Impact Factor
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Prolog
Introduction to...
Main Aspect 1: Clinical...
Main Aspect 2: Preclinical...
Main Aspect 3: First Clinical...
Epilog
Abbreviations
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(5):1336-1347. doi:10.7150/thno.31806 This issue Cite
Review
The Search for an Alternative to [68Ga]Ga-DOTA-TATE in Neuroendocrine Tumor Theranostics: Current State of 18F-labeled Somatostatin Analog Development
Christopher M. Waldmann1,2, Andreea D. Stuparu1,2, R. Michael van Dam2,3, Roger Slavik1,2 ![]()
1. Ahmanson Translational Imaging Division, David Geffen School of Medicine, University of California, Los Angeles, CA 90095
2. Department of Molecular & Medical Pharmacology, David Geffen School of Medicine, University of California, Los Angeles, CA 90095
3. Crump Institute for Molecular Imaging, David Geffen School of Medicine, University of California, Los Angeles, CA 90095
Received 2018-11-26; Accepted 2019-1-14; Published 2019-2-14
Abstract

The trend to inform personalized molecular radiotherapy with molecular imaging diagnostics, a concept referred to as theranostics, has transformed the field of nuclear medicine in recent years. The development of theranostic pairs comprising somatostatin receptor (SSTR)-targeting nuclear imaging probes and therapeutic agents for the treatment of patients with neuroendocrine tumors (NETs) has been a driving force behind this development. With the Neuroendocrine Tumor Therapy (NETTER-1) phase 3 trial reporting encouraging results in the treatment of well-differentiated, metastatic midgut NETs, peptide radioligand therapy (RLT) with the 177Lu-labeled somatostatin analog (SSA) [177Lu]Lu-DOTA-TATE is now anticipated to become the standard of care. On the diagnostics side, the field is currently dominated by 68Ga-labeled SSAs for the molecular imaging of NETs with positron emission tomography-computed tomography (PET/CT). PET/CT imaging with SSAs such as [68Ga]Ga-DOTA-TATE, [68Ga]Ga-DOTA-TOC, and [68Ga]Ga-DOTA-NOC allows for NET staging with high accuracy and is used to qualify patients for RLT. Driven by the demand for PET/CT imaging of NETs, a commercial kit for the production of [68Ga]Ga-DOTA-TATE (NETSPOT) was approved by the U.S. Food and Drug Administration (FDA). The synthesis of 68Ga-labeled SSAs from a 68Ge/68Ga-generator is straightforward and allows for a decentralized production, but there are economic and logistic difficulties associated with these approaches that warrant the search for a viable, generator-independent alternative. The clinical introduction of an 18F-labeled SSTR-imaging probe can help mitigate the shortcomings of the generator-based synthesis approach, but despite extensive research efforts, none of the proposed 18F-labeled SSAs has been translated past prospective first-in-humans studies so far. Here, we review the current state of probe-development from a translational viewpoint and make a case for a clinically viable, 18F-labeled alternative to the current standard [68Ga]Ga-DOTA-TATE.
Keywords: neuroendocrine tumors, SSTR2, 18F-Labeling, PET imaging
Prolog
The diagnostic arm of the theranostic approach to managing patients with neuroendocrine tumors (NETs) utilizes β+-emitter-labeled somatostatin receptor (SSTR)-targeting ligands for tumor localization, staging, and therapy selection with positron emission tomography-computed tomography (PET/CT) [1]. With the FDA-approval of the commercial kit NETSPOT, [68Ga]Ga-DOTA-TATE has become the de facto standard for NET-imaging in the US [2]. The synthesis of [68Ga]Ga-DOTA-TATE from a 68Ge/ 68Ga-generator is straightforward, reliable and allows for the decentralized production of the radionuclide and probe without the need of a cyclotron on site. A further advantage is that the identical chemical precursor can be used as the therapeutic companion when labeled with 177Lu or other therapeutic nuclides. The simplicity of the 68Ga-labeling chemistry has likely been an important factor in helping several novel compounds advance through the clinical development cycle.
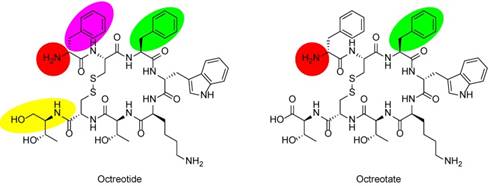
Chemical structures of SSAs octreotide and octreotate. In octreotide, the C-terminal threonine (Thr) is reduced to the amino alcohol (yellow oval). Labeling moieties of all probes covered in this review are attached to the N-termini (red circles). In TOC and TATE, phenylalanine (Phe) of octreotide and octreotate (green ovals) are exchanged for tyrosine (Tyr). In NOC, Phe1 in octreotide (purple oval) is exchanged for naphthylalanine (Nal).
Despite these merits, the generator-based approach holds certain economic and logistic difficulties that can hinder the widespread availability of NET scans. Smaller facilities with low patient throughput might struggle with the profitability of the generator (up to ~$ 100,000 USD per generator per year). Where demand is high, the probe production is complicated by the short half-life of 68Ga (68 min), and the low activity amounts that can be obtained from a single generator elution (typically 1.2 - 1.8 GBq from a fresh generator). Furthermore, driven by the introduction of the prostate cancer imaging probe 68Ga-PSMA-11 and others [3], the popularity of 68Ga-labeled imaging probes has sharply increased, such that the demand for 68Ga exceeds the supply. As a consequence, many facilities are now facing long waiting times to acquire 68Ge/68Ga-generators; a situation that is likely to be aggravated in the coming years. Although a method for the production of 68Ga from a cyclotron has been reported that could eventually address some of these issues, further process-validation and activity yield (AY) optimization are needed to make this process clinically viable [4]. Notni and Wester have recently provided a comprehensive overview on supply and demand issues of various radioiosotopes [5]. The availability of an 18F-labeled, cyclotron-derived alternative to [68Ga]Ga-DOTA-TATE could help mitigate most of the challenges of generator-derived probe production. Large amounts of [18F]fluoride (typically 40 - 700 GBq) can be produced at a comparably low cost, and a well-established distribution network of 18F-labeled probes is already in place [6].
Despite the many years spent developing 18F-labeled SSAs, what prevents these probes from being used as a viable alternative to [68Ga]Ga-DOTA-TATE in the clinic? Following an introductory overview to 18F-labeled SSAs, the current state of probe development is reviewed. We emphasize the three main aspects that are relevant from a translational viewpoint: 1) Clinical suitability of the synthesis protocol, 2) Preclinical imaging performance, and 3) Validity of the probes as assessed in first-in-human studies.
Introduction to 18F-Labeled SSAs
SSTRs belong to the G-protein-coupled receptor family and are frequently upregulated in grade 1-2, well-differentiated NETs. Out of the five subtypes that have been evaluated (SSTR1-SSTR5), SSTR2 is predominantly overexpressed in these tumors, and SSTR2-targeting ligands have been the focus of attention in the last decades of probe development [7]. Most clinically used radioligands are derived from the octapeptide octreotide (Figure 1) which comprises a high affinity for SSTR2 (IC50 = 2.0 nM) and an appropriate elimination half-life (72 to 98 minutes) [8]. The first widely applied SSTR scintigraphy agent used in humans was the single-photon emission computed tomography (SPECT) probe 111In-diethylenetriaminepentaacetic acid-octreotide (OctreoScan) [9, 10]. Later, other octreotide derivatives like Tyr3-octreotide (TOC), Tyr3-octreotate (TATE), and 1-Nal3-octreotide (NOC) that bear a 1,4,7,10-tetraazacyclododecane- 1,4,7,10-tetraacetic acid (DOTA)-chelated 68Ga-metal ion for PET/CT arrived on the scene and improved diagnosis and staging of NETs compared to the 111In-labeled counterpart [11]. Among the technical advantages of PET/CT over SPECT are the higher spatial resolution and image quantification capabilities. This and the proven usefulness of 68Ga-labeled SSAs in NET-theranostics [12] has triggered research aiming at the development of probes labeled with other β+-emitters such as 64Cu [13], 44Sc [14] and 18F. The focus of this review will lie on the latter since 18F is the most widely available PET-nuclide and its half-life (110 min), as well as low positron energy (maximum β+ energy = 635 keV), make it a close-to-ideal choice. More than 150 operating cyclotrons in the US ensure the availability of [18F]fluoride as well as 18F-labeled probes in a large area [6]. Although upfront costs for cyclotrons and the associated laboratory equipment are high (~$ 1,000,000 - 3,000,000 USD), operating costs for the production of [18F]fluoride are comparably low and often financially supported by the distribution of high-demand probes like 2-deoxy-2-[18F]fluoro-D-glucose ([18F]FDG).
Seven different approaches to 18F-labeled SSAs have been described in the literature using a variety of synthetic strategies. Approach 1: Early efforts in the development of 18F-labeled SSAs by Guhlke et al. in 1994 focused on [18F]fluoroacylation of octreotide analogs using the prosthetic group (±)-2-[18F]fluoropropionic acid-4-nitrophenyl ester ([18F]FP) [15, 16]. Later, it was shown by Wester et al. that reducing the lipophilicity with the addition of carbohydrates improved the overall imaging performance [17-19]. Approach 2: A two-step process consisting of the synthesis of [18F]fluorobenzaldehyde ([18F]FBA) followed by labeling of the aminooxy-functionalized peptide was developed by Schottelius et al. [20, 21]. As in the first approach, the addition of sugar moieties was used to increase hydrophilicity and improve pharmacokinetic properties. Approach 3: A click-chemistry approach based on copper(I) catalyzed azide-alkyne cycloaddition (CuAAC) was used in the workgroup of Aboagye to prepare several [18F]fluoroethyltriazole-TATE analogs of which one candidate was investigated in a first-in-human study [22-24]. Approach 4: Similarly, the click-chemistry synthesis of 18F-fluoroglycosylated TATE was reported by Maschauer et al. [25]. Approach 5: In an attempt to shift away from the more classic prosthetic group-labeling approach, Laverman et al. developed a straightforward synthesis based on chelation of aluminum mono-[18F]fluoride cation ([18F]AlF2+) in 1,4,7-triazacyclononae-1,4,7-triacetic acid (NOTA)-octreotide [26]. Approach 6: Using yet another one-step synthesis strategy, Wängler et al. achieved labeling of peptide silicon-fluoride-acceptors (SiFA) by 19F-18F isotopic exchange reactions (IEX) [27]. The high lipophilicity of the SiFA-group was later countered by the introduction of charged linkers, amino acids and sugar moieties [28, 29]. Pending the positive results of an ongoing toxicity assessment, the most promising candidate is lined up for evaluation in a first-in-human study [30]. Approach 7: Perrin's group championed one-step 19F-18F IEX on trifluoroborates such as trialkylammoniomethyl-BF3 (AMBF3)-TATE [31, 32]. A recent addition to the field of SSTR imaging is the introduction of SSTR antagonists that are reported to have superior imaging and therapy properties compared to agonists despite the lack of probe internalization [33]. The only fluorinated example to date is 18F-AMBF3-LM3 [32]. Table 1 depicts the lead candidates of all seven approaches to 18F-labeled SSAs that represent the basis for this comparative review.
Main Aspect 1: Clinical Suitability of 18F-labeled SSA Syntheses
A well-established and reliable synthesis protocol is a fundamental prerequisite for the development of a PET-probe and its clinical acceptance. In this section, the reported synthesis procedures for the 18F-fluorinated SSAs listed in Table 1 are compared and evaluated from a translational viewpoint. The following factors are taken into consideration: The synthetic strategy should ideally allow for a fully automated probe production on a commercial synthesizer yielding multiple patient doses from a single batch of [18F]fluoride. Other clinical demands that should be met are an easily accessible (e.g. commercially available) precursor, minimized workload for the radiochemist, and reasonable overall costs for each batch production. From a regulatory viewpoint, the production of the probe must comply with current good manufacturing practice (cGMP)-regulations as set by the respective regulatory authorities, e.g., the U.S. Food and Drug Administration (FDA). Important denominators of a cGMP-conforming synthesis outcome are sufficient radiochemical yield (RCY), high radiochemical purity (RCP), high chemical purity (CP), molar activity (Am) appropriate for SSTR2 receptor imaging, and chemical stability of the formulated probe.
Approach 1
The key step in the synthesis of [18F]FP-Gluc-TOCA is the acylation of an N-(1-(4,4-dimethyl-2,6-dioxocyclohexylidene)ethyl) (Dde)-protected TATE-derivative with the prosthetic group 4-nitrophenyl 2-[18F]fluoropropionate [17, 20]. The precursor (Nα-(1-deoxy-D-fructosyl)-[Lys0-Tyr3-Lys(Dde)5]-octreotate and the non-radioactive reference FP-Gluc-TOCA are commercially available (ABX Advanced Biochemical Compounds). For each production run, a total of five synthetic steps and two HPLC-purification steps are required. The preparation of the prosthetic group proceeds in three steps followed by HPLC-purification [34]. The following steps are acylation of the TATE-derivative, in situ deprotection of the Dde-moiety and a final HPLC-purification. The resulting synthesis time of about 3h is the longest of all 18F-labeled SSA compared here, and the overall workload for the radiochemist is high. Automation was not reported but would require non-standard laboratory equipment that allows for the implementation of five synthetic steps and two HPLC-purifications. RCYs range from 20% to 30% [17], RCP is >98%, and Am is assumed to be high since non-radioactive compound formed is reported to be below the UV-detection limit [20]. Starting activities, AY and CP of the formulation are not disclosed. Since the probe was used in first-in-human studies [18], it is assumed that the probe-formulation meets clinical requirements regarding purity, sterility, and stability.
18F-labeled SSAs discussed in this review.
| App. | Cpd. | Chemical Structure | IC50 [nM] | Ref. |
|---|---|---|---|---|
| 1 | [18F]FP-Gluc-TOCA | 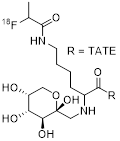 | SSTR1 > 10,000 SSTR2 = 2.8±0.4 SSTR3 > 1,000 SSTR4 = 437±84 SSTR5 = 123±8.8 | [17, 18] |
| 2 | Cel-S-Dpr([18F]FBOA)TOCA | 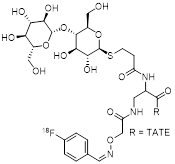 | SSTR1 > 1,000 SSTR2 = 1.8±1.1 SSTR3 = 707±30 SSTR4 = 339±163 SSTR5 = 45±1 | [20, 21] |
| 3 | [18F]FET-βAG-TOCA | 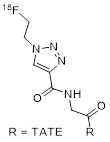 | SSTR2 = 6.9 SSTR4 = 5.4 | [22-24] |
| 4 | [18F]FGlc-TATE | 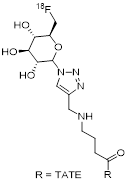 | 4.2 (AR42J cells) | [25] |
| 5 | 18F-IMP466 | 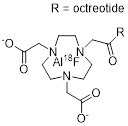 | 3.6±0.6 (AR42J cells) | [26] |
| 6 | [18F]SiFAlin-Glc-Asp2-PEG1-TATE | 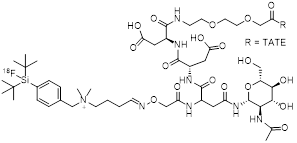 | 14.4±1.2 (AR42J cells) | [28, 29] |
| 7 | [18F]AMBF3-TATE | 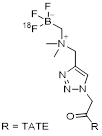 | (Ki values:) SSTR1 > 1,000 SSTR2 = 0.13±0.03 SSTR3 = 28.4±8.6 SSTR5 = 11.6±2.8 | [31, 32] |
Approach 2
The synthesis of Cel-S-Dpr([18F]FBOA)TOCA consists of the production of 4-[18F]fluorobenzaldehyde ([18F]FBA) and subsequent oxime formation with an aminooxy-functionalized TATE-derivative that is not commercially available [20, 21]. The one-step production of [18F]FBA is well established on various commercial synthesizers and only requires cartridge-purification. The oxime-formation proceeds in a single step under pH-controlled conditions and produces the final probe in a RCY-range of 40% to 60% within 50 minutes after HPLC-purification. Only 110 nmol of the peptide precursor are required thereby reducing overall costs. Although automation was not reported, its implementation should be straightforward with minor changes to the protocol to avoid the addition of small reagent volumes, which is challenging on most commercial radiosynthesizers. As for [18F]FP-Gluc-TOCA, RCP is >98%, and Am is estimated to be high. AY is not disclosed, but automated multidose production of Cel-S-Dpr([18F]FBOA)TOCA from high doses of [18F]FBA is conceivable [35].
Approach 3
[18F]FET-βAG-TOCA is synthesized in a two-step process that comprises the formation of 1-azido-[18F]fluoroethane ([18F]FEA) followed by copper(I)-catalyzed azide-alkyne cycloaddition (CuAAC) with the peptide precursor (4 mg per run; obtained under contract from ABX Advanced Biochemical Compounds) [24]. [18F]FEA is purified by distillation, which is not a standard operation on most commercial synthesizers and might require customization as well as the implementation of safety precautions for processing of volatile radioactive compounds. The final product is purified by HPLC. A disposable cassette that allows for full automation of the process on a FASTlab synthesizer (GE Healthcare), including the distillation step, was developed (U.S. Patent 20140097078 A1). [18F]FET-βAG-TOCA is obtained in non-decay-corrected RCY of 6.2% ± 2.9% within 100 minutes. RCP is reported to be 100%, and Am is observed in a range of 224 to 562 GBq/µmol. AY or staring activities are not disclosed, but the mean administered activity was 156 ± 8 MBq. Quality control is performed according to European Pharmacopoeia guidelines.
Approach 4
The production of [18F]FGlc-TATE is conducted following a three-step, two-pot protocol [25, 36]. The prosthetic group 6-deoxy-6-[18F]fluoroglucosyl azide is synthesized in two steps and requires HPLC-purification of the intermediate product. The final product is formed via CuAAC between the prosthetic group and the peptide precursor followed by a second HPLC-purification. The peptide precursor is not commercially available but exhibits structural similarities to the alkyne-TATE derivative from Approach 3. The manual protocol includes small-volume additions of crucial reagents (~10 µL) that are difficult to implement on certain synthesizers and therefore requires modifications. As in the case of Approach 1, the automation of two HPLC-purifications per single production-run is non-standard and warrants special laboratory equipment. [18F]FGlc-TATE is obtained in non-decay-corrected RCY of 19%-22% within 70 minutes and Am in the range of 32-106 GBq/µmol. AY of 95-110 MBq from 500 MBq of [18F]fluoride are reported, but neither RCP nor CP is disclosed.
Approach 5
18F-IMP466 is synthesized by the chelation of [18F]AlF2+ with NOTA-conjugated octreotide and represents the only 18F-labeled SSA prepared by this method [26]. [18F]AlF2+ is produced by reacting [18F]KF with AlCl3 under controlled pH-conditions. The reaction of [18F]AlF2+ with the custom-made precursor leads to the formation of the final product which is purified by HPLC. The synthesis was performed manually starting with 2-6 GBq of [18F]fluoride. Automated synthesis of a different [18F]AlF-labeled peptide in AY of up to 30 GBq was reported elsewhere [37]. 18F-IMP466 is obtained in 50% RCY within 45 minutes and Am of 45 GBq/µmol. After purification, two inseparable radioactive peptide peaks are observed, indicating the formation of two stereoisomers. Based on analytical HPLC, free [18F]AlF2+ is found to be absent in the final formulation, and CP is estimated to be high.
Approach 6
[18F]SiFAlin-Glc-Asp2-PEG1-TATE is prepared by applying a manual 19F-by-18F-IEX protocol [28]. Only 25 nmol of the custom-made peptide precursor are used per run. For the synthesis, [18F]fluoride is activated by aqueous-free elution from a strong anion-exchange cartridge using a complex consisting of cryptand 222 and KOH [38]. For the IEX to proceed in satisfactory yields, the strongly basic mixture needs to be partially neutralized by the addition of a precise amount of oxalic acid. A cartridge-based workup yields the final product in RCY of 49.8% ± 5.9% (n = 20) within 20-25 minutes. RCP is ≥98%, and Am is observed in a range of 44-63 GBq/µmol when starting from 3.3-6.7 GBq of [18F]fluoride. Notably, in IEX the precursor is inseparable from the labeled product, and higher AY is therefore linked to increased Am. Since the authors were aiming at a kit-like production protocol, automation of the procedure that would allow for the production of higher AY is not described. The crucial neutralization step requires precise additions of small oxalic-acid volumes, which complicates automation of the procedure on certain synthesizers. By using commonly-used azeotropic-drying protocols, the neutralization step is made obsolete thereby facilitating automation [27].
Approach 7
[18F]AMBF3-TATE is synthesized by an IEX protocol from a custom-made peptide precursor [31]. The operations of the manual procedure are performed remotely from outside the hot cell, which allows for using starting activities of up to 37 GBq. The AMBF3-TATE precursor (50 nmol) in pyridazine-HCl buffer (pH=2) is reacted with aqueous [18F]fluoride at elevated temperature followed by a cartridge-based workup yielding the final product in injectable form. The procedure yields more than 7.4 GBq of [18F]AMBF3-TATE from 29.6-37 GBq of [18F]fluoride within 25 minutes (decay-corrected RCY > 30%). RCP is >99%, and Am is reported to be >111 GBq/µmol. Full automation of the protocol on common synthesizers will require adjustment of reagent volumes but is feasible, since uncommon operations are not involved.
Which approach is most suitable for clinical production?
Seven different synthetic approaches to 18F-labeled SSA were investigated for their clinical suitability. Approaches 1 to 4 rely on established prosthetic group strategies that require multiple steps, whereas Approaches 5 to 7 are one-step procedures that potentially expedite the production of the injectable probe. Within the prosthetic-group category, the synthesis of Cel-S-Dpr([18F]FBOA)TOCA (Approach 2) stands out owing to its straightforward two-step-strategy that does not require specialized laboratory equipment and appears to be implementable on a wide array of commercially available synthesizers. Overall synthesis time is short, the involved steps are well established, and the production of the injectable probe in high AY is conceivable. In the one-step reaction category, the production of [18F]AMBF3-TATE (Approach 7) is achieved by applying the most straightforward synthesis protocol in the entire field. High AY of the injectable probe can be produced in only 25 minutes, which minimizes the workload for the radiochemist. Associated costs are low since no expensive reagents are required, and only low amounts of the precursor are used. Another prerequisite for the usefulness of the 18F-labeled SSA analogs is their stability in the final product formulation over an extended period of time and at high activity concentrations. At present, there is no stability data available for the approaches summarized in this review; however, it is likely that all probes will exhibit at least sufficient stability for imaging studies. The addition of radical scavengers and pH adjustments can potentially secure the long-term stability to allow the safe distribution of probes to even far distant sites [39]. Overall, the synthesis of [18F]AMBF3-TATE appears to be most suitable for the clinical production of 18F-labeled SSA.
Main Aspect 2: Preclinical Imaging Performance of 18F-labeled SSAs
Along with the clinical suitability of the probe synthesis, the demonstration of satisfactory in vivo imaging properties is a vital denominator in the search for an 18F-labeled SSA for clinical translation. The seven approaches to 18F-labeled SSAs are reviewed with focus on the following desired in vivo properties: Rapid and sufficient uptake of the probe into tumor lesions; Fast clearance from non-target organs to allow for imaging shortly after injection; High target-to-background ratio for efficient detection of metastatic lesions; Minimal physiological uptake in target organs to reduce interference with the lesion identification in those organs; And sufficient probe stability against undesired metabolism, decomposetion, and defluorination (i.e. loss of labeling tag).
Approach 1
Compared to its non-carbohydrated predecessor (2-[18F]fluoropropionyl-D-phe1)-octreotide) which showed low tumor uptake and retention and unfavorable biokinetics in rodents [15, 16], [18F]FP-Gluc-TOCA demonstrates more favorable biodistribution in both AR42J tumor-bearing mice as well as CA20948 tumor-bearing rats [17]. The probe accumulates in the tumor with a percent injected dose per gram tissue (%ID/g) of 13.5 ± 1.47 peaking at around 1 h post injection (p.i.). The predominant renal clearance results in a high uptake in the kidney (8.69 ± 1.09 %ID/g) and a low tumor-to-kidney ratio of 1.56. All other organs demonstrate sufficiently high tumor-to-non-tumor organ ratios. With %ID/g of 0.72 ± 0.14, the uptake in the liver, an organ with frequent NET metastases, is lower than that of other probes such as the non-carbohydrated (2-[18F]fluoropropionyl-D-phe1)-octreotide)[17]. (2-[18F]Fluoropropionyl-D-phe1)-octreotide exhibits a liver uptake of 11.1 ± 4.6 %ID/g in non-tumor bearing NMRI mice, demonstrating the stark impact of the hydrophilic carbohydrate-moiety on the biodistribution. The stability of [18F]FP-Gluc-TOCA towards defluorination is adequate as reflected by a low bone uptake of 0.7 ± 1.47 %ID/g and a tumor-to-bone ratio of 20.
Approach 2
In contrast to the promising pharmacokinetic properties of [18F]FP-Gluc-TOCA (Approach 1) stands its time-consuming and low-yielding synthesis. To improve the synthesis aspect while keeping the apparent pharmacokinetic benefits of the carbohydration, Wester et al. developed the probe Cel-S-Dpr([18F]FBOA)TOCA which benefits from a fast and high-yielding 18F-labeling via oxime formation. Compared to other probes in the field, Cel-S-Dpr([18F]FBOA)TOCA exhibits the highest tumor uptake of 24.04 ± 2.54 %ID/g at 1 h p.i in AR42J tumor bearing mice [20]. At this time-point, fast tumor uptake and a favorable renal clearance result in a high tumor-to-kidney ratio of 3.21. Similar to [18F]FP-Gluc-TOCA, liver uptake is low (0.88 ± 0.12 %ID/g). The comparably low uptake in non-target organs leads to promising tumor-to-organ ratios of 27, 200, and 28 for liver, muscle, and bone, respectively. A low femur uptake of 0.64 ± 0.04 %ID/g suggests good in vivo stability of the probe towards defluorination.
Approach 3
[18F]F-FET-βAG-TOCA, the most clinically advanced 18F-labeled SSA probe in the field, was selected from a series of similar compounds due to its favorable pharmacokinetic properties [22]. In pre-clinical investigations, the derivative [18F]F-FET-G-TOCA showed the highest absolute tumor uptake (17.05 ± 2.77 %ID/g), but the derivative comprising the βAG linker showed a lower background uptake while maintaining a sufficient tumor uptake of 11.58 ± 0.67 %ID/g. It was therefore chosen as the lead. Further characteristics of the probe are a kidney uptake of about 25 %ID/g accompanied by fast renal clearance. [18F]F-FET-βAG-TOCA appears to be stable in plasma as demonstrated by the lack of other metabolites in the radio-HPLC trace of mouse plasma collected 30 min p.i.. Uptake in the liver is low and in vivo stability against defluorination was confirmed by negligible bone uptake. In a head to head comparison with [68Ga]Ga-DOTA-TATE, tumor uptake of [18F]F-FET-βAG-TOCA was found to be higher at the important 1 h p.i. timepoint [22].
Approach 4
Similarly to [18F]FP-Gluc-TOCA and Cel-S-Dpr([18F]FBOA)TOCA, [18F]FGlc-TATE also features a carbohydrate moiety to aid the pharmacokinetic profile, but in this case the carbohydrate also contains the 18F-label. Tumor uptake of [18F]FGlc-TATE peaks around 30 min at %ID/g of 7.87 ± 1.11 in AR42J tumor bearing mice; earlier than most other probes compared here. In combination with the comparably rapid renal clearance (9.0 %ID/g at 30 min p.i.; 3.4 %ID/g at 60 min p.i.; 0.8 %ID/g at 120 min p.i.), the kinetic profile of this probe potentially allows for an earlier imaging post tracer injection, thereby reducing the duration of stay for patients in the clinic [25]. At 1 h p.i., the time point at which tumor uptake of most other probes peaks, tumor uptake of [18F]FGlc-TATE has decreased to 5.62 ± 1.63%ID/g. Non-target organ uptake values at 1 h p.i. are low (0.91 ± 0.25 %ID/g, 0.46 ± 0.78 %ID/g, and 0.58 ± 0.34 %ID/g for liver, muscle, and femur, respectively). Notably, the comparatively low tumor uptake causes the tumor-to-organ ratios to remain only moderate at 1.6, 6.2, 12.2 and 9.7 for kidney, liver, muscle and bone, respectively.
Approach 5
The chemical structure 18F-IMP466 resembles that of other radiometal-containing probes including [68Ga]Ga-DOTA-TATE. Despite the structural uniqueness of 18F-IMP466 among the 18F-labeled SSAs compared here, its biodistribution properties are not remarkably different to the rest of the field. The probe exhibits a noticeably high tumor uptake of 28.3 ± 5.2 %ID/g 2 h p.i. in an AR42J mouse model [40]. Compared to its 68Ga-labeled counterpart, 68Ga-IMP466 exhibits a lower kidney retention of about 10% ID/g, which is nevertheless higher than that of other 18F-labeled SSAs. The bone uptake is negligible (0.33 ± 0.07 %ID/g 2 h p.i.), demonstrating the chemical stability of the [18F]AlF2+-chelation. In comparison, injection of free [18F]AlF2+ results in a bone uptake of 36.9 ± 5 %ID/g. The tumor uptake of 18F-IMP466 in mouse models was compared to that of [18F]F-FET-βAG-TOCA (Approach 3) and shown to be similar at the 1 h p.i. time-point [22].
Approach 6
[18F]SiFAlin-Glc-Asp2-PEG1-TATE exhibits improved pharmacokinetic properties compared to previous SiFA-TATE derivatives which suffered from high liver uptake caused by the high lipophilicity of the SiFA-moiety [27]. This is achieved by combining the positively charged SiFAlin building block with hydrophilic aspartate residues as well as carbohydration. The probe [18F]SiFAlin-Glc-Asp2-PEG1-TATE was compared head-to-head with [68Ga]Ga-DOTA-TATE, giving valuable insight into how 18F-labeled SSAs compare to their radiometal counterparts. In an AR42J mouse model, [18F]SiFAlin-Glc-Asp2-PEG1-TATE shows a higher tumor uptake (18.51 ± 4.89 %ID/g) vs. that of [68Ga]Ga-DOTA-TATE (14.1 ± 4.84 %ID/g) at 1 h p.i. However, the latter has a lower uptake in non-tumor organs giving it a higher tumor-to-background ratio resulting in higher quality PET images [28]. In vivo bone uptake was measured to be 1.31 ± 0.31 %ID/g at 90 min p.i., which is slightly higher than the bone uptake of other 18F-labeled SSAs.
Approach 7
In an AR42J mouse model, [18F]AMBF3-TATE exhibits fast tumor uptake that plateaus around 30 min p.i. which resembles the kinetic characteristic of [18F]FGlc-TATE (Approach 4). PET images at 60 min p.i. show rapid elimination of the probe through the kidneys and accumulation in the bladder [31]. Hepatobiliary excretion causes tracer accumulation in the gut and the gallbladder. The clearance from non-target organs is fast, leading to high tumor-to-non-tumor organ ratios of 26.2 ± 0.8, 25.1 ± 1.0, 89.0 ± 3.1, and 21.3 ± 3.6 for liver, blood, muscle, and bone, respectively. [18F]AMBF3-TATE shows sufficient metabolic stability over the investigated time course of up to 120 min as determined by plasma stability measurements with radio-HPLC.
Which approach shows the most promising imaging performance in the pre-clinical setting?
All 18F-labeled SSAs reviewed here demonstrate favorable in vivo imaging properties in the AR42J mouse model. Overall, the probes show high tumor uptake, fast, primarily renal, clearance, and low non-target organ uptake. An overview of published organ uptake values is listed in Table 2. Where not originally included, tumor-to-organ ratios were calculated based on available data. At 1 h p.i., Cel-S-Dpr([18F]FBOA)TOCA (Approach 2) displays the highest tumor uptake of 24.04 ± 2.54 %ID/g, whereas 18F-IMP466 (Approach 5) shows the highest uptake of 28.3 ± 5.2 %ID/g at 2 h p.i.. Taking into account the higher tumor-to-kidney ratio of Cel-S-Dpr([18F]FBOA)TOCA, the probe appears to be an attractive candidate for clinical translation. Other than the extent of tumor uptake, a rapid accumulation of PET-probes in the target tissue is a desired feature from a patient management perspective, since it decreases the period of stay for patients in the nuclear medicine facility. The available data shows that [18F]FGlc-TATE (Approach 4) and [18F]AMBF3-TATE (Approach 7) show faster tumor uptakes than the other probes. Tumor accumulation for these probes plateaus around 30 min p.i. as opposed to 1 h p.i. for other probes. Since the tumor uptake of [18F]AMBF3-TATE is higher than that of [18F]FGlc-TATE, Approach 7 appears to be a suitable candidate for clinical translation if early imaging post injection is desired. A better comparison of imaging performances in vivo is hampered by the lack of direct comparisons with the clinical standard [68Ga]Ga-DOTA-TATE for most of the 18F-labeled SSAs. Without this direct comparison to a known reference standard, inter-laboratory variability cannot be readily quantified although it is generally regarded to be significant. The absolute values as presented in Table 2 should, therefore, be taken with caution. Unfortunately, none of the probes were tested in a more clinically relevant metastatic mouse model of NETs.
Main Aspect 3: First Clinical Experiences With 18F-labeled SSAs
Three of the probes reviewed herein, [18F]FP-Gluc-TOCA (Approach 1), [18F]F-FET-βAG-TOCA (Approach 3), and 18F-IMP466 (Approach 5) are currently being evaluated in early clinical trials. A phase I/II trial for 18F-IMP466 (NCT03511768) is at an early stage as of the time of writing and limited data is available. These first in-human studies give valuable insight regarding the clinical validity of 18F-labeled SSAs for imaging of NETs. In the following section, the available data is summarized with a focus on the comparison between the imaging properties of 18F-labeled SSAs and other clinically established PET probes.
Summary of preclinical biodistribution data of the seven approaches compared to the current gold standard [68Ga]Ga-DOTA-TATE.
| App. | Compound | Time Point | Tumor [%ID/g] | Bone [%ID/g] | Tumor/ Kidney | Tumor/ Liver | Tumor/ Muscle | Tumor/ Bone | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| 1 | [18F]FP-Gluc-TOCA | 1h | 13.54 | 0.68 | 1.56 | 19 | 56 | 20 | [17] |
| 2 | Cel-S-Dpr([18F]FBOA)TOCA | 1h | 24.04 | 0.64 | 3.21 | 27 | 200 | 38 | [20] |
| 3 | [18F]FET-βAG-TOCA | 1h | 11.58 | - | - | - | 24.12 | - | [22] |
| 4 | [18F]FGlc-TATE | 1h | 5.62 | 0.58 | 1.64 | 6.2 | 12 | 9.7 | [25] |
| 5 | 18F-IMP466 | 2h | 28.3 | 0.33 | 2.83 | - | - | 86 | [26] |
| 1h | 12.73 | - | - | - | 37.44 | - | [22] | ||
| 6 | [18F]SiFAlin-Glc-Asp2-PEG1-TATE | 1h | 18.51 | 1.31 (1.5h) | 1.21 (65m) | 8.97 (65m) | 39.35 (65m) | - | [28, 29] |
| 7 | [18F]AMBF3-TATE | 1h | 10.11 | 0.46 | 2.06 | 26 | 92 | 22 | [31] |
| - | [68Ga]Ga DOTATATE | 1h | 24.1 | - | 6.2 | 48 | 80 | - | [47] |
| 1h | 1.37 | - | - | - | 2.28 | - | [22] | ||
| 1h | 14.1 | - | - | - | - | - | [28] |
Approach 1
In a comparative study in humans, [18F]FP-Gluc-TOCA was evaluated against OctreoScan [18]. [18F]FP-Gluc-TOCA shows high uptake in metastatic lesions in the liver and fast blood clearance, predominantly by renal clearance but also by hepatobiliary transport as shown by the late time point uptake in the gallbladder. The tumor-to-non-tumor contrast is high which allows for clear delineation of the liver lesions as opposed to OctreoScan. Superiority of [18F]FP-Gluc-TOCA over OctreoScan is confirmed by the detection of more than double the number of lesions. Predominant factors for this superiority are the technical advantages of PET over SPECT such as increased spatial resolution, noise reduction, and attenuation correction.
Approach 3
In a first-in-human study, [18F]F-FET-βAG-TOCA shows high uptake in tumor lesions and a favorable tumor-to-background contrast [24]. The tumor-to-background ratio in the liver is similar to that of previously reported values for 68Ga-labeled probes. The probe shows fast uptake into the tumor (<1 h) and clears rapidly from non-target organs, except for the gallbladder due to increased hepatobiliary excretion over time. No bone uptake is observed, confirming stability against defluorination in vivo. In a first report from a phase I clinical trial (EudraCT number 2013-003152-20) comparing [18F]F-FET-βAG-TOCA with [68Ga]Ga-DOTA-TATE in 32 patients diagnosed with NETs as per ENETS criteria, [18F]F-FET-βAG-TOCA was shown to have higher sensitivity (92.8% vs 87.5%) and detect more lesions (209 vs 197 lesions) compared to [68Ga]Ga-DOTA-TATE. [18F]F-FET-βAG-TOCA detected additional bone and lymph node lesions in three of the patients. In addition, [18F]F-FET-βAG-TOCA was safe and well-tolerated in the patients [41].
Approach 5
The safety profile of 18F-IMP466 was studied in three healthy volunteers and was well tolerated. 18F-IMP466 shows physiological uptake in pituitary, thyroid, liver, spleen, adrenals, pancreatic uncinate process, stomach, as well as small and large bowel. It is primarily excreted through the kidney but also partially through the gallbladder [42]. In a study comparing [18F]FDG and 18F-IMP466 in twelve neuroendocrine neoplasms (NENs) patients (three grade 1, four grade 2, and five grade 3), it showed higher maximum standardized uptake value (SUVmax) in grade 1 and 2 patients, but a lower SUVmax in grade 3 patients. Among the twelve first patients, 18F-IMP466 detected lesions in eleven patients (92%) whereas [18F]FDG only detected lesions in eight of the patients (66%). Although the patient number in this study is low, a similar trend was found in other comparison studies between [68Ga]Ga-DOTA-TATE and [18F]FDG [43, 44]. 18F-IMP466 had a higher uptake in grade 1 and 2 patients as opposed to [18F]FDG which had higher uptake in grade 3 patients. This is similar to [68Ga]Ga-DOTA-TATE which also shows higher uptake than [18F]FDG in lower-grade tumors and lower uptake in higher-grade tumors. Unfortunately, no comparative SST2 evaluation of 18F-IMP466 to OctreoScan or [68Ga]Ga-DOTA-TATE was included in this study.
Are 18F-labeled probes a viable alternative to [68Ga]Ga-DOTA-TATE in clinical studies?
First-in-human studies are a critical first step towards initiating phase I/II clinical trials and ultimately getting the probes approved for routine clinical service. For clinical approval, 18F-labeled SSAs need to be safe and tolerable, have favorable biodistribution characteristics, and demonstrate high lesion detection rates. To compete with [68Ga]Ga-DOTA-TATE in routine clinical care, 18F-labeled SSAs should have similar, preferably higher, lesion detection rates.
The 18F-labeled SSAs currently translated into humans are all safe and tolerable. Whereas the tumor uptakes and biodistribution of [18F]FP-Gluc-TOCA, 18F-IMP466, and [18F]F-FET-βAG-TOCA resemble that of [68Ga]Ga-DOTA-TATE, the 18F-labeled probes show an increased uptake in the gallbladder due to a more prevalent biliary excretion. However, we do not expect this to be detrimental to lesion detection. The tumor-to-background ratios in the liver of [18F]FP-Gluc-TOCA and [18F]F-FET-βAG-TOCA are 4.2 ± 2 and 4.3 ± 2.69, respectively, and, therefore, higher than that of [68Ga]Ga-DOTA-TATE (2.0; interquartile range 1.4-2.7) [45]. No data is currently publicly available for 18F-IMP466. [18F]F-FET-βAG-TOCA, the only 18F-labeled SSA compared head-to-head with [68Ga]Ga-DOTA-TATE to date, shows a higher lesion detection rate. Taken together, the available clinical data suggests that 18F-labeled SSAs are a clinically viable alternative to [68Ga]Ga-DOTA-TATE.
Epilog
The ever-increasing understanding of underlying mechanisms in oncology has led to the development of non-invasive diagnostic PET-probes that have become indispensable in clinical patient care. Among those diagnostic probes, [18F]FDG, a probe assessing abnormal glucose metabolism, is the most commonly used. However, [18F]FDG is not well suited for imaging tumors with low glycolytic activity and therefore there is a need for the development of specific, targeted PET-probes in oncology. SST2 is a well-established target for imaging and therapy of NETs. The development and extensive study of the SST receptor targeting peptides NOC, TOC and TATE eventually led to market approval of [68Ga]Ga-DOTA-TATE (Netspot®) and [177Lu]Lu-DOTA-TATE (Lutathera®) for diagnosis and treatment of NETs, respectively.
Due to the increased demand of 68Ga-labeled PET-probes on the one hand, and difficulties in meeting the supply with 68Ge/68Ga-generators on the other, the clinical introduction of 18F-labeled SSAs has become a necessity. Although it has been proposed that theranostic pairs should ideally consist of chemically identical compounds (i.e. compounds that only differ by the chelated radiometal), this paradigm is being questioned. In the case of the theranostic pair [177Lu]Lu-DOTA-TATE and [68Ga]Ga-DOTA-TATE, for example, diagnostic imaging essentially helps localizing the exact sites of subsequent treatment with the therapeutic agent. In this case, dosimetry could arguably help to individualize treatment activity levels. However, in clinical routine, many patients treated with [177Lu]Lu-DOTA-TATE are often diagnosed using structurally different imaging probes such as 111In-labeled OctreoScan, [68Ga]Ga-DOTA-TOC, or [68Ga]Ga-DOTA-NOC and treatment activity levels are mostly defined based on kidney and liver function assays. Another prominent theranostic pair that consists of structurally different compounds are the imaging probe [68Ga]Ga-PSMA-11 and the therapeutic agent [177Lu]Lu-PSMA-617 used for treatment of patients with metastatic, castration-resistant prostate cancer. Prediction of treatment outcome using diagnostic imaging has been shown to be difficult and the evidence suggests that PET-imaging will likely not contribute significantly to prognosis. The most important criteria that can be assessed by imaging for treatment planning is the evaluation of target expression. Arguably, any diagnostic probe with suitable pharmacokinetic properties as well as high affinity and selectivity for the same target as the therapeutic compound can be used for that. We, therefore, expect that the 18F-labeled SSAs compared here will be suitable for this task.
However, among the numerous published options, most 18F-labeled SSAs have not yet been evaluated in humans and no candidate has been evaluated in a prospective phase II/III trial so far.
In an effort to select the candidates that have the potential to become a widely used routine SST2 PET imaging agent, we have summarized the current state of the art. Considering all available data, which compound should be chosen to undergo the clinical development process? The probes compared herein all exhibit high binding affinities to SSTR2 and perform well in preclinical imaging studies. With the highest tumor uptake at 1 h p.i. and a favorable tumor-to-kidney ratio, Cel-S-Dpr([18F]FBOA)TOCA performed exceptionally well in a preclinical setting. If an earlier time-point for imaging is desired, [18F]AMBF3-TATE stands out with the highest tumor uptake at 30 min p.i.
The similar characteristics of the various 18F-labeled probes in preclinical investigations adds importance to the synthesis aspect. A successful 18F-labeled SSA candidate will have to feature a cheap, straightforward, and reliable synthesis procedure that requires no dedicated equipment or specialized process experience. The synthesis of Cel-S-Dpr([18F]FBOA)TOCA meets most of these requirements and should be amenable to implement on commercial synthesis modules without much efforts. Although standard equipment in most radiopharmacies, expensive laboratory equipment such as the radiosynthesizer and HPLC equipment are required. [18F]AMBF3-TATE on the other hand can be produced in high RCY and Am in a single step and does not require HPLC purification. Taking into account its favorable pharmacokinetic properties that potentially allows for imaging at earlier time-points p.i., a widespread acceptance of the probe in clinical patient care is conceivable.
In the reality of a nowadays highly regulated probe development cycle, scientific considerations are not the only driving force in establishing a diagnostic probe as the standard of care. Despite its more challenging production characteristics and slightly inferior imaging properties in preclinical studies, [18F]F-FET-βAG-TOCA currently is the most clinically advanced probe in the field; A fact that must be attributed to the remarkable efforts of the scientists and caregivers involved in its development. The probe has shown promising results in a phase I trial including 32 patients [41]. These efforts are crucial since they underline the feasibility of applying 18F-labeled SSAs in patients. It remains to be seen if an agreement on the future standard of care will be reached based on practical assumptions or a dedicated research consortium making the case for their respective approach. In any way, the development of 18F-labeled SSAs will be of benefit to the patients since it will reduce costs and increase the availability of probes. More patients will benefit from the theranostic approach towards the state of the art treatment of NETs.
Abbreviations
Am: Molar activity; AY: Activity yield; cGMP: Current good manufacturing practice; C: Chemical purity; CT: Computed tomography; IEX: Isotopic exchange reactions; NET: Neuroendocrine tumor; PET: Positron emission tomography; RCP: Radiochemical yield; RLT: Radioligand therapy; SPECT: Single-photon emission computed tomography; SSA: Somatostatin analog; SSTR: Somatostatin receptor.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Smit Duijzentkunst DA, Kwekkeboom DJ, Bodei L. Somatostatin Receptor 2-Targeting Compounds. J Nucl Med. 2017;58:54S-60S
2. Hope TA, Bergsland EK, Bozkurt MF, Graham M, Heaney AP, Herrmann K. et al. Appropriate Use Criteria for Somatostatin Receptor PET Imaging in Neuroendocrine Tumors. J Nucl Med. 2018;59:66-74
3. Kopka K, Benesova M, Barinka C, Haberkorn U, Babich J. Glu-Ureido-Based Inhibitors of Prostate-Specific Membrane Antigen: Lessons Learned During the Development of a Novel Class of Low-Molecular-Weight Theranostic Radiotracers. J Nucl Med. 2017;58:17S-26S
4. Pandey MK, Byrne JF, Jiang H, Packard AB, DeGrado TR. Cyclotron production of (68)Ga via the (68)Zn(p,n)(68)Ga reaction in aqueous solution. Am J Nucl Med Mol Imaging. 2014;4:303-10
5. Notni J, Wester HJ. Re-thinking the role of radiometal isotopes: Towards a future concept for theranostic radiopharmaceuticals. J Labelled Comp Radiopharm. 2018;61:141-53
6. Ikotun O, Clarke B, Sunderland J. A snapshot of United States PET cyclotron and radiopharmaceutical production operations and locations. J Nucl Med. 2012;53:1085
7. Reubi J, Waser B, Schaer J-C, Laissue JA. Somatostatin receptor sst1-sst5 expression in normal and neoplastic human tissues using receptor autoradiography with subtype-selective ligands. Eur J Nucl Med. 2001;28:836-46
8. Katz MD, Erstad BL. Octreotide, a new somatostatin analogue. Clin Pharm. 1989;8:255-73
9. Krenning EP, Kwekkeboom DJ, Bakker WH, Breeman WAP, Kooij PPM, Oei HY. et al. Somatostatin receptor scintigraphy with [111In-DTPA-d-Phe1]- and [123I-Tyr3]-octreotide: the Rotterdam experience with more than 1000 patients. Eur J Nucl Med. 1993;20:716-31
10. Levine R, Krenning EP. Clinical History of the Theranostic Radionuclide Approach to Neuroendocrine Tumors and Other Types of Cancer: Historical Review Based on an Interview of Eric P. Krenning by Rachel Levine. J Nucl Med. 2017;58:3S-9S
11. Deppen SA, Blume J, Bobbey AJ, Shah C, Graham MM, Lee P. et al. 68Ga-DOTATATE Compared with 111In-DTPA-Octreotide and Conventional Imaging for Pulmonary and Gastroenteropancreatic Neuroendocrine Tumors: A Systematic Review and Meta-Analysis. J Nucl Med. 2016;57:872-8
12. Fendler WP, Barrio M, Spick C, Allen-Auerbach M, Ambrosini V, Benz M. et al. 68Ga-DOTATATE PET/CT Interobserver Agreement for Neuroendocrine Tumor Assessment: Results of a Prospective Study on 50 Patients. J Nucl Med. 2017;58:307-11
13. Marciniak A, Brasun J. Somatostatin analogues labeled with copper radioisotopes: current status. J Radioanal Nucl Chem. 2017;313:279-89
14. Domnanich KA, Muller C, Farkas R, Schmid RM, Ponsard B, Schibli R. et al. (44)Sc for labeling of DOTA- and NODAGA-functionalized peptides: preclinical in vitro and in vivo investigations. EJNMMI Radiopharm Chem. 2017;1:8
15. Guhlke S, Wester HJ, Bruns C, Stocklin G. (2-[18F]fluoropropionyl-(D)phe1)- octreotide, a potential radiopharmaceutical for quantitative somatostatin receptor imaging with PET: synthesis, radiolabeling, in vitro validation and biodistribution in mice. Nucl Med Biol. 1994;21:819-25
16. Wester HJ, Brockmann J, Rosch F, Wutz W, Herzog H, Smith-Jones P. et al. PET-pharmacokinetics of 18F-octreotide: a comparison with 67Ga-DFO- and 86Y-DTPA-octreotide. Nucl Med Biol. 1997;24:275-86
17. Wester HJ, Schottelius M, Scheidhauer K, Meisetschlager G, Herz M, Rau FC. et al. PET imaging of somatostatin receptors: design, synthesis and preclinical evaluation of a novel 18F-labelled, carbohydrated analogue of octreotide. Eur J Nucl Med Mol Imaging. 2003;30:117-22
18. Meisetschlager G, Poethko T, Stahl A, Wolf I, Scheidhauer K, Schottelius M. et al. Gluc-Lys([18F]FP)-TOCA PET in patients with SSTR-positive tumors: biodistribution and diagnostic evaluation compared with [111In]DTPA-octreotide. J Nucl Med. 2006;47:566-73
19. Wieder H, Beer AJ, Poethko T, Meisetschlaeger G, Wester H-J, Rummeny E. et al. PET/CT with Gluc-Lys-([18F]FP)-TOCA: correlation between uptake, size and arterial perfusion in somatostatin receptor positive lesions. Eur J Nucl Med Mol Imaging. 2008;35:264-71
20. Schottelius M, Poethko T, Herz M, Reubi JC, Kessler H, Schwaiger M. et al. First (18)F-labeled tracer suitable for routine clinical imaging of sst receptor-expressing tumors using positron emission tomography. Clin Cancer Res. 2004;10:3593-606
21. Poethko T, Schottelius M, Thumshirn G, Hersel U, Herz M, Henriksen G. et al. Two-step methodology for high-yield routine radiohalogenation of peptides: (18)F-labeled RGD and octreotide analogs. J Nucl Med. 2004;45:892-902
22. Leyton J, Iddon L, Perumal M, Indrevoll B, Glaser M, Robins E. et al. Targeting somatostatin receptors: preclinical evaluation of novel 18F-fluoroethyltriazole-Tyr3-octreotate analogs for PET. J Nucl Med. 2011;52:1441-8
23. Iddon L, Leyton J, Indrevoll B, Glaser M, Robins EG, George AJ. et al. Synthesis and in vitro evaluation of [18F]fluoroethyl triazole labelled [Tyr3]octreotate analogues using click chemistry. Bioorg Med Chem Lett. 2011;21:3122-7
24. Dubash SR, Keat N, Mapelli P, Twyman F, Carroll L, Kozlowski K. et al. Clinical Translation of a Click-Labeled 18F-Octreotate Radioligand for Imaging Neuroendocrine Tumors. J Nucl Med. 2016;57:1207-13
25. Maschauer S, Heilmann M, Wangler C, Schirrmacher R, Prante O. Radiosynthesis and Preclinical Evaluation of 18F-Fluoroglycosylated Octreotate for Somatostatin Receptor Imaging. Bioconjug Chem. 2016;27:2707-14
26. Laverman P, McBride WJ, Sharkey RM, Eek A, Joosten L, Oyen WJ. et al. A novel facile method of labeling octreotide with (18)F-fluorine. J Nucl Med. 2010;51:454-61
27. Wängler C, Waser B, Alke A, Iovkova L, Buchholz H-G, Niedermoser S. et al. One-Step 18F-Labeling of Carbohydrate-Conjugated Octreotate-Derivatives Containing a Silicon-Fluoride-Acceptor (SiFA): In Vitro and in Vivo Evaluation as Tumor Imaging Agents for Positron Emission Tomography (PET). Bioconjug Chem. 2010;21:2289-96
28. Niedermoser S, Chin J, Wangler C, Kostikov A, Bernard-Gauthier V, Vogler N. et al. In Vivo Evaluation of (1)(8)F-SiFAlin-Modified TATE: A Potential Challenge for (6)(8)Ga-DOTATATE, the Clinical Gold Standard for Somatostatin Receptor Imaging with PET. J Nucl Med. 2015;56:1100-5
29. Litau S, Niedermoser S, Vogler N, Roscher M, Schirrmacher R, Fricker G. et al. Next Generation of SiFAlin-Based TATE Derivatives for PET Imaging of SSTR-Positive Tumors: Influence of Molecular Design on In Vitro SSTR Binding and In Vivo Pharmacokinetics. Bioconjug Chem. 2015;26:2350-9
30. Bernard-Gauthier V, Lepage ML, Waengler B, Bailey JJ, Liang SH, Perrin DM. et al. Recent Advances in (18)F Radiochemistry: A Focus on B-(18)F, Si-(18)F, Al-(18)F, and C-(18)F Radiofluorination via Spirocyclic Iodonium Ylides. J Nucl Med. 2018;59:568-72
31. Liu Z, Pourghiasian M, Benard F, Pan J, Lin KS, Perrin DM. Preclinical evaluation of a high-affinity 18F-trifluoroborate octreotate derivative for somatostatin receptor imaging. J Nucl Med. 2014;55:1499-505
32. Liu Z, Pourghiasian M, Radtke MA, Lau J, Pan J, Dias GM. et al. An organotrifluoroborate for broadly applicable one-step 18F-labeling. Angew Chem Int Ed Engl. 2014;53:11876-80
33. Fani M, Nicolas GP, Wild D. Somatostatin Receptor Antagonists for Imaging and Therapy. J Nucl Med. 2017;58:61S-6S
34. Guhlke S, Coenen HH, Stöcklin G. Fluoroacylation agents based on small n.c.a. [18F]fluorocarboxylic acids. Appl Radiat Isot. 1994;45:715-27
35. Waldmann CM, Gomez A, Marchis P, Bailey ST, Momcilovic M, Jones AE. et al. An Automated Multidose Synthesis of the Potentiometric PET Probe 4-[(18)F]Fluorobenzyl-Triphenylphosphonium ([(18)F]FBnTP). Mol Imaging Biol. 2018;20:205-12
36. Maschauer S, Haubner R, Kuwert T, Prante O. 18F-Glyco-RGD Peptides for PET Imaging of Integrin Expression: Efficient Radiosynthesis by Click Chemistry and Modulation of Biodistribution by Glycosylation. Mol Pharm. 2014;11:505-15
37. Kersemans K, De Man K, Courtyn J, Van Royen T, Piron S, Moerman L. et al. Automated radiosynthesis of Al[(18)F]PSMA-11 for large scale routine use. Appl Radiat Isot. 2018;135:19-27
38. Wessmann SH, Henriksen G, Wester HJ. Cryptate mediated nucleophilic 18F-fluorination without azeotropic drying. Nuklearmedizin. 2012;51:1-8
39. Scott PJ, Hockley BG, Kung HF, Manchanda R, Zhang W, Kilbourn MR. Studies into radiolytic decomposition of fluorine-18 labeled radiopharmaceuticals for positron emission tomography. Appl Radiat Isot. 2009;67:88-94
40. Laverman P, McBride WJ, Sharkey RM, Goldenberg DM, Boerman OC. Al(18) F labeling of peptides and proteins. J Labelled Comp Radiopharm. 2014;57:219-23
41. Dubash SR, Barwick T, Mauri FA, Kozlowski K, Frilling A, Valle JW. et al. [18F]FET-βAG-TOCA versus [68Ga]DOTATATE PET/CT in functional imaging of neuroendocrine tumours. ASCO Annual Meeting. Chicago, IL: J Clin Oncol. 2018 p. e24193
42. Long T, Yang N, Li Z, Hu S. A primitive study for clinical application of 18F-AlF-NOTA-octreotide PET/CT in combination with 18F-FDG PET/CT for imaging neuroendocrine neoplasms. SNMMI Annual Meeting. Philadelphia PA: Society of Nuclear Medicine. 2018
43. Kayani I, Conry BG, Groves AM, Win T, Dickson J, Caplin M. et al. A comparison of 68Ga-DOTATATE and 18F-FDG PET/CT in pulmonary neuroendocrine tumors. J Nucl Med. 2009;50:1927-32
44. Kayani I, Bomanji JB, Groves A, Conway G, Gacinovic S, Win T. et al. Functional imaging of neuroendocrine tumors with combined PET/CT using 68Ga-DOTATATE (DOTA-DPhe1,Tyr3-octreotate) and 18F-FDG. Cancer. 2008;112:2447-55
45. Wild D, Bomanji JB, Benkert P, Maecke H, Ell PJ, Reubi JC. et al. Comparison of 68Ga-DOTANOC and 68Ga-DOTATATE PET/CT within patients with gastroenteropancreatic neuroendocrine tumors. J Nucl Med. 2013;54:364-72
46. Hofsli E, Thommesen L, Nørsett K, Falkmer S, Syversen U, Sandvik AK. et al. Expression of chromogranin A and somatostatin receptors in pancreatic AR42J cells. Mol Cell Endocrinol. 2002;194:165-73
47. Schottelius M, Šimeček J, Hoffmann F, Willibald M, Schwaiger M, Wester H-J. Twins in spirit - episode I: comparative preclinical evaluation of [68Ga]DOTATATE and [68Ga]HA-DOTATATE. EJNMMI Res. 2015;5:22
Author contact
![]() Corresponding author: Roger Slavik, Ph.D., Address: 780 Westwood Plaza, 90095 Los Angeles. Tel.: +1 310-749-47638; RSlavikucla.edu
Corresponding author: Roger Slavik, Ph.D., Address: 780 Westwood Plaza, 90095 Los Angeles. Tel.: +1 310-749-47638; RSlavikucla.edu