Impact Factor
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Methods
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(4):1115-1124. doi:10.7150/thno.29622 This issue Cite
Research Paper
Genome-wide profiling of Epstein-Barr virus integration by targeted sequencing in Epstein-Barr virus associated malignancies
Miao Xu1,*, Wei-Long Zhang2,*, Qing Zhu2,*, Shanshan Zhang1, You-yuan Yao1, Tong Xiang1, Qi-Sheng Feng1, Zhe Zhang3, Rou-Jun Peng1, Wei-Hua Jia1, Gui-Ping He1, Lin Feng1, Zhao-Lei Zeng1, Bing Luo4, Rui-Hua Xu1, Mu-Sheng Zeng1, Wei-Li Zhao5, Sai-Juan Chen5, Yi-Xin Zeng1,2, ![]() , Yuchen Jiao2,
, Yuchen Jiao2, ![]()
1. State Key Laboratory of Oncology in South China, Collaborative Innovation Center for Cancer Medicine, Sun Yat-sen University Cancer Center, Guangzhou, China.
2. State Key Lab of Molecular Oncology, National Cancer Center/Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College; Collaborative Innovation Center for Cancer Medicine, Beijing, China.
3. Department of Otolaryngology/Head and Neck Surgery, First Affiliated Hospital of Guangxi Medical University, Nanning, China.
4. Department of Medical Microbiology, Qingdao University Medical College, Qingdao, China.
5. State Key Laboratory of Medical Genomics, Shanghai Institute of Hematology, Rui Jin Hospital, Shanghai Jiao Tong University (SJTU) School of Medicine and Collaborative Innovation Center of Systems Biomedicine, Shanghai, China.
* These authors contributed equally to the work.
Received 2018-8-31; Accepted 2019-1-18; Published 2019-1-30
Abstract

Rationale: Epstein-Barr virus (EBV) is associated with multiple malignancies with expression of viral oncogenic proteins and chronic inflammation as major mechanisms contributing to tumor development. A less well-studied mechanism is the integration of EBV into the human genome possibly at sites which may disrupt gene expression or genome stability.
Methods: We sequenced tumor DNA to profile the EBV sequences by hybridization-based enrichment. Bioinformatic analysis was used to detect the breakpoints of EBV integrations in the genome of cancer cells.
Results: We identified 197 breakpoints in nasopharyngeal carcinomas and other EBV-associated malignancies. EBV integrations were enriched at vulnerable regions of the human genome and were close to tumor suppressor and inflammation-related genes. We found that EBV integrations into the introns could decrease the expression of the inflammation-related genes, TNFAIP3, PARK2, and CDK15, in NPC tumors. In the EBV genome, the breakpoints were frequently at oriP or terminal repeats. These breakpoints were surrounded by microhomology sequences, consistent with a mechanism for integration involving viral genome replication and microhomology-mediated recombination.
Conclusion: Our finding provides insight into the potential of EBV integration as an additional mechanism mediating tumorigenesis in EBV associated malignancies.
Keywords: Epstein-Barr virus (EBV), nasopharyngeal carcinoma (NPC), DNA integration
Introduction
Epstein-Barr virus (EBV) is one of the first described human cancer viruses. EBV is associated with ~ 1% of cancers worldwide, including Burkitt lymphoma, nasopharyngeal carcinoma (NPC), Hodgkin lymphomas, NK/T cell lymphomas, and a subset of gastric carcinomas [1, 2]. The EBV genome typically exists as an episome in infected cells. The most well-described EBV carcinogenic mechanisms are mediated through EBV viral protein effects or EBV infection. Expression of EBV proteins, EBNA-1, EBNA-2, EBNA-3A/3B/3C, LMP-1 and LMP-2, causes B cell and epithelial cell proliferation, increases viability of Burkitt lymphoma and NPC cells, and induces DNA damage and genomic instability [3-6], while EBV infection promotes chronic inflammation and reduces anti-tumor immune surveillance in the epithelium [4].
The first reports of the integration of the EBV genome into host genomes date back to the 1980s [7-10]. Subsequent studies confirmed the frequent integration of full-length EBV genomes as well as DNA fragments in EBV-positive lymphoma and epithelial carcinomas including NPC and gastric carcinoma [11-21]. These findings suggest that integrated and episomal EBV DNA coexist in tumor cells in vivo and in vitro. The significance of viral integrations as they may relate to host genome abnormalities and ultimately the development of cancer is not well understood [22]. Thus, it is important to determine whether EBV randomly integrates into human genomes or not [23-27]. The discoveries that integration sites occasionally co-localize with regions containing cancer associated genes JAK2, PD-L1 and PD-L2 in gastric carcinomas and BACH2, REL and BCL-11A in Burkitt lymphoma cells raise the possibility that EBV integration can promote carcinogenesis [20, 28, 29]. However, these studies are limited by a small sample size and the absence of a systematic investigation of the EBV integration landscape on a genome-wide scale. To provide systematic insight into EBV integration in associated malignancies, we performed EBV-targeted ultra-deep sequencing and conducted a comprehensive survey of EBV integration in a variety of human malignancies. This work provides the first unbiased, genome-wide analysis of EBV integrations, and reveals the involvement of novel inflammation-related genes in NPC.
Results
To perform comprehensive profiling of EBV integration, we conducted EBV-targeted ultra-deep sequencing on 177 NPCs, 39 gastric carcinomas, 25 NK/T cell lymphomas, 11 Hodgkin lymphomas, one nasopharyngitis tissue and the EBV-positive NPC cell line C666-1. A total of 197 EBV integration breakpoints were identified from 33 tumors and the C666-1 cell line (Table 1 and Figure S1). The integration rates were higher in the gastric carcinomas (25.6%; 95% confidence interval (CI): 13.0 - 42.1%) than in the NPC tumors (9.6%; 95% CI: 5.7 - 14.9%). We observed slightly more EBV integration positive samples in late-stage NPC tumors (stage III-IV) and large-size gastric cancers (> 5 cm; Table S1). The EBV integration counts in positive tumor samples varied widely among tumor types and individual cases. Twenty-seven of the 34 positive samples harbored 1-2 breakpoints. The remaining positive samples (n = 7) contained more than two with one gastric cancer harboring an especially large number (118) of integration breakpoints. At least 2 EBV integration breakpoints were consistent between matched primary and metastatic NPC tumors from the same patient (Figure S2).
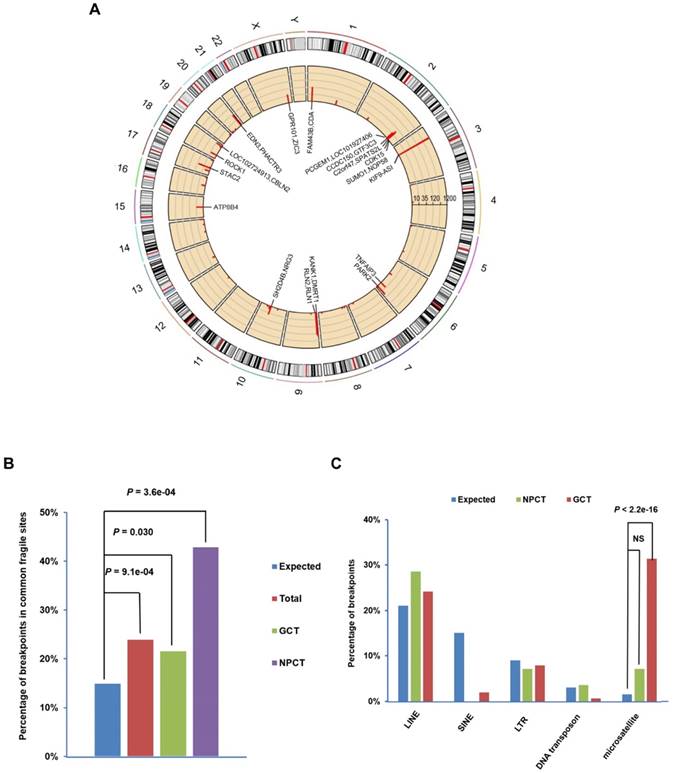
The 197 breakpoints were distributed over all 23 human chromosomes (Figure 1A). EBV showed a strong tendency to integrate near common fragile sites in both NPC and gastric cancer samples (Figure 1B). Similarly, two of the six breakpoints identified in NK/T cell lymphoma samples were located at the same common fragile site (Table S2). EBV also tended to integrate into microsatellite repeats in gastric carcinomas, but avoided SINE repeats in NPC and gastric carcinomas (Figure 1C; For details, see Figure S3 and Table S2). Common fragile regions and microsatellite repeats are vulnerable to DNA damage, which increases the chance for EBV DNA insertion into host genomes through microhomology-mediated DNA repair.
We also tested the relative position of EBV integrations to genes in the human genome. We found that 75 breakpoints (38.1%) were located within known UCSC-annotated genes (Figure S4). The integration sites were slightly skewed toward gene body and promoter regions (26 breakpoints; 13.2%; Figure S4). Unlike HBV and HPV integrations, no associations were found between EBV integration sites and CpG islands, the repetitive elements other than microsatellite repeats, or the binding sites of the genome architecture regulator CCCTC-binding factor (CTCF; Figure S5).
EBV integrations detected in EBV-associated malignancies
| No. of integration positive samples (total samples) | Integration rate (95% confidence interval) | Total No. of breakpoints detected | No. of breakpoints per sample | |
|---|---|---|---|---|
| Gastric carcinoma | 10 (39) | 25.6% (13.0 - 42.1%) | 153 | 0 - 118 |
| Hodgkin lymphoma | 2 (11) | 18.2% (2.3 - 51.8%) | 8 | 0 - 5 |
| NK/T cell lymphoma | 4 (25) | 16.0% (4.5 - 36.1%) | 6 | 0 - 2 |
| NPC | 17 (177) | 9.6% (5.7 - 14.9%) | 28 | 0 - 6 |
| Nasopharyngitis | 0 (1) | |||
| C666-1 cell line | 1 | 2 |
Distribution of EBV integration breakpoints. (A) Distribution of the 197 integration breakpoints across the human genome. For each integration breakpoint, each bar represents the total number of supporting reads at a specific locus in the human genome. Gene annotations for 23 breakpoints in 10 cancers and the C666.1 cell line supported by ≥ 9 EBV-DNA chimeric read pairs are labeled. (B) Distribution of 197 breakpoints in common fragile regions. The expected (assuming uniform and random distribution, blue) and observed ratios of EBV integration breakpoints detected in all samples (n=34, red, total), gastric carcinomas (n=10, green, GCT) and NPC (n=17, purple, NPCT) in common fragile regions are shown. P-values were calculated using the binomial exact test. (C) Significant enrichment of integration breakpoints with microsatellite repeats in gastric carcinomas. The expected and observed ratios of breakpoints co-localized with repeat elements LINE, SINE, LTR, DNA transposon and microsatellite in NPC (green, NPCT) and gastric carcinoma (red, GCT) are shown (for detailed frequencies, see Supplementary Figure. 3). P-values were calculated using the binomial exact test. NS, non-significant; LINE, long interspersed nuclear element; SINE, long interspersed nuclear element; LTR, long terminal repeat.
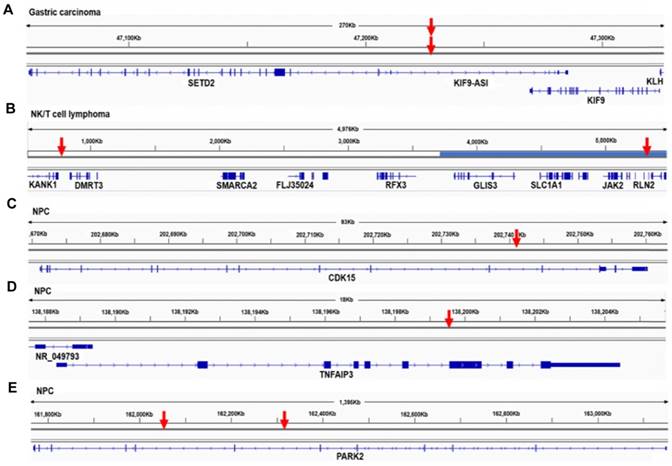
We detected EBV integration in the proximity of tumor suppressor genes: KANK1 in one NK/T cell lymphoma, RB1CC1 in one Hodgkin lymphoma, and DLEC1 in one NPC tumor (Table S2). Integrations in gastric carcinoma samples were associated with tumor suppressor genes SETD2, KISS1, FHIT, PTEN and TET2 (Table S2). The integration breakpoints associated with the histone methyltransferase, SETD2, were located in a region 22 kb upstream of the gene (Figure 2A). In one NK/T cell lymphoma, a breakpoint was found 27 kb downstream of the tumor suppressor gene KANK1. The other EBV integration breakpoint in the same sample was identified 204 kb downstream of JAK2. Amplification was also detected in this region (Figure 2B). Common fragile regions were often co-localized with tumor suppressor genes; for example, the common fragile site FRA3B lies within the tumor suppressor gene FHIT. A number of studies have shown that tumor suppressor genes and common fragile regions are frequent targets of viral DNA integration [30, 31]. The breakpoints associated with tumor suppressor genes in our study showed significantly higher coverage in the targeted sequencing, which indicates that they were more likely to be clonal in the tumorigenesis (P < 0.0001, unpaired, two-sided t-test). Integration could alter the expression or function of tumor suppressor genes and provide host cells with a selective advantage during tumorigenesis.
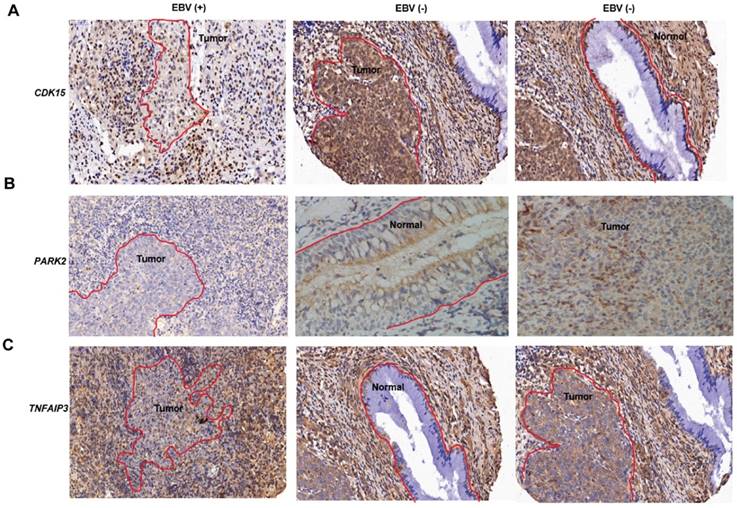
We also identified EBV integrations located within the introns of CDK15 in the primary (Figures S2A-B) and metastatic (Figures 2C, S2C and D) NPC tumors from a single patient, and TNFAIP3 and PARK2 in two additional NPC tumors from two other patients (Figures 2D-E). These breakpoints were all supported by a high number of sequencing reads, suggesting clonal expansion of cancer cells after EBV integration (Table S2). Notably, TNFAIP3, CDK15 and PARK2 are all inflammation-related genes involved in the regulation of TNF-alpha-induced apoptosis/NF-κB pathways [32-34], and dysregulation of these pathways contributes to the development of EBV-associated cancers, including NPC [35]. We performed the immunohistochemistry staining of CDK15, TNFAIP3 and PARK2 proteins using the integrated and non-integrated NPC samples. We found that the protein levels of CDK15, TNFAIP3 and PARK2 were lower in the samples harboring EBV integrations into the introns of the respective genes. (Figures 3A-C). Using qPCR of NF-κB targeted genes and a luciferase reporter gene assay, we found that NF-κB activity was up-regulated in NPC cells with TNFAIP3 knockdown (Figure S6A), confirming its role as an inhibitor of NF-κB pathway. In contrast, NF-κB activity was down-regulated, and nuclear localization of p65 after TNF-α treatment was diminished in NPC cells with CDK15 knockdown (Figure S6B), indicating that CDK15 is positively related to the activation of the NF-κB pathway.
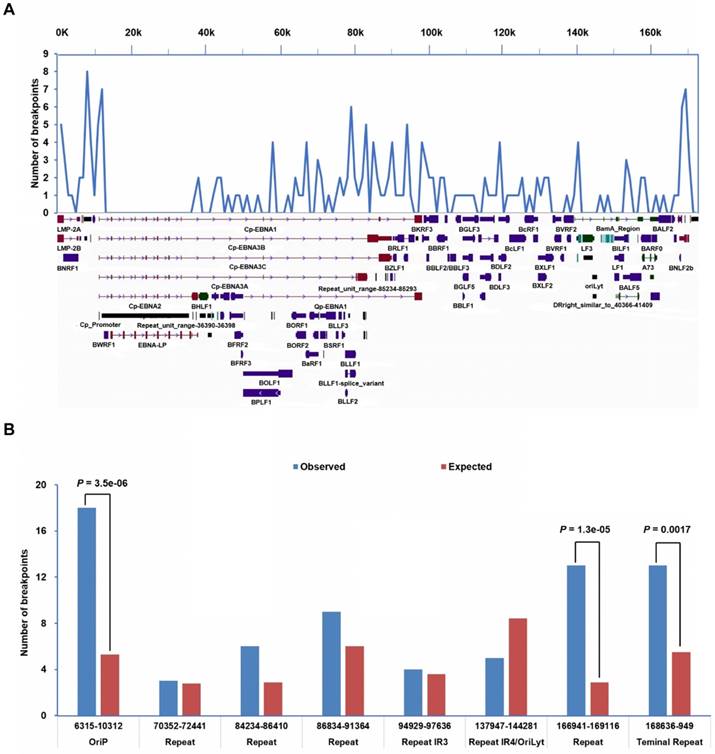
To investigate the mechanisms of EBV integration, we surveyed the distribution of the 197 EBV integration breakpoints in the EBV genome. EBV breakpoints were spread over the entire viral genome with multiple hotspots (Figure 4A). Breakpoints were enriched in the proximity of oriP and terminal repeats, while no breakpoints were detected within the two long internal repeats (Figure 4B). The tendency for EBV breakpoints to localize around oriP and terminal repeats indicated that EBV integration was related to viral genome replication. We further analyzed the microhomology (MH) sequences in the regions flanking integration sites. We found frequent microhomologies between the human genome and the EBV genome near integration breakpoints (Figure S2 and Figure S7). Insertions of 2-10 bp were also observed near the EBV integration breakpoints (Figures S2 and Figure S7). Two EBV integrations containing MH sequences were observed in matched primary and metastatic NPC tumors from a single patient (Figure S2).
Mapping of EBV integration breakpoints in proximity of tumor suppressor and inflammation-related genes. (A-E) The EBV integration breakpoints located in proximity of tumor suppressor genes: SETD2 in one gastric carcinoma (A), KANK1 in one NK/T cell lymphoma (B), and inflammation-related genes CDK15 (C), TNFAIP3 (D) and PARK2 (E) in NPC tumors. Red arrows indicate a transcription factor binding site near the EBV integration breakpoint. Three of the transcription factor binding sites are in the promoter, and one is in the EBV integration breakpoint.
Gene expression in normal epithelium and NPC tumors. (A-C) Immunohistochemical images of CDK15 (A), PARK2 (B), TNFAIP3 (C) expression in normal epithelium and NPC tumors with and without EBV integration. The red curve marks the neoplasm or epithelium, the rest is inflammatoryinfiltration. EBV (+) indicates positive for EBV integration; EBV (-) indicates negative for EBV integration.
Methods
Ethics statement
This study was approved by the institutional ethics committees of the Sun Yat-sen University Cancer Center (Guangzhou, China), the First Affiliated Hospital of Guangxi Medical University (Nanning, China), the Affiliated Hospital of Qingdao University (Qingdao, China), and Rui Jin Hospital (Shanghai, China). Written informed consent was obtained from each study participant.
Sample collection and DNA extraction
Frozen samples were collected for isolation of genomic DNAs from the following tissue types: NPC (n = 177; the Sun Yat-sen University Cancer Center and the First Affiliated Hospital of Guangxi Medical University); gastric carcinoma (n = 39; the Sun Yat-sen University Cancer Center and the Affiliated Hospital of Qingdao University); NK/T cell lymphoma (n = 25; the Sun Yat-sen University Cancer Center and Rui Jin Hospital); Hodgkin lymphoma (n = 11; the Sun Yat-sen University Cancer Center); and nasopharyngitis (n = 1; the Sun Yat-sen University Cancer Center). All histopathological diagnoses were performed according to WHO classifications and were reviewed by two pathologists. The clinicopathological characteristics of the study subjects are listed in Table S1.
Genomic DNAs were extracted from the frozen tissue samples using the DNeasy Blood and Tissue Kit (Qiagen; Germantown, MD, USA).
EBV DNA capture and sequencing
Genomic DNA was subjected to hybrid capture using an EBV-targeting single-stranded DNA probe developed by MyGenostics (Beijing, China). Sequencing libraries were constructed by shearing genomic DNA into 150-200 bp fragments, followed by DNA purification, end blunting and adaptor ligation according to the instructions provided by Illumina (San Diego, CA, USA). The library concentrations were evaluated with a Bioanalyzer 2100 (Agilent Technologies; Santa Clara, CA, USA). EBV DNA was captured from the genomic DNA following the MyGenostics GenCap Target Enrichment Protocol (GenCap Enrichment, MyGenostics). The libraries were hybridized with EBV probes at 65°C for 24 h and then washed to remove uncaptured DNA. The eluted DNA fragments were amplified through 18 PCR cycles to generate libraries for sequencing. The libraries were subsequently quantified and subjected to paired-end sequencing (2 × 100 or 150 bp) on an Illumina HiSeq 2000 sequencer according to the manufacturer's instructions (Illumina).
Distribution of integration breakpoints in the EBV genome. (A) Distribution of breakpoints across the EBV genome. Histogram of the frequency of breakpoints was constructed for 1000 bp intervals. EBV genome annotation is shown. (B) Breakpoints enriched in oriP and terminal repeats in the EBV genome. The observed (blue) and expected (red) frequencies of breakpoints within fragments are shown. P-values were calculated using the binomial exact test.
Integration detection, validation and annotation
Quality assessment of the raw reads was conducted using TrimGalore to remove adaptor sequences and low-quality reads. High-quality reads were aligned to the human (NCBI build 37, hg19) and EBV genomes (NC_007605.1) using the Burrows-Wheeler Aligner (BWA, version 0.7.5a) [36]. Alignments were converted from a sequence alignment map format to sorted and indexed binary alignment map (BAM) files [37]. The Picard tool was used to remove duplicate reads. We developed a bioinformatic method based on LUMPY to identify EBV-human chimeric reads [38]. Briefly, paired-end reads mapping solely to the human or EBV reference genomes were removed. Next, two types of integration-supportive signals were extracted by LUMPY as follows: 1) read pairs, in which one of the paired reads was mapped to the EBV genome and the other to the human genome and 2) chimeric reads, in which one read covered both the EBV genome sequence and the human genome sequence. Integration events supported by ≥ 3 read pairs or chimeric reads were retained. ANNOVAR [39] and the UCSC table browser [40] were used to annotate the breakpoints using fragile regions [41, 42] and the UCSC hg19 CpG island and RepeatMasker database. We randomly selected 12 integrations and performed PCR and Sanger sequencing to validate. Ten (83.3%) breakpoints were validated with Sanger sequencing, while PCR for two breakpoints failed to generate products (Table S3).
Copy number variation and gene expression
CNV data for three NK/T cell lymphomas were retrieved from the study by Jiang et al. [43]. Publicly available gene expression data for normal epithelium (n = 10) and NPC tumors (n = 31) obtained on the Affymetrix Human Genome U133 Plus 2.0 Array were retrieved from the NCBI GEO database (GSE12452). Probeset measures of all 41 arrays were calculated by robust multiarray averaging. The relative RNA expression value was log-transformed using log2. Data were analyzed with the unpaired t-test and presented as the mean ± SEM. A P-value of < 0.05 was considered to be statistically significant.
siRNA transfection
CDK15 and TNFAIP3 siRNAs were designed and synthesized by RIBOBIO (Guangzhou, China). Non-targeting siRNA duplexes are denoted as “scr”. Knockdown efficiency was confirmed with qPCR. Cells (7 × 105) were seeded into 6 well plates, and after incubation overnight, transfected with the indicated siRNA duplexes using Lipofectiamine RNAiMAX Transfection Reagent (Thermo Fisher Scientific; Waltham, MA, USA), according to the manufacturer's instructions. Sequences of the CDK15 siRNAs were the following:
CDK15-siRNA#1: 5'-GAGGAAGGAGTCCCATTTA-3'.
CDK15-siRNA#2: 5'-CCTCAGAACTTACTCATCA-3'.
TNFAIP3-siRNA#1: 5'-GCGGAAAGCTGTGAAGATA-3'.
TNFAIP3-siRNA#2: 5'-CAACTCACTGGAAGAAATA-3'.
Quantitative Real-time PCR
Total RNA was isolated using Trizol reagent (Thermo Fisher Scientific), and cDNA was prepared from RNA (1 µg) using the PrimeScript RT Reagent Kit (TaKaRa; Tokyo, Japan) according to the manufacturer's instructions. Quantitative real-time PCR was performed using the Platinum SYBR Green qPCR SuperMix (Thermo Fisher Scientific) on a Light cycler 480 Real-time system (Roche; Indianapolis, IN, USA).
The primers sequences used were the following:
CDK15-F: 5'-TATGCGACAGTTTACAAGG-3'.
CDK15-R: 5'-TCATGCAGGAGCACAATA-3'.
IKBα-F: 5'-CTCCGAGACTTTCGAGGAAATAC-3'.
IKBα-R: 5'-GCCATTGTAGTTGGTAGCCTTCA-3'.
IL6-F: 5'-ACTCACCTCTTCAGAACGAATT-3'.
IL6-R: 5'-CCATCTTTGGAAGGTTCAGGTTG-3'.
MCP1-F: 5'-CAGCCAGATGCAATCAATGC-3'.
MCP1-R: 5'-TGGAATCCTGAACCCACTTCT-3'.
TNF-F: 5'-CCTCTCTCTAATCAGCCCTCTG-3'.
TNF-R: 5'-GAGGACCTGGGAGTAGATGAG-3'.
GAPDH-F: 5'-TTGCCATCAATGACCCCTTCA-3'.
GAPDH-R: 5'-CGCCCCACTTGATTTTGGA-3'.
Western blot analysis
Western blot analysis was performed as previously described [44]. Cells were lysed in RIPA lysis buffer containing protease inhibitors (Roche). The subcellular fractionation was performed as previously described [45]. The proteins were separated with 10% SDS-PAGE and transferred to PVDF membranes (Thermo Fisher Scientific). Membranes were blocked with 5% BSA for 1 h and incubated with primary antibodies overnight in 4°C. Antibodies against p65 (ab53465; Abcam; Cambridge, MA, USA), ß-actin (#3700; Cell Signaling Technology; Danvers, MA, USA), and GAPDH (60004-1-1 g; Protein-tech; Chicago, IL, USA) were used for western blot analysis.
Luciferase reporter assay
(CAGA)12-Luc and the control vector pRL-TK (Promega) encoding Renilla luciferase were cotransfected into HEK293T or NPC cells using PEI. Luciferase activity was measured 24 h after transfection using the Dual-Luciferase Reporter Assay System (Promega). The firefly luciferase activity values were normalized to those of Renilla, and the ratios of firefly/Renilla activities were determined. The experiments were independently performed in triplicate.
Discussion
EBV integration during tumorigenesis has not yet been systematically investigated using rigorous methods. In this study, we conducted the first large-scale analysis of EBV integration in multiple malignancies using EBV genome-targeted sequencing. Our method, combining EBV genome capture and ultra-deep sequencing, efficiently detected integrated EBV sequences from background “noise” introduced by nuclear EBV episomes. Our results indicate that EBV can integrate into host genomes at a significant rate in multiple tumor types. We observed that EBV integration frequencies varied among tumor types as well as the number of integrated EBV genomes among tumor samples. The heterogeneity of these tumor genomes may underlie the observed variation of EBV integration in these tumors.
Our study revealed that common fragile regions were preferred sites for EBV integration. Common fragile regions are genomic hotspots for DNA damage and are susceptible to genome rearrangement, thereby increasing the chance for EBV DNA insertion through microhomology-mediated DNA repair, which has an important role in the integration of other tumorigenic viruses, HBV and HPV [30, 31]. We observed EBV integrations into or near tumor suppressor genes that were often colocalized with common fragile regions. Integration in the proximity of tumor suppressor genes may provide host cells with a selective advantage. Moreover, integration distribution in gastric carcinomas correlated with microsatellite repeats which are vulnerable to DNA damage, one of the samples which has the 118 integration breakpoints, the whole-exome sequencing showed the mutation including XRCC2, PARP3, SLX4, and PMS2, which are involved in DNA repair, further suggesting that host genome stability has a strong impact on EBV integration. Although genome instability and microhomology-mediated DNA repair are involved in the integration of EBV, HPV and HBV DNA into the host genome, why does EBV integration occur at a relatively lower rate (25.6% in GC, 9.6% in NPC) than HPV integration in cervical cancer (76.3%) and head and neck squamous cell carcinoma (HNSCC; 60.7%), and HBV in hepatocellular carcinoma (HCC; 92.6%)? [31, 46-49]. There are several possible reasons. First, mechanisms underlying tumorigenesis and genetic backgrounds differ greatly between these tumor types associated with the different viruses. The tumor suppressors, TP53 and RB, are inactivated by the expression of HPV-encoded oncogenes E6 and E7 in HPV-associated cervical cancer and HNSCC. Dysfunction of the TP53 pathway has also been frequently observed in HBV-associated HCC (~ 18-51.8%) [50-52]. The impairment of the TP53 pathway leads to increased genomic instability and accumulation of somatic mutations, possibly also contributing to the high rate of HPV and HBV integration. However, the TP53 pathway is not mutated as frequently in NPC (~ 7-10%) and EBV-associated gastric cancer (rarely) as in HCC [53-55]. Moreover, large-scale whole-genome surveys indicate that NPC and EBV-associated gastric carcinomas tend to have relatively stable genomes, compared to many other carcinomas including cervical cancer, HNSCC and HCC [53]. Second, life cycles and genomic features of the viruses themselves may also affect their integration into the host genome. In latent infection state, EBV episomes are replicated along with chromosomes in host cells and therefore are relatively stable. EBV has a much larger genome than HPV or HBV, which may make its integration by microhomology-mediated DNA recombination more difficult.
In NPC, three integration events were localized to introns of the inflammation-related genes PARK2, TNFAIP3 and CDK15, which regulate the TNF-alpha-induced apoptosis/NF-κB pathways. PARK2 deficiency promotes inflammation and genome instability and has an important role in the development of lung cancer [56]. Dysregulation of NF-κB activation contributes to the development of various EBV-associated cancers, including NPC [35]. We found lower expression of PARK2, TNFAIP3 and CDK15 proteins and also dysregulated NF-κB activity in the integrated NPC tumors. Our results suggest that integration of EBV into these genes may disrupt their function and contribute to tumorigenesis through the TNF-alpha-induced apoptosis/NF-κB pathways. The identification of integrations with a high number of supporting reads associated with inflammatory genes indicates that such EBV integration events are potentially selected for during tumor development. If these genes are involved in NPC development, they may be frequent targets of somatic mutation in NPC. TNFAIP3 gene as a mutation hot spot has been already confirmed by previous studies in NPC [53-55]. We also searched our unpublished data and confirmed that about 5% of NPC tumors harbor copy number variation at CDK15 and PARK2 loci.
In the EBV genome, the breakpoints identified in this study were concentrated around oriP and the terminal repeats. During latency, EBNA1 binding to oriP can recruit host cell replication machinery to facilitate the formation of an efficient origin of replication for the EBV episome [57]. The terminal repeats are responsible for the circularization of the EBV genome after it enters the nucleus and cleavage/encapsulation of EBV DNA. Both EBV genome circularization and cleavage involve recombination events. The microhomology sequences around breakpoints indicate that EBV integration involves microhomology-mediated DNA repair pathways. These integrations may be triggered by genomic vulnerability/fragility during genome replication and the physical proximity of the oriP repeats to the host DNA bridged by EBNA1 and DNA recombination, which underlies the EBV integration mechanism.
Till now, we still do not know the size of the EBV sequence integrated into the host genome. Due to the limit of the read length in the second or third generation sequencing, we can only identify a few hundred base pair to around 10 Kb of EBV sequence fused into the host genome. Compared with the large size of the EBV genome, it is difficult to determine whether a portion or the full EBV genome is integrated into human genome. Future development of sequencing technology could help to map the landscape of EBV genome integrated into the human genome.
In summary, our work provides an unbiased large-scale genome-wide analysis of the EBV integration landscape in multiple malignancies. EBV integration occurs preferentially within unstable chromosomal regions of the host genome, surrounding oriP or terminal repeats of the EBV genome. Several integration sites were located in the proximity of tumor suppressor genes that are frequently disrupted during cancer progression. We detected multiple integrations into genes regulating TNF-alpha-induced apoptosis/NF-κB pathways in NPC. These pathways are closely related to EBV-associated diseases and indicate that EBV integration disrupts the function of crucial genes, leading to the development of cancer in some cases of latent EBV infection.
Abbreviations
EBV: Epstein-Barr virus; NPC: nasopharyngeal carcinoma; CI: confidence interval; MH: microhomology.
Supplementary Material
Supplementary figures.
Supplementary tables.
Acknowledgements
We would like to thank all the participants recruited for this study. We thank Dr. E.D. Kieff for helpful discussions and suggestions on this manuscript. This work was supported by the National Natural Science Foundation of China (81872228 and 81430059), the National Key R&D Program of China (No. 2016YF0902000) and the CAMS Innovation Fund for Medical Sciences (2016-I2M-1-001).
Author contributions
Y.-X.Z. and Y.-C.J. were the overall principal investigators who conceived the study and obtained financial support. Y.-X.Z., M.X. and Y.-C. J designed and oversaw the study. M.X., W.-L.Z., S.Z. and Z.Q. performed sample preparation, sequencing and statistical analysis and validation. Y.Y., T.X., S.-J.C., W-L.Z., R.-H.X., Z.Z. and B.L. contributed to sample collection. Q.Z. edited the tables and figures. The manuscript was drafted by M.X. and W.-L.Z. under the supervision of Y.-X.Z. and Y.-C.J. All authors critically reviewed the article and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Parkin DM. The global health burden of infection-associated cancers in the year 2002. Int J Cancer. 2006;118:3030-44
2. Zur Hausen H, de Villiers EM. Reprint of: cancer "causation" by infections-individual contributions and synergistic networks. Semin Oncol. 2015;42:207-22
3. Arvey A, Tempera I, Tsai K, Chen HS, Tikhmyanova N, Klichinsky M. et al. An atlas of the Epstein-Barr virus transcriptome and epigenome reveals host-virus regulatory interactions. Cell Host Microbe. 2012;12:233-45
4. Tsao SW, Tsang CM, To KF, Lo KW. The role of Epstein-Barr virus in epithelial malignancies. J Pathol. 2015;235:323-33
5. Li R, Liao G, Nirujogi RS, Pinto SM, Shaw PG, Huang TC. et al. Phosphoproteomic Profiling Reveals Epstein-Barr Virus Protein Kinase Integration of DNA Damage Response and Mitotic Signaling. PLoS Pathog. 2015;11:e1005346
6. Shumilov A, Tsai MH, Schlosser YT, Kratz AS, Bernhardt K, Fink S. et al. Epstein-Barr virus particles induce centrosome amplification and chromosomal instability. Nat Commun. 2017;8:14257
7. Henderson A, Ripley S, Heller M, Kieff E. Chromosome site for Epstein-Barr virus DNA in a Burkitt tumor cell line and in lymphocytes growth-transformed in vitro. Proc Natl Acad Sci U S A. 1983;80:1987-91
8. Matsuo T, Heller M, Petti L, O'Shiro E, Kieff E. Persistence of the entire Epstein-Barr virus genome integrated into human lymphocyte DNA. Science. 1984;226:1322-5
9. Lawrence JB, Villnave CA, Singer RH. Sensitive, high-resolution chromatin and chromosome mapping in situ: presence and orientation of two closely integrated copies of EBV in a lymphoma line. Cell. 1988;52:51-61
10. Anvret M, Karlsson A, Bjursell G. Evidence for integrated EBV genomes in Raji cellular DNA. Nucleic Acids Res. 1984;12:1149-61
11. Delecluse HJ, Bartnizke S, Hammerschmidt W, Bullerdiek J, Bornkamm GW. Episomal and integrated copies of Epstein-Barr virus coexist in Burkitt lymphoma cell lines. J Virol. 1993;67:1292-9
12. Wolf J, Pawlita M, Klevenz B, Frech B, Freese UK, Muller-Lantzsch N. et al. Down-regulation of integrated Epstein-Barr virus nuclear antigen 1 and 2 genes in a Burkitt lymphoma cell line after somatic cell fusion with autologous EBV-immortalized lymphoblastoid cells. Int J Cancer. 1993;53:621-7
13. Hurley EA, Agger S, McNeil JA, Lawrence JB, Calendar A, Lenoir G. et al. When Epstein-Barr virus persistently infects B-cell lines, it frequently integrates. J Virol. 1991;65:1245-54
14. Kripalani-Joshi S, Law HY. Identification of integrated Epstein-Barr virus in nasopharyngeal carcinoma using pulse field gel electrophoresis. Int J Cancer. 1994;56:187-92
15. Chang Y, Cheng SD, Tsai CH. Chromosomal integration of Epstein-Barr virus genomes in nasopharyngeal carcinoma cells. Head Neck. 2002;24:143-50
16. Zhang HY, Qu G, Deng ZW, Yao TH, Glaser R. Epstein-Barr virus DNA in nasopharyngeal biopsies. Virus Res. 1989;12:53-9
17. Xiao K, Yu Z, Li X, Li X, Tang K, Tu C. et al. Genome-wide Analysis of Epstein-Barr Virus (EBV) Integration and Strain in C666-1 and Raji Cells. J Cancer. 2016;7:214-24
18. Morissette G, Flamand L. Herpesviruses and chromosomal integration. J Virol. 2010;84:12100-9
19. Cao S, Strong MJ, Wang X, Moss WN, Concha M, Lin Z. et al. High-throughput RNA sequencing-based virome analysis of 50 lymphoma cell lines from the Cancer Cell Line Encyclopedia project. J Virol. 2015;89:713-29
20. Gulley ML. Genomic assays for Epstein-Barr virus-positive gastric adenocarcinoma. Exp Mol Med. 2015;47:e134
21. Ohshima K, Suzumiya J, Kanda M, Kato A, Kikuchi M. Integrated and episomal forms of Epstein-Barr virus (EBV) in EBV associated disease. Cancer Lett. 1998;122:43-50
22. Ohshima K, Suzumiya J, Ohga S, Ohgami A, Kikuchi M. Integrated Epstein-Barr virus (EBV) and chromosomal abnormality in chronic active EBV infection. Int J Cancer. 1997;71:943-7
23. Lestou VS, De Braekeleer M, Strehl S, Ott G, Gadner H, Ambros PF. Non-random integration of Epstein-Barr virus in lymphoblastoid cell lines. Genes Chromosomes Cancer. 1993;8:38-48
24. Jox A, Rohen C, Belge G, Bartnitzke S, Pawlita M, Diehl V. et al. Integration of Epstein-Barr virus in Burkitt's lymphoma cells leads to a region of enhanced chromosome instability. Ann Oncol. 1997;8(Suppl 2):131-5
25. Wuu KD, Chen YJ, Wuu SW. Frequency and distribution of chromosomal integration sites of the Epstein-Barr virus genome. J Formos Med Assoc. 1996;95:911-6
26. Oh JH, Kim YJ, Moon S, Nam HY, Jeon JP, Lee JH. et al. Genotype instability during long-term subculture of lymphoblastoid cell lines. J Hum Genet. 2013;58:16-20
27. Gao J, Luo X, Tang K, Li X, Li G. Epstein-Barr virus integrates frequently into chromosome 4q, 2q, 1q and 7q of Burkitt's lymphoma cell line (Raji). J Virol Methods. 2006;136:193-9
28. Takakuwa T, Luo WJ, Ham MF, Sakane-Ishikawa F, Wada N, Aozasa K. Integration of Epstein-Barr virus into chromosome 6q15 of Burkitt lymphoma cell line (Raji) induces loss of BACH2 expression. Am J Pathol. 2004;164:967-74
29. Luo WJ, Takakuwa T, Ham MF, Wada N, Liu A, Fujita S. et al. Epstein-Barr virus is integrated between REL and BCL-11A in American Burkitt lymphoma cell line (NAB-2). Lab Invest. 2004;84:1193-9
30. Zhao LH, Liu X, Yan HX, Li WY, Zeng X, Yang Y. et al. Genomic and oncogenic preference of HBV integration in hepatocellular carcinoma. Nat Commun. 2016;7:12992
31. Hu Z, Zhu D, Wang W, Li W, Jia W, Zeng X. et al. Genome-wide profiling of HPV integration in cervical cancer identifies clustered genomic hot spots and a potential microhomology-mediated integration mechanism. Nat Genet. 2015;47:158-63
32. Muller-Rischart AK, Pilsl A, Beaudette P, Patra M, Hadian K, Funke M. et al. The E3 ligase parkin maintains mitochondrial integrity by increasing linear ubiquitination of NEMO. Mol Cell. 2013;49:908-21
33. Park MH, Kim SY, Kim YJ, Chung YH. ALS2CR7 (CDK15) attenuates TRAIL induced apoptosis by inducing phosphorylation of survivin Thr34. Biochem Biophys Res Commun. 2014;450:129-34
34. Wertz IE, Newton K, Seshasayee D, Kusam S, Lam C, Zhang J. et al. Phosphorylation and linear ubiquitin direct A20 inhibition of inflammation. Nature. 2015;528:370-5
35. Sun SC, Cesarman E. NF-kappaB as a target for oncogenic viruses. Curr Top Microbiol Immunol. 2011;349:197-244
36. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754-60
37. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078-9
38. Layer RM, Chiang C, Quinlan AR, Hall IM. LUMPY: a probabilistic framework for structural variant discovery. Genome Biol. 2014;15:R84
39. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164
40. Karolchik D, Hinrichs AS, Furey TS, Roskin KM, Sugnet CW, Haussler D. et al. The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 2004;32:D493-6
41. Fungtammasan A, Walsh E, Chiaromonte F, Eckert KA, Makova KD. Corrigendum: A genome-wide analysis of common fragile sites: What features determine chromosomal instability in the human genome? Genome Res. 2016;26:1451
42. Fungtammasan A, Walsh E, Chiaromonte F, Eckert KA, Makova KD. A genome-wide analysis of common fragile sites: what features determine chromosomal instability in the human genome? Genome Res. 2012;22:993-1005
43. Jiang L, Gu ZH, Yan ZX, Zhao X, Xie YY, Zhang ZG. et al. Exome sequencing identifies somatic mutations of DDX3X in natural killer/T-cell lymphoma. Nat Genet. 2015;47:1061-6
44. Ma W, Feng L, Zhang S, Zhang H, Zhang X, Qi X. et al. Induction of chemokine (C-C motif) ligand 5 by Epstein-Barr virus infection enhances tumor angiogenesis in nasopharyngeal carcinoma. Cancer Sci. 2018;109:1710-22
45. Holden P, Horton WA. Crude subcellular fractionation of cultured mammalian cell lines. BMC Res Notes. 2009;2:243
46. Koneva LA, Zhang Y, Virani S, Hall PB, McHugh JB, Chepeha DB. et al. HPV Integration in HNSCC Correlates with Survival Outcomes, Immune Response Signatures, and Candidate Drivers. Mol Cancer Res. 2018;16:90-102
47. Sung WK, Zheng H, Li S, Chen R, Liu X, Li Y. et al. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat Genet. 2012;44:765-9
48. Zhao LH, Liu X, Yan HX, Li WY, Zeng X, Yang Y. et al. Genomic and oncogenic preference of HBV integration in hepatocellular carcinoma. Nat Commun. 2016;7:12992
49. Kawai-Kitahata F, Asahina Y, Tanaka S, Kakinuma S, Murakawa M, Nitta S. et al. Comprehensive analyses of mutations and hepatitis B virus integration in hepatocellular carcinoma with clinicopathological features. J Gastroenterol. 2016;51:473-86
50. Guichard C, Amaddeo G, Imbeaud S, Ladeiro Y, Pelletier L, Maad IB. et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet. 2012;44:694-8
51. Fujimoto A, Totoki Y, Abe T, Boroevich KA, Hosoda F, Nguyen HH. et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat Genet. 2012;44:760-4
52. Cleary SP, Jeck WR, Zhao X, Chen K, Selitsky SR, Savich GL. et al. Identification of driver genes in hepatocellular carcinoma by exome sequencing. Hepatology. 2013;58:1693-702
53. Lin DC, Meng X, Hazawa M, Nagata Y, Varela AM, Xu L. et al. The genomic landscape of nasopharyngeal carcinoma. Nat Genet. 2014;46:866-71
54. Zheng H, Dai W, Cheung AK, Ko JM, Kan R, Wong BW. et al. Whole-exome sequencing identifies multiple loss-of-function mutations of NF-kappaB pathway regulators in nasopharyngeal carcinoma. Proc Natl Acad Sci U S A. 2016;113:11283-8
55. Li YY, Chung GT, Lui VW, To KF, Ma BB, Chow C. et al. Exome and genome sequencing of nasopharynx cancer identifies NF-kappaB pathway activating mutations. Nat Commun. 2017;8:14121
56. Lee S, She J, Deng B, Kim J, de Andrade M, Na J. et al. Multiple-level validation identifies PARK2 in the development of lung cancer and chronic obstructive pulmonary disease. Oncotarget. 2016;7:44211-23
57. Hung SC, Kang MS, Kieff E. Maintenance of Epstein-Barr virus (EBV) oriP-based episomes requires EBV-encoded nuclear antigen-1 chromosome-binding domains, which can be replaced by high-mobility group-I or histone H1. Proc Natl Acad Sci U S A. 2001;98:1865-70
Author contact
![]() Corresponding authors: E-mail: zengyxorg.cn (Y.-X.Z.), jiaoyuchenac.cn (Y.-C.J.)
Corresponding authors: E-mail: zengyxorg.cn (Y.-X.Z.), jiaoyuchenac.cn (Y.-C.J.)