Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Materials and Methods
Acknowlegement
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(5):1369-1384. doi:10.7150/thno.32451 This issue Cite
Research Paper
Absence of GdX/UBL4A Protects against Inflammatory Diseases by Regulating NF-кB Signaling in Macrophages and Dendritic Cells
Chunxiao Liu1*, Yifan Zhou2*, Mengdi Li1, Ying Wang1, Liu Yang1, Shigao Yang1, Yarui Feng1, Yinyin Wang1, Yangmeng Wang1, Fangli Ren1, Jun Li3, Zhongjun Dong2, Y. Eugene Chin4, Xinyuan Fu5, Li Wu2 ![]() , Zhijie Chang1
, Zhijie Chang1 ![]()
1. State Key Laboratory of Membrane Biology, School of Medicine, Tsinghua University, Beijing (100084), China.
2. Institute for Immunology, School of Medicine, Tsinghua University, Beijing (100084), China.
3. Institute of Immunology, PLA, The Third Military Medical University, Chongqing, (400038), China.
4. Institute of Health Sciences, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, Shanghai Jiaotong University School of Medicine, Shanghai (200025), China.
5. Department of Biochemistry, South University of Science and Technology of China, Shenzhen (518055), China.
*These authors contributed equally to this work.
Received 2018-12-20; Accepted 2018-12-21; Published 2019-2-14
Abstract

Nuclear factor-kappa B (NF-κB) activation is critical for innate immune responses. However, cellular-intrinsic regulation of NF-κB activity during inflammatory diseases remains incompletely understood. Ubiquitin-like protein 4A (UBL4A, GdX) is a small adaptor protein involved in protein folding, biogenesis and transcription. Yet, whether GdX has a role during innate immune response is largely unknown.
Methods: To investigate the involvement of GdX in innate immunity, we challenged GdX-deficient mice with lipopolysaccharides (LPS). To investigate the underlying mechanism, we performed RNA sequencing, real-time PCR, ELISA, luciferase reporter assay, immunoprecipitation and immunoblot analyses, flow cytometry, and structure analyses. To investigate whether GdX functions in inflammatory bowel disease, we generated dendritic cell (DC), macrophage (Mφ), epithelial-cell specific GdX-deficient mice and induced colitis with dextran sulfate sodium.
Results: GdX enhances DC and Mφ-mediated innate immune defenses by positively regulating NF-κB signaling. GdX-deficient mice were resistant to LPS-induced endotoxin shock and DSS-induced colitis. DC- or Mφ- specific GdX-deficient mice displayed alleviated mucosal inflammation. The production of pro-inflammatory cytokines by GdX-deficient DCs and Mφ was reduced. Mechanistically, we found that tyrosine-protein phosphatase non-receptor type 2 (PTPN2, TC45) and protein phosphatase 2A (PP2A) form a complex with RelA (p65) to mediate its dephosphorylation whereas GdX interrupts the TC45/PP2A/p65 complex formation and restrict p65 dephosphorylation by trapping TC45.
Conclusion: Our study provides a mechanism by which NF-κB signaling is positively regulated by an adaptor protein GdX in DC or Mφ to maintain the innate immune response. Targeting GdX could be a strategy to reduce over-activated immune response in inflammatory diseases.
Keywords: GdX/UBL4A, NF-κB, p65/RelA, inflammation, dendritic cells, macrophages
Introduction
Nuclear factor-kappa B (NF-κB) signaling is important in innate immune responses, mediated by innate immune cells, including dendritic cells (DCs) and macrophages (Mφ). DCs and Mφ express toll-like receptors (TLRs) to activate NF-κB, leading to the production of pro-inflammatory cytokines, including IL-1β, IL-6, and TNF-α during microorganism infection [1, 2]. Dysregulation of NF-κB signaling is associated with a variety of human diseases, such as inflammatory bowel diseases (IBDs) [3].
The NF-κB signaling pathway is activated by TNF-α and TLR agonists [4]. In both situations, TNF receptor-associated factor 6 (TRAF6) passes the baton to the inhibitor of κB kinase (IKK) for activation, leading to phosphorylation and ubiquitination-proteasome dependent degradation of the inhibitor of NF-κB (IκBα). Subsequently, IκB-released p65 and p50 form a dimer and translocate into the nucleus where targeted gene transcription is initiated [5]. During this process, both IκΒ and p65 are phosphorylated by IKK. p65 phosphorylation at Ser276 and Ser536 by IKK or cAMP-dependent protein kinase (PKAc) in the cytoplasm or by mitogen- and stress-activated protein kinase 1 and 2 (MSK1/2) or ribosomal S6 kinase 1 (RSK-1) in the nucleus stabilizes their IκB free activation status [6]. Conversely, NF-κB signaling must be downregulated after its activation. One example by which NF-κB signaling is shut down during inflammation is through microorganisms that may trigger protein tyrosine phosphatases (PTPs) in immune cells [7].
PTPs trigger p65 dephosphorylation to terminate its transcriptional activity [8]. Serine/threonine protein phosphatase 1 (PP1), PP2A, PP4 and WIP1 [9-12] have been identified to dephosphorylate p65. In particular, PP2A was identified as a specific regulator of NF-κB signaling by either suppressing the NF-κB transcriptional activity or reducing its binding ability to DNA. Dephosphorylation of NF-κB by PP2A leads to inhibition of NF-κB transcriptional activity, which is involved in chemokines or cytokines induction in astrocytes [11]. Recently, natural compounds isoliensine [13], rographolide [14], and the hydrophilic alpha-tocopherol derivative, hydrophilic α-tocopherol derivative, 2,2,5,7,8-pentamethyl-6-hydroxychromane (PMC) [15], are found to inhibit NF-κB activity by regulating its dephosphorylation through activation of PP2A. Overall, manipulation of PP2A activity regulates NF-κB activity in immune cells.
GdX is an X-linked gene in the G6PD cluster at Xq28, also named UBL4A (Ubiquitin-like protein 4A) that encodes a small protein with an N-terminal ubiquitin-like domain [16, 17]. GdX has also been reported to regulate ER stress responses for protein folding [18, 19] and functions in tail-anchored protein biogenesis by interacting with BCL2-associated athanogene 6 (BAG6) [20]. GdX also promotes Akt activation through interacting with actin related protein 2/3 (Arp2/3) in the cytoplasm [21]. While studying the regulation of signal transducer and activator of transcription factor 3 (STAT3), we discovered GdX mediates tyrosine phosphatase TC45 recruitment for STAT3 dephosphorylation [17] and regulates osteoblastogenesis and chondrogenesis [22]. Interestingly, in this study, we found that GdX-deficient mice [23] were resistant to lipopolysaccharides (LPS)-induced endotoxin shock and dextran sulfate sodium (DSS)-induced colitis, suggesting a critical effect of GdX in NF-κB signaling pathway modulation. Here, we describe the mechanism of GdX in the regulation of p65 dephosphorylation and maintenance of p65 in a hyper-active status during inflammatory responses.
Results
GdX positively regulates innate immune responses
To investigate the role of GdX in innate immune responses, a lethal dosage of LPS (30 mg/kg) was injected intraperitoneally to GdX+/Y and GdX-/Y mice [24]. A significantly higher survival rate was observed in GdX-/Y mice than in control mice (GdX+/Y) (Fig. 1A). We further compared gene expression profiles of splenocytes from GdX+/Y and GdX-/Y mice upon LPS stimulation. Interestingly, RNA-sequencing analysis indicated a number of NF-κB targeting genes, such as IL-6, IL-1a and IL-12, have dramatically higher expressions in the splenocytes of GdX+/Y mice than that of GdX-/Y mice (Fig. 1B, S1A). Moreover, gene ontology (GO) analysis of GdX function in LPS-treated mice showed one of the most significantly enriched biological processes is related to inflammatory response (Fig. S1B). These results were further validated by testing the serum levels of IL-6 (Fig. 1C) and TNF-α (Fig. 1D), which are two important NF-κB targeted genes involved in innate immune responses [24]. Correspondingly, lower mRNA levels of IL-6, TNF-α and IL-1β were detected in the splenocytes from GdX-/Y mice compared to those from control mice (Fig. S1C-F), and was also validated in the thymus (Fig. S1G) and liver (Fig. S1H). These results suggest that GdX-/Y mice were resistant to LPS challenge, implying a positive role for GdX in regulating innate immune responses.
GdX deletion attenuates LPS-induced inflammatory responses in vivo. (A) GdX-deficient mice (GdX-/Y) were resistant to LPS challenge. Survival rates of GdX-/Y and their wildtype (GdX+/Y) littermates (n=15) were observed after challenged with a lethal dosage of LPS (30 mg/kg body weight, ip). (B) A number of NF-κB target genes had significantly lower expressions in GdX-/Y mice than control mice after LPS injection. The heat map of downregulated NF-κB target genes (list of 32 genes, absolute log2 >1, false discovery rate <0.05, p<0.05) in GdX-/Y mice (n=2) and wildtype littermates (n=2) injected with LPS. The mice were injected with LPS (20 mg/kg body weight, ip), and 6 hrs after injection, RNA was extracted from their splenocytes and performed the RNA-Seq analysis. (C-D) GdX deletion decreased the serum concentrations of pro-inflammatory cytokines under acute inflammation. Levels of IL-6 and TNF-α in serum from GdX+/Y and GdX-/Y mice (n≥6) were examined by ELISA after LPS challenge (20 mg/kg body weight; ip). (E) GdX is abundantly expressed in myeloid cells. mRNA levels of GdX were measured by real-time PCR in different immune cells. (F) GdX is highly expressed in RAW264.7 and DC2.4 cell lines. (G-I) Different subtypes of FLDCs from GdX+/Y and GdX-/Y mice were used to measure the productions of IL-6 in response to indicated TLR ligand stimulation. Results were presented as mean ± SEM from at least two repeats *, p < 0.05; **, p < 0.01; ***, p < 0.001.
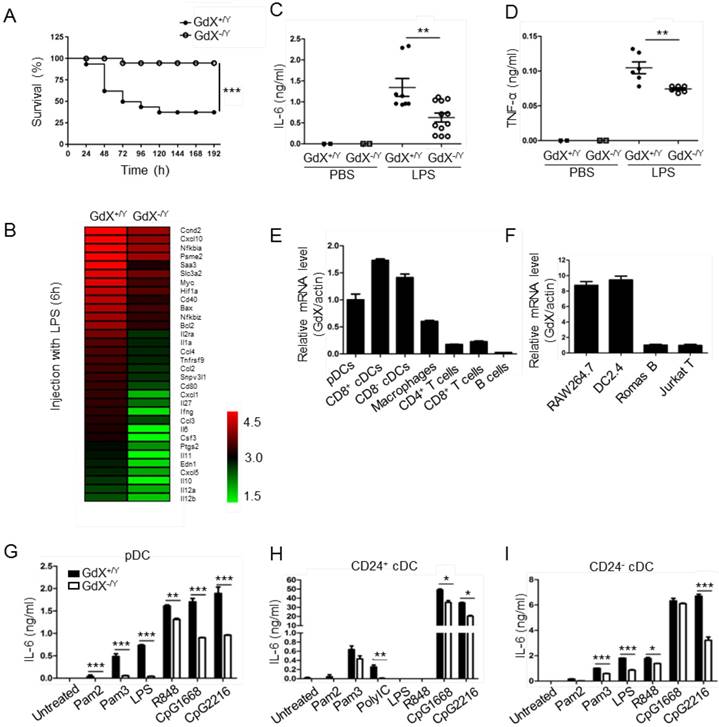
To address whether the resistance of GdX-/Y mice was due to a change of immune cell homeostasis, we compared immune cell profiles of GdX-/Y mice and their control littermates. The results showed that percentages of DCs, Mφ, B cells and T cells were similar in lymphoid organs of GdX+/Y and GdX-/Y mice (Fig. S1I). These results indicate that GdX is dispensable for immune cell development, suggesting GdX might play a role in regulating the function of immune cells.
GdX promotes the production of inflammatory cytokines by DCs and Mφ
To reveal the role of GdX in immune cells, we compared the mRNA level of GdX in different immune cell types. The results showed that GdX was expressed abundantly in DCs (pDCs and cDCs) from the spleen, Mφ from BM, but weakly in T cells (CD4+ and CD8+), and rarely in B cells from the spleen (Fig. 1E). Consistently, GdX was highly expressed in RAW264.7 and DC2.4 cell lines, but low in Romas B and Jurkat T cells (Fig. 1F and S1J). These results suggest that GdX might play a role in myeloid cells, in particular, DCs and Mφ.
We then investigated whether GdX deficiency affected inflammatory cytokine production by DCs and Mφ. BM-derived DCs in the presence of Fms-like tyrosine kinase 3 ligand (Flt3L) (FLDCs, including pDCs, CD24+ cDCs and CD24- cDCs, equivalent to splenic CD8α+ cDCs and CD8α- cDCs respectively in the steady state) were stimulated with a panel of TLR ligands in vitro and examined for IL-6, IL-12, and other cytokines and chemokines. The results showed that the pDCs, CD24+ cDCs and CD24- cDCs from GdX-deficient mice produced considerably lower amounts of IL-6 (Fig. 1G-I) and IL-12 (Fig. S1K-M) than that from WT mice in response to different stimulations. Simultaneously, the production of IFN-λ was dramatically decreased in pDCs from GdX-deficient mice in response to CpG2216 (Fig. S1N). In addition, GdX deficiency slightly impaired the production of MIP-1α and RANTES by CD24- cDCs (Fig. S1O and S1P). In line with these findings, deletion of GdX significantly decreased LPS-induced secretion of IL-6 and TNF-α by GMDCs (Granulocyte-macrophage colony-stimulating factor-induced DCs) (Fig. S1Q and S1R) and BMDMs (bone marrow-derived Mφ) (Fig. S1S and S1T). These results suggest that GdX deficiency attenuates the production of inflammatory cytokines by myeloid cells.
Additionally, we confirmed that deletion of GdX had no significant effect on the cell viability (Fig. S1U), expression of TLR2/4/9 (data not shown), or antigen presentation ability of the myeloid cells (data not shown). We speculated that the reduced inflammatory cytokine production by DCs and Mφ in GdX-deficient mice might be due to the regulation of TLR signaling.
GdX facilitates the NF-κB signaling by promoting p65 activation
The NF-κB signaling pathway is considered as a typical pro-inflammatory pathway downstream of TLR activation. The regulatory role of GdX on the NF-κB signaling was examined by transfecting the NF-κB luciferase reporter (κB-luc) along with MyD88, TRAF6, IKKβ or p65, key components in the NF-κB signaling pathway, in HEK293T cells. The cell viability was not significantly affected by transfection reagent (Fig. S2A). The results showed that luciferase activities were further increased by over-expression of GdX when MyD88 (Fig. 2A), TRAF6 (Fig. 2B), IKKβ (Fig. 2C) or p65 (Fig. 2D) was co-transfected. Over-expression of GdX also promoted NF-κB activation by TNF-α (Fig. S2B) or LPS/TLR4 (Fig. S2C), but inhibited STAT3 activity as we reported previously (Fig. S2D) [17]. In contrast, depletion of GdX using siRNAs inhibited NF-κB activation (Fig. S2E-2I). These results strongly suggest that GdX directly regulates p65 rather than the upstream components of the NF-κB signaling pathway.
GdX positively regulates NF-κB signaling by targeting p65. (A-D) GdX promoted the transcriptional activity of NF-κB. HEK293T cells were transfected with NF-κB response luciferase reporter (NF-κB-luc), together with MyD88 (A), TRAF6 (B), IKKβ (C), or p65 (D), along with different dosages of GdX. Luciferase activity was measured at 36 h after transfection. (E) The level of p-p65 was significantly lower in splenocytes of GdX-/Y mice than that of GdX+/Y mice. Individual mouse was labeled with # and number. (F and G) Deletion of GdX resulted in decreased phosphorylation of p65 in response to LPS. Levels of indicated proteins in GMDCs (F) and BMDMs (G) after LPS treatments were examined by WB. (H and I) Over-expression of GdX increased the levels of p-p65. DC2.4 (H) or RAW264.7 (I) cells infected with an adenovirus expressing GFP (Ad-GFP) or Myc-GdX (Ad-Myc-GdX) were stimulated with LPS (100 ng/mL). Results were presented as mean ± SEM from three repeats. *, p < 0.05; **, p < 0.01; ***, p < 0.001
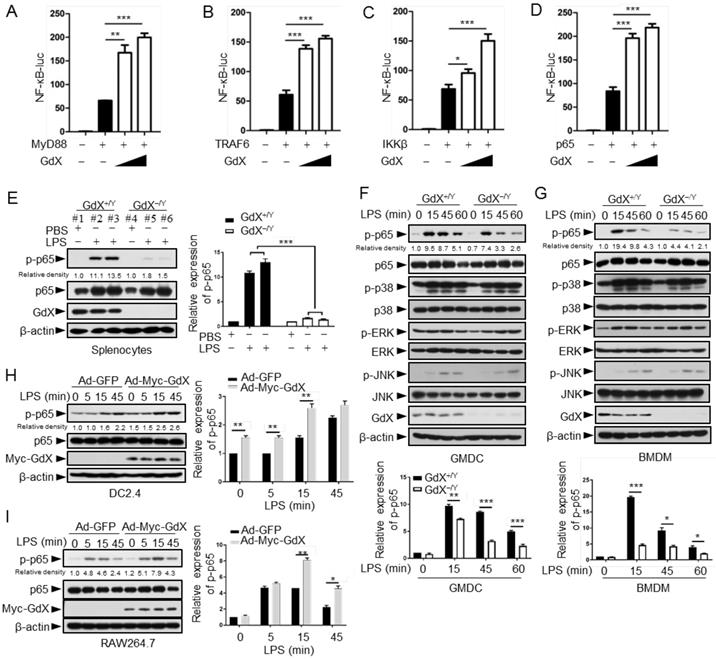
To investigate the effect of GdX on p65, the splenocytes from GdX-/Y mice, challenged with LPS for 16 h, were collected for western blot analyses. The results showed that p65 serine 536 phosphorylation [25] was dramatically decreased in the splenocytes obtained from GdX-/Y mice, compared with that in GdX+/Y mice (Fig. 2E). We then treated GMDCs and BMDMs of GdX+/Y (WT) or GdX-/Y mice with LPS, and examined the activation of different signaling pathways involved in TLR activation. The p65 phosphorylation intensity was dramatically decreased in both GMDCs (Fig. 2F, top panel) and BMDMs (Fig. 2G, top panel) of GdX-/Y mice. However, levels of phospho-p38, phospho-ERK and phospho-JNK were comparable (Fig. 2F, 2G, middle panels). These results suggest that GdX specifically regulates the phosphorylation of p65 in DCs and Mφ. Additionally, p65 phosphorylation induced by LPS was increased in GdX over-expressed DC2.4 and RAW264.7 cells (Fig. 2H, 2I). These results suggest that GdX participates in TLR-activated NF-κB pathway, up-regulating the phosphorylation of p65 in both in vitro and in vivo models, promoting the pro-inflammatory cytokine production by DCs and Mφ.
GdX disrupts the TC45/p65 complex formation
Since GdX had no direct interaction with p65 (Fig. S3A), we hypothesized that GdX regulates the phosphorylation status of p65 by controlling a protein kinase or phosphatase. We focused on IKKα, IKKβ, IKKε, canonical and non-canonical kinases known to be important for the phosphorylation of p65 [26, 27]; PP2A, a serine phosphatase that dephosphorylates p-p65 (phosphorylated p65) [28, 29]; WIP1, a PP2C family number [12], and TC45, a tyrosine phosphatase that interacts with GdX [17]. Immunoprecipitation (IP) experiments demonstrated that GdX interacted only with TC45 among these kinases and phosphatases (Fig. 3A), suggesting TC45 might associate with the regulation of p65 phosphorylation by GdX.
GdX blocks the interaction of TC45 with p65. (A) GdX specific interacted with TC45. Myc-GdX was co-expressed with Flag-tagged IKKα, IKKβ, IKKε, WIP1, PP4 or HA-tagged PP1, PP2A and TC45 in HEK293T cells. Immunoprecipitation (IP) experiments were performed using an anti-Flag or HA antibody. (B) HA-TC45 interacted with Flag-p65. HEK293T cells were transfected for IP with an anti-Flag antibody. (C) The association of endogenous TC45 and p65 was detected in immune cell lines. Lysates from DC2.4 or RAW264.7 cells were subjected to IP experiments with an antibody against TC45. (D) Flag-p65 interacted with GST-TC45 purified from E. coli. (E) The interaction of Flag-p65 and HA-TC45 was increased under TNF-α stimulation (10 ng/mL) for 15 min. (F) The interaction of endogenous TC45 and p65 was increased under LPS or TNF-α stimulation. IP experiments were performed by an anti-TC45 antibody. (G) Over-expression of GdX disrupted the association of TC45 and p65. HEK293T cells were used for IP experiments after transfected with HA-TC45 in the presence or absence of Myc-GdX. (H) The association of endogenous TC45 and p65 was increased in GdX-deficient (KO) splenocytes comparing with that in the wildtype (WT) cells. (I) GdX(L29P) mutant failed to block the interaction of p65 and TC45. (J-K) A molecular docking analysis shows the interaction surface of TC45 with the N-terminus of p65 (J) and GdX (K). F183 in TC45 maintains a core residue for the interaction with p65 and GdX although GdX shifts slightly to one side in the interaction (K).
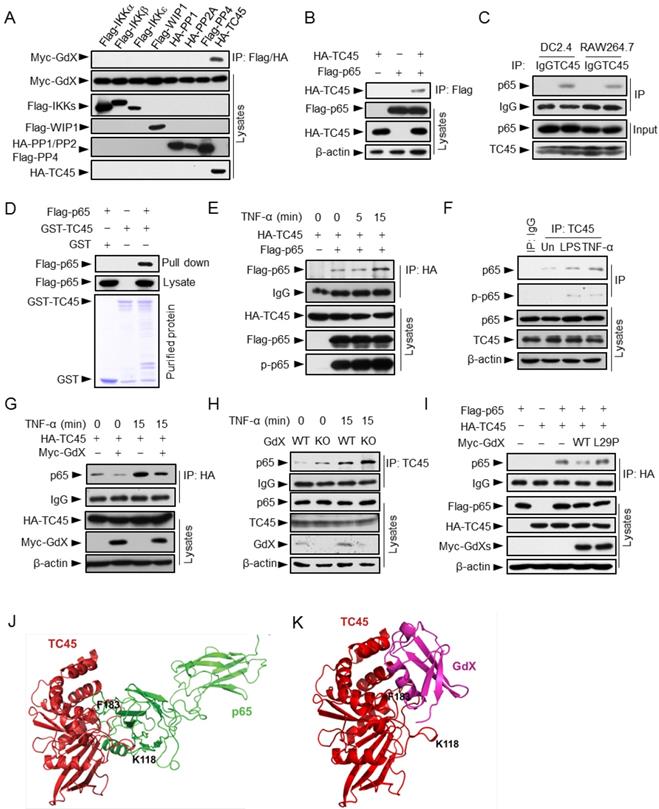
We then questioned whether TC45 could directly interact with p65. Co-IP analyses revealed that Flag-tagged p65 was able to precipitate with HA-tagged TC45 (Fig. 3B), and reciprocally, HA-TC45 pulled down Flag-tagged p65 (Fig. S3B). Consistently, the endogenous TC45 protein associated with p65 in both DC2.4 and RAW264.7 cells (Fig. 3C). In addition, purified proteins GST-TC45 pulled down Flag-p65, suggesting a direct interaction of TC45 with p65 (Fig. 3D). Furthermore, the interaction of p65 and TC45 was enhanced after TNF-α stimulation in both the over-expression (Fig. 3E) and endogenous conditions (Fig. 3F). Interestingly, when Myc-GdX, HA-TC45 and Flag-p65 were co-expressed together, we observed that HA-TC45 precipitated down both Myc-GdX and Flag-p65, but Myc-GdX failed to precipitate down Flag-p65 (although Myc-GdX precipitated down HA-TC45), and Flag-p65 failed to precipitate down Myc-GdX (Fig. S3C). These results indicated that TC45 exclusively forms a complex with p65 or GdX.
To analyze whether GdX affects the association of TC45 and p65, IP experiments were performed using HA-TC45 and endogenous p65 protein in the absence or presence of Myc-GdX in 293T cells. The results showed that the interaction of p65 and HA-TC45 was significantly decreased in the presence of Myc-GdX in conditions with or without TNF-α stimulation (Fig. 3G). Consistently, we observed that the interaction of endogenous TC45 with p65 was dramatically increased in the splenocytes from GdX-/Y mice (Fig. 3H). Similar results were obtained in GMDCs (Fig. S3D) and BMDMs (Fig. S3E) and suggest that GdX inhibits TC45 binding to p65.
To reveal whether GdX-impaired interaction of TC45 and p65 is due to the interaction of GdX with TC45, we recruited a mutant GdX, GdX(L29P), a leucine residue mutation in UBL domain, which lacks the ability to interact with TC45 [17]. IP experiments demonstrated that GdX(L29P) failed to disrupt the interaction of TC45 and p65 (Fig. 3I), suggesting that UBL domain of GdX plays a critical role in disrupting the interaction of TC45 with p65.
Furthermore, we performed a molecular docking analysis to show the detailed interaction of TC45 with p65 (Fig. 3J) and GdX (Fig. 3K). The results showed that the surface of TC45 for the interaction with GdX is the same as it interacts with p65 although GdX interacts on a slight shift site. In particular, residue F183 in TC45 faces to the interacting sites of p65 and GdX (Fig. 3J and 3K). Therefore, the interaction of GdX with TC45 occupies F183 and disrupts the interaction of p65 with TC45 (Fig. S3F). This structure base of interaction explains the mechanism for the exclusive interaction of TC45 with GdX or p65.
GdX prolongs p65 phosphorylation
Since GdX inhibits TC45 binding to p65, we speculated that the altered p-p65 level by GdX might be due to an altered dephosphorylation process. To test this possibility, we stimulated the GMDCs and BMDMs with LPS, and then we withdrew LPS and allowed cells to undergo starvation to induce protein dephosphorylation. Western blot analyses demonstrated that the level of p-p65 was slightly decreased in GMDCs and BMDMs from WT mice but quickly decreased in the cells from GdX-/Y mice when the cells were subject to starvation for different times (Fig. 4A, 4B). Similar results were observed in the splenocytes from WT and GdX-/Y mice (Fig. S4A). Reciprocally, when GdX was over-expressed by an adenovirus in DC2.4 and Raw264.7 cells, which were challenged by LPS for 30 min, high level of p-p65 remained for 60 min after starvation (Fig. 4C, 4D). Over-expression of GdX also increased the level of p-p65 after TNF-α stimulation in 293T cells (Fig. S4B). These results demonstrated that deletion of GdX shortened, while over-expression of GdX extended, the maintaining time of p-p65. In consistency with the prolonged p-p65 levels, we further observed that p65 occupied the promoter of IL-6 for a longer time (Fig. S4C, S4D) and remained in the nucleus after starvation for 120 min when GdX was over-expressed, while it redistributed into the cytoplasm after starvation for 60 min in the control cells (Fig. 4E). These results suggested that GdX abrogates the dephosphorylation process of p-p65.
GdX maintains the phosphorylation of p65 by counteracting TC45. (A and B) Depletion of GdX led to increased dephosphorylation of p65 in GMDC (A) and BMDM (B) cells. Cells derived from GdX+/Y or GdX-/Y mice were treated with LPS (100ng/mL) and then subjected to starvation at different times for dephosphorylation. (C and D) GdX sustained the level of p-p65 in DC2.4 (C) and RAW264.7 (D) cell lines. Cells were treated with LPS (100ng/mL) after infection with an adenovirus expressing GFP or GdX and then subjected to starvation for indicated times. (E) HeLa cells were infected with either Ad-GFP (an adenovirus expressing GFP) or Ad-GdX (an adenovirus expressing GdX) for 36 h, and then treated with TNF-α for indicated times. Subcellular localization of p65 was analyzed by immunofluorescence confocal microscopy with a rabbit anti-p65 antibody (red). DAPI was used to stain the nucleus. The percentage of nuclear staining of p65 was quantified. Scale bar, 10 μm. (F) Dephosphorylation of p-p65 was inhibited in TC45-depleted cells. p-p65 levels were examined in MEFTC45+/+ and MEFTC45-/- cells treated with or without TNF-α (10 ng/mL) for 15 min, followed by starvation for 30 min before harvesting. (G) TC45 accelerated p65 dephosphorylation. p-p65 levels were examined in HEK293T cells, which were treated with TNF-α for 15 min and then subjected to starvation, in the presence or absence of HA-TC45. (H) GdX failed to maintain the p-p65 level in TC45-deficient cells.
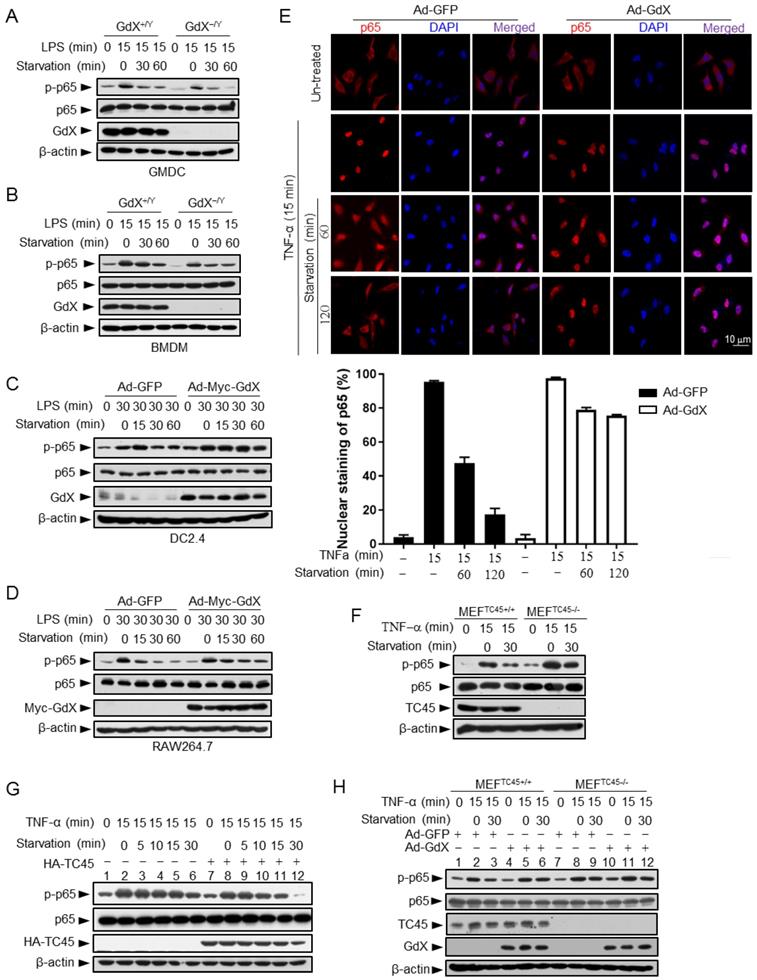
Given that TC45 interacts with p65, we speculated that TC45 might mediate the dephosphorylation of p65. Indeed, deletion of TC45 in MEFs showed a significantly prolonged phosphorylation of p65 (Fig. 4F) while over-expression of TC45 dramatically decreased the level of p-p65 after starvation for 30 min (Fig. 4G, lanes 12 and 6). Since GdX interrupts the interaction of TC45 with p65, we questioned whether GdX affects the dephosphorylation of p65 via blocking TC45. For this purpose, we used MEFs with a TC45 deletion, under the over-expression of GdX. We observed that, over-expression of GdX maintained the level of p-p65 after starvation for 30 min in the WT cells (Fig. 4H, comparing lane 6 to lane 3), confirming that GdX impairs the dephosphorylation of p-p65. However, over-expression of GdX failed to further increase the level of p-p65 under the starvation condition when TC45 was deleted (Fig. 4H, comparing lanes 12 to 9), indicating that GdX is unable to regulate the p-p65 level without TC45. These results suggest that GdX regulates the dephosphorylation of p65 via TC45. Taken together, we conclude that GdX-elevated phosphorylation of p65 is due to the interruption of TC45 from binding to p65.
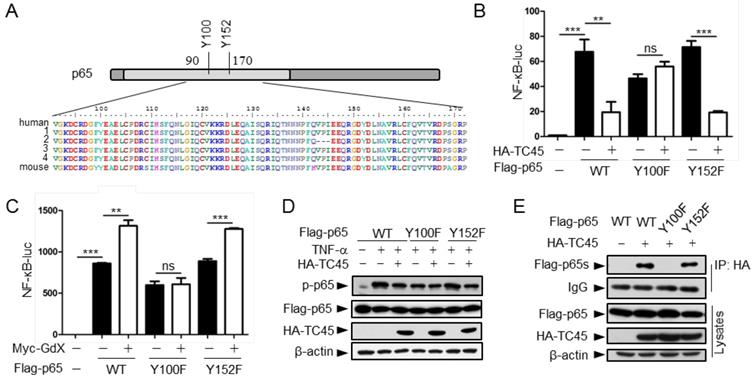
Residue Y100 in p65 is critical for TC45 to mediate p65 dephosphorylation
We next mapped the region for the interaction of p65 with TC45. IP experiments (Fig. S5A) showed that truncated p65-n6 (where 1-90 amino acids remained) failed to interact with TC45, whereas other truncated forms of p65 maintained strong interactions with TC45 (Fig. S5B). These data suggested that the region from amino acid 90 to 170 is essential for p65 to interact with TC45.
Since TC45 is a tyrosine phosphatase, we checked the conserved residues in this region and identified two tyrosine residues Y100 and Y152 (Fig. 5A). We speculated that these two tyrosine residues might be critical for TC45-mediated p65 dephosphorylation. To examine this hypothesis, we mutated these two residues into phenylalanine (F). Luciferase experiments indicated that p65(Y100F) had a decreased activity on the NF-κB reporter whereas p65(Y152F) remained the same activity as wild type p65 (Fig. 5B, black columns). Interestingly, over-expression of TC45 failed to inhibit p65(Y100F)-induced luciferase activity but remained to inhibit p65(Y152F)-induced activity (Fig. 5B). These results suggest that TC45 inhibits the activity of p65 through Y100. Simultaneously, we observed that over-expression of GdX elevated the reporter activity mediated by p65 and p65(Y152F) but had no effect on p65(Y100F)-induced activation (Fig. 5C). Consistent with these observations, over-expression of TC45 appeared to have no effect on the phosphorylation level of p65(Y100F), which though appeared lower than that of p65 (WT) and p65(Y152F) (Fig. 5D). We further deciphered that p65(Y100F) lost the interaction with TC45 but retained the interaction with p65(Y152F) (Fig. 5E). A molecular structure docking analysis suggests that this Y100 forms a link with K118 at TC45 to maintain a surface for the interaction of p65 with TC45 (Fig. S5C, S5D). These results indicate that Y100 is a key residue for the interaction of p65 with TC45 and thereafter the regulation of p65 dephosphorylation by TC45.
Tyrosine-100 in p65 is critical for TC45 function. (A) A schematic diagram showing two tyrosine (Y) residues in p65 in the region of amino acids 90-170. Both human and mouse sequences are showed. Numbers on the left indicated variants of p65 in human. (B) Y100 in p65 is critical for TC45 to suppress NF-κB signaling. Y: tyrosine; F: phenylalanine. (C) Y100 in p65 is critical for GdX to promote the activation of NF-κB signaling. Results were presented as mean ± SEM from three independent repeats. ns: no significant difference; *, p < 0.05; **, p < 0.01; ***, p < 0.001. (D) TC45 failed to promote the dephosphorylation of p65(Y100F) mutant. HEK-293T cells were transfected with the Flag-tagged p65, p65(Y100F), or p65(Y152F), in the presence or absence of HA-TC45. (E) TC45 failed to interact with p65(Y100F). HEK-293T cells were transfected with indicated plasmids. IP experiments were performed by using an anti-HA antibody.
TC45 recruits PP2A to dephosphorylate p65 at S536
To determine whether TC45 recruits a serine phosphatase to reduce p65 S536 phosphorylation, we screened several p65 S536 phosphatases. The results showed that over-expression of TC45 further decreased the phosphorylation of p65 based on PP2A, but not on other phosphatases including PP1, PP4 and WIP1 (Fig. 6A), which implied that TC45 might mediate the dephosphorylation of p65 through PP2A. PP2A is the main source of phosphatase activity in the cell, which has been reported to interact with p65 [10]. We then questioned whether TC45 affects the interaction of PP2A with p65. Intriguingly, we observed that TC45 enhanced the interaction of PP2A with p65 (Fig. 6B). On the other hand, the interaction of PP2A with p65 was dramatically impaired when TC45 was knocked out (Fig. 6C) or depleted by siRNA (Fig. S6A). Moreover, we observed that over-expression of PP2A failed to mediate the dephosphorylation of p65 in TC45-deficient MEFs (Fig. 6D, lanes 11-12), but strongly decreased the phosphorylation level in the WT cells (Fig. 6D, lanes 5-6). These results suggest that TC45 is required for PP2A-mediated dephosphorylation of p-p65.
TC45 mediates PP2A to dephosphorylation p-p65, which is interrupted by GdX. (A) TC45 and PP2A synergistically dephosphorylate p-p65. HEK293T cells were co-transfected with different phosphatases and Flag-tagged p65 plasmids. (B) Over-expression of TC45 enhanced the interaction of PP2A and p65. (C) The endogenous interaction of PP2A and p65 was decreased in TC45-deficient cells. (D) PP2A failed to dephosphorylate p-p65 without TC45. p-p65 levels were examined in MEFTC45+/+ and MEFTC45-/- cells infected with Ad-GdX or Ad-PP2A and treated with TNF-α (10 ng/mL) for 15 min, followed by TNF-α withdrawal for 30 min before harvesting. (E) Y100 is critical for the interaction of PP2A and p65. IP assays were performed after HEK-293T cells were transfected with the Flag-tagged p65, p65(Y100F), or p65(Y152F), as well as HA-PP2A for 24 h. (F) A molecular docking model showed TC45/p65/PP2A complex formation and disassociation. (a) The C-terminus of p65 is hided at the N-terminus of p65 to maintain an active form of p65. Without association of TC45 with p65 at its N-terminus, p65 remains a coated confirmation where the C-terminus, in particular S538, is hided. (b) TC45 starts to bind to the C-terminus of p65 and release the N-terminus of p65 from hiding. (c) PP2A gets a chance to associate with the released C-terminus and the heter-trimer complex of TC45/p65/PP2A is formed. In this complex, PP2A dephosphorylates S536 and finally maintains p65 unphosphorylated status (d). (G) GdX inhibited the interaction of p65 and PP2A. (H) GdX(L29P) mutant failed to decrease the interaction of PP2A and p65. HEK293T cells were transfected with the indicated plasmids for the IP experiment. (I) A model of the competition of GdX with TC45 to interact with p65. When TC45 interacts with the N-terminus of p65, the C-terminus of p65 is exposed to interact with PP2A. GdX interacts with TC45 and blocks its interaction with p65. In this way p65 maintains at its active form without interaction of PP2A.
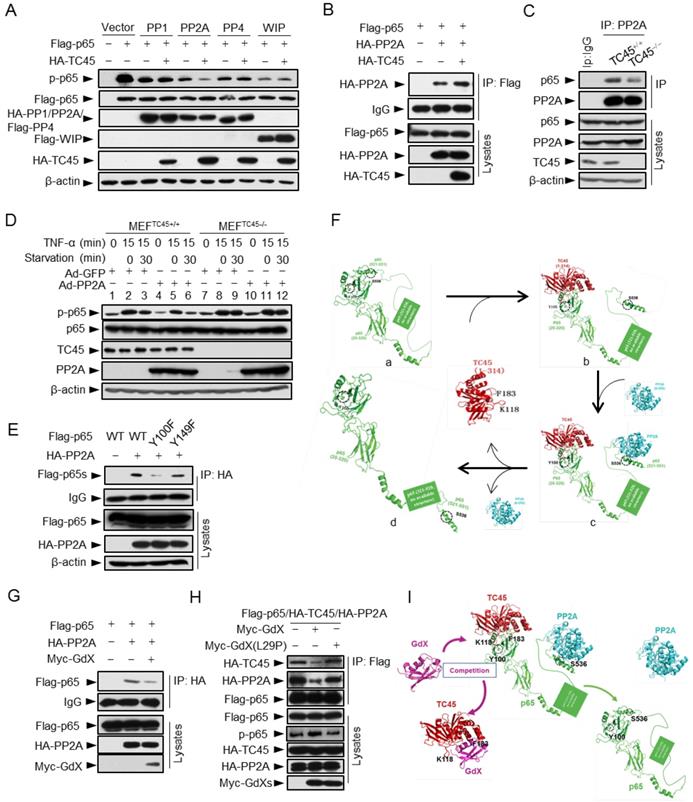
Further results revealed that TC45, PP2A and p65 forms a hetero-trimer complex (Fig. S6B). Interestingly, p65 (Y100F) showed much less interaction with PP2A (Fig. 6E), suggesting that Y100 is critical for p65 to interact with both PP2A and TC45. Molecular docking analyses provided the structural base for the hetero-trimer complex, where TC45 binds to the N-terminus, and PP2A binds to the C-terminus separately (Fig. 6Fc). In phosphoylated p65, the C-terminus is covered by the N-terminus (Fig. 6Fa). When the C-terminus of p65 is released by TC45 (Fig. 6Fb), PP2A associates with it and initiates the dephosphorylation of S536 (Fig. 6Fc), leading to p65 dephosphorylation (Fig. 6Fd).
Next, we determined whether GdX affects the interaction of PP2A with p65. Interestingly, IP experiments demonstrated that over-expression of Myc-GdX inhibited the interaction of HA-PP2A with Flag-p65 (Fig. 6G). Consistently, we observed that the interaction of PP2A and p65 was dramatically increased when GdX was deleted in GMDCs (Fig. S6C) and BMDMs (Fig. S6D). To valid that the inhibitory role of GdX on the interaction of PP2A and p65 is due to its interaction with TC45, we used mutant GdX(L29P), which lost the ability to associate with TC45. The results showed that GdX(L29P) had a lesser influence on the interaction of PP2A and p65 (Fig. S6E) and also the interaction of PP2A with TC45 (Fig. S6F), similar to that as observed for its effect on the interaction of TC45 with p65 (Fig. 3I). Consistent with the alteration of p65 phosphorylation (Fig. 6A), we confirmed that TC45 further inhibited the NF-κB transcriptional activity based on over-expression of PP2A (Fig. S6G). Furthermore, we observed that over-expression of Myc-GdX abrogated the complex of TC45/PP2A/p65 (Fig. 6H). This rescued the p-p65 level, which was decreased by over-expression of HA-PP2A (Fig. S6H), and was consistent with the results that GdX, but not GdX (L29P), rescued the NF-κB transcriptional activity inhibited by both TC45 (Fig. S6I) and PP2A (Fig. S6J). Based on the biochemical results, we proposed a model by molecular docking analysis. TC45 associates with p65 at the N-terminus via Y100, which leads to the release of the C-terminus of p65 to interact with PP2A. In this way, TC45, PP2A and p65 form a complex to dephosphorylate S536 of p65. When GdX sequestered TC45, the complex of TC45/PP2A/p65 is impaired and dephosphorylation of p65 is abrogated (Fig. 6I).
Specific deletion of GdX in DCs or Mφ protects the mice from colitis
The dysregulation of NF-κB signaling and innate immunity are closely associated with IBDs; thus we further examined the role of GdX in acute colitis. To evaluate the outcome of acute colitis in GdX-/Y mice and their littermates, we used a model in which the mice were administrated with 3% DSS in drinking water for 6 days. GdX-/Y mice exhibited a significantly decrease in body weight loss (Fig. 7A), a longer colon length (Fig. 7E, S7A), and a markedly lower disease activity index (DAI) (Fig. 7I) in comparison of control WT mice. The DAI is based on the magnitudes of body weight loss, diarrhea, and hemorrhage [30]. Furthermore, histological analyses of the bowel tissue from the WT mice administered with DSS showed manifestations of inflammatory colitis, including loss of crypts, mucosal erosion, ulcers, and infiltration of inflammatory cells (Fig. 7M, comparing b, c to a). In contrast, the specimens from the DSS-administered GdX-/Y mice showed lower levels of inflammation, with marginal infiltration in the mucosa (Fig. 7M, comparing e, f to d; also, comparing e to b and f to c).
GdX-deficient mice show a reduced inflammatory colitis. Mice of GdX germline deletion (GdX-/Y), specific deletion in DC cells (GdXΔDC), macrophages (GdXΔMφ), epithelial cells (GdXΔIEC ) and their WT (Cre-negative) littermates were treated with 3% DSS in drinking water for 6 days. (A) Body weight of GdX-/Y mice was decreased in comparison with that of GdX+/Y mice at day 5 after DSS treatment. Body weight was measured daily and presented as means ± SEM. p value on day 5 was significant, p < 0.05 (n=6 for DSS-treated WT group, and n=12 for KO group). (B and C) Body weights of GdXΔDC (B) and GdXΔMφ (C) mice were decreased in comparison with that of WT mice after DSS treatment. (D) Body weight of GdXΔIEC mice was unchanged compared with that of WT mice after DSS treatment. (E-H) The effect of GdX deletion on the colon lengths. The length of colon from GdX-/Y (E), GdXΔDC (F), GdXΔMφ (G) and GdXΔIEC (H) mice and their WT (Cre-negative) littermates were determined on day 6 after DSS treatment. (I-L) The disease activity index (DAI) was determined on day 6. The DAI of GdX-/Y (I), GdXΔDC (J), GdXΔMφ (K) and GdXΔIEC (L) mice were compared with their WT (Cre-negative) littermates. (M and N) Microscopic views of HE-stained colon sections. Tissue sections from GdX-/Y (M), GdXΔDC, GdXΔMφ and GdXΔIEC (N) mice and their WT (Cre-negative) littermates were subject to HE-staining. (O-R) The production of IL-6 was decreased in GdX-/Y, GdXΔDC and GdXΔMφ mice but not in GdXΔIEC mice. The serum IL-6 concentrations of the mice were compared with their WT (Cre-negative) littermates by ELISA. (S) Deletion of GdX resulted in decreased phosphorylation of p65 in IECs. Immuno-histochemical analyses of p-p65 (upper) and p65 (lower) in paraffin-embedded colon sections of GdX+/Y (n=5) and GdX-/Y mice (n=5) at day 6 after treatment with 3% DSS; nuclei were counterstained with hematoxylin (blue). Scale bars, 100 μm. (T) Examinations of p-p65 levels and GdX depletion efficiency in the spleen DCs of GdXΔDC mice and Mφ of GdXΔMφ mice. The DCs, Mφ were sorted by FACS and then protein levels were determined by WB.
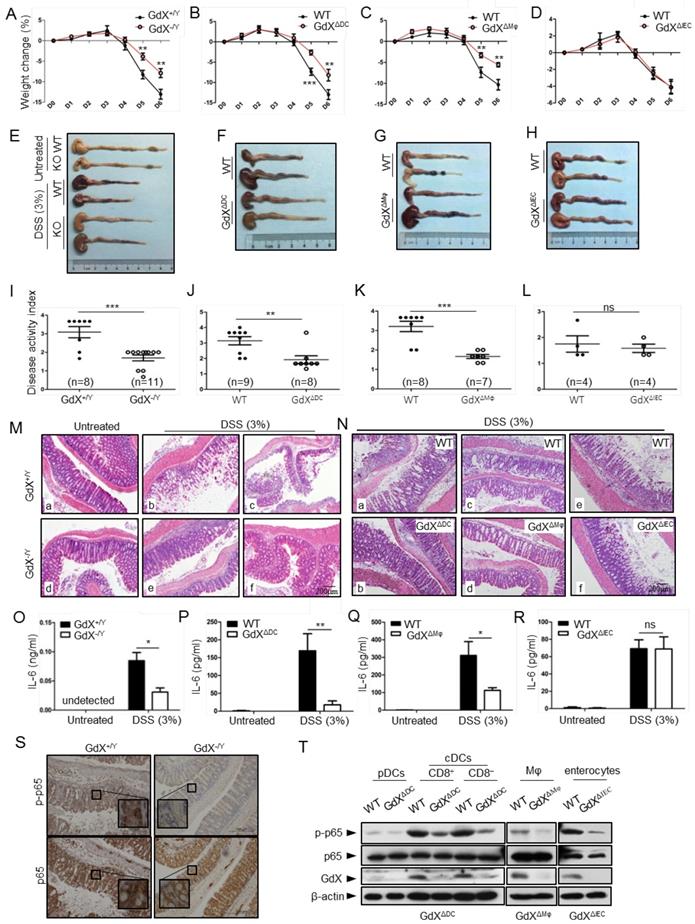
To further investigate the whether GdX regulate IBD development by innate immune cell-intrinsic mechanisms, we generated GdX conditional knockout mice in DCs (GdXΔDC), Mφ (GdXΔMφ) and intestine epithelial cells (IECs) (GdXΔIEC) by crossing the CD11c-Cre, LysM-Cre or Villin-Cre mice to GdXflox/flox mice. After administration with 3% DSS in drinking water for 6 days, GdXΔDC, and GdXΔMφ mice lost less weight than littermate controls (Fig. 7B, 7C), exhibited longer colons lengths (Fig. 7F, 7G, S7B, S7C), lower disease activity index (DAI) (Fig. 7J, 7K), and significantly lower levels of inflammation observed via histological damage (Fig. 7N, comparing b to a, d to c). The phenotypes of GdXΔDC, and GdXΔMφ mice during acute colitis is similar with that observed in GdX-/Y mice, suggesting deletion of GdX in DC and Mφ attenuate intestinal inflammation. However, GdXΔIEC mice did not show decreased intestinal inflammation (Fig. 7D,7H, S7D, 7L, 7N, e and f showed similar severe damages), suggesting the deletion of GdX in IECs was not responsible for the decreased severity of colitis in GdX-/Y mice. In addition, GdX-/Y (Fig. 7O), GdXΔDC (Fig. 7P) and GdXΔMφ (Fig. 7Q) mice showed significantly decreased levels of IL-6 in serum compared with the control mice, whereas GdXΔIEC mice maintained comparable levels of serum IL-6 to that of the control mice (Fig. 7R) during DSS-induced colitis. These data suggest that the alleviated DSS-induced colitis in GdX-/Y mice is largely due to functional changes of DCs and Mφ.
Inflammatory disorders in the gut are usually associated with disrupted homeostasis of T regulatory (Treg) and T helper 17 (Th17) cells, which can be induced by three types of myeloid cells including CD11chiCD11b+CD103+ DCs, CD11chiCD11b-CD103+DCs and F4/80+CD11cintCD11b+CD103- Mφ [31]. However, we did not find any changes in the frequencies of the intestinal Treg, Th17 cells and myeloid cell populations in GdXΔDC mice compared to WT mice after DSS treatment for 6 days (Fig. S7E). These results supported the notion that the reduced severity of gut inflammation in GdX-deficient mice was largely due to the impaired production of inflammatory cytokine in the myeloid cells (DCs and Mφ), rather than the consequence of defects in myeloid cell development and Treg/Th17 cell balance.
As our aforementioned results suggested that GdX regulates dephosphorylation of p65, we examined the levels of p-p65 in the colons of the mice after DSS treatment. Immunohistochemical analyses demonstrated that p-p65 staining was much stronger in the nucleus in the colon section from WT mice than that from GdX-/Y mice treated with DSS (Fig. 7S, upper panels), while the non-phosphorylated p65 remained mainly in the cytoplasm of the cells from GdX-/Y mice (Fig. 7S, bottom panels). These results suggested that deletion of GdX impaired the activation of NF-κB in colon tissue. Consistently, we observed that specific deletion of GdX in DCs and Mφ significantly decreased the p-p65 levels during acute colitis (Fig. 7T). Taken together, these data suggested that GdX deficiency alleviates the colon inflammation through regulation of NF-κB activity in DCs and Mφ.
Discussion
In this study, we revealed a previously unrecognized regulatory mechanism of NF-κB signaling in innate immune cells. GdX forms a complex with tyrosine phosphatase TC45, traps PP2A by blocking its termination to warranty a sufficient activation of NF-κB (Fig. 6I). GdX deletion impaired the production of pro-inflammatory cytokines by DCs and Mφ, protected mice against septic shock and acute colitis (Fig. 8). Immune system requires a mechanism to fight against antigens rapidly and efficiently, where GdX functions as a 'bodyguard' in DCs and Mφ to maintain their sensitivity to pathogenic stimuli.
A model of the regulation of dephosphorylation of p65 by GdX. GdX maintained the level of p-p65 by blocking the complex formation of TC45 and PP2A with p65, leading to abrogated dephosphorylation of p65. Without GdX, TC45 brings PP2A to associate with p-p65 and mediates dephosphorylation of p-p65 for the termination of NF-kB signaling. In the presence of GdX, GdX interacts with TC45 and competes TC45 from interaction of PP2A and p-p65, leading to impaired dephosphorylation of p-p65. In such a way, GdX guards TC45 and PP2A from interaction with p65 to maintain DCs and Mφs alert to pathogen attacks.
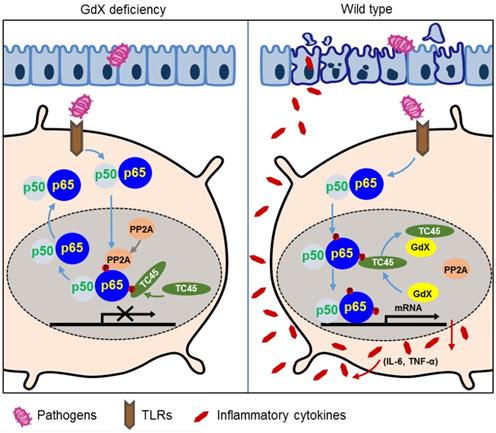
Although NF-κB signaling is tightly associated with IBDs, the cell-type specific mechanisms of NF-κB is poorly understood. Our finding implied that the NF-κB signaling in DCs and Mφ is critical for tissue inflammation and damage. We found that deletion of GdX in IECs had no effect on DSS-induced colitis, but GdXΔDC and GdXΔMφ mice displayed alleviated mucosal inflammation. Hence, we conclude that GdX mainly functions in DCs and Mφ to regulate the acute colitis. This notion is supported by other studies that SCID mice developed intestinal inflammation similar to that developed in immunocompetent Balb/c mice after DSS-treatment [32], and adoptive transfer of GMDCs exacerbated DSS-induced colitis while ablation of DCs ameliorated the colitis [33]. Our results are also supported by the depletion of Mφ [34], inhibition of myeloid cells by an antibody against CD11b/CD18 (Mac-1) [35] or deletion of IL-6 reduced intestinal inflammation [36]. Mechanistically, GdX deficiency led to reduced phosphorylation of p65 in colon sections during mucosal inflammation, which resulted in a less severe colitis. Consistent with our studies, the level of p65 has been reported increased in the nuclear extracts of intestinal lamina propria biopsy from IBD patients [37]. Moreover, activated p65 was found in either Mφ or epithelial cells from inflamed mucosa but was almost absent in normal mucosa [38]. Opposite to NF-κB, STAT3 signaling is considered as an anti-inflammation pathway [39]. We previously reported GdX repressed tumorigenesis through dephosphorylation of STAT3. We also observed that IECs from GdX-/Y mice had a greater proliferative ability than these from WT mice, implying that GdX endowed an enhanced ability to maintain mucosal integrity, which is due to enhanced STAT3 activity [17]. However, in this study, the phosphorylation of STAT3 could not be detected in either LPS-stimulated myeloid cells, or the colon tissues from GdXΔDC, and GdXΔMφ conditional KO mice treated by DSS. Simultaneously we observed that the NF-κB pathway has a dominant role during acute colitis. Although NF-κB is also involved in tumorigenesis, the effect of intrinsic NF-κB in myeloid cells on tumorigenesis remains still unclear. Furthermore, we observed that the role of GdX on the regulation of NF-κB mainly occurred in DC and macrophages but not in epithelial cells, as a Villin-Cre-driven deletion of GdX had no phenotype on NF-κB signaling. We speculate that the regulation of GdX on STAT3 and NF-κB may occur in different cells, depending on the dominance of the signals. Nevertheless, our findings echo the observations that DCs and Mφ are direct contributors in response to acute mucosal damage during intestinal inflammation after DSS treatment [40].
p65 phosphorylation is critical for activation of NF-κB-dependent transcription. Our data showed that GdX prolonged LPS-induced phosphorylation of p65 by trapping TC45 and PP2A. Consequently, p65 phosphorylation is sustained by GdX. Mechanistically, we observed that TC45 was critical for GdX-maintained p65 phosphorylation on serine 536. As dephosphorylation of serine 536 of p65 is induced by PP2A [14], we were considering that TC45 might function as a co-factor to bridge PP2A to p-p65, similar to the function of PHF20 [41]. However, TC45 failed to interact with PP2A. Therefore, we proposed a model the TC45 interacts with the N-terminus of p65 via Y100 and releases the coating of the C-terminus of p65 for PP2A association. Our functional results further demonstrated that mutant p65 (Y100F) impaired the interaction of TC45 to inhibit the transcriptional activity of p65. These results suggested that Y100 is critical for TC45 interaction and GdX functions to complete via Y100. However, further works are required to analyze the whole structure of p65, and how the interaction of TC45 with the N-terminus of p65 mediates the interaction of PP2A at the C-terminus of p65 remains to be elucidated.
In summary, our study provided the evidence that GdX promote the pro-inflammatory phenotypes of DCs and Mφ by activating NF-κB signaling. Targeting GdX in innate immune cells might offer therapeutic benefits for colitis, particularly for the development of DC- or Mφ-based therapeutic strategies for acute inflammation.
Materials and Methods
Mice
C57BL/6J mice were housed in a specific pathogen-free (SPF) facility at Tsinghua University. GdX-/Y and GdXfl/fl mice were generated as described previously [23], and have been backcrossed to the C57BL/6J background for at least eight generations. Floxed GdX mice were bred with CD11c-Cre (Jackson stock 008068, B6.Cg-Tg (Itgax-cre) 1-1Reiz/J), LysM-Cre (Jackson stock 004781, B6.129P2-Lyz2tm1(cre)Ifo/J) or Villin-Cre (Jackson stock 018963, B6N.Cg-Tg (Vil-cre) 997Gum/J). Cre-negative littermates were used as controls.
Assessment of the biological effects of LPS
To compare the survival rate under endotoxin shock, mice were injected with 30 mg/kg of LPS from Escherichia coli 055:B5 (Sigma, St. Louis, MO, USA) [24, 42]. Mice were injected with 20 mg/kg LPS for induction of acute inflammation and serums were collected for cytokine measurement by using ELISA assay.
RNA sequencing
The WT and GdX-deficient mice were injected (i.p.) with 20 mg/kg LPS, and then the splenocytes were collected after 1.5 or 6 h. RNAs were purified and reverted into cDNA libraries. High-throughput sequencing was performed by BGISEQ-500 (Beijing Genomics Institute, BGI). The RNA-seq was carried out with two biological replicates. The RNA-seq reads were mapped to mm10 genome by HISAT2 v2.0.4. The differentially expressed gene (DEG) reads were analyzed by DEGseq. The genes with absolute log2 >1 fold changes and a threshold q value<0.05 were regarded as DEGs. GO analysis was performed using differentially expressed genes against gene sets from DAVID GO database (https://david.ncifcrf.gov/) and KEGG pathway database (http://www.genome.jp/kegg/pathway.html). Sequencing data have been deposited in GEO under accession code GSE116956.
Cell culture
For Fms-like tyrosine kinase 3 ligand (Flt3L)-supplemented BM-cultured DCs (FLDCs) preparation, bone marrow (BM) cells were cultured in RPMI-1640 complete medium (RPMI-1640-10% FBS-1% P/S) in the presence of recombinant murine Flt3L (200 ng/mL) for 7-8 days, and then sorted for pDCs, CD24+ cDCs and CD24- cDCs [43]. For granulocyte-macrophage colony-stimulating factor-induced DCs (GMDCs) preparation, BM cells were incubated with GM-CSF (20 ng/mL) and IL-4 (20 ng/mL) for 6-7 days. For bone marrow derived macrophages (BMDMs), BM cells were cultured in macrophage colony-stimulating factor (M-CSF) conditional medium (DMEM-10% FBS-1% P/S with the supplement of L929 cell supernatant) for 5-6 days. All of the cytokines were purchased from PeproTech (Rocky Hill, NJ, USA). For isolation of peritoneal Mφ, mice were injected with 4% thioglycollate medium in a total volume of 0.8 ml, and then peritoneal Mφ were isolated 3 days later.
Cytokine secretion analyses
FLDCs were seeded in 96-well plates and treated with TLR agonists, including 50 nM Pam2CSK4 (InvivoGen, San Diego, CA, USA, tlrl-pm2s-1), 50 nM Pam3CSK4 (InvivoGen, tlrl-pms), 100 µg/ml Poly (I:C) (InvivoGen, tlrl-pic), 100 ng/mL LPS (Sigma), 1 µg/ml R848 (InvivoGen, tlrl-r848), 10nM ODN 1668 (AdipoGen, San Diego, CA, USA, IAX-200-001) and 1uM ODN 2216 (AdipoGen, IAX-200-005), respectively for 16 hrs. Supernatants were then collected for ELISA analyses of IL-6, TNF-α, IL-12p40, IL-12p70, MIP-1α, RANTES and IFN-λ respectively. Recombinant mouse RANTES (R&D, Minneapolis, MN, USA, 478-MR), MIP-1α (R&D, 450-MA), IFN-λ3 (R&D, 1789-ML), IL-12p70 (eBioscience, Palo Alto, CA, USA, 14-8121), TNF-α (eBio, 39-8321-65) and IL-6 (eBio, 39-8061-65) were used as internal standard protein controls. Antibodies against mouse RANTES (R&D, MAB4781), MIP-1α (R&D, AF-450-NA), IFN-λ2/3 (R&D, MAB17892), IL-12p70 (BioLegend, San Diego, CA, USA, 511802), IL-12/IL-23p40 (eBio, 14-7125-85), IL-6 (eBio, 14-7061-81) and IFN-α (PBL, 22100-1) were used as capture antibodies. Biotinylated monoclonal antibodies against mouse RANTES (R&D BAF478), MIP-1α (R&D BAF450), IFN-λ3 (R&D BAM17891), IL-12/IL-23p40 (eBio 13-7123), TNF-α (eBio14-7423-81), IL-6 (eBio13-7062-81) were used in combination with streptavidin-HRP (Amersham, Little Chalfont, UK, RPN4401) for ELLSA analyses.
Transfection and luciferase reporter assay
HEK293T cells were transfected with plasmids encoding κB-luciferase reporter, pRL-TK Renilla luciferase, and different expression vectors using Lipofectamine-2000 (Invitrogen, Carlsbad, CA, USA). The κB-luciferase reporter activity was determined by a dual luciferase assay kit (Promega, Madison, WI, USA) as previously reported [44].
Antibodies, immunoprecipitation and immunoblot analyses
Total cell lysates were prepared after transfection or stimulation for the experiments. For IP experiments, cell extracts were incubated with indicated antibodies together with Protein A/G beads (Pierce, Rockford, IL, USA) overnight. Beads were washed four times with lysis buffer, and immunoprecipitates were eluted with SDS loading buffer (Cell Signaling Technology, Danvers, MA, USA) and resolved in SDS-PAGE gels. Antibodies used were rabbit polyclonal antibodies against p65, p-p65, IkBα, p-IkBα, ERK, p-ERK, p38, p-p38, JNK, and p-JNK (from Cell Signaling) and mouse monoclonal antibodies against Myc (9E10), HA (Santa Cruz, Dallas, Texas, USA), Flag (M2, Sigma). Anti-GdX antibodies were generated in this lab, and the specificity was examined [23]. WB and IP experiments were performed according to previous protocols [44].
Quantitative RT-PCR
Total RNA was isolated with the RNeasy kit (Qiagen, Germantown, MD, USA), and cDNA was synthesized with SuperScript RT III (Invitrogen). The mRNA levels of IL-6, IL-1β, TNF-α, GdX and GAPDH were measured by real-time PCR performed in SYBR Green I on 7900 real-time PCR detection system (Applied Biosystems, Grand Island, NY, USA). Primer sequences were listed as follows: mouse IL-1β (forward: 5′-CTCCATGAGCTTTGTACAAGG-3′, reverse: 5′- TGCTGATGTACCAGTTGGGG -3′), mouse TNF-α (forward: 5′- CGGACTCCGCAAAGTCTAAG-3′, reverse: 5′-ACGGCATGGATCTCAAAGAC-3′), mouse IL-6 (forward: 5′- GGAAATTGGGGTAGGAAGGA -3′, reverse: 5′- CCGGAGAGGAGACTTCACAG -3′), mouse GdX (forward: 5′-AGCACCTGGTCTCGGATAAG -3′, reverse: 5′-GCCCAATGTTGTAATCTGACAG-3′) mouse GAPDH (forward: 5′-TGTGTCCGTCGTGGATCTGA-3′, reverse: 5′-CCTGCTTCACCACCTTCTTGA-3′). PCR was carried out for 35 cycles using the following conditions: denaturation at 95°C for 20 s, annealing at 58°C for 20 s, and elongation at 72°C for 20 s.
Structural Analysis
All protein interacting models were predicted by Z-Dock (v.3.0.2) [7]. The proteins accession numbers used for docking models are 2ie3 (PP2A, 1-309), 2n22 (p65, 521-551), 1nfi (p65, 20-320), 1l8k (TC45, 1-314) and 2dzi (Gdx, 1-74), which were obtained from RCSB Protein Data Bank [45-48]. The results of the models were visualized and processed by the PyMOL Molecular Graphics System (Version 1.8 Schrödinger, LLC).
DSS-induced colitis
Mice were subjected to acceptance of 3% (wt/vol) DSS (molecular weight, 36,000-50,000; MP Biomedicals Santa Ana, CA, USA) in drinking water ad libitum for 6 days. Body weight and stool were monitored daily starting from day 0 of treatment. Colons were collected after mice were sacrificed, fixed in 10% (vol/vol) formaldehyde, and sectioned for immunohistological staining with indicated antibodies and for H&E staining according to protocols used previously [17].
Statistical analyses
All of the in vitro experiments were repeated at least three times whereas all in vivo experiments were performed at least twice. All statistical analyses were calculated using GraphPad Prism 5. Data were presented as mean ± SEM. Statistical significance was determined with the two-tailed unpaired Student's t test. Differences were considered to be statistically significant when p < 0.05.
Acknowlegement
This work was supported by grants from the Chinese National Major Scientific Research Program (2016YFA0500301, 2015CB943200), grants from the National Natural Science Foundation of China (81830092, 81372167, 81301700, 81572729, 31330027, 81872244, 81372372), and the Tsinghua Science Foundation (20121080018, 20111080963). We thank Dr. Andrew Larner to provide TC45 KO cells. We thank Dr. Xinquan Wang and Dongli Wang for providing the assistance in the molecular docking analyses. We thank Taylor T. Chrisikos and Rachel Babcock for revising the manuscript.
Author Contribution
Liu C., Zhou Y., Wu L. and Chang Z. contributed to experimental design and experiments. Li M., Wang Y., Yang L., Yang S., Feng Y., Wang YY., Wang YM., and Ren F. performed experiments, Li J., Dong Z., Fu X. and Chin Y.E. contributed to experimental design, Liu C., Zhou Y., Wu L. and Chang Z. wrote the paper.
Supplementary Material
Supplementary figures.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34(5):637-50
2. Aderem A. and R.J. Ulevitch, Toll-like receptors in the induction of the innate immune response. Nature. 2000;406(6797):782-7
3. Molodecky NA. et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology. 2012;142(1):46-54 e42; quiz e30
4. Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132(3):344-62
5. Ahmad SF. et al. Regulation of TNF-alpha and NF-kappaB activation through the JAK/STAT signaling pathway downstream of histamine 4 receptor in a rat model of LPS-induced joint inflammation. Immunobiology. 2015;220(7):889-98
6. Oeckinghaus A, Ghosh S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol. 2009;1(4a):000034
7. Pierce BG, Hourai Y, Weng Z. Accelerating protein docking in ZDOCK using an advanced 3D convolution library. PLoS One. 2011;6(9e):24657
8. Ghosh S, Hayden MS. New regulators of NF-kappaB in inflammation. Nat Rev Immunol. 2008;8(11):837-48
9. Li HY. et al. Deactivation of the kinase IKK by CUEDC2 through recruitment of the phosphatase PP1. Nat Immunol. 2008;9(5):533-41
10. Yang J. et al. Protein phosphatase 2A interacts with and directly dephosphorylates RelA. J Biol Chem. 2001;276(51):47828-33
11. Li S. et al. RNAi screen in mouse astrocytes identifies phosphatases that regulate NF-kappaB signaling. Mol Cell. 2006;24(4):497-509
12. Chew J. et al. WIP1 phosphatase is a negative regulator of NF-kappaB signalling. Nat Cell Biol. 2009;11(5):659-66
13. Shu G. et al. Isoliensinine induces dephosphorylation of NF-kB p65 subunit at Ser536 via a PP2A-dependent mechanism in hepatocellular carcinoma cells: roles of impairing PP2A/I2PP2A interaction. Oncotarget. 2016
14. Hsieh C.Y. et al. Andrographolide enhances nuclear factor-kappaB subunit p65 Ser536 dephosphorylation through activation of protein phosphatase 2A in vascular smooth muscle cells. J Biol Chem. 2011;286(8):5942-55
15. Hsieh CY. et al. PMC, a potent hydrophilic alpha-tocopherol derivative, inhibits NF-kappaB activation via PP2A but not IkappaBalpha-dependent signals in vascular smooth muscle cells. J Cell Mol Med. 2014;18(7):1278-89
16. Toniolo D, Persico M, Alcalay M. A "housekeeping" gene on the X chromosome encodes a protein similar to ubiquitin. Proc Natl Acad Sci U S A. 1988;85(3):851-5
17. Wang Y. et al. GdX/UBL4A specifically stabilizes the TC45/STAT3 association and promotes dephosphorylation of STAT3 to repress tumorigenesis. Mol Cell. 2014;53(5):752-65
18. Xu Y. et al. SGTA recognizes a noncanonical ubiquitin-like domain in the Bag6-Ubl4A-Trc35 complex to promote endoplasmic reticulum-associated degradation. Cell Rep. 2012;2(6):1633-44
19. Xu Y. et al. A ubiquitin-like domain recruits an oligomeric chaperone to a retrotranslocation complex in endoplasmic reticulum-associated degradation. J Biol Chem. 2013;288(25):18068-76
20. Kuwabara N. et al. Structure of a BAG6 (Bcl-2-associated athanogene 6)-Ubl4a (ubiquitin-like protein 4a) complex reveals a novel binding interface that functions in tail-anchored protein biogenesis. J Biol Chem. 2015;290(15):9387-98
21. Zhao Y. et al. Ubl4A is required for insulin-induced Akt plasma membrane translocation through promotion of Arp2/3-dependent actin branching. Proc Natl Acad Sci U S A. 2015;112(31):9644-9
22. Liang J. et al. GdX/UBL4A null mice exhibit mild kyphosis and scoliosis accompanied by dysregulation of osteoblastogenesis and chondrogenesis. Cell Biochem Funct. 2018;36(3):129-136
23. Wang Y. et al. Generation of mice with conditional null allele for GdX/Ubl4A. Genesis. 2012;50(7):534-42
24. Scheibel M. et al. IkappaBbeta is an essential co-activator for LPS-induced IL-1beta transcription in vivo. J Exp Med. 2010;207(12):2621-30
25. Zhang L. et al. Function of phosphorylation of NF-kB p65 ser536 in prostate cancer oncogenesis. Oncotarget. 2015;6(8):6281-94
26. Sugimoto N. et al. Points of control exerted along the macrophage-endothelial cell-polymorphonuclear neutrophil axis by PECAM-1 in the innate immune response of acute colonic inflammation. J Immunol. 2008;181(3):2145-54
27. Hoesel B, Schmid JA. The complexity of NF-kappaB signaling in inflammation and cancer. Mol Cancer. 2013;12:86
28. London NR. et al. Targeting Robo4-dependent Slit signaling to survive the cytokine storm in sepsis and influenza. Sci Transl Med. 2010;2(23):23ra19
29. Nathan C. Points of control in inflammation. Nature. 2002;420(6917):846-52
30. Murano M. et al. Therapeutic effect of intracolonically administered nuclear factor kappa B (p65) antisense oligonucleotide on mouse dextran sulphate sodium (DSS)-induced colitis. Clin Exp Immunol. 2000;120(1):51-8
31. Iwasaki A. Mucosal dendritic cells. Annu Rev Immunol. 2007;25:381-418
32. Tlaskalova-Hogenova H. et al. Involvement of innate immunity in the development of inflammatory and autoimmune diseases. Ann N Y Acad Sci. 2005;1051:787-98
33. Bernd BE. et al. The role of dendritic cells in the development of acute dextran sulfate sodium colitis. J Immunol. 2007;179(9):6255-62
34. Watanabe N. et al. Elimination of local macrophages in intestine prevents chronic colitis in interleukin-10-deficient mice. Dig Dis Sci. 2003;48(2):408-14
35. Palmen M.J. et al. Anti-CD11b/CD18 antibodies reduce inflammation in acute colitis in rats. Clin Exp Immunol. 1995;101(2):351-6
36. Naito Y. et al. Reduced intestinal inflammation induced by dextran sodium sulfate in interleukin-6-deficient mice. Int J Mol Med. 2004;14(2):191-6
37. Ardite E. et al. Effects of steroid treatment on activation of nuclear factor kappaB in patients with inflammatory bowel disease. Br J Pharmacol. 1998;124(3):431-3
38. Roesky HW. et al. A Facile Route to Group 13 Difluorodiorganometalates. Angew Chem Int Ed Engl. 2000;39(1):171-173
39. Chrisiko TT. et al. Molecular regulation of dendritic cell development and function in homeostasis, inflammation, and cancer. Mol Immunol. 2018
40. Coombes JL, Powrie F. Dendritic cells in intestinal immune regulation. Nat Rev Immunol. 2008;8(6):435-46
41. Zhang T. et al. PHF20 regulates NF-kappaB signalling by disrupting recruitment of PP2A to p65. Nat Commun. 2013;4:2062
42. Li Q. et al. Inhibition of autophagy with 3-methyladenine is protective in a lethal model of murine endotoxemia and polymicrobial sepsis. Innate Immun. 2018;24(4):231-239
43. Zhou Y. et al. p38alpha has an important role in antigen cross-presentation by dendritic cells. Cell Mol Immunol. 2018;15(3):246-259
44. Liu C. et al. p15RS/RPRD1A (p15INK4b-related sequence/regulation of nuclear pre-mRNA domain-containing protein 1A) interacts with HDAC2 in inhibition of the Wnt/beta-catenin signaling pathway. J Biol Chem. 2015;290(15):9701-13
45. Xing Y. et al. Structure of protein phosphatase 2A core enzyme bound to tumor-inducing toxins. Cell. 2006;127(2):341-53
46. Lecoq L. et al. Structural characterization of interactions between transactivation domain 1 of the p65 subunit of NF-kappaB and transcription regulatory factors. Nucleic Acids Res. 2017;45(9):5564-5576
47. Jacobs MD, Harrison SC. Structure of an IkappaBalpha/NF-kappaB complex. Cell. 1998;95(6):749-58
48. Iversen LF. et al. Structure determination of T cell protein-tyrosine phosphatase. J Biol Chem. 2002;277(22):19982-90
Author contact
![]() Corresponding authors: Zhijie Chang Tel: (86-10)62785076; Fax: (86-10)62773624; E-mail: zhijiecedu.cn and Li Wu Tel: (86-10)62794835; Fax: (86-10)62794835;E-mail: wuliedu.cn
Corresponding authors: Zhijie Chang Tel: (86-10)62785076; Fax: (86-10)62773624; E-mail: zhijiecedu.cn and Li Wu Tel: (86-10)62794835; Fax: (86-10)62794835;E-mail: wuliedu.cn