Impact Factor
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Rodent models of hepatoma
Chemotherapy and molecular...
Summary and perspectives
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(5):1426-1452. doi:10.7150/thno.31683 This issue Cite
Review
Evaluation of Polymer Nanoformulations in Hepatoma Therapy by Established Rodent Models
Qilong Wang1,2, Ping Zhang1, ![]() , Zhongmin Li2,5,
, Zhongmin Li2,5, ![]() , Xiangru Feng2,4, Chengyue Lv2,4, Huaiyu Zhang2,5, Haihua Xiao3, Jianxun Ding2,4,
, Xiangru Feng2,4, Chengyue Lv2,4, Huaiyu Zhang2,5, Haihua Xiao3, Jianxun Ding2,4, ![]() , Xuesi Chen2,4
, Xuesi Chen2,4
1. Department of Hepatobiliary and Pancreatic Surgery, The First Hospital of Jilin University, Changchun 130021, P. R. China
2. Key Laboratory of Polymer Ecomaterials, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, P. R. China
3. Beijing National Laboratory for Molecular Sciences, State Key Laboratory of Polymer Physics and Chemistry, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, P. R. China
4. Jilin Biomedical Polymers Engineering Laboratory, Changchun 130022, P. R. China
5. Department of Gastrointestinal Colorectal and Anal Surgery, China-Japan Union Hospital of Jilin University, Changchun 130033, P. R. China
Received 2018-11-21; Accepted 2019-1-8; Published 2019-2-20
Abstract

Hepatoma is one of the most severe malignancies usually with poor prognosis, and many patients are insensitive to the existing therapeutic agents, including the drugs for chemotherapy and molecular targeted therapy. Currently, researchers are committed to developing the advanced formulations with controlled drug delivery to improve the efficacy of hepatoma therapy. Numerous inoculated, induced, and genetically engineered hepatoma rodent models are now available for formulation screening. However, animal models of hepatoma cannot accurately represent human hepatoma in terms of histological characteristics, metastatic pathways, and post-treatment responses. Therefore, advanced animal hepatoma models with comparable pathogenesis and pathological features are in urgent need in the further studies. Moreover, the development of nanomedicines has renewed hope for chemotherapy and molecular targeted therapy of advanced hepatoma. As one kind of advanced formulations, the polymer-based nanoformulated drugs have many advantages over the traditional ones, such as improved tumor selectivity and treatment efficacy, and reduced systemic side effects. In this article, the construction of rodent hepatoma model and much information about the current development of polymer nanomedicines were reviewed in order to provide a basis for the development of advanced formulations with clinical therapeutic potential for hepatoma.
Keywords: hepatoma, rodent model, polymer nanoparticle, drug delivery, chemotherapy, molecular targeted therapy
Introduction
Hepatoma ranks fifth in the incidence of solid tumors worldwide with a mortality rate ranking fourth among all cancer subtypes. Even if hepatoma can be surgically resected, the 5-year overall survival (OS) rate is only 30% - 40%, and the 2-year recurrence rate is as high as 50% [1]. Recently, chemotherapy is not recommended for the therapy of advanced hepatoma in many countries because of the poor efficacies of cytotoxic drugs, and molecular targeted drugs rank first-line and second-line treatment modalities in the clinic [2, 3]. However, the outcomes of molecular targeted therapy in some clinical trials were not satisfactory. With the recent progress in emerging polymer nanomedicines, this situation is expected to change. The nanoformulated polymer drugs are usually prepared from natural or synthetic polymer materials with conventional chemical methods [4]. The chemotherapeutic agents and the molecular targeted drugs are loaded inside or conjugated on the surface of the nanoparticles using encapsulation, intercalation, adsorption, polymerization, condensation or coupling reactions [5]. As a drug delivery system, nanoparticles typically have excellent biocompatibility [6, 7], stable physical and chemical properties [8, 9], surface modifications [10, 11], and reticuloendothelial system (RES) escape [12]. In addition, some nanoformulations exhibit unique optical, thermal, mechanical, or magnetic properties to facilitate controlled drug release, making them more effective for cancer chemotherapy [13]. With these apparent advantages, nanoscale drug delivery systems have become a hotspot of targeted chemotherapy and molecular targeted therapy for various malignancies [14], including hepatoma.
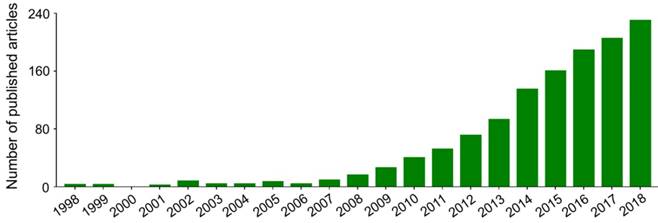
Nanoparticles with specific advantages, such as adjustable size and transformable surface, prolong the blood circulation and improve the intratumoral accumulation of antineoplastic agents [15]. However, in most cases, after intravenous administration, nanoparticles in the blood are coated with plasma proteins, glycoproteins, or other components, and then consumed by the mononuclear macrophage system (MPS) [16], specifically those that gather in the macrophage-rich organs, including the liver, spleen, and bone marrow [17]. In addition, the antineoplastic drugs lacking specific affinity are widely distributed in the body, which may cause damage to normal tissues. These side effects are intolerable and mostly limit the clinical efficacy of antineoplastic drugs. Fortunately, these problems can be readily solved to some extent by the application of targeted nanoparticles, either by passive or by active targeting of tumor cells [18]. Passively targeted nanoparticles can increase the concentration in target tumors by changing the surface charges, chemical groups, hydrophilicity (or hydrophobicity), and other physicochemical factors while reducing their accumulation in normal tissues. The surface modifiers are commonly used for passive targeting include poly(ethylene glycol) (PEG) and polysaccharide. Active targeting refers to modifying the surface of nanoparticles with targeted molecules or antibodies to make them highly selective and specific to tumors. Glycyrrhetinic acid [19], lactobionic acid [20], anti-CD147 antibody [21], anti-GPC3 antibody [22], and other ligands have been used to enhance the targeting of nanoparticles to hepatoma cells. As shown in Figure 1, researches on the treatment of hepatoma with polymer nanoformulations have increased dramatically in recent years. The development of nanoscale drug delivery systems has opened up an effective way to treat hepatoma.
For the screening of polymer nanoformulations, the critical step is to construct ideal animal models of hepatoma with similar pathological characteristics to clinical tumor tissues. With the help of animal models, it is effective to explore the mechanisms of pathogenesis and progression of hepatoma, as well as the development of various drug formulations. The selections of hepatoma animal models mainly depend on the purposes of studies. At present, there are various animal hepatoma models constructed by different approaches, including subcutaneously and orthotopically inoculated hepatoma models, chemically and virally induced hepatoma models, and genetically engineered hepatoma models. The most common type of human hepatoma is hepatocellular carcinoma (HCC), which accounts for 70% - 85% of primary hepatoma [23]. Due to the high morbidity and high mortality of HCC, animal models of hepatoma are mainly used to study HCC. Rodents are the most commonly used HCC models because of their small size, similarity to the human genome, and similar pathogenesis.
In this article, we make a comprehensive review on the construction of rodent hepatoma models and the development of polymer nanomedicine for enhancing hepatoma chemotherapy and molecular targeted therapy, and predict their future directions (Scheme 1).
Rodent models of hepatoma
Implantation models of hepatoma
Implantation models are currently widely used for hepatoma formation in mice in most cases. The xenogeneic and allogeneic hepatoma cells or tumor tissues are implanted into immunocompromised recipient mice by ectopic or orthotopic transplantation. Ectopic transplantation of tumor cells or tissues is usually performed subcutaneously. For tissue implantation, the fragments with size at approximately 1.0 mm3 are obtained from mouse subcutaneous liver tumor or human surgical hepatoma specimens [23]. In this section, we mainly focus our discussion on the subcutaneous and orthotopic liver transplantation models.
Subcutaneously inoculated models of hepatoma
Among various types of hepatoma models, the inoculated models established by subcutaneous injection of hepatoma cells are most commonly used [24]. These subcutaneously inoculated hepatoma models maintain the significant characteristics of primary tumors, such as tumor microenvironment, morphology, metastatic potential, and reaction to antitumor agents. Moreover, this kind of model is easy to reproduce and suitable for the evaluation of antitumor activity, but the tumors often grow locally without distant metastasis, which is different from primary tumors [25].
The subcutaneous environments of mice provide appropriate “soil” for the tumors, so they can grow and interact with the host. The subcutaneous models might differ in various factors including tumor type, origin, and the anatomical site [26]. Specifically, the anatomical site of the tumor plays a vital role in determining whether the tumor develops as a discrete nodule or disseminates at the beginning. The establishment of hepatoma models has remarkable utility in the basic and preclinical researches [27]. If more severe immunosuppression is needed to increase the chance of engraftment, SCID, NOD/ SCID, and CB17 mice are used [28].
Schematic illustration of rodent hepatoma models of various types and therapy using different polymer-based nanoplatforms.
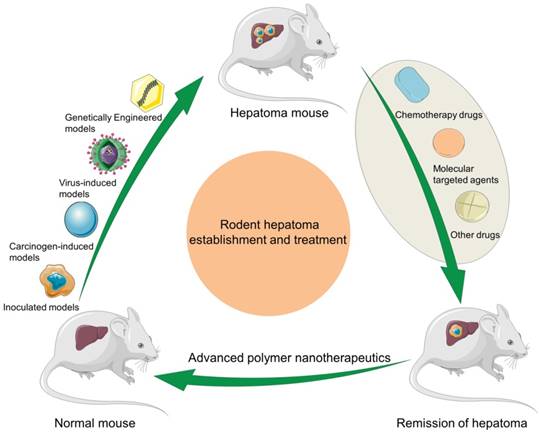
However, it is noteworthy that the physiological interaction between the tumor and the inoculated model is insufficient, partly because the damaged immune system does not allow the tumor to use the escape mechanisms that exist in humans. The tumor microenvironment is closely related to the occurrence and development of malignant tumors. The interaction between tumor cells and liver-specific factors, including endothelial cells, fibroblasts, and inflammatory cells, etc., is very important for the progression of hepatoma. This interaction is absent in the subcutaneous transplantation model, so spontaneous distant metastasis does not occur. In the xenograft models, the histological appearance of human tumors can be well preserved [29]. Therefore, the method of subcutaneous implantation of hepatoma in rodents is not sufficient.
Orthotopically implanted models of hepatoma
The surgical orthotopically inoculated hepatoma models are the most clinically relevant because they mimic human hepatomas in terms of the tumor microenvironment, morphology, metastatic potential, and the reaction to antitumor agents [30, 31]. Moreover, the pathological processes, including local invasion and angiogenesis, can be tested in the normal microenvironment. However, orthotopic implantation of hepatoma grafts exhibits some disadvantages, such as high cost and difficult surgical inoculation procedure. Furthermore, the growth and response of tumors cannot be detected as easily as in ectopic transplantation models. Currently, immunocompromised mice are used to inoculate human liver cell lines or tissue masses. Athymic nude mice are the most widely used recipients, and they show a functional deficiency in T and B cells [32].
Among various methods delivering cancer cells to the liver, the most effective way is through the hepatic artery, as it allows cells to be infused directly into the artery bed and provides ample blood supply for the tumor growth [33]. Injecting hepatoma cells into the liver results in the progressive growth of tumor cells in the immunodeficient rodents [34]. The orthotopic hepatoma models require surgical methods to implant tumor cells into the liver and require serum biomarkers and sophisticated imaging technologies to monitor the progression of tumors [35]. The studies using orthotopic models provide more credible data to the clinicians because tumors grow in their native environments in such models [36]. The positive findings of studies using orthotopic models promote the development of treatments in the clinical trials. Therefore, the subcutaneous tumor models are mainly used to evaluate the antitumor effect of new drugs. In contrast, the orthotopic transplantation models are used to study the mechanism of distant metastasis of hepatoma, the interaction between tumor and host, and the effect of immunotherapy.
Chemically induced hepatoma models
It has been recognized that many chemical reagents are associated with the occurrence of hepatoma. The mechanism of human hepatoma caused by chemical agents has been researched in many epidemiological studies [37]. Although some of these chemical agents have carcinogenic effects on the mouse, they generally do not cause hepatoma. Cirrhosis and hepatoma are not observed in mice receiving specific chemical agents [38]. Few mouse hepatoma carcinogens have carcinogenic effects on humans, with the exceptions of aflatoxin B1 and oral contraceptives. The view that oral contraceptives increase the incidence of hepatoma in mice and humans has become a common understanding of academia. Nevertheless, the carcinogen-induced mouse model is still widely used in some studies on hepatoma [39]. The carcinogen-induced hepatoma mouse models are used to reveal the relationship between exposure to carcinogens and specific gene alterations [40]. There are currently two types of carcinogens: (i) genotoxic agents that can directly induce tumorigenesis, such as N-nitrosodiethylamine (DEN), peroxisome proliferator, and aflatoxin B (AFB); (ii) accelerators that promote tumor formation when combined with genotoxic agents, such as thioacetamide (TAA), carbon tetrachloride (CCl4), and choline-deficient diet (CDD) [41]. In this section, we will introduce several types of carcinogen-induction models.
N-Nitrosodiethylamine
Hepatoma models can be established by administering DEN to mice [42]. The carcinogenicity of DEN has been demonstrated to involve two parallel pathways: alkylation of DNA structures that cause cell degeneration and DNA damage, and production of reactive oxygen species (ROS) by activation of cytochrome P450 [43]. Compared with other models, the DEN-induced hepatoma model has unique requirements, including the time and dosage of administration [44]. DEN is usually used in B6C3F1 mice at a concentration of 5.0 µg g-1 (once, i.p.). The tumorigenesis induced by DEN varies with mouse strain, sex, and age [45]. A single time intravenous injection (i.v.) of DEN is sufficient to induce tumorigenesis in a variety of mouse models. However, 100% tumor development is difficult to achieve through a single injection, but long-term DEN administration has been shown to cause nearly 100% tumor formation. In addition, the models may be negatively influenced by repeated DEN injection and the experiments may be delayed by the long administration period.
Carbon tetrachloride
CCl4 is classified in Group 2B (possibly carcinogenic to humans) on the list of carcinogens by the International Agency for Research on Cancer (IARC), meaning there is insufficient evidence that it can cause human hepatoma [46]. However, CCl4 is a recognized liver-toxic substance frequently applied to induce hepatoma in rodents [47]. Two different factors contribute to the hepatotoxicity of CCl4. First, the level of ROS is increased by cytochrome P450 [48]. Second, chemokines, cytokines, and other proinflammatory factors are produced by Kupffer cells. Both processes induce an inflammatory response [49]. It initiates a repeated cycle of injury, inflammation, and repair which finally leads to fibrosis and hepatoma. In one study, researchers administered DEN and CCl4 into mice and provided alcohol through drinking water, which led to hepatoma after five months [50].
Thioacetamide
TAA is also classified in Group 2B in the list of carcinogens by IARC [51]. TAA is a type of hepatotoxin used to induce hepatoma in rodents either by drinking or by intraperitoneal (i.p.) injection [24]. Many studies have shown that TAA can cause hepatic fibrosis in mice in 10 - 15 weeks [52]. TAA is also related to the formation of oxidative stress, and the increase of ROS in the liver can progressively lead to the damage of DNA and the development of hepatoma [53].
Trends in published articles on application of polymer nanoformulations in treatment of hepatoma from 1998 to 2018 included in Web of Science Core Collection. The search topic is ((liver cancer OR liver carcinoma OR hepato* carcinoma OR hepatoma) AND (nano*) AND (polym* OR macromolecul*) AND (*therapy)).
Peroxisome proliferator
It was reported that mice fed on peroxisome proliferator (PP) diet developed hepatoma [54]. Currently, it is widely accepted that long-term exposure to PPs can induce mouse hepatoma [55]. PPs stimulate liver cell growth and inhibit the apoptosis of cancerous cells [56]. The peroxisome proliferator-activated receptors (PPARs) are ligand-inducible nuclear receptors that, when combined with fatty acid-derived ligands, activate the transcription of genes regulating lipid metabolism [57]. PPAR ligands can promote hepatoma development through the activation of peroxisomal oxidase, inducing ROS formation [58]. Mouse models fed with a PP diet have specific characteristics, including trabecular histological patterns, metastasis in one-fifth to two-fifths of cases, and the induction of gene mutations. However, it is unclearly known whether long-term exposure to PPs is also harmful to human [24]. One should be cautious in using this model for interpretation of human disease, because the hepatoma induced by PPs may be species-specific, as the genetic features of many PP models are significantly different from those of human hepatoma.
Aflatoxin
Only a small number of studies have used aflatoxin for hepatoma modeling in mouse [59]. In this method, hepatotoxicity is mainly caused by Aspergillus fungi. These fungi are mainly present in corn, rice, and peanuts under moist conditions. In China and West Africa, the high rate of AFB leads to a high prevalence of hepatoma [60]. Carcinogenic activity of AFB is mainly due to chromosomal strand breaks, chromosomal aberrations, the formation of DNA-adducts, micronuclei, and unregulated DNA syntheses [61]. A study showed that 7-day-old mice receiving 6.0 mg per kg body weight (mg (kg BW)-1) of AFB developed into hepatoma at a rate of nearly 100% in 52 weeks [62]. Some reports suggested that hepatoma models could be established in DBA/2J and C57BL/N mice using AFB. However, the hepatoma-induction rate in C57BL/N mice was quite low [63]. The difference in susceptibility between DBA/2J and C57BL/N mice may attribute to the differences in several AFB sites.
Choline-deficient diet
Many studies have shown that hepatoma can be induced by the application of a CDD. This method was first used to induce the formation of steatohepatitis, liver fibrosis, and liver cirrhosis in rats and mice [64]. Recently, it was found that mice receiving CDD developed into hepatoma in 50 - 52 weeks [65]. Similarly, a large proportion of rats that received CDD developed into hepatoma. The primary mechanisms of hepatoma development induced by CDD are associated with the stimulation of oval cells, resulting in increased oxidative stress, genetic mutations, modifications, and DNA damage. The carcinogenic mechanisms of CDD might be similar to the carcinogenicity of chemotoxic compounds such as DEN and CCl4 [66]. In addition, the ethionine administration combined with a CDD can enhance the stimulation to oval cells and increase carcinogenicity [67]. Similarly, CDD combined with DEN can further stimulate the induction of hepatoma and shorten the period of tumor formation compared with CDD alone [68].
Virally induced hepatoma models
More than 80% of human hepatoma patients are caused by the hepatitis B virus (HBV) and hepatitis C virus (HCV) [69]. Because of the rigorous human tropism of these viruses, one problem with the HBV or HCV-associated hepatoma mouse model is that HBV or HCV requires human hepatocyte to induce hepatitis [70]. For example, cirrhosis is one of the characteristic manifestations of HBV and HCV, but sometimes it does not necessarily occur in rodents [71]. Cirrhosis in rodents was observed only in a few cases, but cirrhosis is very common in human hepatoma patients [72]. The development of hepatoma induced by HBV or HCV infection may take more than 20 years because this process requires many steps of genetic alterations. It is difficult to detect the molecular mechanisms of all these steps using cell culture or non-genetic animal models. Therefore, various animal models have been designed to investigate viral hepatitis. The two standard models for HBV-induced hepatoma are woodchucks (Marmota monax) and ground squirrels (Spermophilus beecheyi), which are induced by long-term woodchuck hepatitis virus (WHV) and chronic Squirrel Hepatitis Virus, respectively [73]. The woodchuck model of hepatoma is a valuable tool to test the effect of nucleoside analogs in the long-term treatment of hepatoma [74].
Hepatitis B virus
The high incidence of hepatoma is closely related to the large proportion of people infected with HBV, especially in developing countries. HBV infection causes liver fibrosis and cirrhosis, eventually leading to hepatoma. This pathological evolution is often referred to as the hepatitis B liver trilogy. The control of the HBV plays an important role in the prevention of hepatoma. The HBV genome encodes a variety of proteins, the most carcinogenic of which is X-protein (HBx). There is growing evidence that viral genes, particularly HBx-encoding genes, may cause uncontrolled cell growth and viability, making liver cells sensitive to exogenous and endogenous carcinogens [75]. In 1985, two research teams developed the first HBV-related transgenic mouse model [76]. Many HBV-related transgenic animals express HBx gene, which is closely related to the change of liver function and the occurrence and development of hepatoma [77]. HBx transgenic mice are more susceptible to hepatoma than their non-transgenic counterparts after a single DEN injection [78].
Hepatitis C virus
Approximately 30% of hepatoma cases are associated with chronic HCV infection, which is the second most common cause of hepatoma [69]. The possibility for HCV-infected patients to develop cirrhosis is as high as 35% [70]. The cumulative risk of developing hepatoma in these patients with liver cirrhosis is 1% to 7% per year. Considering hepatoma is the most common cause of death in the HCV patients [79], it is necessary to study the mechanism of HCV-induced hepatoma and learn how to intervene in its progression. Preclinical models are often performed in rodents. For example, mouse models of HCV-infected hepatoma developed by Lerat et al. are similar to HCV-infected hepatoma in human [80]. These models could help to study the carcinogenic mechanisms of HCV-associated hepatoma and the possible causes of HCV infection in hepatoma [81]. There are still some models that do not develop hepatoma, which may be explained by the difference in expression levels of HCV RNA or protein and the genetic differences in mice. It is difficult to establish an ideal mouse model due to the different pathological features between mouse hepatoma and human hepatoma and the heterogeneity of hepatoma itself. However, they still have many advantages and broad applications in preclinical and clinical studies of hepatoma.
Genetically engineered hepatoma models
A transgenic mouse model constructed in the early 1980s was used to study the molecular characteristics of human malignant tumors in vivo [82]. A variety of transgenic animal models have been studied, of which the most widely studied is the transgenic Simian Virus 40 (SV40) T-antigen mouse model. The genome of SV40 is a well-known DNA tumor virus, which encodes the large and small oncogenic T antigens, T-Ag, and t-Ag, collectively known as T-Ag [83]. After infection with the virus, the large T-Ag causes malignant transformation of host cells mainly through the deactivation of p53 and Rb [84]. Another such transgenic mouse model was constructed by Murakami et al. [85]. They hybridized transgenic Alb/c-Myc mice (overexpressing c-Myc, guided by the albumin promoter) and transgenic MT/TGF-α mice (overexpressing TGF-α, guided by the metallothionein 1 promoter) to produce double transgenic mice that overexpressed c-Myc and TGF-α in the liver. These traditional transgenic mouse models have been frequently used to reveal the role of a particular gene in the development of hepatoma and to study the development of multiple individual stages of hepatocellular carcinogenesis [86, 87]. Recently, conditional mouse models have been developed by inducing the genetic alterations in a unique time-controlled, tissue-specific manner. For example, based on the fact that mice do not express TVA receptor of subgroup A avian leucosis sarcoma virus (ALSV-A), Lewis et al. used the retroviral transduction strategy to transfer oncogenes to liver cells in situ [88].
Recently, there are many breakthroughs in genetically engineered hepatoma mouse models. However, so far these models have been used to study the effects of gene changes, e.g., mutations, deletions, or overexpression, during the onset of hepatoma, rather than at the point of hepatocarcinogenesis [89]. A significant breakthrough in the application of mouse models to study hepatoma in this area is the development of ribonucleic acid (RNA) microarray technology. It helps us to estimate which subclass a particular hepatoma tissue belongs to and to verify the most divergent genes among subclasses. Lee and colleagues found that c-Myc/TGF-α mice were more likely to reproduce the prognosis of advanced hepatoma, and the hepatoma in E2f1, c-Myc, and c-Myc/E2f1 transgenic mice was similar to a set of hepatoma with good prognosis [90]. The results were further confirmed by the analyses of the chromosomal change pattern and the existence of β-catenin mutations obtained from the corresponding transgenic animal hepatoma [91].
In conclusion, various animal models of hepatoma have been constructed, and they are helpful for screening new drugs for the treatment of hepatoma, studying the interactions between tumor and host, discovering possible carcinogens of hepatoma, and exploring the molecular mechanism of hepatoma. Researchers should choose appropriate animal models according to the characteristics of each animal model and research purposes. As shown in Table 2, we summarized the advantages and disadvantages of different hepatoma models. Among them, we believe that the most promising model is the genetically engineered hepatoma model, which can help to investigate the mechanisms of hepatoma from the genetic perspective and provide a basis for individualized and precise treatment of hepatoma. With the rapid development of genome editing technology, more ideal hepatoma models will be established, which can effectively guide the diagnosis and treatment of hepatoma.
Chemotherapy and molecular targeted therapy of hepatoma with nanoformulated drugs
As mentioned above, we know that an essential role of animal models of hepatoma is to screen new therapeutic drugs, including chemotherapy drugs, molecular targeted agents, immunotherapy drugs, such as nivolumab and pembrolizumab, and gene therapy drugs, and the most commonly used models are subcutaneously inoculated hepatoma models. Chemotherapy and molecular targeted therapy are currently the primary treatment for advanced hepatoma, but the effect is often unsatisfactory. With the development of nanotechnology, many nanoformulated drugs have been developed for the treatment of hepatoma. However, the antitumor efficacies and side effects of these drugs need to be thoroughly evaluated before they can be applied to clinical treatment. Therefore, in this part, we will review the application and progress of nanoformulated drugs in the treatment of hepatoma in combination with relevant animal models.
Hepatoma is a deadly malignant tumor with high morbidity worldwide. In recent years, due to the rapid development of early diagnosis, the resection rate of hepatoma has increased significantly [92]. However, hepatoma is prone to metastasis, and a majority of patients have already progressed to the intrahepatic or distant metastasis at the time of detection. As a result, a large number of hepatoma patients miss the chance to receive surgical resection. Therefore, nonsurgical treatments of hepatoma, especially chemotherapeutics, including doxorubicin (DOX), 5-fluorouracil (5-FU), gemcitabine (GEM), cisplatin (CDDP), and mitoxantrone (MX), and molecular targeted drugs, including sorafenib (SF), lenvatinib (LT), regorafenib (RF), and cabozantinib (CT), play an essential role in the clinical treatment of hepatoma.
Generally, hepatoma is resistant to chemotherapeutic agents, and the therapy efficacy is relatively limited. The rapid development of nanomedicines has dramatically improved the therapeutic effects of many small molecule cytotoxic drugs used in the clinic [93, 94]. Compared with traditional chemotherapeutics, antineoplastic drug-loaded polymer nanoparticles have significant advantages in several aspects: (i) Prolonged blood circulation. The encapsulation of polymer nanoparticles increases the half-life of drugs in the blood. (ii) Enhanced tumor accumulation. The nanosized platforms facilitate the localization of drugs in tumor tissues through the enhanced permeability and retention (EPR) effect and/or tumor targeting ligand-mediated active targeting [95]. (iii) Tumor microenvironment-specific drug release profile. The on-demand drug delivery is triggered by the specific endogenous intracellular stimuli (e.g., pH, redox environment, ROS, or enzyme) or exogenous excitations (e.g., light, temperature, or voltage) [96-99]; (iv) Synergetic therapy. Different drugs with various antitumor mechanisms achieve synergistic effects through encapsulation in polymer nanoparticles with well-designed release profiles [100]; (v) Crossing biological barriers. Antineoplastic drug-loaded polymer nanoparticles can cross the blood-brain barrier [101] and even escape from intracellular autophagy. Due to these advantages, polymer nanoparticles significantly improve the antitumor efficacies and reduce the side effects of chemotherapeutic drugs. In addition, the combined chemotherapy is widely used to improve the antitumor efficacy and overcome drug resistance to tumors [102]. Different drugs can induce cell apoptosis at different cell cycle stages, which requires careful consideration of their therapeutic pathways to reduce side effects.
Recently, molecular targeted therapy has shown effective efficacy in the treatment of advanced and refractory hepatoma, providing a promising strategy for improving the survival of patients without effective therapy [103]. At present, according to the latest results in clinical trials, SF and LV are the first-line treatment for hepatoma, and RF and CT are the second-line treatment [104]. In the advanced therapy, molecular targeted drugs are developed to inhibit specific pathways, including epidermal growth factor receptor (EGFR), vascular endothelial growth factor receptor (VEGFR), phosphoinositol 3-kinase (PI3K)/ mammalian target of rapamycin (mTOR), Ras/ extracellular signal-regulated kinase, hepatocyte growth factor (HGF)/mesenchymal-epithelial transition factor (MET), Hedgehog, Wnt, and apoptotic signaling. In addition, the combination of different molecular targeted drugs, and the combination of molecularly targeted drugs with other antineoplastic agents show great potential in the clinical therapy of advanced hepatoma.
Nanoformulations of chemotherapeutic drugs
Doxorubicin-loaded nanoparticles
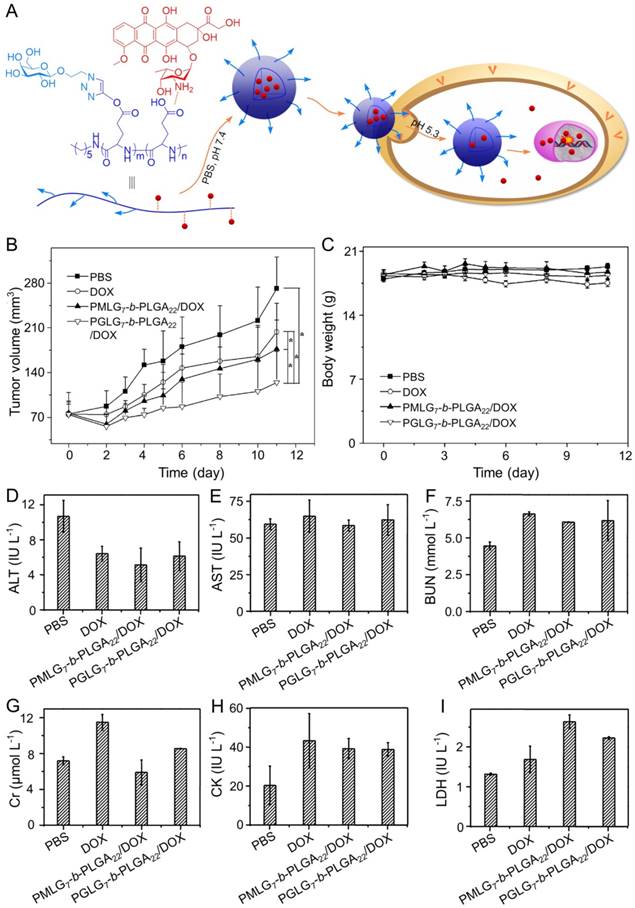
DOX is one of the most common chemotherapeutic drugs to treat a variety of cancers [105], including hepatoma [106]. However, its clinical application is often hindered by severe side effects, especially cardiotoxicity [107]. Recently, different nanoscale drug delivery systems have been developed to decrease the side effects of DOX. For this reason, our group has carried out much work in the field of smart antitumor drug delivery with smart polymer nanocarriers. Ding et al. fabricated a series of glycopolypeptide-based micelles for intracellular DOX delivery in hepatoma chemotherapy using the five-week-old BALB/c nude mice subcutaneously inoculated with HepG2 cells, as shown in Figure 2A [108, 109]. The hepatoma-targeted DOX-loaded galactosylated polypeptide (PGLG-b-PLGA) nanoplatforms (i.v.) were more effective to inhibit the tumor growth as compared to a formulation that substituted galactose with oligo(ethylene glycol) (PMLG-b-PLGA) (Figure 2B). It was attributed to its enhanced cellular uptake of tumor cells. Moreover, no significant body weight loss was observed during this treatment period. In contrast, mice treated with free DOX showed mild weight loss due to its poor toleration and minor acute plasma DOX concentration, which was shown in Figure 2C. As shown in Figure 2D - 2I, the parameters of the liver, kidney, and heart in all treatment groups were at normal levels, indicating that free DOX and DOX-loaded nanomedicines didn't cause severe toxicity to mice.
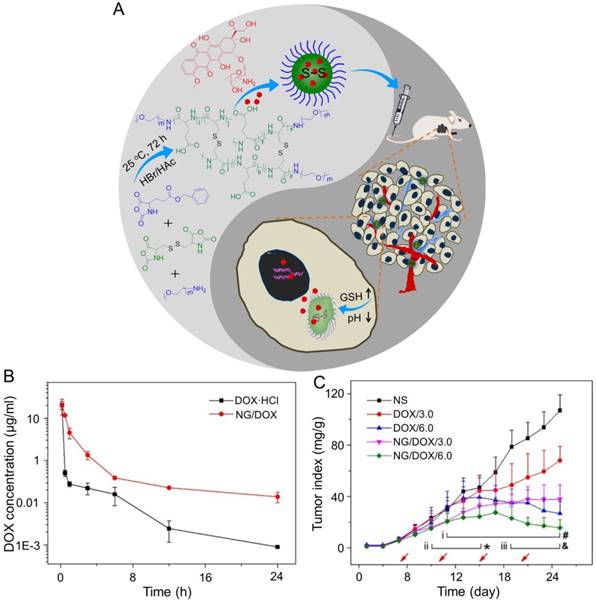
In the study of Ding's group, a methoxy poly(ethylene glycol)-poly(L-phenylalanine-co-L-cystine) (mPEG-P(LP-co-LC)) nanogel with reduction-responsiveness was prepared by one step ring-opening polymerization of L-cystine N-carboxyanhydride (LC NCA) and L-phenylalanine N-carboxyanhydride (LP NCA), as depicted in Figure 3A [96, 97]. At 6 and 12 h after administration (i.v.), the accumulation of DOX-loaded nanogel (NG/DOX) at the tumor site was higher than that of free DOX·HCl at a dose of 6.0 mg (kg BW)-1. The results were due to the EPR effect of NG/DOX in the tumor tissues (Figure 3B). Compared with the free drug, NG/DOX showed the improved antitumor effect on human HepG2 hepatoma-xenografted BALB/c nude mouse model (Figure 3C and D). In the same group, a pH and reduction dual-responsive polypeptide nanogel methoxy poly(ethylene glycol)-poly(L-glutamic acid-co-L-cystine) (mPEG-P(LG-co-LC)) was developed for DOX delivery in hepatoma chemotherapy, as shown in Figure 4A [110]. The NG/DOX showed enhanced capability for inhibiting proliferation of human hepatoma HepG2 and H22 cells compared with free DOX·HCl in vitro. Moreover, NG/DOX showed a lower half-maximal inhibitory concentration (IC50) than free DOX·HCl, suggesting efficient endocytosis and intracellular microenvironment-responsive release of NG/DOX. As shown in Figure 4B, the plasma concentration of free DOX·HCl decreased rapidly due to its fast elimination. In contrast, the level of NG/ DOX (i.v.) in the plasma was much higher than that of free DOX·HCl. As a result, the antitumor efficacy of NG/DOX on H22 hepatoma-bearing BALB/c mouse model was better than that of free DOX·HCl, as depicted in Figure 4C. Altogether, NG/DOX showed great potential in clinical chemotherapy of malignancy.
Synopsis of leading experimental features of preclinical mouse models of hepatoma.
| Model | Promoter | Steatosis | Injury | Inflammation | Fibrosis | References |
|---|---|---|---|---|---|---|
| DEN | PB | + | + | + | - | [86] |
| PP | - | - | + | + | - | [176] |
| Aflatoxin B1 | - | - | - | -/+ | + | [177] |
| CCl4 | - | - | + | + | + | [178] |
| TAA | - | - | - | + | - | [52] |
| CDD | Methionine | + | + | + | + | [179] |
| HBV transgenic | X protein | + | + | - | - | [87] |
| HCV transgenic | Polyprotein | + | + | - | - | [80] |
+: available; -: not available.
Synopsis of the advantages and disadvantages of different hepatoma models.
| Hepatoma models | Advantages | Disadvantages | Applied research |
|---|---|---|---|
| Subcutaneously inoculated hepatoma models | Easy to establish Rapid tumor formation (5 - 20 weeks) Easy to monitor size of tumor Low cost Highly reproducible | Lacking interaction between tumors and liver tissues Lacking tumor-host interactions (such as metastasis and angiogenesis) | Screening of new drugs |
| Orthotopically implanted hepatoma models | Rapid tumor formation Metastases can be observed Replicating the tumor microenvironment Tumor-host interactions can be tested | Difficult to establish Complex procedures High cost Difficult to monitor the size of tumor | Screening of new drugs Investigating tumor-host interactions |
| Chemically and virally-induced hepatoma models | The lesion-fibrosis-malignant cycle similar to humans can be observed | Slow tumor formation Causing severe liver damage There are species differences in response to carcinogens between humans and mice | Identifying possible carcinogens of hepatoma Investigating the relationship between carcinogen exposure and specific genetic changes |
| Genetically engineered hepatoma models | Specific to the liver or hepatocytes Temporary or permanent regulation May be reversible sometimes | Difficult technique High cost Transgenes are expressed in all hepatocytes Genetic alterations exist throughout the embryogenesis process | Studying the effect of genetic alterations Investigating the molecular mechanism of hepatoma |
Preparation process of DOX-loaded PMLG7-b-PLGA22 and PGLG7-b-PLGA22 micelles and evaluation of the antitumor efficacy [109]. (A) Preparation and targeted delivery of micelles assembled from glycopeptide and DOX. (B) Antitumor efficacies in vivo, (C) and body weight changes treated with PBS, DOX, and micelles from PMLG7-b-PLGA22/DOX and PGLG7-b-PLGA22/DOX. Evaluation of (D) ALT, (E) AST, (F) BUN, (G) Cr, (H) CK, and (I) LDH levels after all the treatments of PBS, DOX, and nanomedicines from PMLG7-b-PLGA22/DOX and PGLG7-b-PLGA22/DOX. Copyright 2013. Reproduced with permission from Elsevier Ltd.
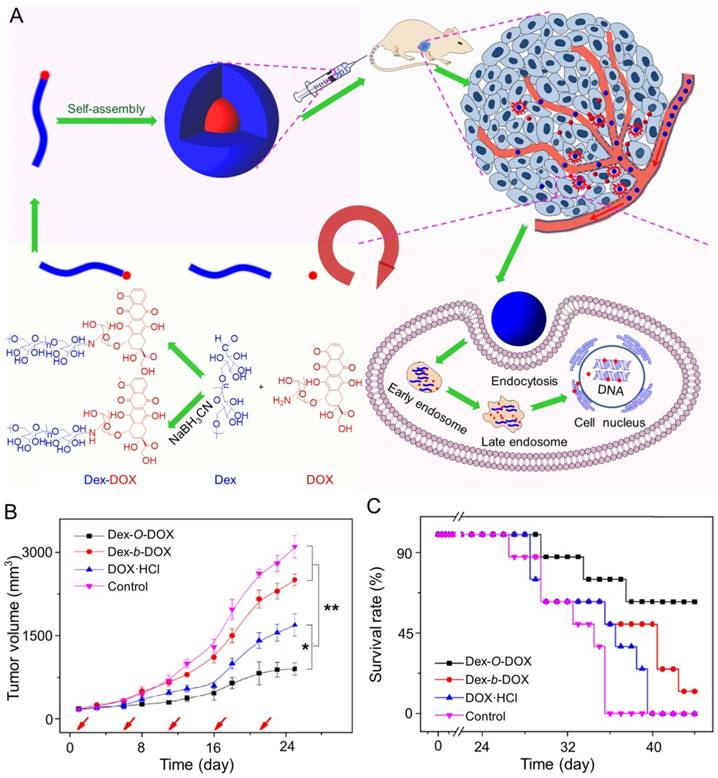
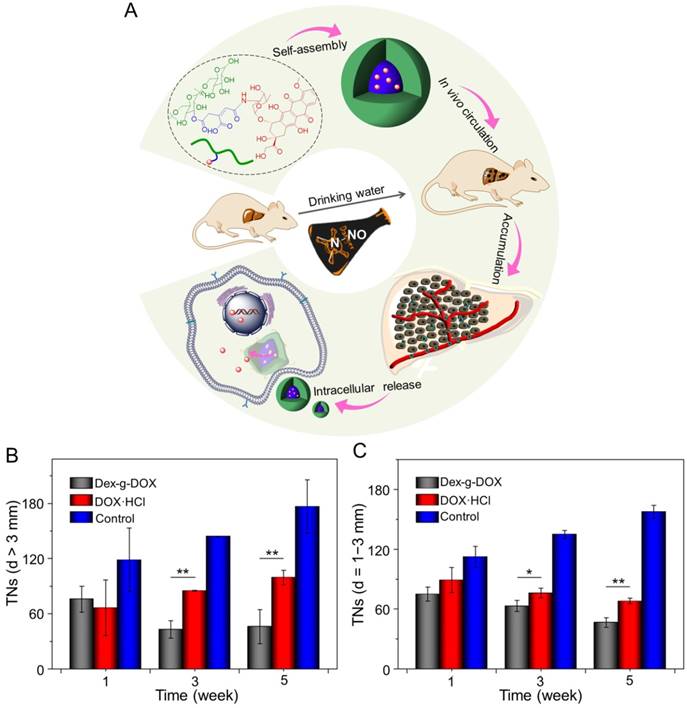
In addition, Ding and coworkers synthesized an acid-sensitive dextran-doxorubicin conjugate (Dex-O-DOX) for hepatoma therapy (Figure 5A) [111]. All DOX formulations were given to Kunming mice with H22 tumors by intravenous injection. As expected, the antitumor activities of these groups were ranked in the following order: Dex-O-DOX > free DOX·HCl > Dex-b-DOX (Figure 5B). This finding can be attributed to the selective DOX release of Dex-O-DOX in tumor cells (HepG2 cells). The body weights of mice receiving Dex-O-DOX and Dex-b-DOX increased more significantly than those of mice receiving free DOX·HCl, suggesting that the conjugation of DOX to Dex could efficiently reduce the drug toxicity. As shown in Figure 5C, the Dex-O-DOX group showed an extended survival period compared with the control group, mainly because the sustained DOX release enhanced its antitumor effect. These findings suggested that Dex-O-DOX could effectively suppress the growth of hepatoma in vivo and significantly reduce the systemic side effects. In the same group, an acid-sensitive Dex-DOX prodrug (Dex-g-DOX) was facilely synthesized for chemotherapy of the DEN-induced orthotopic hepatoma in rats, as depicted in Figure 6A [112]. As shown in Figure 6B, the number of large tumor nodules (TNs) (>3 mm) in rats treated using Dex-g-DOX (i.v.) was lower than that in rats treated using free DOX·HCl. Meanwhile, Dex-g-DOX also showed significantly fewer TNs (1-3 mm) and higher antitumor efficacy (Figure 6C). Overall, Dex-g-DOX significantly boosted the therapeutic efficacy in orthotopic hepatoma in rats. These studies demonstrate that polymer-drug conjugates have great potential in clinical chemotherapy of hepatoma.
Preparation of mPEG-P(LP-co-LC) nanogel and determination of the antitumor effect [96]. (A) Synthetic pathway for mPEG-P(LP-co-LC) nanogel, illustrations of DOX encapsulation by nanogel, and its circulation, intratumoral accumulation, endocytosis, and targeting intracellular DOX release after intravenous injection. (B) Ex vivo DOX fluorescence images of major visceral organs and tumor isolated at 6 or 12 h post-injection of NS, free DOX·HCl, or NG/DOX at a dose of 6.0 mg DOX·HCl equivalent per kg body weight toward BALB/c nude mice bearing a HepG2 tumor. (C) In vivo antitumor efficacies of NS, free DOX·HCl, and NG/DOX at a dose of 3.0 and 6.0 mg DOX·HCl equivalent per kg body weight. Copyright 2015. Reproduced with permission from Elsevier Ltd.
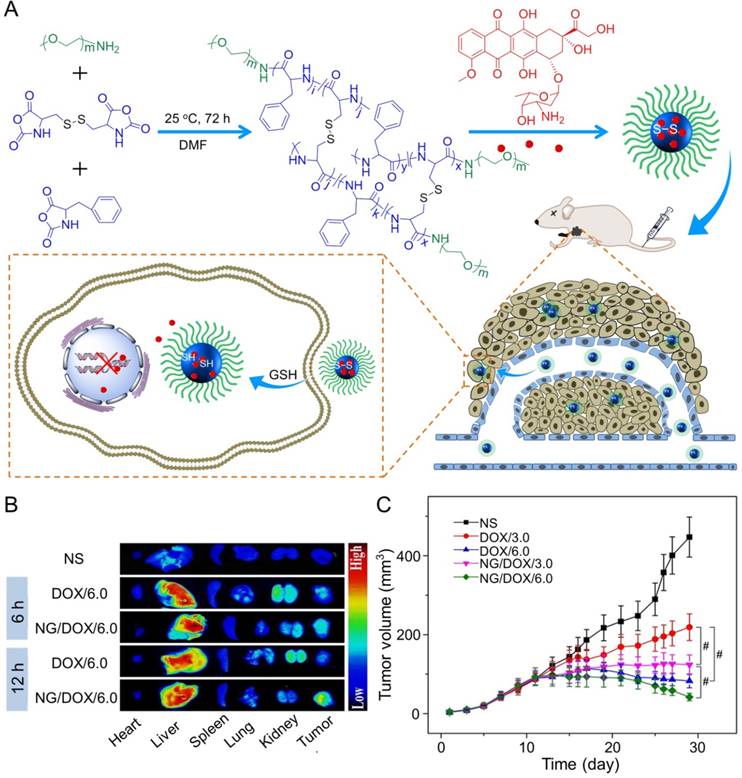
NG/DOX characterizations and DOX encapsulation, cell proliferation inhibition, and pharmacokinetics in vivo [110]. (A) Synthetic pathway for mPEG-P(LG-co-LC) nanogel, DOX encapsulation by nanogel, and its characterization. (B) In vivo pharmacokinetic profiles after injection of DOX and NG/DOX in rats. (C) In vivo antitumor efficacy of NS, or of free DOX·HCl or NG/DOX at a dosage of 3.0 and 6.0 mg DOX equivalent per kg body weight toward H22-hepatoma-grafted BALB/c mouse model. The arrows indicated the treatment times. Each set of data was represented as mean ± SD (n = 10; * P < 0.05, & P < 0.01, # P < 0.001; i, DOX/3.0 vs NG/DOX/3.0; ii and iii, DOX/6.0 vs NG/DOX/6.0). Copyright 2017. Reproduced with permission from the Ivyspring International Publisher.
The above-mentioned smart polymer nanoparticles exploited in the Ding's group showed good passive and/or active targeting to the liver. Notably, the cardiac toxicity of DOX was also decreased in the groups of DOX-incorporated nanoparticles. These findings suggested that the DOX-loaded nanoparticles could effectively inhibit the growth of hepatoma and significantly reduce the systemic side effects. In addition to Ding and his colleagues' work, a variety of other DOX-loaded nanoparticles have also been developed. Increasing the targeting of DOX to tumor cells is one strategy of nanoformulations. Wang et al. designed and prepared CD147-targeted DOX-loaded immunoliposomes (anti-CD147 ILs-DOX) [21]. Because CD147 is an important marker expressed on the surface of hepatoma cells, anti-CD147 ILs-DOX (i.v.) could specifically and efficiently deliver DOX to CD147-overexpressing hepatoma cells, resulting in enhanced antitumor effects and lower side effects in Huh-7 xenograft mice models. Similarly, in order to enhance the efficacy and safety of doxorubicin, Xu et al. designed hepatoma-targetable DOX-encapsulating nanoparticles (tNP-PLA-DOX) by a modular assembly approach [113]. At first, they synthesized DOX-derived polymeric prodrug (PLA-DOX) by attaching DOX to a polylactide building block. Then PLA-DOX coassembled with 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy poly(ethylene glycol) 2000] (DSPE-PEG2000) to form injectable nanomedicine. Finally, a hepatoma-specific homing ligand was decorated on the surface of nanomedicine. The tNP-PLA-DOX was characterized by good stability, low toxicity, and high selectivity to tumor cells. In vivo antitumor efficacy experiment showed the tumor weights of HCC-LM3 xenograft-bearing nude mice treated with tNP-PLA-DOX (i.v.) were lower than those of the mice in control groups. Therefore, nanoparticles can improve the therapeutic index of DOX and provide a promising direction for the advanced treatment of hepatoma.
Fabrication of Dex-DOX conjugates and the assessments of antitumor activity and security [111]. (A) Syntheses and self-assembly of Dex-DOX conjugates and characterization. (B) Tumor volumes and (C) survival rates of mice treated with Dex-O-DOX, Dex-b-DOX, or free DOX·HCl with NS as a control. Copyright 2015. Reproduced with permission from Elsevier Ltd.
Although the above nanoparticles all increased the tumor targeting and antitumor efficacy of DOX and reduced its side effects to varying degrees, we can see that there are some apparent differences between them. First of all, different kinds of nanocarriers are used in these nanoformulations, including polypeptides (PLGA), polysaccharides (e.g., Dex), polypeptide nanogels, polylactide (PLA), and liposomes. These nanocarriers have some unique physical and chemical properties, such as biocompatibility, biodegradability, high drug loading capability, pH sensitivity and reduction reactivity, which contribute to the delivery and release of nanomedicines. For example, the reduction-responsive polypeptide nanogels enabled NG/DOX to release DOX triggered by the intracellular microenvironment rapidly. Dex made Dex-g-DOX have the property of inherent self-targeting, allowing it to accumulate highly in the liver. Secondly, different ligands are modified on the surface of the nanoparticles to increase the targeting of the nanoformulations to the cancers cells. The galactose ligand can recognize the asialoglycoprotein receptor (ASGP-R) of HepG2 cells, which significantly promoted the uptake of PGLG-b-PLGA and PMLG-b-PLGA. Anti-human CD147 antibody and HCC-specific homing ligand (SP94) significantly increased the active targeting of anti-CD147 ILs-DOX and tNP-PLA-DOX on hepatoma, respectively. Finally, the structure and synthesis of nanoparticles are different. PMLG-b-PLGA and PGLG-b-PLGA were prepared by self-assembly of DOX and galactopeptide, and Dex-O-DOX was synthesized by the versatile oximation reaction. NG/DOX was synthesized by sequential dispersion and dialysis technique, which made it highly efficient in drug loading. In contrast, tNP-PLA-DOX was synthesized by multiple components through a modular assembly approach, which was cost-effective. In summary, we can see that different nanoformulations have their own characteristics and advantages. With the development of nanotechnology, the properties and anticancer effects of nanoformulations are continually improving, and they will become an effective means for treating hepatoma in the future.
5-Fluorouracil nanoformulations
As one of the most widely used antitumor drugs, 5-FU is broadly used to treat many solid tumors, such as colon cancer, gastric cancer, and hepatoma [114]. As a pyrimidine analog, 5-FU inhibits DNA synthesis and thymidylate synthase, leading to cell cycle arrest and apoptosis. However, its clinical applications and efficacy are impacted mainly by poor pharmacokinetic properties. Moreover, compared with other chemotherapeutic drugs used clinically, 5-FU can cause bone marrow depression, gastrointestinal irritation, leucopenia, and thrombocytopenia, which may be due to unstable drug concentration in the blood and systemic nonspecific distribution of free drugs [115]. Therefore, it is necessary to develop effective drug carriers that can be used for controlled release of 5-FU.
Self-assembly, characterization, and antitumor efficacies of Dex-g-DOX [112]. (A) Schematic illustration for some characterizations of Dex-g-DOX. (B) TNs with diameters > 3 mm; (C) TNs with diameters = 1 - 3 mm. Copyright 2016. Reproduced with permission from the American Chemical Society.
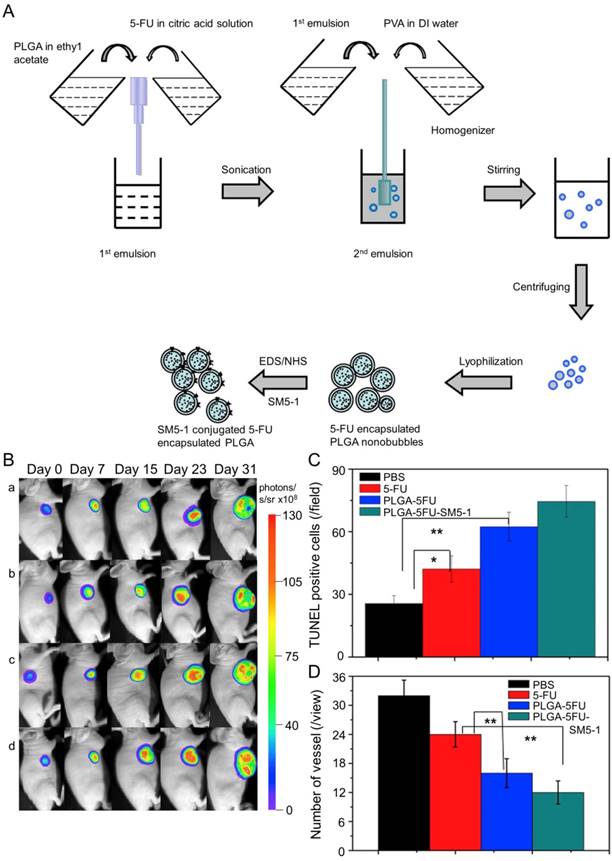
In Huang's work, the N-galactosylated-chitosan-5-FU (GC-FU) acetic acid conjugate was evaluated by different experiments with HepG2 and A549 cells in vitro and in vivo [116]. The half-life of GC-FU-NPs (i.v.) in blood circulation was longer than that of free 5-FU. GC-FU-NPs could specifically and effectively recognize ASGPR receptors on the surface of HepG2 cells, resulting in lower cytotoxicity than free 5-FU. Therefore, GC-FU-NP was a promising targeted system for hepatoma therapy. Cheng and coworkers synthesized GC/5-FU-NPs through a combination of GC and 5-FU, and examined its efficacy in treating hepatoma in vitro and in vivo [117]. GC/5-FU (i.v.) could naturally inhibit tumor growth in orthotropic hepatoma models. GC/5-FU caused higher cytotoxicity to hepatoma cells than to other cells, thus reducing the side effects of 5-FU. Compared with free 5-FU, GC/5-FU could significantly reduce tumor weight and prolong survival time. Ma et al. prepared a humanized mouse antibody SM5-1-conjugated poly(D,L-lactide-co-glycolide) (PLGA) nanobubble containing 5-FU (PLGA-5FU-SM5-1) to treat HCC-LM3-fLuc orthotopic hepatoma tumors in mice (Figure 7A) [118]. PLGA-5FU-SM5-1 (i.v.) had a prolonged circulation and could target the tumor cells specifically, thereby reducing the toxic side effects and increasing the antitumor activity. The tumor sizes of mice treated with PLGA-5FU-SM5-1 were well controlled compared with other groups. In addition, PLGA-5FU-SM5-1 posed a significantly higher inhibition rate toward HCC mice than other groups on day 31 (Figure 7B and C). Another essential condition for advanced tumor growth is angiogenesis, so the number of vessels was counted to evaluate the antiangiogenic effect of the drugs. The results exhibited that PLGA-5FU-SM5-1 could significantly down-regulate tumor angiogenesis compared with other groups (Figure 7D).
Gemcitabine-loaded nanosystems
GEM is an antitumor nucleoside analog that interferes with cellular replication in various solid tumors. Its cell internalization mainly relies on the nucleoside transporter hENT1, the downregulation of which may lead to GEM resistance because it is too hydrophilic to pass through the plasma membrane passively. To this end, the GEM-loaded nanoparticles can partially avoid hENT1 because they can be internalized through endocytosis [119]. Some studies have reported that the combination of chlorambucil (CHL) and GEM can improve the therapeutic efficacy [120, 121]. CHL (4-[bis(2-chloroethyl) amino] benzenebutanoic acid) is a type of lipophilic DNA alkylating agent, but the toxic side effects limit its therapeutic performance. It was reported that delivering CHL in tumor-targeted nanocarriers was a practical approach to reduce toxicity [122].
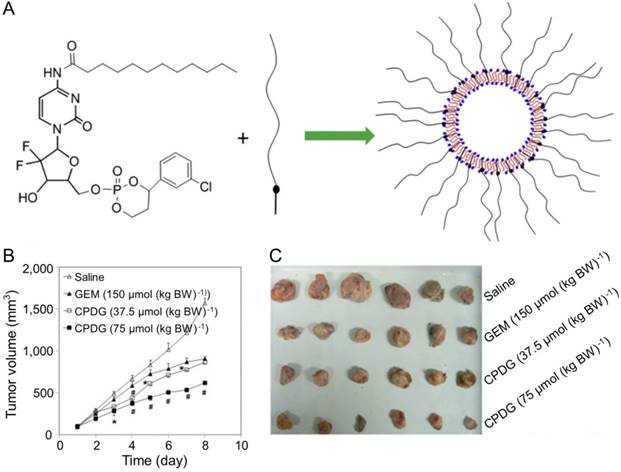
Furthermore, CHL combined with GEM was a promising strategy for treating hepatoma. Fan and coworkers designed and synthesized chlorambucil gemcitabine (CHL-GEM) conjugates nanomedicine [120]. Because of the amphiphilic characteristic, CHL-GEM conjugates self-assemble into nanoparticles and then accumulate in tumor tissues via the EPR effect. This conjugate formulation showed significant improvement in the treatment of hepatoma in SMMC-7721 tumor-bearing nude mice. More studies have been conducted to study the combined effect of nanoparticles and GEM. Du et al. synthesized cyclic phosphoryl N-dodecanoyl GEM (CPDG) and then synthesized the long-circulating CPDG nanoassemblies composed of CPDG and a long-circulating material—cholesteryl hemisuccinate PEG1500 (CHSPEG1500) for the treatment of H22-beared hepatoma (Figure 8A) [123]. The CPDG nanoassemblies were intravenous injected into the tumor-bearing mice at doses of 37.5 and 75.0 mmol per kg body weight (mmol (kg BW)-1) while the control group was treated with free GEM (150.0 mmol (kg BW)-1). The group treated with long-circulating CPDG NPs (75 mmol (kg BW)-1) showed the highest tumor inhibition rate among all treatment groups (75 mmol (kg BW)-1) (Figure 8B and C). However, similar to many other antitumor drugs, GEM is prone to induce the resistance of hepatoma, which ultimately compromises its therapeutic effect in hepatoma patients [124].
Therefore, it is necessary to conduct further studies to investigate the molecular mechanism of its drug resistance. Although GEM has certain defects, it is a promising antitumor drug for the treatment of hepatoma whether given alone or combined with other therapies.
Cisplatin-loaded nanoplatforms
CDDP is a widely used platinum-based antitumor drug in the treatment of solid malignancies, working as a cell cycle phase-nonspecific drug [125-127]. After the CDDP enters the body, the chlorine atoms are gradually replaced by water molecules to form [Pt(H2O)2(NH3)2]+ [128]. Platinum crosslinks with two bases on the DNA to form a closed chain or inter-chain adduct, which inhibits the DNA replication process of cancer cells, leading to apoptosis [129-132]. At the cellular level, CDDP could impact various cellular components through nucleophilic sites, then results in cell malfunction and death [124]. Moreover, intracellular CDDP can react with nuclear DNA to produce DNA-protein crosslinks and intrastrand DNA crosslinks [133]. CDDP can target not only genomic DNA but also other cellular components. For example, CDDP can interact with phospholipids and phosphatidylserines in membranes, bind to mitochondrial DNA and disrupt the cytoskeleton [134]. Cytotoxicity of CDDP is also observed in DNA repair-deficient cells. These cells will die at concentrations of CDDP that do not inhibit DNA synthesis in normal cells and then get blocked in the S phase [135].
However, the use of CDDP always causes severe toxicity to normal tissues or organs, such as gastrointestinal toxicity, hepatotoxicity, nephrotoxicity, and neurotoxicity. In addition, the treatment with CDDP usually induces drug resistance, which always leads to treatment failure. At present, there are many studies on the mechanism of CDDP resistance. Current hypotheses include the enhancement of detoxification of CDDP, the inhibition of apoptosis, and the enhancement of DNA repair ability.
Fabrication process of PLGA, PLGA-5FU, and PLGA-5FU-SM5-1, and inhibition of tumor growth [118]. (A) Schematic illustration of the fabrication process of PLGA, PLGA-5FU, and PLGA-5FU-SM5-1. (B) Serial bioluminescent images of the HCC-LM3-fLuc tumor-bearing nude mice that underwent PLGA-5FU-SM5-1 (a) PLGA-5FU (b) 5-FU (c) saline (d) treatment. (C) The quantitative results of cell apoptosis and (D) angiogenesis. Copyright 2014. Reproduced with permission from Elsevier Ltd.
Preparation of the long-circulating CPDG nanoassemblies and inhibition of tumor growth [123]. (A) The long-circulating CPDG nanoassemblies synthesis and preparation process. (B) Growth profiles of tumor volume after i.v. injection of GEM solution and long-circulating CPDG nanoassemblies into the mice. (C) Tumor images following i.v. administration of GEM and long-circulating CPDG nanoassemblies to the mice. Copyright 2016. Reproduced with permission from Elsevier Ltd.
Nanoparticles can effectively enhance the transport and aggregation of CDDP in tumor cells through EPR effect and enhance the targeting of CDDP to tumor cells. Therefore, the combination of CDDP and polymer nanocarriers reduces the side effects on normal organs, reverses the CDDP-resistance to tumor cells, and improves the antitumor efficacies [136, 137]. Li and coworkers first delivered CDDP by the micelles of amphiphilic block copolymers methoxy poly(ethylene glycol)-block-poly(ε-caprolactone) (mPEG-b-PCL) [138]. The laden micelle delivered intratumorally (i.t.) exhibited cytotoxicity comparable to that of free CDDP against BGC823 and H22 cells, and it could improve the antitumor effect and reduce side effects in vivo. Subsequently, Ding et al. developed the gelatin-poly(acrylic acid) nanoparticles to deliver CDDP [139]. The CDDP-loaded nanoparticles delivered i.p. exhibited improved antitumor efficacy against the mouse H22 hepatoma-allografted mouse model, and the treatment with CDDP nanomedicine prolonged the survival time of mice.
Mitoxantrone-loaded nanoparticles
MX, dihydroxy anthracene dione, is a tricyclic planar chromophore with two basic side chains [124]. It possesses antitumor activity against many kinds of tumors, including hepatoma [140]. MX was initially developed as a simplified DOX with lower cardiotoxicity. Later, the US Food and Drug Administration (FDA) approved it for the treatment of prostate cancer, multiple sclerosis, and acute myeloid leukemia [141]. The antitumor mechanism of MX is to inhibit cell division through inserting into DNA, causing DNA condensation and preventing DNA replication as well as RNA synthesis. Apart from its effect on cell division, MX also kills non-proliferating cells, suggesting that it may function in a cell-cycle-independent way. MX binds to chromatin to form a complex that inhibits the release of histone proteins, with an affinity to chromatin that is much higher than DNA [142]. Moreover, MX suppresses the topoisomerase II enzyme [143]. In HepG2 and Hep3B cell lines, MX stimulates the accumulation of apoptotic and tumor suppressor proteins, upregulates the levels of p53, p73, and p63, and induces cell apoptosis in a dose-and-time-dependent manner [144].
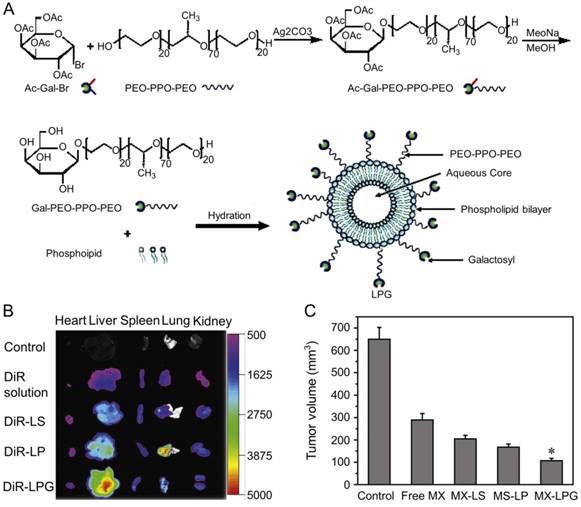
Zhang et al. designed MX-loaded dual-functional liposome (MX-LPG) with a synthetic polymer nano-biomaterial (Gal-P123) and evaluated the uptake and cytotoxicity of MX-LPG in hepatoma Huh-7 cells (Figure 9A) [145]. The in vitro cumulative MX release from different formulations indicated an absence of MX burst release suggesting that MX was stably added to the aqueous core of the LPG. There was a significantly increased accumulation of 30-tetramethylindotricarbocyanine iodide (DiR) Gal-P123-modified liposome (DiR-LPG) in the liver compared to other formulations, demonstrating that LPG could selectively target the liver and hepatoma (Figure 9B). LPG prolonged the circulation time and increased the antitumor activity against BALB/c mice bearing orthotopic xenograft hepatoma of MX in MX-LPG. As shown in Figure 9C, MX-LPG (i.v.) showed the best tumor inhibition efficacy compared with the untargeted platform and free drugs. It is believed that LPG would provide a reliable basis for clinical treatment of hepatoma in the future. The group of Zhang conducted a multicenter randomized controlled phase II clinical trial using MX-loaded polybutylcyanacrylate nanoparticles (DHAD-PBCA-NPs) to treat individuals with unresected hepatoma [146]. This study included 108 patients, 57 of whom received DHAD-PBCA-NPs therapy and 51 received MX therapy. The mean diameter of DHAD-PBCA-NPs was 55.11 ± 5.8 nm, and the drug loading was 46.77%. After dilution with normal saline or 5% glucose, DHAD-PBCA-NPs and MX were intravenously injected into patients of treatment and control groups at a dose of 12 mg/m2, respectively. Both drugs were given every three weeks, and each patient should receive at least two cycles of treatment. The results demonstrated that all patients receiving MX injection died within a year (median survival period = 3.23 months), while five patients receiving MX-NPs survived for 16 - 19 months (median survival period = 5.46 months). This clinical trial showed that using nanoparticles as a delivery system enhanced the MX efficacy against hepatoma. The present results demonstrated that the MX-loaded nanoparticles had great potential in the treatment of unresected advanced hepatoma.
Nanoformulations of molecular targeted drugs
Sorafenib-loaded nanosystems
SF is a kind of multikinase inhibitor and an effective therapeutic agent for advanced hepatoma, which is widely used in many experimental and clinical studies [147]. SF inhibits the activities of serine, threonine kinases Raf-1, and B-Raf, as well as the TK activities of PDGFR-II and VEGFR 1-3, thereby blocking RAS/RAF/MEK/ERK signaling pathways and inhibiting tumor cell proliferation [148]. SF can also interfere with the activities of multiple tyrosine, serine, and threonine kinases to induce apoptosis and inhibit tumor angiogenesis [149]. The main side effects of SF include nausea, diarrhea, arterial hypertension, rash, fatigue, and weight loss, the most common of which is diarrhea.
SF can effectively improve the survival time of patients with advanced hepatoma and has shown encouraging clinical effect in the treatment of hepatoma [150]. However, during clinical application, SF shows low tumor response rates in a majority of hepatoma patients and is beneficial to only about 30% of patients [151]. Moreover, for most patients who initially responded to SF, tumor refractory responses and progression would occur after a few months of SF therapy [152]. Therefore, it is necessary and urgent to find an approach to improve the therapeutic efficacy of SF on hepatoma.
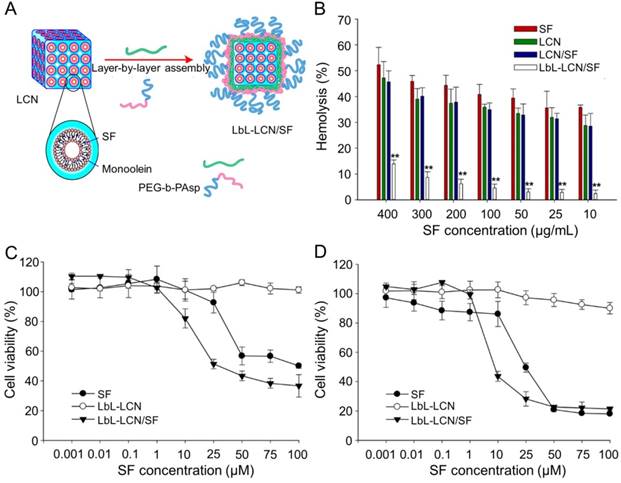
Thapa et al. synthesized layer-by-layer polymer-assembled SF-loaded LCNs (LbL-LCN/SF) (Figure 10A) [153]. LbL-LCN/SF showed significantly reduced hemolysis compared with blank liquid crystalline nanoparticles (LCN) and SF-loaded LCNs (LCN/SF) (Figure 10B). In contrast, the in vitro cytotoxicity of LbL-LCN/SF toward HepG2 cells was remarkably enhanced compared to free SF and blank LbL-LCN (Figure 10C and D). The findings confirmed that the synthesized LbL-LCN/SF had an excellent antitumor effect with reduced toxicity.
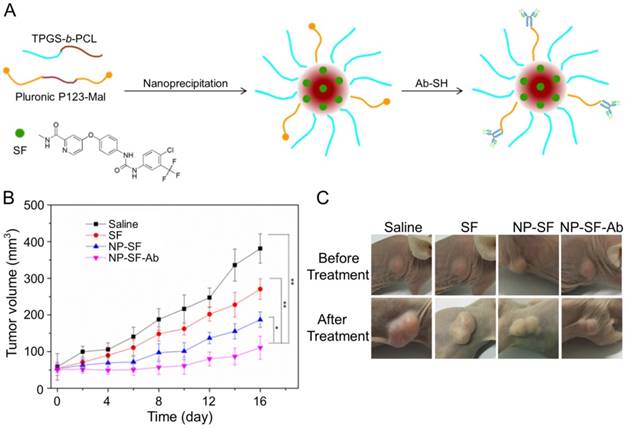
As shown in Figure 11A, Gan et al. engaged anti-GPC3 antibody to modify the interface of the SF-encapsulated nanoparticle with the matrices of biodegradable D-α-tocopheryl poly(ethylene glycol) 1000 succinate-block-poly(ε-caprolactone) (TGPS-b-PCL) and Pluronic P123 [154]. The SF-loaded anti-GPC3 antibody-modified nanoformulation (NP-SF-Ab) intravenous injected via the tail vein showed the best antitumor efficacy compared with free SF and untargeted NP-SF, and the human HepG2 hepatoma in the NP-SF-Ab group only grew to about 110 mm3 16 days post-treatment, as depicted in Figure 11B and C. It has been proved that SF can induce autophagy, which can cause hepatoma resistance to SF. MicroRNA-375 can inhibit autophagy in hepatoma by targeting autophagy-related gene ATG7. Zhao and coworkers prepared miR-375/Sf-LCC NPs by loading miR-375 and SF into lipid-coated calcium carbonate nanoparticles [155]. MiR-375/Sf-LCC NPs (i.v.) significantly inhibited autophagy and enhanced the antitumor effect of SF on HepG2 xenograft tumor mouse model. The synergistic effect of chemotherapeutic agent DOX and molecular targeted drug SF was evaluated by Duan and coworkers [156]. In this study, the SF-loaded NAcGal-DOX lipid nanoparticle (NAcGal-DOX/SF LNP) was intravenously injected into HepG2 hepatoma-bearing BALB/c mice, and it showed a higher antitumor efficiency than the single drug-loaded LNPs toward HepG2 hepatoma in vitro and in vivo. In addition, NAcGal-DOX/SF LNP demonstrated better tolerance and less toxicity than free drugs.
Preparation and fluorescence images of nanomaterials in different treatment groups [145]. (A) Schematic presentation of the synthesis of Gal-P123 and preparation of LPG-modified Gal-P123 modified LPG. (B) Fluorescence images of organs excised at 12 h post injection of DIR solution, DiR-labeled liposome (DiR-LS), DiR-labeled Pluronic P123 modified liposome (DiR-LP) and DiR-labeled Gal-P123 modified liposome (DiR-LPG). (C) Tumor volume of the mice. Copyright 2012. Reproduced with permission from Elsevier Ltd.
Preparation process of LbL-LCN/SF and safety comparison of different treatments [153]. (A) Schematic representation of fabrication process of LbL-LCN/SF. (B) Effects of free SF, LCN, LCN/SF, and LbL-LCN/SF on hemolytic toxicity. In vitro cytotoxicity of control (blank LbL-LCN), free SF, and LbL-LCN/SF on HepG2 cell lines following 24 h (C) and 72 h incubation (D). Copyright 2015. Reproduced with permission from the American Chemical Society.
Preparation process of SF-loaded targeted polymeric nanoparticle (NP-SF-Ab) and tumor volume comparison of different formulations [154]. (A) Schematic representation of the NP-SF-Ab fabricated from SF, TPGS-b-PCL, and Pluronic P123-Mal by nanoprecipitation method followed with conjugating anti-GPC3 antibody. (B) Tumor volume changes after treatment with saline, SF, NP-SF, and NP-SF-Ab. (C) Tumor images of groups treated with saline, SF, NP-SF, and NP-SF-Ab before and after treatment at day 14. Copyright 2018. Reproduced with permission from Elsevier Ltd.
Gefitinib-loaded nanoparticles
Gefitinib (GT) is an orally administered, reversible, and selective tyrosine kinase inhibitor (TKI) in the clinic, which inhibits tumor growth, angiogenesis, and distant metastasis, and increases tumor cell apoptosis through blocking adenosine triphosphate (ATP) from binding to epidermal growth factor receptor tyrosine kinase (EGFR-TK) activation [157]. GT was first approved for the treatment of metastatic and locally advanced non-small cell lung cancer (NSCLC) that had been previously treated with chemotherapy [158, 159]. Recently, GT was widely used for the treatment of hepatoma in the basic and clinical studies, while the drug-resistance after a period of treatment restricted the antitumor efficacy of GT toward malignancies, ending up with tumor progression by 6 - 15 months of therapy [160, 161]. The controlled delivery by polymer nanocarriers may reverse the GT resistance and prolong the progression-free survival of hepatoma. Zheng and coworkers developed the multidrug resistance 1 (MDR1) antibody-decorated chitosan nanoparticle for delivery of GT and autophagy inhibitor chloroquine (CQ) [162]. Chitosan nanoparticles were prepared by the method of ionic crosslinking. Chitosan was first dissolved in the glacial acetic acid solution, and then sodium polyphosphate was added in the mixed solution. Chitosan nanoparticles were formed by the combination of positively charged chitosan and negatively charged polyphosphate. Finally, chitosan nanoparticles were modified with a monoclonal antibody against MDR1 (mAb MDR1) by electrostatic attraction and loaded with GT and CQ. GT/CQ mAb MDR1-NP showed their multi-targeted potential to achieve both selective tumor targeting and the expected antitumor effects by blocking MDR1 on the cell surface and inhibiting autophagy toward human SMMC-7721 hepatoma cells.
Vandetanib-loaded nanoformulations
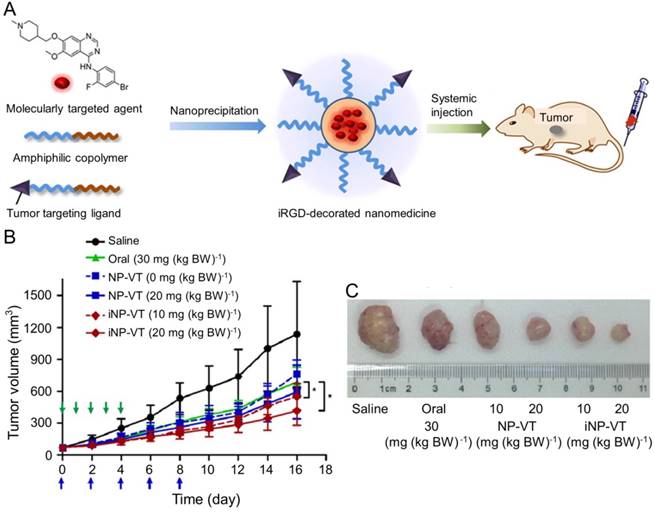
Vandetanib (VT) is another orally active small molecule multi-targeted TKI, which inhibits endothelial cell migration, proliferation, survival, and neovascularization through suppressing the tyrosine kinase receptors, including EGFR, VEGFR-2, and the rearranged during transfection tyrosine kinase receptor [163]. Currently, VT has been approved to treat unresectable, locally advanced, metastatic or progressive medullary thyroid cancer (MTC) in Europe and USA based on the appealing clinical results [164-166], which demonstrated the objective response rates of 16% [164] and 20% [166] and the stable disease rate of 53% [164, 166]. As reported, VT was demonstrated to exhibit excellent inhibition efficacy toward the xenograft human hepatoma mouse model with prolonged survival of tumor-bearing mice and inconspicuous serious adverse events [167], The delivery with polymer nanoparticle will improve the selectivity and efficacy of VT toward hepatoma therapy. Wang et al. explored the VT nanoformulation with the matrices of poly(ethylene glycol)-block-poly(D,L-lactic acid) (PEG-PLA) and iRGD-modified PEG-PLA, as shown in Figure 12A [168]. Mice bearing HCC BEL-7402 xenografts tumor were intravenously injected with VT-Loaded NPs every other day for 10 days. The iRGD, a tumor-homing and tumor-penetrating motif, functionalized nanoparticle loaded with VT (iNP-VT) inhibited the tumor growth by 60% in 16 days, which was more effective compared with other groups (Figure 12B and C). The results indicated that the polymer nanoformulations of molecular targeted drugs exhibited great potential in the molecular targeted therapy of hepatoma.
Nanoformulations of other therapeutic agents
In addition to molecular targeted therapy and systemic chemotherapy, other treatments for advanced hepatoma include immunotherapy and locoregional therapy (such as ablation, arterially directed therapies and radiation therapy). In recent years, due to its safety and effectiveness, immunotherapy has become a potential method to treat many advanced malignant tumors, such as malignant melanoma, NSCLC and Hodgkin's lymphoma. CheckMate 040 trial, a phase I/II nonrandomized multi-institution trial, assessed the efficacy of nivolumab in the treatment of advanced hepatoma [169]. The trial included 214 patients in a dose-expansion phase and 48 patients in a dose-escalation phase. The results showed the objective response rates of patients in the dose-expansion phase and in the dose-escalation phase treated with nivolumab were 20% and 15%, respectively. The FDA has approved Nivolumab for the treatment of advanced hepatoma in 2017. Locoregional therapy is mainly used for advanced hepatoma and patients with hepatoma who cannot tolerate surgery. In recent years, hepatic artery infusion chemotherapy (HAIC) has attracted much attention because of its high response rate and excellent long-term survival rate in the treatment of advanced hepatoma. A prospective, multicenter, phase II study conducted by Lyu and coworkers showed hepatic artery infusion (HAI) of oxaliplatin plus fluorouracil/leucovorin (FOLFOX) was effective and well tolerated for advanced hepatoma [170]. The 6-month and 12-month survival rates of patients receiving HAI of FOLFOX (HAIF) were 71.4% and 55.1%, respectively. Lyu's group further compared the efficacy of HAIF and SF for the treatment of advanced hepatoma in a retrospective study [171]. The results showed the median OS of the HAIF group (14.5 months) was significantly longer than that of the SF group (7.0 months). The efficacy of HAIC needs to be validated by more randomized controlled trials. Nanoformulations for immunotherapy [172], HAIC and other treatments are also under study, and we expect more types of nanoformulated drugs to be used in the treatment of hepatoma.
Summary and perspectives
Hepatoma is one of the most severe malignant tumors with high mortality that severely endanger public health worldwide, especially in China. Although the therapeutic strategies for hepatoma based on surgical resection and molecular targeted therapy have been improved, the long-term survival rates of hepatoma patients are still not satisfactory. On the one hand, most hepatoma patients show hidden symptoms, which make it somewhat tricky for early diagnosis; On the other hand, hepatoma is characterized by a long incubation period followed by rapid tumor growth. Even after treatment, drug resistance, tumor recurrence, and tumor metastasis always lead to death. Therefore, it is an urgent challenge to achieve early diagnosis and effective therapy in the clinic.
Features of nanocarriers in references.
| Excipient | Nanoparticle | Model drug | Diameter or hydrodynamic radius (Rh) (nm) | LC and EE (wt%) | Cell type | Therapeutic effect | Reference |
|---|---|---|---|---|---|---|---|
| PEG-PLE | Nanoparticle | BDNF | 191 - 246 | - | NIH 3T3 cells | The nanoformulation improved BDNF delivery throughout the brain and displayed a preferable regional distribution pattern. Furthermore, Nano-BDNF had superior neuroprotective effects in the mouse brain with lipopolysaccharides-induced inflammation. | [9] |
| PEG-b-TCL | Micelle | PZn3 | 110 | - | HMEC, 4T1 cells | PEG-b-TCL-based micelles demonstrated favorable characteristics for further development for minimally invasive imaging of breast tumors. | [180] |
| PEG-b-PLLA/PDLA-CHOL | Micelle | DOX | 84.1 - 107 | - | A549 cells | The micelles possessed excellent abilities in drug release, cell internalization as well as proliferation inhibitory effect toward human A549 lung cancer cells. | [181] |
| PEG | Micelle | DOX | 48.5 ± 8.8 | - | HepG2 cells | CAD-PEG-CAD exhibited more efficient cellular uptake and potent cytotoxicity in vitro, as well as improved tissue distribution and superior tumor suppression in vivo. More importantly, the PEGylated DOX exhibited favorable security in vivo. | [14] |
| HES-CHO | Micelle | DOX | HESQDOX1.7: 73.4 ± 5.3; HESQDOX3.3: 63.9 ± 5.5; HESQDOX5.9: 51.9 ± 8.5; | LC: 5.4, EE: 68; LC: 9.9, EE: 66; LC: 16.5, EE: 59; | B16F10 cells | The HESQDOX micelles selectively released DOX in the endosome and/or lysosome after cellular uptake, and exhibited excellent proliferation inhibition. Furthermore, the antitumor efficacy was upregulated. | [182] |
| mPEG-P(LG-co-LC) | Nanogel | DOX | 58.8 ± 2.9 | LC: 16.1, EE: 96.7; | HepG2, H22 cells | NG/DOX showed excellent safety and great potential for on-demand delivery of antitumor drug. | [110] |
| Dex-O-DOX | Micelle | DOX | 90 ± 14 | LC: 9.98 | HepG2, H22 cells | Dex-O-DOX exhibited higher antitumor activity and lower toxicity and exhibited great potential in the clinical chemotherapy of malignancy. | [111] |
| mPEG-P(LP-co-LC) | Nanogel | DOX | 56.1 ± 3.5 | LC: 10.2, EE: 56.8; | HepG2 cells | NG/DOX exhibited upregulated intratumoral accumulation and improved antitumor efficacy. | [96] |
| PEG-PPLG | Micelle | DOX | 54 | LC:9.53, EE:69.18; | HeLa cells | Cross-linked micelles were biocompatible, and DOX-loaded micelles showed higher cellular proliferation inhibition. | [183] |
| PEG-poly(amino acid)s | Nanogel | DOX | 168 ± 7.9, 193 ± 4.8, 234 ± 4.1; | LC:2.86, EE:14.72; LC: 8.64, EE: 47.29; LC: 12.34, EE: 70.39; | HeLa cells | The reduction-responsive PEG poly(amino acid)s nanogels efficiently delivered antitumor drugs into tumor cells and inhibited cell proliferation, rendering highly promising for targeted intracellular delivery of operative chemotherapeutic drugs in tumor therapy. | [97] |
| mPEG-b-P(LGA-co-CELG) | Nanogel | DOX | 49.2 ± 1.8, 4.1 ± 2.3; | LC: 10.7, EE: 60.2; LC: 13.2, EE: 75.8; | HepG2 cells | DOX-loaded nanogels exhibited enhanced antitumor efficacies andsecurities. | [98] |
| Copolypeptide | Micelle | DOX | 40.5 - 91.6 | LC: 4.08 - 12.37, EE: 21.27 - 70.58; | HepG2, L929 cells | The nanomedicine retained much higher antitumor activity and possessed great promising for hepatoma-targeted chemotherapy. | [109] |
| Dex-DOX | Micelle | DOX | 22.9 ± 4.2 | - | B16F10 cells | The newly-constructed Dex-DOX promoted the pH-dependent drug release, highlight the cellular uptake efficiency, and strengthen the antitumor ability toward mouse B16F10 melanoma. | [107] |
| Dex-g-DOX | Micelle | DOX | 102.0 ± 6.2 | LC: 12, EE: 78.2; | - | Dex-g-DOX exhibited ultraselective accumulation in cancerous liver tissue and high antitumor efficacy. | [112] |
| Liposomes | Liposomes | DOX | 90.97 ± 0.91 | - | HepG2, Huh-7, PLC/PRF/5, Hep3B cells | Anti-CD147 ILs-DOX showed long circulation time, efficient accumulation in tumors and superior antitumor effects. | [21] |
| PLA | Nanoparticles | DOX | 75.3 ± 9.6 | EE: 88.77% ± 3.79% | HCC-LM3, BEL-7402, HL-7702, NCI-H1299 | tNP-PLA-DOX showed long-term stability, high selectivity toward cancer cells alleviated drug toxicity. | [113] |
| Fib-graft-PNVCL | Nanogel | 5-FU | 110 ± 55 | LC: 3.1, EE: 62 | L929, MCF-7 cells | The multidrug loaded fib-graft-PNVCL NGs showed enhanced toxicity, apoptosis, and uptake by MCF-7 cells. The in vivo assessment showed sustained release of 5-FU, confirming the therapeutic efficiency of the formulation. | [100] |
| GC-FU | Nanoparticle | 5-FU | 163.2 | LC: 21.25 ± 2.3 | HepG2, A549 cells | GC-FU-NPs played great function in killing cancer cells for the cell endocytosis mediated by the asialoglycoprotein receptor. The drug-loaded nanoparticles had a much longer half-time and a long circulation effect than free 5-FU. | [116] |
| PLA-Cy7-SM5-1 | Nanoparticle | 5-FU | - | LC: 9.87 ± 0.58, EE: 8.97 ± 0.94; | HCC-LM3-fLuc cells | PLA-5FU-SM5-1 efficiently inhibited the tumor rapid progression. | [118] |
| SQ-gem/isoCA-4 | Nanoparticle | GEM | 142 ± 6 | LC: 27.3 | LS174-T, HUVECs cells | SQ-gem/isoCA-4 NAs displayed comparable antiproliferative and cytotoxic effects than free GEM. | [184] |
| GemSQ | Liposome | GEM | 113 ± 24 | - | L1210wt cells | The PEGylated liposomal formulation did not exhibit superior antitumor activity over the non-PEGylated liposomal formulation in the tumor model chosen. | [185] |
| CPDG/CHS-PEG1500 | Nanoparticle | GEM | 54.4 ± 0.73 | - | H22 cells | The nanoassemblies had a much higher antitumor effect, and it will be promising nanomedicines to treat hepatoma. | [123] |
| Dual-functional liposome | Liposome | MX | 100.57 ± 0.75 | LC: 1.37 ± 0.10, EE: 97.33 ± 0.37; | Huh-7 cells | MX-LPG increased antitumor activity and improved selectivity in hepatoma tumors. | [145] |
| PLH-PEG-biotin | Nanoparticle | SF | 181.4 ± 3.4 | LC: 2.38 ± 0.04, EE: 95.02 ± 1.47; | HepG2, H22 cells | PTN showed a similar antitumor effect against HepG2 cells. In vivo antitumor studies, TPTN showed a significantly higher antitumor effect in H22 tumor-bearing mice. | [6] |
| LbL-LCN | Nanoparticle | SF | 160.2 ± 1.1 | LC: 1.2 - 2.5, EE: 47 - 100; | HepG2 cells | Higher cellular uptake and greater apoptotic effects of LbL-LCN/SF indicated superior antitumor effects. | [153] |
| Calcium carbonate | Nanoparticle | SF | 100.7 ± 12.1 | - | HepG2 cells | miR-375/Sf-LCC NPs exhibited pH-dependent drug release and potent cytotoxicity and showed greatly enhanced therapeutic efficacy. | [155] |
+: available, -: not available.
Preparation of the iNP-VT nanoassemblies and inhibition of tumor growth [168]. (A) Schematic illustration showing the reformulation and self-assembly of VT into PEG-PLGA NPs. (B) Tumor growth curves of different groups. NP-VT and iNP-VT were i.v. injected on day 0, 2, 4, 6, and 8. The blue and green arrows represent the day on which the i.v. and p.o. injections were performed, respectively. (C) Representative images of BEL-7402 after 16 days of treatment. Copyright 2016. Reproduced with permission from the American Chemical Society.
In order to reveal the pathogenesis of hepatoma and to screen therapeutic agents, various animal hepatoma models have been established. Typically, various types of rodent models with distinct features have been successfully constructed. Each model has its pros and cons, and no model can genuinely reflect the full characteristics of human carcinogenesis. The sex, age, and genetic background of mice are fundamentally different from those of humans. When hepatoma occurs, the response of the human body to disease is different from that of rodents, so mice models do not fully develop the pathological process of human hepatoma. Considering the antitumor effect of certain drugs in hepatoma patients, the predictive value of these models is often less accurate or worse than expected. To credibly mimic the human carcinogenesis, the following rodent models are more attractive: (i) patient liver tumor tissues or cells inoculated hepatoma models, that is, patient-derived xenograft hepatoma models; (ii) genetically engineered hepatoma models with similar gene mutations to human patients; (iii) HBV or HCV induced hepatoma models in humanized mouse models; (iv) hepatoma models evolved from cirrhosis. With the development of rodent hepatoma models, we can more accurately and efficiently reveal the pathogenesis mechanisms of hepatoma and evaluate the safety and antitumor efficacy of advanced formulations of cytostatic and molecular targeted drugs.
In recent years, the rise of nanotechnology has brought extensive opportunities for the hepatoma therapy. As a promising representative, the polymer nanoparticle-based nanoformulations of cytostatic and molecular targeted agents exhibit great potential in the effective therapy of hepatoma and even advanced hepatoma. The tailor-made polymer nanoparticles exhibit excellent biocompatibility, long blood circulation cycle, passive and active targeting, and controlled release of payload, so the polymer nanoformulations show better antitumor efficacy and fewer side effects compared with free therapeutic drugs.
Although some researches on the treatment of hepatoma with nanoformulations have achieved some exciting results, most of them are still limited to the level of cell or animal experiments. Few nanoformulations can be applied to human clinical trials, and their development still faces many challenges. Firstly, the genetic background, immune system and tumor development process of experimental animals are quite different from those of humans, so the drug distribution and antitumor effect of nanoformulations may also be different. In order to fully evaluate the properties of nanoformulations, it is better to develop and use patient-derived xenograft models. Secondly, the side effects of nanoformulations and their uncertain long-term safety limit their clinical application. Currently, only a few ingredients (such as PEG and albumin) are considered safe for the human body. It is necessary to develop more effective methods to evaluate the safety of nanoformulations. In addition, the tumor microenvironment is very complex and closely related to the occurrence and metastasis of tumors. It is necessary to improve the permeability of nanoparticles and their tolerance to hypoxic and low-acid environments. Finally, the current prognosis of advanced hepatoma is still weak, and one treatment method often fails to achieve a satisfactory therapeutic effect, requiring a combination of multiple treatment methods. Some other treatments based on nanoparticles have been studied to treat hepatomas, such as immunotherapy, photothermal therapy [173], microwave ablation [174], microwave thermal therapy (MTT) and microwave dynamic therapy (MDT) [175]. For example, Fu and colleagues attempted to treat hepatoma with the combination of MDT and MTT using manganese-zirconium metal-organic framework nanospheres (Mn-ZrMOF NCs) [175]. The results showed that Mn-ZrMOF NCs could effectively inhibit the tumor growth of H22-bearing mice by improving thermal effects and producing ROS. Therefore, the combination therapy based on nanoparticles may provide a new effective method for the treatment of hepatoma.
To further improve the performance and to ultimately realize the clinical transformation of polymer formulations, the following aspects will be further developed: (i) the FDA-approved polymers will be used as the matrices of nanocarriers, such as polylactide (PLA), poly(lactic-co-glycolic acid) (PLGA), and polypeptides; (ii) the molecular targeted agents will be the preferred drugs for on-demand delivery; (iii) the in situ administrated formulations will be applied; (iv) the emerging nanotechnology such as supramolecular chemotherapy, DNA nanorobot, and tumor imprisonment will be further studied and utilized; (v) the molecular targeted agents will be combined with other effective management, including radiotherapy and immunotherapy. It is believed that with the continuous development and maturity of polymer nanoformulations, they will play an essential role in the targeted therapy of hepatoma.
Abbreviations
5-FU: 5-fluorouracil; AFB: aflatoxin B; ALSV-A: avian leucosis sarcoma virus subgroup A; Anti-CD147 ILs-DOX: CD147-targeted doxorubicin-loaded immunoliposomes; ASGP-R: asialoglycoprotein receptor; ATP: adenosine triphosphate; CCl4: carbon tetrachloride; CDD: choline deficient diet; CDDP: cisplatin; CHL: chlorambucil; CHL-GEM: chlorambucil gemcitabine; CHSPEG1500: cholesteryl hemisuccinate PEG1500; CPDG: cyclic phosphoryl N-dodecanoyl GEM; CQ: chloroquine; CT: cabozantinib; DEN: N-nitrosodiethylamine; Dex: dextran; Dex-g-DOX: dextran-doxorubicin prodrug; Dex-O-DOX: dextran-doxorubicin conjugate; DHAD-PBCA-NPs: MX-loaded polybutylcyanacrylate nanoparticles; DiR: 30-tetramethylindotricarbocyanine iodide; DiR-LPG: 30-tetramethylindotricarbocyanine iodide Gal-P123 modified liposome; DOX: doxorubicin; DSPE-PEG2000: 1,2-distearoyl-sn- glycero-3-phosphoethanolamine-N-[methoxy poly (ethylene glycol) 2000]; EGFR: epidermal growth factor receptor; EGFR-TK: epidermal growth factor receptor tyrosine kinase; EPR: enhanced permeability and retention; FDA: Food and Drug Administration; FOLFOX: oxaliplatin plus fluorouracil/leucovorin; GC-FU: N-galactosylated-chitosan-5-FU; GEM: gemcitabine; GT: Gefitinib; HAI: hepatic artery infusion; HAIC: hepatic artery infusion chemotherapy; HAIF: HAI of FOLFOX; HBV: hepatitis B virus; HBx: X-protein; HCC: Hepatocellular carcinoma; HCV: hepatitis C virus; HGF: hepatocyte growth factor; IARC: International Agency for Research on Cancer; IC50: half-maximal inhibitory concentration; iNP-VT: the iRGD, a tumor-homing and tumor-penetrating motif, functionalized nanoparticle loading with VT; i.p.: intraperitoneal injection; i.t.: intratumoral injection; i.v.: intravenous injection; LbL-LCN/SF: layer-by-layer polymer-assembled sorafenib-loaded LCN; LCN: liquid crystalline nanoparticle; LC NCA: L-cystine N-carboxyanhydride; LCN/SF: sorafenib -loaded LCN; LP NCA: L-phenylalanine N-carboxyanhydride; LT: lenvatinib; mAb MDR1: antibody against MDR1; MDR1: multidrug resistance 1; MDT: microwave dynamic therapy; MET: mesenchymal-epithelial transition factor; Mn-ZrMOF NC: manganese-zirconium metal-organic framework nanosphere; mPEG-b-PCL: methoxy poly(ethylene glycol)-block-poly(ε-caprolactone); mPEG-P(LG-co- LC): methoxy poly(ethylene glycol)-poly(L-glutamic acid-co-L-cystine); mPEG-P(LP-co-LC): methoxy poly (ethylene glycol)-poly(L-phenylalanine-co-Lcystine); MPS: mononuclear macrophage system; MTC: medullary thyroid cancer; mTOR: mammalian target of rapamycin; MTT: microwave thermal therapy; MX: Mitoxantrone; MX-LPG: MX-loaded dual-functional liposome; NAcGal-DOX/SF LNP: sorafenib-loaded NAcGal-DOX lipid nanoparticle; NG/DOX: DOX-loaded nanogel; NP: nanoparticle; NP-SF-Ab: SF-loaded anti-GPC3 antibody-modified nanoformulation; NSCLC: non- small cell lung cancer; OS: overall survival; PEG: poly(ethylene glycol); PEG-PLA: poly(ethylene glycol)- block-poly(D,L-lactic acid); PGLG-b-PLGA: DOX- loaded galactosylated polypeptide; PI3K: phosphoinositol 3-kinase; PLA-DOX: poly(lactic acid)-DOX; PLGA: poly(D,L-lactide-co-glycolide); PLGA-5FU- SM5-1: SM5-1-conjugated poly(D,L-lactide-co-glycolide) nanobubble containing 5-FU; PMLG-b-PLGA: galactose with oligo(ethylene glycol); PP: peroxisome proliferator; PPARs: peroxisome proliferator- activated receptors; RES: reticuloendothelial system; RET: rearranged during transfection; RF: regorafenib; RNA: ribonucleic acid; ROS: reactive oxygen species; SF: sorafenib; SV40: Simian Virus 40; TAA: Thioacetamide; TGPS-b-PCL: D-α-tocopheryl poly(ethylene glycol) 1000 succinate-block-poly(ε-caprolactone); TKI: tyrosine kinase inhibitor; TNs: tumor nodules; VEGFR: vascular endothelial growth factor receptor; WHV: woodchuck hepatitis virus.
Acknowledgements
This study was financially supported by grants from the National Key Research and Development Program of China (Grant No. 2016YFC1100701), the National Natural Science Foundation of China (Grant Nos. 51873207, 51803006, 51673190, 51603204, 51673187, 51473165, and 51520105004), and the Science and Technology Development Program of Jilin Province (Grant Nos. 20190201068JC and 20160204015SF).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Thomas MB, Zhu AX. Hepatocellular carcinoma: The need for progress. J Clin Oncol. 2005;23:2892-9
2. Vogel A, Cervantes A, Chau I, Daniele B, Llovet J, Meyer T. et al. Hepatocellular carcinoma: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2018;29:238-55
3. Omata M, Cheng AL, Kokudo N, Kudo M, Lee JM, Jia J. et al. Asia-Pacific clinical practice guidelines on the management of hepatocellular carcinoma: A 2017 update. Hepatol Int. 2017;11:317-70
4. Chen J, Ding J, Xiao C, Zhuang X, Chen X. Emerging antitumor applications of extracellularly reengineered polymeric nanocarriers. Biomater Sci. 2015;3:988-1001
5. Ding J, Shi F, Li D, Chen L, Zhuang X, Chen X. Enhanced endocytosis of acid-sensitive doxorubicin derivatives with intelligent nanogel for improved security and efficacy. Biomater Sci. 2013;1:633-46
6. Liu Y, Feng L, Liu T, Zhang L, Yao Y, Yu D. et al. Multifunctional pH-sensitive polymeric nanoparticles for theranostics evaluated experimentally in cancer. Nanoscale. 2014;6:3231-42
7. Zhang Y, Cai L, Li D, Lao YH, Liu D, Li M. et al. Tumor microenvironment-responsive hyaluronate-calcium carbonate hybrid nanoparticle enables effective chemotherapy for primary and advanced osteosarcomas. Nano Res. 2018;11:4806-22
8. Chan YC, Hsiao M. Protease-activated nanomaterials for targeted cancer theranostics. Nanomedicine. 2017;12:2153-9
9. Jiang Y, Fay JM, Poon CD, Vinod N, Zhao Y, Bullock K. et al. Nanoformulation of brain-derived neurotrophic factor with target receptor-triggered-release in the central nervous system. Adv Funct Mater. 2018;28:1703982
10. Shen K, Li D, Guan J, Ding J, Wang Z, Gu J. et al. Targeted sustained delivery of antineoplastic agent with multicomponent polylactide stereocomplex micelle. Nanomedicine (NY, NY, US). 2017;13:1279-88
11. Li S, Zhang T, Xu W, Ding J, Yin F, Xu J. et al. Sarcoma-targeting peptide-decorated polypeptide nanogel intracellularly delivers shikonin for upregulated osteosarcoma necroptosis and diminished pulmonary metastasis. Theranostics. 2018;8:1361-75
12. Chang H, Yhee JY, Jang GH, You DG, Ryu JH, Choi Y. et al. Predicting the in vivo accumulation of nanoparticles in tumor based on in vitro macrophage uptake and circulation in zebrafish. J Controlled Release. 2016;244:205-13
13. Naseema NawazishMehdi S, Hussain MM Basha SK, Samad MA. Heat enhancement of heat exchanger using aluminium oxide (Al2O3), copper oxide (CuO) nano fluids with different concentrations. Mater Today Proc. 2018;5:6481-8
14. Sun D, Ding J, Xiao C, Chen J, Zhuang X, Chen X. pH-responsive reversible PEGylation improves performance of antineoplastic agent. Adv Healthcare Mater. 2015;4:844-55
15. Liang S, Yang XZ, Du XJ, Wang HX, Li HJ, Liu WW. et al. Optimizing the size of micellar nanoparticles for efficient siRNA delivery. Adv Funct Mater. 2015;25:4778-87
16. Liu Y, Wang Z, Liu Y, Zhu G, Jacobson O, Fu X. et al. Suppressing nanoparticle-mononuclear phagocyte system interactions of two-dimensional gold nanorings for improved tumor accumulation and photothermal ablation of tumors. ACS Nano. 2017;11:10539-48
17. Huang M, Wu W, Qian J, Wan DJ, Wei XL, Zhu JH. Body distribution and in situ evading of phagocytic uptake by macrophages of long-circulating poly(ethylene glycol) cyanoacrylate-co-n-hexadecyl cyanoacrylate nanoparticles. Acta Pharm Sin. 2005;26:1512-8
18. Jadia R, Kydd J, Rai P. Remotely phototriggered, transferrin-targeted polymeric nanoparticles for the treatment of breast cancer. Photochem Photobiol. 2018;94:765-74
19. Wu F, Li X, Jiang B, Yan J, Zhang Z, Qin J. et al. Glycyrrhetinic acid functionalized nanoparticles for drug delivery to liver cancer. J Biomed Nanotechnol. 2018;14:1837-52
20. Alonso S. Exploiting the bioengineering versatility of lactobionic acid in targeted nanosystems and biomaterials. J Controlled Release. 2018;287:216-34
21. Wang J, Wu Z, Pan G, Ni J, Xie F, Jiang B. et al. Enhanced doxorubicin delivery to hepatocellular carcinoma cells via CD147 antibody-conjugated immunoliposomes. Nanomedicine (NY, NY, US). 2018;14:1949-61
22. Tang X, Chen L, Li A, Cai S, Zhang Y, Liu X. et al. Anti-GPC3 antibody-modified sorafenib-loaded nanoparticles significantly inhibited HepG2 hepatocellular carcinoma. Drug Delivery. 2018;25:1484-94
23. Leenders MWH, Nijkamp MW, Rinkes IHMB. Mouse models in liver cancer research: A review of current literature. World J Gastroenterol. 2008;14:6915-23
24. Heindryckx F, Colle I, Van Vlierberghe H. Experimental mouse models for hepatocellular carcinoma research. Int J Exp Pathol. 2009;90:367-86
25. Marx J. Medicine: Building better mouse models for studying cancer. Science. 2003;299:1972-5
26. Naumov GN, Akslen LA, Folkman J. Role of angiogenesis in human tumor dormancy-animal models of the angiogenic switch. Cell Cycle. 2006;5:1779-87
27. Waern JM, Yuan Q, Rüdrich U, Becker PD, Schulze K, Strick-Marchand H. et al. Ectopic expression of murine CD47 minimizes macrophage rejection of human hepatocyte xenografts in immunodeficient mice. Hepatology. 2012;56:1479-88
28. Li ZY, Ni SH, Yang XL, Kiviat N, Lieber A. Xenograft models for liver metastasis: Relationship between tumor morphology and adenovirus vector transduction. Mol Ther. 2004;9:650-7
29. Shimosato Y, Kameya T, Nagai K, Hirohashi S, Koide T, Hayashi H. et al. Transplantation of human tumors in nude mice. J Natl Cancer Inst. 1976;56:1251-60
30. Bibby MC. Orthotopic models of cancer for preclinical drug evaluation: Advantages and disadvantages. Eur J Cancer. 2004;40:852-7
31. Ding J, Li D, Zhuang X, Chen X. Self-assemblies of pH-activatable PEGylated multiarm poly(lactic acid-co-glycolic acid)-doxorubicin prodrugs with improved long-term antitumor efficacies. Macromol Biosci. 2013;13:1300-7
32. Pelleitier M, Montplaisir S. The nude mouse: A model of deficient T-cell function. Methods Achiev Exp Pathol. 1975;7:149-66
33. Nakashima Y, Nakashima O, Hsia CC, Kojiro M, Tabor E. Vascularization of small hepatocellular carcinomas: Correlation with differentiation. Liver. 1999;19:12-8
34. Garlanda C, Parravicini C, Sironi M, De Rossi M, Wainstok de Calmanovici R, Carozzi F. et al. Progressive growth in immunodeficient mice and host cell recruitment by mouse endothelial cells transformed by polyoma middle-sized T antigen: Implications for the pathogenesis of opportunistic vascular tumors. Proc Natl Acad Sci USA. 1994;91:7291-5
35. Li J, Xu W, Li D, Liu T, Zhang YS, Ding J. et al. Locally deployable nanofiber patch for sequential drug delivery in treatment of primary and advanced orthotopic hepatomas. ACS Nano. 2018;12:6685-99
36. Sham JG, Kievit FM, Grierson JR, Miyaoka RS, Yeh MM, Zhang M. et al. Glypican-3-targeted Zr-89 PET imaging of hepatocellular carcinoma. J Nucl Med. 2014;55:799-804
37. Hann B, Balmain A. Building 'validated' mouse models of human cancer. Curr Opin Cell Biol. 2001;13:778-84
38. Wogan GN. Impacts of chemicals on liver cancer risk. Semin Cancer Biol. 2000;10:201-10
39. Seitz HK, Stickel F. Molecular mechanisms of alcohol-mediated carcinogenesis. Nat Rev Cancer. 2007;7:599-612
40. Hirst GL, Balmain A. Forty years of cancer modelling in the mouse. Eur J Cancer. 2004;40:1974-80
41. Pitot HC, Dragan YP. Facts and theories concerning the mechanisms of carcinogenesis. FASEB J. 1991;5:2280-6
42. Gray R, Peto R, Brantom P, Grasso P. Chronic nitrosamine ingestion in 1040 rodents: The effect of the choice of nitrosamine, the species studied, and the age of starting exposure. Cancer Res. 1991;51:6470-91
43. Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact. 2006;160:1-40
44. Zimmers TA, Jin X, Gutierrez JC, Acosta C, McKillop IH, Pierce RH. et al. Effect of in vivo loss of GDF-15 on hepatocellular carcinogenesis. J Cancer Res Clin Oncol. 2008;134:753-9
45. Henderson JM, Zhang HE, Polak N, Gorrell MD. Hepatocellular carcinoma: Mouse models and the potential roles of proteases. Cancer Lett. 2017;387:106-13
46. Carbon tetrachloride. IARC Monogr Eval Carcinog Risks Hum. 1999:71 Pt 2: 401-32
47. Weisburger EK. Carcinogenicity studies on halogenated hydrocarbons. Environ Health Perspect. 1977;21:7-16
48. Jackson AF, Williams A, Recio L, Waters MD, Lambert LB, Yank CL. Case study on the utility of hepatic global gene expression profiling in the risk assessment of the carcinogen furan. Toxicol Appl Pharmacol. 2014;274:63-77
49. Sheweita SA, Abd El-Gabar M, Bastawy M. Carbon tetrachloride-induced changes in the activity of phase II drug-metabolizing enzyme in the liver of male rats: Role of antioxidants. Toxicology. 2001;165:217-24
50. Xin B, Cui Y, Wang Y, Wang L, Yin J, Zhang L. et al. Combined use of alcohol in conventional chemical-induced mouse liver cancer model improves the simulation of clinical characteristics of human hepatocellular carcinoma. Oncol Lett. 2017;14:4722-8
51. Overall evaluations of carcinogenicity: An updating of IARC monographs volumes 1 to 42. IARC Monogr Eval Carcinog Risks Hum. 1987;7:1-440
52. Palacios RS, Roderfeld M, Hemmann S, Rath T, Atanasova S, Tschuschner A. et al. Activation of hepatic stellate cells is associated with cytokine expression in thioacetamide-induced hepatic fibrosis in mice. Lab Invest. 2008;88:1192-203
53. Ghosh S, Sarkar A, Bhattacharyya S, Sil PC. Silymarin protects mouse liver and kidney from thioacetamide induced toxicity by scavenging reactive oxygen species and activating PI3K-Akt pathway. Front Pharmacol. 2016;7:481
54. Elcock FJ, Chipman JK, Roberts RA. The rodent nongenotoxic hepatocarcinogen and peroxisome proliferator nafenopin inhibits intercellular communication in rat but not guinea-pig hepatocytes, perturbing S-phase but not apoptosis. Arch Toxicol. 1998;72:439-44
55. Calfee-Mason KG, Lee EY, Spear BT, Glauert HP. Role of the p50 subunit of NF-kappa B in vitamin E-induced changes in mice treated with the peroxisome proliferator, ciprofibrate. Food Chem Toxicol. 2008;46:2062-73
56. Ma X, Stoffregen DA, Wheelock GD, Rininger JA, Babish JG. Discordant hepatic expression of the cell division control enzyme p34cdc2 kinase, proliferating cell nuclear antigen, p53 tumor suppressor protein, and p21Waf1 cyclin-dependent kinase inhibitory protein after WY14,643 (4-chloro-6-(2,3-xylidino)-2-pyrimidinylthio acetic acid) dosing to rats. Mol Pharmacol. 1997;51:69-78
57. Hasmall SC, James NH, Macdonald N, Gonzalez FJ, Peters JM, Roberts RA. Suppression of mouse hepatocyte apoptosis by peroxisome proliferators: Role of PPAR-α and TNF-α. Mutat Res Fundam Mol Mech Mutagen. 2000;448:193-200
58. Zhao W, Iskandar S, Kooshki M, Sharpe JG, Payne V, Robbins ME. Knocking out peroxisome proliferator-activated receptor (PPAR)-α inhibits radiation-induced apoptosis in the mouse kidney through activation of NF-κB and increased expression of IAPs. Radiat Res. 2007;167:581-91
59. Hulla JE, Chen ZY, Eaton DL. Aflatoxin B1 induced rat hepatic hyperplastic nodules do not exhibit a site-specific mutation within the P53 Gene. Cancer Res. 1993;53:9-11
60. Vartanian V, Minko IG, Chawanthayatham S, Egner PA, Lin YC, Earley LF. et al. NEIL1 protects against aflatoxin-induced hepatocellular carcinoma in mice. Proc Natl Acad Sci USA. 2017;114:4207-12
61. Woo LL, Egner PA, Belanger CL, Wattanawaraporn R, Trudel LJ, Croy RG. et al. Aflatoxin B1 DNA adduct formation and mutagenicity in livers of neonatal male and female B6C3F1 mice. Toxicol Sci. 2011;122:38-44
62. McGlynn KA, Hunter K, LeVoyer T, Roush J, Wise P, Michielli RA. et al. Susceptibility to aflatoxin B1 related primary hepatocellular carcinoma in mice and humans. Cancer Res. 2003;63:4594-601
63. Ghebranious N, Sell S. The mouse equivalent of the human p53ser249 mutation p53ser246 enhances aflatoxin hepatocarcinogenesis in hepatitis B surface antigen transgenic and p53 heterozygous null mice. Hepatology. 1998;27:967-73
64. Knight B, Yeoh GCT, Husk KL, Ly T, Abraham LJ, Yu CP. et al. Impaired preneoplastic changes and liver tumor formation in tumor necrosis factor receptor type 1 knockout mice. J Exp Med. 2000;192:1809-18
65. Nakano T, Cheng YF, Lai CY, Hsu LW, Chang YC, Deng JY. et al. Impact of artificial sunlight therapy on the progress of non-alcoholic fatty liver disease in rats. J Hepatol. 2011;55:415-25
66. de Lima VMR, Oliveira CPMS, Alves VAF, Charnmas MC, Oliveira EP, Stefano JT. et al. A rodent model of NASH with cirrhosis, oval cell proliferation and hepatocellular carcinoma. J Hepatol. 2008;49:1055-61
67. Gogoi-Tiwari J, Köhn-Gaone J, Giles C, Schmidt-Arras D, Gratte FD, Elsegood CL. et al. The murine choline-deficient, ethionine-supplemented (CDE) diet model of chronic liver injury. J Visualized Exp. 2017;128:e56138
68. Guest I, Ilic Z, Sell S. Age dependence of oval cell responses and bile duct carcinomas in male fischer 344 rats fed a cyclic choline-deficient, ethionine-supplemented diet. Hepatology. 2010;52:1750-7
69. Parkin DM. The global health burden of infection-associated cancers in the year 2002. Int J Cancer. 2006;118:3030-44
70. Kremsdorf D, Brezillon N. New animal models for hepatitis C viral infection and pathogenesis studies. World J Gastroenterol. 2007;13:2427-35
71. Fattovich G, Stroffolini T, Zagni I, Donato F. Hepatocellular carcinoma in cirrhosis: Incidence and risk factors. Gastroenterology. 2004;127(Suppl 1):S35-S50
72. Jeong DH, Jeong WI, Chung JY, An MY, Jung CY, Lee GJ. et al. Hepatic cirrhosis occurring in a young woodchuck (Marmota monax) due to vertical transmission of woodchuck hepatitis virus (WHV). J Vet Sci. 2003;4:199-201
73. Kramer MG, Hernandez-Alcoceba R, Qian C, Prieto J. Evaluation of hepatocellular carcinoma models for preclinical studies. Drug Discovery Today: Dis Models. 2005;2:41-9
74. Bilbao R, Gerolami R, Bralet MP, Qian C, Tran PL, Tennant B. et al. Transduction efficacy, antitumoral effect, and toxicity of adenovirus-mediated herpes simplex virus thymidine kinase/ganciclovir therapy of hepatocellular carcinoma: The woodchuck animal model. Cancer Gene Ther. 2000;7:657-62
75. Wu CG, Salvay DM, Forgues M, Valerie K, Farnsworth J, Markin RS. et al. Distinctive gene expression profiles associated with hepatitis B virus X protein. Oncogene. 2001;20:3674-82
76. Chisari FV, Pinkert CA, Milich DR, Filippi P, McLachlan A, Palmiter RD. et al. A transgenic mouse model of the chronic hepatitis B surface antigen carrier state. Science. 1985;230:1157-60
77. Xiong J, Yao YC, Zi XY, Li JX, Wang XM, Ye XT. et al. Expression of hepatitis B virus X protein in transgenic mice. World J Gastroenterol. 2003;9:112-6
78. Zheng Y, Chen WI, Louie SG, Yen TSB, Ou JHJ. Hepatitis B virus promotes hepatocarcinogenesis in transgenic mice. Hepatology. 2007;45:16-21
79. Koike K, Tsutsumi T, Fujie H, Shintani Y, Moriya K. Molecular mechanism of viral hepatocarcinogenesis. Oncology. 2002;62:29-37
80. Lerat H, Honda M, Beard MR, Loesch K, Sun J, Yang Y. et al. Steatosis and liver cancer in transgenic mice expressing the structural and nonstructural proteins of hepatitis C virus. Gastroenterology. 2002;122:352-65
81. Kamegaya Y, Hiasa Y, Zukerberg L, Fowler N, Blackard JT, Lin WY. et al. Hepatitis C virus acts as a tumor accelerator by blocking apoptosis in a mouse model of hepatocarcinogenesis. Hepatology. 2005;41:660-7
82. Shuldiner AR. Transgenic animals. N Engl J Med. 1996;334:653-5
83. Vilchez RA, Jauregui MP, Hsi ED, Novoa-Takara L, Chang CC. Simian virus 40 in posttransplant lymphoproliferative disorders. Hum Pathol. 2006;37:1130-6
84. Ahuja D, Saenz-Robles MT, Pipas JM. SV40 large T antigen targets multiple cellular pathways to elicit cellular transformation. Oncogene. 2005;24:7729-45
85. Murakami H, Sanderson ND, Nagy P, Marino PA, Merlino G, Thorgeirsson SS. Transgenic mouse model for synergistic effects of nuclear oncogenes and growth factors in tumorigenesis: Interaction of c-myc and transforming growth factor α in hepatic oncogenesis. Cancer Res. 1993;53:1719-23
86. Shiota G, Harada K, Ishida M, Tomie Y, Okubo M, Katayama S. et al. Inhibition of hepatocellular carcinoma by glycyrrhizin in diethylnitrosamine-treated mice. Carcinogenesis. 1999;20:59-63
87. Koike K, Moriya K, Iino S, Yotsuyanagi H, Endo Y, Miyamura T. et al. High-level expression of hepatitis B virus HBx gene and hepatocarcinogenesis in transgenic mice. Hepatology. 1994;19:810-9
88. Lewis BC, Klimstra DS, Socci ND, Xu S, Koutcher JA, Varmus HE. The absence of p53 promotes metastasis in a novel somatic mouse model for hepatocellular carcinoma. Mol Cell Biol. 2005;25:1228-37
89. Ju HL, Han KH, Lee JD, Ro SW. Transgenic mouse models generated by hydrodynamic transfection for genetic studies of liver cancer and preclinical testing of anticancer therapy. Int J Cancer. 2016;138:1601-8
90. Lee JS, Chu IS, Mikaelyan A, Calvisi DF, Heo J, Reddy JK. et al. Application of comparative functional genomics to identify best-fit mouse models to study human cancer. Nat Genet. 2004;36:1306-11
91. Calvisi DF, Factor VM, Ladu S, Conner EA, Thorgeirsson SS. Disruption of β-catenin pathway or genomic instability define two distinct categories of liver cancer in transgenic mice. Gastroenterology. 2004;126:1374-86
92. Sandhu DS, Tharayil VS, Lai JP, Roberts LR. Treatment options for hepatocellular carcinoma. Expert Rev Gastroenterol Hepatol. 2008;2:81-92
93. Ladju RB, Pascut D, Massi MN, Tiribelli C, Sukowati CHC. Aptamer: A potential oligonucleotide nanomedicine in the diagnosis and treatment of hepatocellular carcinoma. Oncotarget. 2018;9:2951-61
94. Cong YW, Xiao HH, Xiong HJ, Wang ZG, Ding JX, Li C. et al. Dual drug backboned shattering polymeric theranostic nanomedicine for synergistic eradication of patient-derived lung cancer. Adv Mater. 2018;30:1706220
95. Kanapathipillai M, Brock A, Ingber DE. Nanoparticle targeting of anticancer drugs that alter intracellular signaling or influence the tumor microenvironment. Adv Drug Delivery Rev. 2014;79-80:107-18
96. Huang K, Shi B, Xu W, Ding J, Yang Y, Liu H. et al. Reduction-responsive polypeptide nanogel delivers antitumor drug for improved efficacy and safety. Acta Biomater. 2015;27:179-93
97. Ding J, Shi F, Xiao C, Lin L, Chen L, He C. et al. One-step preparation of reduction-responsive poly(ethylene glycol)-poly(amino acid)s nanogels as efficient intracellular drug delivery platforms. Polym Chem. 2011;2:2857-64
98. Ding J, Xu W, Zhang Y, Sun D, Xiao C, Liu D. et al. Self-reinforced endocytoses of smart polypeptide nanogels for "on-demand" drug delivery. J Controlled Release. 2013;172:444-55
99. Chen W, Zeng K, Liu H, Ouyang J, Wang L, Liu Y. et al. Cell membrane camouflaged hollow prussian blue nanoparticles for synergistic photothermal-/chemotherapy of cancer. Adv Funct Mater. 2017;27:1605795
100. Rejinold NS, Baby T, Chennazhi KP, Jayakumar R. Multidrug loaded thermo-responsive fibrinogen-graft-poly(N-vinyl caprolactam) nanogels for breast cancer drug delivery. J Biomed Nanotechnol. 2015;11:392-402
101. Kolter M, Ott M, Hauer C, Reimold I, Fricker G. Nanotoxicity of poly(N-butylcyano-acrylate) nanoparticles at the blood-brain barrier, in human whole blood and in vivo. J Controlled Release. 2015;197:165-79
102. Prasad P, Shuhendler A, Cai P, Rauth AM, Wu XY. Doxorubicin and mitomycin C co-loaded polymer-lipid hybrid nanoparticles inhibit growth of sensitive and multidrug resistant human mammary tumor xenografts. Cancer Lett. 2013;334:263-73
103. Llovet JM, Bruix J. Molecular targeted therapies in hepatocellular carcinoma. Hepatology. 2008;48:1312-27
104. Nault JC, Galle PR, Marquardt JU. The role of molecular enrichment on future therapies in hepatocellular carcinoma. J Hepatol. 2018;69:237-47
105. Feng X, Li D, Han J, Zhuang X, Ding J. Schiff base bond-linked polysaccharide-doxorubicin conjugate for upregulated cancer therapy. Mat Sci Eng, C. 2017;76:1121-8
106. Zhang Y, Ding J, Sun D, Sun H, Zhuang X, Chang F. et al. Thermogel-mediated sustained drug delivery for in situ malignancy chemotherapy. Mat Sci Eng, C. 2015;49:262-8
107. Working PK, Newman MS, Sullivan T, Yarrington J. Reduction of the cardiotoxicity of doxorubicin in rabbits and dogs by encapsulation in long-circulating, pegylated liposomes. J Pharmacol Exp Ther. 1999;289:1128-33
108. Xiao CS, Ding JX, He CL, Chen XS. Glycopolypeptides: Synthesis, self-assembly and biomedical applications. Acta Polym Sin. 2018:45-55
109. Ding J, Xiao C, Li Y, Cheng Y, Wang N, He C. et al. Efficacious hepatoma-targeted nanomedicine self-assembled from galactopeptide and doxorubicin driven by two-stage physical interactions. J Controlled Release. 2013;169:193-203
110. Shi B, Huang K, Ding J, Xu W, Yang Y, Liu H. et al. Intracellularly swollen polypeptide nanogel assists hepatoma chemotherapy. Theranostics. 2017;7:703-16
111. Xu W, Ding J, Xiao C, Li L, Zhuang X, Chen X. Versatile preparation of intracellular-acidity-sensitive oxime-linked polysaccharide-doxorubicin conjugate for malignancy therapeutic. Biomaterials. 2015;54:72-86
112. Li D, Xu W, Li P, Ding J, Cheng Z, Chen L. et al. Self-targeted polysaccharide prodrug suppresses orthotopic hepatoma. Mol Pharmaceutics. 2016;13:4231-5
113. Xu L, Xu S, Wang H, Zhang J, Chen Z, Pan L. et al. Enhancing the efficacy and safety of doxorubicin against hepatocellular carcinoma through a modular assembly approach: The combination of polymeric prodrug design, nanoparticle encapsulation, and cancer cell-specific drug targeting. ACS Appl Mater Interfaces. 2018;10:3229-40
114. Wilson B, Ambika TV, Patel RDK, Jenita JL, Priyadarshini SRB. Nanoparticles based on albumin: Preparation, characterization and the use for 5-flurouracil delivery. Int J Biol Macromol. 2012;51:874-8
115. Kodama Y, Fumoto S, Nishi J, Nakashima M, Sasaki H, Nakamura J. et al. Absorption and distribution characteristics of 5-fluorouracil (5-FU) after an application to the liver surface in rats in order to reduce systemic side effects. Biol Pharm Bull. 2008;31:1049-52
116. Huang C, Li NM, Gao P, Yang S, Ning Q, Huang W. et al. In vitro and in vivo evaluation of macromolecular prodrug GC-FUA based nanoparticle for hepatocellular carcinoma chemotherapy. Drug Delivery. 2017;24:459-66
117. Cheng M, Han J, Li Q, He B, Zha B, Wu J. et al. Synthesis of galactosylated chitosan/5-fluorouracil nanoparticles and its characteristics, in vitro and in vivo release studies. J Biomed Mater Res B. 2012;100B:2035-43
118. Ma X, Cheng Z, Jin Y, Liang X, Yang X, Dai Z. et al. SM5-1-Conjugated PLA nanoparticles loaded with 5-fluorouracil for targeted hepatocellular carcinoma imaging and therapy. Biomaterials. 2014;35:2878-89
119. Guo Z, Wang F, Di Y, Yao L, Yu X, Fu D. et al. Antitumor effect of gemcitabine-loaded albumin nanoparticle on gemcitabine-resistant pancreatic cancer induced by low hENT1 expression. Int J Nanomed. 2018;13:4869-80
120. Fan M, Liang X, Li Z, Wang H, Yang D, Shi B. Chlorambucil gemcitabine conjugate nanomedicine for cancer therapy. Eur J Pharm Sci. 2015;79:20-6
121. Huang P, Ao J, Zhou L, Su Y, Huang W, Zhu X. et al. Facile approach to construct ternary cocktail nanoparticles for cancer combination therapy. Bioconjugate Chem. 2016;27:1564-8
122. Yordanov GG, Bedzhova ZA, Dushkin CD. Preparation and physicochemical characterization of novel chlorambucil-loaded nanoparticles of poly(butylcyanoacrylate). Colloid Polym Sci. 2010;288:893-9
123. Du L, Zhang B, Lei Y, Wang S, Jin Y. Long-circulating and liver-targeted nanoassemblies of cyclic phosphoryl N-dodecanoyl gemcitabine for the treatment of hepatocellular carcinoma. Biomed Pharmacother. 2016;79:208-14
124. Shaaban S, Negm A, Ibrahim EE, Elrazak AA. Chemotherapeutic agents for the treatment of hepatocellular carcinoma: Efficacy and mode of action. Oncol Rev. 2014;8:25-35
125. Rosenberg B, VanCamp L, Trosko JE, Mansour VH. Platinum compounds: A new class of potent antitumour agents. Nature. 1969;222:385-6
126. He S, Li C, Zhang Q, Ding J, Liang XJ, Chen X. et al. Tailoring platinum (IV) amphiphiles for self-targeting all-in-one assemblies as precise multimodal theranostic nanomedicine. ACS Nano. 2018;12:7272-81
127. Zhang Y, Wang F, Li MQ, Yu ZQ, Qi RG, Ding JX. et al. Self-stabilized hyaluronate nanogel for intracellular codelivery of doxorubicin and cisplatin to osteosarcoma. Adv Sci. 2018;5:1700821
128. Kelland LR. Preclinical perspectives on platinum resistance. Drugs. 2000;59:1-8
129. Miao YF, Lv T, Wang R, Wu H, Yang SF, Dai J. et al. CpG and transfer factor assembled on nanoparticles reduce tumor burden in mice glioma model. RSC Adv. 2017;7:11644-51
130. Real NE, Castro GN, Dario Cuello-Carrion F, Perinetti C, Roehrich H, Cayado-Gutierrez N. et al. Molecular markers of DNA damage and repair in cervical cancer patients treated with cisplatin neoadjuvant chemotherapy: An exploratory study. Cell Stress Chaperones. 2017;22:811-22
131. Song HQ, Li WL, Qi RG, Yan LS, Jing XB, Zheng MH. et al. Delivering a photosensitive transplatin prodrug to overcome cisplatin drug resistance. Chem Commun. 2015;51:11493-5
132. Ma PA, Xiao HH, Yu C, Liu JH, Cheng ZY, Song HQ. et al. Enhanced cisplatin chemotherapy by iron oxide nanocarrier-mediated generation of highly toxic reactive oxygen species. Nano Lett. 2017;17:928-37
133. Merk O, Speit G. Detection of crosslinks with the comet assay in relationship to genotoxicity and cytotoxicity. Environ Mol Mutagen. 1999;33:167-72
134. Jamieson ER, Lippard SJ. Structure, recognition, and processing of cisplatin-DNA adducts. Chem Rev. 1999;99:2467-98
135. Sorenson CM, Eastman A. Influence of cis-diamminedichloroplatinum (II) on DNA synthesis and cell cycle progression in excision repair proficient and deficient Chinese hamster ovary cells. Cancer Res. 1988;48:6703-7
136. Yu M, Zhang C, Tang Z, Tang X, Xu H. Intratumoral injection of gels containing losartan microspheres and (PLG-g-mPEG)-cisplatin nanoparticles improves drug penetration, retention and antitumor activity. Cancer Lett. 2018;442:396-408
137. Lan X, She J, Lin Da, Xu Y, Li X, Yang Wf. et al. Microneedle-mediated delivery of lipid-coated cisplatin nanoparticles for efficient and safe cancer therapy. ACS Appl Mater Interfaces. 2018;10:33060-9
138. Li X, Li R, Qian X, Ding Y, Tu Y, Guo R. et al. Superior antitumor efficiency of cisplatin-loaded nanoparticles by intratumoral delivery with decreased tumor metabolism rate. Eur J Pharm Biopharm. 2008;70:726-34
139. Ding D, Zhu Z, Liu Q, Wang J, Hu Y, Jiang X. et al. Cisplatin-loaded gelatin-poly(acrylic acid) nanoparticles: Synthesis, antitumor efficiency in vivo and penetration in tumors. Eur J Pharm Biopharm. 2011;79:142-9
140. Shenkenberg TD, Von Hoff DD. Mitoxantrone: A new anticancer drug with significant clinical activity. Ann Intern Med. 1986;105:67-81
141. Fox EJ. Mechanism of action of mitoxantrone. Neurology. 2004;63(Suppl 6):S15-S8
142. Hajihassan Z, Rabbani-Chadegani A. Studies on the binding affinity of anticancer drug mitoxantrone to chromatin, DNA and histone proteins. J Biomed Sci. 2009;16:31
143. Thielmann HW, Popanda O, Gersbach H, Gilberg F. Various inhibitors of DNA topoisomerases diminish repair-specific DNA incision in UV-irradiated human fibroblasts. Carcinogenesis. 1993;14:2341-51
144. Seitz SJ, Schleithoff ES, Koch A, Schuster A, Teufel A, Staib F. et al. Chemotherapy-induced apoptosis in hepatocellular carcinoma involves the p53 family and is mediated via the extrinsic and the intrinsic pathway. Int J Cancer. 2010;126:2049-66
145. Zhang X, Guo S, Fan R, Yu M, Li F, Zhu C. et al. Dual-functional liposome for tumor targeting and overcoming multidrug resistance in hepatocellular carcinoma cells. Biomaterials. 2012;33:7103-14
146. Zhou Q, Sun X, Zeng L, Liu J, Zhang Z. A randomized multicenter phase II clinical trial of mitoxantrone-loaded nanoparticles in the treatment of 108 patients with unresected hepatocellular carcinoma. Nanomedicine (NY, NY, US). 2009;5:419-23
147. Zhao Z, Long J, Zhao Y, Yang J, Jiang W, Liu Q. et al. Adaptive immune cells are necessary for the enhanced therapeutic effect of sorafenib-loaded nanoparticles. Biomater Sci. 2018;6:893-900
148. Saidak Z, Giacobbi AS, Louandre C, Sauzay C, Mammeri Y, Galmiche A. Mathematical modelling unveils the essential role of cellular phosphatases in the inhibition of RAF-MEK-ERK signalling by sorafenib in hepatocellular carcinoma cells. Cancer Lett. 2017;392:1-8
149. Wilhelm SM, Adnane L, Newell P, Villanueva A, Llovet JM, Lynch M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol Cancer Ther. 2008;7:3129-40
150. Forner A, Llovet JM, Bruix J. Hepatocellular carcinoma. Lancet. 2012;379:1245-55
151. Cheng AL, Kang YK, Chen Z, Tsao CJ, Qin S, Kim JS. et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: A phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009;10:25-34
152. Shen YC, Ou DL, Hsu C, Lin KL, Chang CY, Lin CY. et al. Activating oxidative phosphorylation by a pyruvate dehydrogenase kinase inhibitor overcomes sorafenib resistance of hepatocellular carcinoma. Br J Cancer. 2013;108:72-81
153. Thapa RK, Choi JY, Poudel BK, Tran Tuan H, Pathak S, Gupta B. et al. Multilayer-coated liquid crystalline nanoparticles for effective sorafenib delivery to hepatocellular carcinoma. ACS Appl Mater Interfaces. 2015;7:20360-8
154. Gan H, Chen L, Sui X, Wu B, Zou S, Li A. et al. Enhanced delivery of sorafenib with anti-GPC3 antibody-conjugated TPGS-b-PCL/Pluronic P123 polymeric nanoparticles for targeted therapy of hepatocellular carcinoma. Mat Sci Eng C. 2018;91:395-403
155. Zhao P, Li M, Wang Y, Chen Y, He C, Zhang X. et al. Enhancing antitumor efficiency in hepatocellular carcinoma through the autophagy inhibition by miR-375/sorafenib in lipid-coated calcium carbonate nanoparticles. Acta Biomater. 2018;72:248-55
156. Duan W, Liu Y. Targeted and synergistic therapy for hepatocellular carcinoma: Monosaccharide modified lipid nanoparticles for the co-delivery of doxorubicin and sorafenib. Drug Des Dev Ther. 2018;12:2149-61
157. Javle M, Pande A, Iyer R, Yang G, LeVea C, Wilding G. et al. Pilot study of gefitinib, oxaliplatin, and radiotherapy for esophageal adenocarcinoma: Tissue effect predicts clinical response. Am J Clin Oncol. 2008;31:329-34
158. Masago K, Fujita S, Irisa K, Kim YH, Ichikawa M, Mio T. et al. Good clinical response to gefitinib in a non small cell lung cancer patient harboring a rare somatic epidermal growth factor gene point mutation. Jpn J Clin Oncol. 2010;40:1105-9
159. Ferry DR, Anderson M, Beddard K, Tomlinson S, Atherfold P, Obszynska J. et al. A phase II study of gefitinib monotherapy in advanced esophageal adenocarcinoma: Evidence of gene expression, cellular, and clinical Response. Clin Cancer Res. 2007;13:5869-75
160. Desbois-Mouthon C, Cacheux W, Blivet-Van Eggelpoël MJ, Barbu V, Fartoux L, Poupon R. et al. Impact of IGF-1R/EGFR cross-talks on hepatoma cell sensitivity to gefitinib. Int J Cancer. 2006;119:2557-66
161. Jackman DM, Holmes AJ, Lindeman N, Wen PY, Kesari S, Borras AM. et al. Response and resistance in a non small cell lung cancer patient with an epidermal growth factor receptor mutation and leptomeningeal metastases treated with high-dose gefitinib. J Clin Oncol. 2006;24:4517-20
162. Zheng Y, Su C, Zhao L, Shi Y. mAb MDR1-modified chitosan nanoparticles overcome acquired EGFR-TKI resistance through two potential therapeutic targets modulation of MDR1 and autophagy. J Nanobiotechnol. 2017;15:66
163. Frampton JE. Vandetanib. Drugs. 2012;72:1423-36
164. Jr SAW, Robinson BG, Gagel RF, Dralle H, Fagin JA, Santoro M. et al. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: Arandomized, double-blind phase III trial. J Clin Oncol. 2012;30:134-41
165. Robinson BG, Paz-Ares L, Krebs A, Vasselli J, Haddad R. Vandetanib (100 mg) in patients with locally advanced or metastatic hereditary medullary thyroid cancer. J Clin Endocr Metab. 2010;95:2664-71
166. Jr SAW, Gosnell JE, Gagel RF, Moley J, Pfister D, Sosa JA. et al. Vandetanib for the treatment of patients with locally advanced or metastatic hereditary medullary thyroid cancer. J Clin Oncol. 2010;28:767-72
167. Inoue K, Torimura T, Nakamura T, Iwamoto H, Masuda H, Abe M. et al. Vandetanib, an inhibitor of VEGF receptor-2 and EGF receptor, suppresses tumor development and improves prognosis of liver cancer in mice. Clin Cancer Res. 2012;18:3924-33
168. Wang J, Wang H, Li J, Liu Z, Xie H, Wei X. et al. iRGD-decorated polymeric nanoparticles for the efficient delivery of vandetanib to hepatocellular carcinoma: Preparation and in vitro and in vivo evaluation. ACS Appl Mater Interfaces. 2016;8:19228-37
169. El-Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, Hsu C. et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet. 2017;389:2492-502
170. Lyu N, Lin Y, Kong Y, Zhang Z, Liu L, Zheng L. et al. FOXAI: A phase II trial evaluating the efficacy and safety of hepatic arterial infusion of oxaliplatin plus fluorouracil/leucovorin for advanced hepatocellular carcinoma. Gut. 2018;67:395-258
171. Lyu N, Kong Y, Mu L, Lin Y, Li J, Liu Y. et al. Hepatic arterial infusion of oxaliplatin plus fluorouracil/leucovorin vs. sorafenib for advanced hepatocellular carcinoma. J Hepatol. 2018;69:60-9
172. Liu L, Zong ZM, Liu Q, Jiang SS, Zhang Q, Cen LQ. et al. A novel galactose-PEG-conjugated biodegradable copolymer is an efficient gene delivery vector for immunotherapy of hepatocellular carcinoma. Biomaterials. 2018;184:20-30
173. Jin Y, Yang X, Tian J. Targeted polypyrrole nanoparticles for the identification and treatment of hepatocellular carcinoma. Nanoscale. 2018;10:9594-601
174. Tang S, Fu C, Tan L, Liu T, Mao J, Ren X. et al. Imaging-guided synergetic therapy of orthotopic transplantation tumor by superselectively arterial administration of microwave-induced microcapsules. Biomaterials. 2017;133:144-53
175. Fu C, Zhou H, Tan L, Huang Z, Wu Q, Ren X. et al. Microwave-Activated Mn-doped zirconium metal organic framework nanocubes for highly effective combination of microwave dynamic and thermal therapies against cancer. ACS Nano. 2018;12:2201-10
176. Hays T, Rusyn I, Burns AM, Kennett MJ, Ward JM, Gonzalez FJ. et al. Role of peroxisome proliferator-activated receptor-α (PPAR-α) in bezafibrate-induced hepatocarcinogenesis and cholestasis. Carcinogenesis. 2005;26:219-27
177. Vesselinovitch SD, Mihailovich N, Wogan GN, Lombard LS, Rao KV. Aflatoxin B1, a hepatocarcinogen in the infant mouse. Cancer Res. 1972;32:2289-91
178. Farazi PA, Glickman J, Horner J, DePinho RA. Cooperative interactions of p53 mutation, telomere dysfunction, and chronic liver damage in hepatocellular carcinoma progression. Cancer Res. 2006;66:4766-73
179. Ghoshal AK, Farber E. The induction of liver cancer by dietary deficiency of choline and methionine without added carcinogens. Carcinogenesis. 1984;5:1367-70
180. Hofmann CL, O'Sullivan MC, Detappe A, Yu Y, Yang X, Qi W. et al. NIR-emissive PEG-b-TCL micelles for breast tumor imaging and minimally invasive pharmacokinetic analysis. Nanoscale. 2017;9:13465-76
181. Feng XR, Ding JX, Gref R, Chen XS. Poly(β-cyclodextrin)-mediated polylactide-cholesterol stereocomplex micelles for controlled drug delivery. Chin J Polym Sci. 2017;35:693-9
182. Li D, Ding J, Zhuang X, Chen L, Chen X. Drug binding rate regulates the properties of polysaccharide prodrugs. J Mater Chem B. 2016;4:5167-77
183. Cheng Y, He C, Xiao C, Ding J, Ren K, Yu S. et al. Reduction-responsive cross-linked micelles based on PEGylated polypeptides prepared via click chemistry. Polym Chem. 2013;4:3851-8
184. Maksimenko A, Alami M, Zouhiri F, Brion JD, Pruvost A, Mougin J. et al. Therapeutic modalities of squalenoyl nanocomposites in colon cancer: An ongoing search for improved efficacy. ACS Nano. 2014;8:2018-32
185. Pili B, Reddy LH, Bourgaux C, Lepetre-Mouelhi S, Desmaele D, Couvreur P. Liposomal squalenoyl-gemcitabine: Formulation, characterization and anticancer activity evaluation. Nanoscale. 2010;2:1521-6
Author contact
![]() Corresponding authors: Ping Zhang (z_pingedu.cn), Zhongmin Li (lizhongmin1211com), Jianxun Ding (jxdingac.cn)
Corresponding authors: Ping Zhang (z_pingedu.cn), Zhongmin Li (lizhongmin1211com), Jianxun Ding (jxdingac.cn)