story of a transcription factor. Acta Oncol. 2017;56:503-15
29. Lee MJ, Yaffe MB. Protein regulation in signal transduction. Cold Spring Harb Perspect Biol. 2016;8:a005918
30. Kappler M, Taubert H, Schubert J, Vordermark D, Eckert AW. The real face of HIF1α in the tumor process. Cell Cycle. 2012;11:3932-6
31. Schwartz L, Seyfried T, Alfarouk KO, Da Veiga Moreira J, Fais S. Out of Warburg effect: an effective cancer treatment targeting the tumor specific metabolism and dysregulated pH. Semin Cancer Biol. 2017;43:134-8
32. Parks SK, Chiche J, Pouyssegur J. pH control mechanisms of tumor survival and growth. J Cell Physiol. 2011;226:299-308
33. Wang Y, Stark GR. A new STAT3 function: pH regulation. Cell Res. 2018;28:1045
34. Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721-32
35. Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR. et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462-77
36. Mahabir R, Tanino M, Elmansuri A, Wang L, Kimura T, Itoh T. et al. Sustained elevation of Snail promotes glial-mesenchymal transition after irradiation in malignant glioma. Neuro Oncol. 2014;16:671-85
37. Gray GK, McFarland BC, Nozell SE, Benveniste EN. NF-κB and STAT3 in glioblastoma: therapeutic targets coming of age. Expert Rev Neurother. 2014;14:1293-306
38. Bae WJ, Lee SH, Rho YS, Koo BS, Lim YC. Transforming growth factor β1 enhances stemness of head and neck squamous cell carcinoma cells through activation of Wnt signaling. Oncol Lett. 2016;12:5315-20
39. Yao C, Su L, Shan J, Zhu C, Liu L, Liu C. et al. IGF/STAT3/NANOG/Slug signaling axis simultaneously controls epithelial-mesenchymal transition and stemness maintenance in colorectal cancer. Stem Cells. 2016;34:820-31
40. Katoh M, Katoh M. Integrative genomic analyses of ZEB2: transcriptional regulation of ZEB2 based on SMADs, ETS1, HIF1alpha, POU/OCT, and NF-kappaB. Int J Oncol. 2009;34:1737-42
41. Yang HW, Menon LG, Black PM, Carroll RS, Johnson MD. SNAI2/Slug promotes growth and invasion in human gliomas. BMC Cancer. 2010;10:301
42. Qi S, Song Y, Peng Y, Wang H, Long H, Yu X. et al. ZEB2 mediates multiple pathways regulating cell proliferation, migration, invasion, and apoptosis in glioma. PloS One. 2012;7:e38842
43. Zheng Y, Miu Y, Yang X, Yang X, Zhu M. CCR7 mediates TGF-β1-induced human malignant glioma invasion, migration, and epithelial- mesenchymal transition by activating MMP2/9 through the nuclear factor kappaB signaling pathway. DNA Cell Biol. 2017;36:853-61
44. Zhu Y, Yang P, Wang Q, Hu J, Xue J, Li G. et al. The effect of CXCR4 silencing on epithelial-mesenchymal transition related genes in glioma U87 cells. Anat Rec (Hoboken). 2013;296:1850-6
45. Oliveira AI, Anjo SI, Vieira de Castro J, Serra SC, Salgado AJ, Manadas B. et al. Crosstalk between glial and glioblastoma cells triggers the "go-or-grow" phenotype of tumor cells. Cell Commun Signal. 2017;15:37
46. Xie Q, Mittal S, Berens ME. Targeting adaptive glioblastoma: an overview of proliferation and invasion. Neuro Oncol. 2014;16:1575-84
47. Osmanbeyoglu HU, Pelossof R, Bromberg JF, Leslie CS. Linking signaling pathways to transcriptional programs in breast cancer. Genome Res. 2014;24:1869-80
48. Chen YD, Zhang Y, Dong TX, Xu YT, Zhang W, An TT. et al. Hyperthermia with different temperatures inhibits proliferation and promotes apoptosis through the EGFR/STAT3 pathway in C6 rat glioma cells. Mol Med Rep. 2017;16:9401-8
49. Sun J, Guo M, Pang H, Qi J, Zhang J, Ge Y. Treatment of malignant glioma using hyperthermia. Neural Regen Res. 2013;8:2775-82
50. Jin H, Xie X, Hu B, Gao F, Zhou J, Zhang Y. et al. Hyperthermia inhibits the proliferation and invasive ability of mouse malignant melanoma through TGF-beta(1). Oncol Rep. 2013;29:725-34
51. Le Fèvre R, Durand-Dubief M, Chebbi I, Mandawala C, Lagroix F, Valet JP. et al. Enhanced antitumor efficacy of biocompatible magnetosomes for the magnetic hyperthermia treatment of glioblastoma. Theranostics. 2017;7:4618-31
52. Chen Q, Yu H, Wang L, Abdin ZU, Yang X, Wang J. et al. Synthesis and characterization of amylose grafted poly(acrylic acid) and its application in ammonia adsorption. Carbohydr Polym. 2016;153:429-34
53. Chen CH, Zhou Y, Wang NN, Cheng LY, Ding HM. Cu2(OH)PO4/g-C3N4 composite as an efficient visible light-activated photo-fenton photocatalyst. RSC Adv. 2015;5:95523-31
54. Garcia J, Tang T, Louie AY. Nanoparticle-based multimodal PET/MRI probes. Nanomedicine (Lond). 2015;10:1343-59
55. Perlman O, Weitz IS, Azhari H. Copper oxide nanoparticles as contrast agents for MRI and ultrasound dual-modality imaging. Phys Med Biol. 2015;60:5767-83
56. Tsai YC, Vijayaraghavan P, Chiang WH, Chen HH, Liu TI, Shen MY. et al. Targeted delivery of functionalized upconversion nanoparticles for externally triggered photothermal/photodynamic therapies of brain glioblastoma. Theranostics. 2018;8:1435-48
57. Bale SS, Moore L, Yarmush M, Jindal R. Emerging in vitro liver technologies for drug metabolism and inter-organ interactions. Tissue Eng Part B Rev. 2016;22:383-94
58. Knights KM, Rowland A, Miners JO. Renal drug metabolism in humans: the potential for drug-endobiotic interactions involving cytochrome P450 (CYP) and UDP-glucuronosyltransferase (UGT). Br J Clin Pharmacol. 2013;76:587-602
59. Yang CY, Chen YD, Guo W, Gao Y, Song CQ, Zhang Q. et al. Bismuth ferrite-based nanoplatform design: an ablation mechanism study of solid tumor and NIR-triggered photothermal/photodynamic combination cancer therapy. Adv Funct Mater. 2018;28:1706827
60. Zhang C, Ren J, Hua J, Xia L, He J, Huo D. et al. Multifunctional Bi2WO6 nanoparticles for CT-guided photothermal and oxygen-free photodynamic therapy. ACS Appl Mater Interfaces. 2018;10:1132-46
61. Jaque D, Martínez Maestro L, del Rosal B, Haro-Gonzalez P, Benayas A, Plaza JL. et al. Nanoparticles for photothermal therapies. Nanoscale. 2014;6:9494-530
62. Ahmad R, Fu J, He N, Li S. Advanced gold nanomaterials for photothermal therapy of cancer. J Nanosci Nanotechnol. 2016;16:67-80
63. Liu Y, Bhattarai P, Dai Z, Chen X. Photothermal therapy and photoacoustic imaging via nanotheranostics in fighting cancer. Chem Soc Rev. 2018 doi: 10.1039/c8cs00618k
64. Chitgupi U, Qin Y, Lovell JF. Targeted nanomaterials for phototherapy. Nanotheranostics. 2017;1:38-58
65. Jiang Q, Luo Z, Men Y, Yang P, Peng H, Guo R. et al. Red blood cell membrane-camouflaged melanin nanoparticles for enhanced photothermal therapy. Biomaterials. 2017;143:29-45
66. Paviolo C, Stoddart PR. Gold nanoparticles for modulating neuronal behavior. Nanomaterials (Basel). 2017;7:E92
67. Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45:W98-102
68. Pontén F, Jirström K, Uhlen M. The Human Protein Atlas-a tool for pathology. J Pathol. 2008;216:387-93
69. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1
70. Jensen LJ, Kuhn M, Stark M, Chaffron S, Creevey C, Muller J. et al. STRING 8-a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 2009;37:D412-6
Author contact
![]() Corresponding authors: Xiuhua Yang, Email address: yangxiuhuaedu.cn; Tel: +86-13936516597; Fax: +86-0451-85555034 and Zizhuo Li, Email address: lizizhuoedu.cn; Tel: +86-15145114052; Fax: +86-0451-85555033
Corresponding authors: Xiuhua Yang, Email address: yangxiuhuaedu.cn; Tel: +86-13936516597; Fax: +86-0451-85555034 and Zizhuo Li, Email address: lizizhuoedu.cn; Tel: +86-15145114052; Fax: +86-0451-85555033
Citation styles
APA
Chen, Y., Liu, P., Sun, P., Jiang, J., Zhu, Y., Dong, T., Cui, Y., Tian, Y., An, T., Zhang, J., Li, Z., Yang, X. (2019). Oncogenic MSH6-CXCR4-TGFB1 Feedback Loop: A Novel Therapeutic Target of Photothermal Therapy in Glioblastoma Multiforme. Theranostics, 9(5), 1453-1473. https://doi.org/10.7150/thno.29987.
ACS
Chen, Y.; Liu, P.; Sun, P.; Jiang, J.; Zhu, Y.; Dong, T.; Cui, Y.; Tian, Y.; An, T.; Zhang, J.; Li, Z.; Yang, X. Oncogenic MSH6-CXCR4-TGFB1 Feedback Loop: A Novel Therapeutic Target of Photothermal Therapy in Glioblastoma Multiforme. Theranostics 2019, 9 (5), 1453-1473. DOI: 10.7150/thno.29987.
NLM
Chen Y, Liu P, Sun P, Jiang J, Zhu Y, Dong T, Cui Y, Tian Y, An T, Zhang J, Li Z, Yang X. Oncogenic MSH6-CXCR4-TGFB1 Feedback Loop: A Novel Therapeutic Target of Photothermal Therapy in Glioblastoma Multiforme. Theranostics 2019; 9(5):1453-1473. doi:10.7150/thno.29987. https://www.thno.org/v09p1453.htm
CSE
Chen Y, Liu P, Sun P, Jiang J, Zhu Y, Dong T, Cui Y, Tian Y, An T, Zhang J, Li Z, Yang X. 2019. Oncogenic MSH6-CXCR4-TGFB1 Feedback Loop: A Novel Therapeutic Target of Photothermal Therapy in Glioblastoma Multiforme. Theranostics. 9(5):1453-1473.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.