Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Results and discussion
Conclusion
Experimental Section
Abbreviations
Supplementary Material
Acknowledgements
References
Introduction
Results and discussion
Conclusion
Experimental Section
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(4):1047-1065. doi:10.7150/thno.29820 This issue Cite
Research Paper
GSH-sensitive Pt(IV) prodrug-loaded phase-transitional nanoparticles with a hybrid lipid-polymer shell for precise theranostics against ovarian cancer
Hui Huang, Yang Dong, Yanhua Zhang, Dan Ru, Zhihua Wu, Jiali Zhang, Ming Shen, Yourong Duan ![]() , Ying Sun
, Ying Sun ![]()
State Key Laboratory of Oncogenes and Related Genes, Shanghai Cancer Institute, Renji Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai 200032, People's Republic of China.
Received 2018-9-10; Accepted 2019-1-2; Published 2019-1-30
Citation:
Huang H, Dong Y, Zhang Y, Ru D, Wu Z, Zhang J, Shen M, Duan Y, Sun Y. GSH-sensitive Pt(IV) prodrug-loaded phase-transitional nanoparticles with a hybrid lipid-polymer shell for precise theranostics against ovarian cancer. Theranostics 2019; 9(4):1047-1065. doi:10.7150/thno.29820. https://www.thno.org/v09p1047.htm
Other stylesAbstract

Background: Platinum (II) (Pt(II))-based anticancer drugs dominate the chemotherapy field of ovarian cancer. However, the patient's quality of life has severely limited owing to dose-limiting toxicities and the advanced disease at the time of diagnosis. Multifunctional tumor-targeted nanosized ultrasound contrast agents (glutathione (GSH)-sensitive platinum (IV) (Pt(IV)) prodrug-loaded phase-transitional nanoparticles, Pt(IV) NP-cRGD) were developed for precise theranostics against ovarian cancer.
Methods: Pt(IV) NP-cRGD were composed of a perfluorohexane (PFH) liquid core, a hybrid lipid-polymer shell with PLGA12k-PEG2k and DSPE-PEG1k-Pt(IV), and an active targeting ligand, the cRGD peptide (PLGA: poly(lactic-co-glycolic acid), PEG: polyethylene glycol, DSPE: 1,2-distearoyl-sn-glycero-3-phosphoethanolamine, cRGD: cyclic Arg-Gly-Asp). Pt(IV), a popular alternative to Pt(II), was covalently attached to DSPE-PEG1k to form the prodrug, which fine-tuned lipophilicity and improved cellular uptake. The potential of Pt(IV) NP-cRGD as contrast agents for ultrasound (US) imaging was assessed in vitro and in vivo. Moreover, studies on the antitumor efficiency and antitumor mechanism of Pt(IV) NP-cRGD assisted by US were carried out.
Results: Pt(IV) NP-cRGD exhibited strong echogenic signals and excellent echo persistence under an US field. In addition, the GSH-sensitive and US-triggered drug delivery system maximized the therapeutic effect while reducing the toxicity of chemotherapy. The mechanistic studies confirmed that Pt(IV) NP-cRGD with US consumed GSH and enhanced reactive oxy gen species (ROS) levels, which further causes mitochondria-mediated apoptosis.
Conclusion: A multifunctional nanoplatform based on phase-transitional Pt(IV) NP-cRGD with US exhibited excellent echogenic signals, brilliant therapeutic efficacy and limited side effect, suggesting precise theranostics against ovarian cancer.
Keywords: platinum (IV), phase transition, ultrasound contrast agents, mitochondrial apoptosis, ovarian cancer.
Introduction
Ovarian cancer is the most prevalent gynecologic cancer with a high mortality rate in recent decades [1, 2]. At present, platinum (II) (Pt(II))-based anticancer drugs dominate the chemotherapy field of ovarian cancer [3]. However, the patient's quality of life has not improved significantly owing to dose-limiting toxicities, partial antitumor response, drug resistance and many other challenges [4, 5]. Moreover, many ovarian cancer patients have developed advanced disease at the time of diagnosis [6]. Therefore, the development of new theranostic strategies to diagnose and overcome chemotherapy toxicities is highly desirable.
Platinum (IV) (Pt(IV)) prodrug, a new class of molecules, has improved the pharmacological properties of Pt(II)-based anticancer drugs in use [7-9]. Compared with the four-coordinate centers of Pt(II), Pt(IV) prodrug is more resistant to ligand substitution reactions and more stable in blood circulation. In addition, the two additional ligands of Pt(IV) provide a promising way to fine-tune desired biological properties, such as lipophilicity, cancer-cell targeting and improved cellular uptake [10]. Based on our previous work [11], we covalently attached 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[ amino (polyethylene glycol)] (DSPE-PEG) to Pt(IV), which enhanced not only the drug loading efficiency but also the plasma half-life. Moreover, because lipid DSPE interacts favorably with and adsorbs onto the cell membrane, it enables the prodrug to be taken up by the cells through passive diffusion, activated by reducing agents such as glutathione (GSH), and to release the active pharmaceutical ingredient in the cytoplasm [12-14]. Therefore, Pt(IV) prodrugs can improve the therapeutic effect while reducing the toxicity of chemotherapy.
Multifunctional ultrasound contrast agents (UCAs), which give a contrast-enhanced imaging effect, enable real-time imaging and contain therapeutic contents, have been applied for the detection, treatment and monitoring of various diseases on account of its safety for the body, cost-effectiveness and high patient acceptability [15-18]. The microscale size of traditional UCAs restricts them from extravasating out of the vasculature and makes them easily captured by the reticuloendothelial systems (RES), resulting in their short circulation in the blood, low accumulation within the tumors and comprised contrast-enhanced imaging effect [19, 20]. Alternatively, nanosized UCAs containing drugs or genes have been steadily developed as a new generation of UCAs to overcome these limitations and to treat tumors, because of the phase-transition behavior of the nanosized UCAs [21-24]. Nanosized UCAs typically consist of a perfluorocarbon core in the form of a liquid and a shell made from materials such as proteins [25], lipids [26-28], polymers [29, 30], and silica [31, 32]. However, there are still some challenges in the development of UCAs. Generally, UCAs made of lipids or phospholipids have a soft and light membrane that produces strong ultrasonic echo signals, but its low stability makes it difficult for continuous US imaging and long-term observation. In addition, UCAs organized by polymers or silica exhibit superior stability because of a rigid and stable shell structure. However, the rigid polymer layers require higher acoustic energy to enhance the US contrast imaging, which is likely to be harmful to human bodies [15]. To solve these challenges, we developed multifunctional tumor-targeted nanosized UCAs (GSH-sensitive Pt(IV) prodrug-loaded phase-transitional nanoparticles, Pt(IV) NP-cRGD) with a hybrid lipid-polymer shell consisting of 1,2-distearoyl-sn-glycero-3-phosphoethanolamine (DSPE) and poly (lactic-co-glycolic acid) (PLGA). The hybrid shell remained the stable structure of the polymer shell, which can enhance the circulation time for continuous US imaging and improve therapy efficiency. In addition, the introduction of a lipid softened the outer shell and made it lighter so that it did not require much acoustic energy to promptly expand, rupture, reseal, compress, buckle, or re-spread during each acoustic cycle to enhance the ultrasonic echo signals. Therefore, nanoparticles with a hybrid lipid-polymer shell easily responded to ultrasound exposure for improved US imaging and drug delivery. Polyethylene glycol (PEG) was modified on the surface of the hybrid shell in order to escape RES and further prolong blood-circulating duration. Moreover, a cyclic Arg-Gly-Asp (cRGD) was conjugated to the outer shells of the nanoparticles to enable actively targeted accumulation within the tumor tissues. It is known that this cyclic peptide has selective affinity for the αvβ3 and αvβ5 integrins, which are overexpressed on the endothelial cells of tumor angiogenic vessels as well as most malignant tumor cells [33-35].
In this study, multifunctional tumor-targeted nanosized UCAs (Pt(IV) NP-cRGD) composed of a perfluorohexane (PFH) liquid core and a hybrid lipid-polymer shell with PLGA12k-mPEG2k, PLGA12k-PEG2k-cRGD and DSPE-PEG1k-Pt(IV) were prepared. The modification of the DSPE-PEG1k with Pt(IV) fine-tuned lipophilicity and improved cellular uptake. In addition, it was confirmed that the active pharmaceutical ingredient release of the DSPE-PEG1k-Pt(IV) prodrug was sensitive to GSH levels and US stimulation. The Pt(IV) NP-cRGD showed great potential for ovarian tumor therapy as well as contrast-enhanced US imaging in vitro and in vivo. Moreover, studies on the relationship among the drug release, intracellular uptake, cytotoxicity, reactive oxygen species (ROS) levels and mitochondria-mediated apoptosis of the nanoparticles assisted by ultrasound were carried out.
Results and discussion
Synthesis and characterization of Pt(IV) NP-cRGD
Pt(IV) NP-cRGD was prepared by “postconjugating” Cys-containing cRGD peptides onto maleimide-functionalized Pt(IV) NPs (Pt(IV) NP-Mal) obtained from a mixture of PLGA12k-mPEG2k, PLGA12k-PEG2k-Mal and DSPE-PEG1k-Pt(IV) (Figure 1A). The results of 1H NMR and ESI-MS confirmed the successful synthesis of Pt(IV) (Figure S1, 3). DSPE-PEG1k-Pt(IV) was successfully synthesized and isolated in 75.9% (410 mg) yield. As shown in Figure 1B and Figure S1-2, 1H NMR (500 MHz, CDCl3, ppm): δ 0.89 (t, CH3, 6H), 1.25 (s, CH2, 56H), 2.60 (m, CH2-succinic, 8H), 3.65 (s, PEG ~ 92 H), 6.23 (bs, NH3, 6H), 6.68 (bs, NHCOCH2), 6.84 (bs, NHCOOCH2). According to the 1H NMR (500 MHz) spectrum of Pt(IV) NP-cRGD (Figure 1C and Figure S4), the successful conjugation of cRGD onto the solvated PEG shells was verified by the resonances of the phenyl protons of cRGD at 7.15-7.25 ppm and the complete disappearance of the maleimide signal at 6.55 ppm. As described in detail later, Pt(IV) N-1%cRGD (Pt(IV) NP-cRGD) had the best potential for in vitro cytotoxicity and was adopted in the following study.
Figure 1
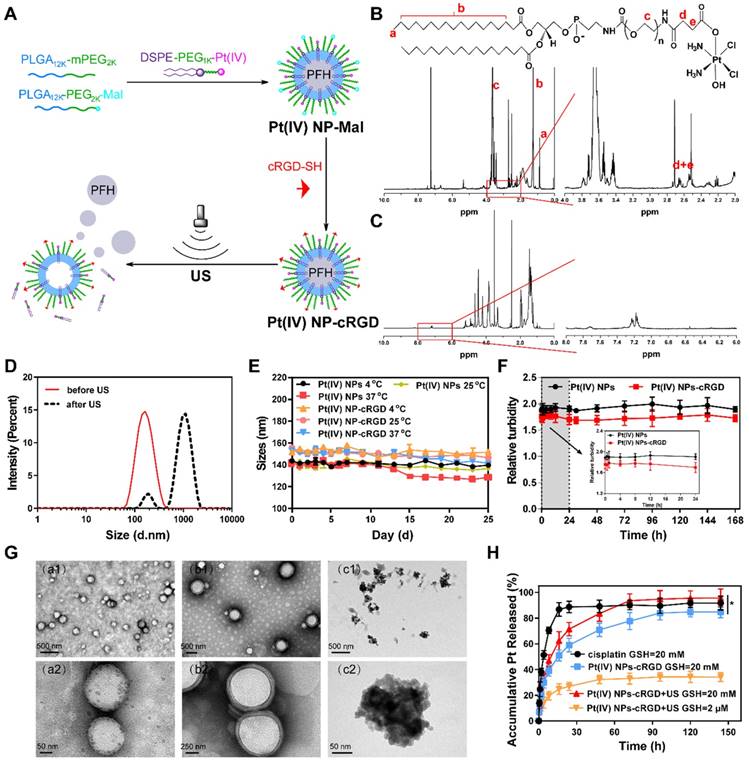
Synthesis and characterization of Pt(IV) NP-cRGD. (A) Synthetic route used to prepare Pt(IV) NP-cRGD. (B) 1H NMR spectra of the DSPE-PEG1k-Pt(IV) in CDCl3. The characteristic peaks are pointed out and magnified (right). (C) 1H NMR spectra of Pt(IV) NP-cRGD in DMSO-d6. The characteristic peaks are pointed out and magnified (right). (D) Size distribution of Pt(IV) NP-cRGD before (red) and after (black) US exposure. (E) Storage stability of Pt(IV) NPs and Pt(IV) NP-cRGD at 4 °C, 25 °C and 37 °C. (F) Serum stability of Pt(IV) NPs and Pt(IV) NP-cRGD. Serum-induced aggregation assay was monitored based on turbidity at the indicated time. (G) TEM image of Pt(IV) NP-cRGD before (a1,a2) and after US treatment at 10 s (b1,b2) and 60 s (c1,c2). (H) Pt release profiles from Pt(IV) NP-cRGD, GSH: glutathione. Data are presented as the mean ± SD of three independent experiments. Statistical significance in (H) was calculated by two-way ANOVA with Sidak's post hoc test. *P < 0.05, **P < 0.01, ***P < 0.005, NS indicates P > 0.05.
Table 1
Characterization of Pt(IV) NPs with different cRGD ligand densities.
| Nanoparticles | cRGD (%) | Size (nm)a) | PDIa,b) | Zeta potential (mV)a) | Drug loading efficiencies (%)c) |
|---|---|---|---|---|---|
| Pt(IV) NPs | 0 | 148.8 ± 0.9 | 0.17 ± 0.02 | -5.97 ± 0.42 | 2.12 ± 0.14% |
| Pt(IV) NP-0.25%cRGD | 0.25 | 149.6 ± 1.3 | 0.21 ± 0.01 | -6.17 ± 0.89 | 2.17 ± 0.21% |
| Pt(IV) NP-0.5%cRGD | 0.5 | 153.7 ± 0.7 | 0.20 ± 0.02 | -5.93 ± 1.27 | 2.24 ± 0.17% |
| Pt(IV) NP-cRGD | 1 | 151.1 ± 1.3 | 0.22 ± 0.01 | -5.27 ± 0.38 | 2.37 ± 0.11% |
| Pt(IV) NP-2%cRGD | 2 | 158.1 ± 1.1 | 0.24 ± 0.03 | -5.42 ± 0.60 | 2.32 ± 0.25% |
a) Determined by DSL; b) Polydispersity; c) Determined by ICP-MS; Data were represented as mean ± SD (n = 3).
The average size of Pt(IV) NP-cRGD was measured as 151.1 ± 1.3 nm, which was slightly higher than that of the Pt(IV) NPs, determined as 148.8 ± 0.9 nm (Figure 1D, Figure S5A and Table 1). This might be attributed to the modification of cRGD on the hybrid shell of the Pt(IV) NPs. The zeta potential analysis demonstrated that the surface charge of the Pt(IV) NPs was -5.97 ± 0.42 mV in aqueous solution (Figure S5B). After modification with cRGD, the zeta potential increased slightly to -5.27 ± 0.38 mV (Figure S6A). Besides, the drug loading efficiencies (DL%) of the Pt(IV) NPs and Pt(IV) NP-cRGD were 2.12 ± 0.14% and 2.37 ± 0.11%, respectively. The average sizes of the Pt(IV) NPs and Pt(IV) NP-cRGD did not change significantly within 25 days at 4 °C, 25 °C and 37 °C, suggesting good storage stability (Figure 1E). In addition, the serum stability of Pt(IV) NPs and Pt(IV) NP-cRGD were evaluated by a serum-induced aggregation assay. The turbidity of Pt(IV) NP-cRGD kept stable for 7 days, indicating that Pt(IV) NP-cRGD resisted the serum-induced aggregation and remained stable in the blood circulation (Figure 1F). These properties were beneficial for applications in the drug delivery considering the passively tumor-targeting mechanism based on enhanced permeability and retention effect (EPR).
Liquid PFH is a typical highly biocompatible phase-shift material that can be converted into gas when the temperature approaches its boiling point (≈56 °C) and is often encapsulated in nanoparticles to form UCAs for tumor therapy and ultrasound imaging [36, 37]. The optical microscopic images demonstrated that the Pt(IV) NP-cRGD were transformed from liquid to gas after being exposed to high temperatures (Figure S7). Besides, the average size of Pt(IV) NP-cRGD was measured as 962.7 ± 4.8 nm after US exposure (Figure 1D and Figure S6B). To further assess the phase-transition behavior of the Pt(IV) NP-cRGD under US exposure, transmission electron microscopy (TEM) was used to determine whether US exposure could trigger their structural expansion and collapse. The TEM images revealed nearly spherical morphologies of the Pt(IV) NP-cRGD and condensed PFH before US exposure (Figure 1G (a1-a2)). Interestingly, structural expansion was clearly observed after US exposure for 10 s (Figure 1G (b1-b2)). Meanwhile, after ultrasound exposure for 60 s, the TEM image showed extensive irregularly shaped particles that were likely produced by the fragmentation of the hybrid shell during nanoparticle expansion (Figure 1G (c1-c2)). The results confirmed that the morphology and structure of the Pt(IV) NP-cRGD expanded and collapsed under US stimulation due to the phase-transition behavior of PFH. Thus, we further hypothesized that US exposure could trigger the release of Pt(IV) prodrugs from lipid-polymer hybrid shells.
Our previous work has confirmed that GSH can competitively coordinate with platinum and release it from the prodrug complexes [11]. To further evaluate the GSH-sensitive and US-triggered drug release of Pt(IV) NP-cRGD, an in vitro Pt release experiment was performed at 37°C in different concentrations of GSH under US exposure (Figure 1H). Compared to free cisplatin, the cumulative release of platinum from Pt(IV) NP-cRGD with or without US at 20 mM GSH was estimated as 71.39 ± 5.20% and 58.99 ± 5.33%, respectively, after 24 h, indicating controllable drug release of the nanoparticles. In addition, it clearly demonstrated that US promoted platinum release due to US cavitation. Compared with the 83.62 ± 6.11% obtained at 20 mM GSH, the cumulative release of drug from the Pt(IV) NP-cRGD with US at 2 μM GSH was 32.33 ± 3.25% after 48 h, indicating GSH-sensitive drug release. The concentration of GSH in the tumor cells is approximately 4-fold higher than that of normal cells [38], thus Pt(IV) NP-cRGD can avoid the leakage of the drug in the blood circulation and rapidly release drug in the tumor tissue and tumor cells, which greatly enhanced the platinum concentration in the tumor tissue and cells, leading to a better tumor inhibition effect. The above results provided a foundation for the GSH-sensitive and US-triggered drug release of Pt(IV) NP-cRGD in vivo.
US imaging in vitro and in vivo
The potential of Pt(IV) NP-cRGD as contrast agents for ultrasound imaging was assessed in vitro using a homemade model. As shown in Figure 2A (a-d), the ultrasound images of different concentrations of Pt(IV) NP-cRGD demonstrated that the echo signal intensity of Pt(IV) NP-cRGD was indeed enhanced after US exposure and the ultrasound signal intensity was strongly correlated with Pt(IV) NP-cRGD concentration. In addition, the ultrasound signal intensity gradually increased with increasing Pt(IV) NP-cRGD concentrations. According to Figure 2A (e-h), the B-mode and CEUS images of the nanoparticles clearly exhibited increased echo signal and contrast enhancement after 5 min. However, the ultrasound signals decreased as time continued to increase because of the lack of sufficient microbubbles. Figure 2B-C demonstrated that the acoustic signal intensity value changed at different concentrations and times after ultrasound exposure. These measurements suggested that Pt(IV) NP-cRGD exhibited strong echogenic signals and excellent echo persistence under US exposure in vitro.
In vivo, no contrast-enhanced ultrasound imaging was observed in the group whose tumors were given a saline preinjection (Figure 2D(a)) and postinjection (Figure 2D(b)) under the contrast mode. The ultrasound images of the tumors of the nude mice from the Pt(IV) NP-cRGD and Sonovue groups clearly exhibited echo signal and contrast enhancement under the contrast mode after injection (Figure 2D(c-f)). The above results indicated that the Pt(IV) NP-cRGD exhibited excellent contrast enhancement under US exposure in vivo.
Figure 2
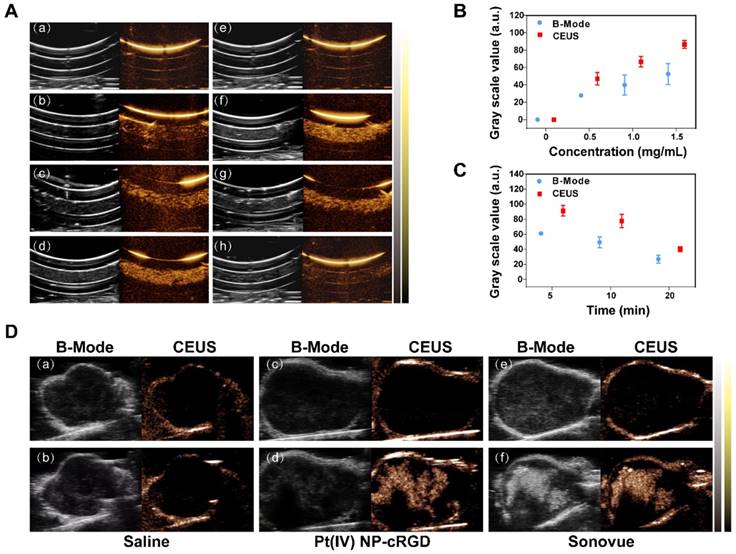
US imaging in vitro and in vivo. (A) Evaluation of US imaging performance in vitro. US imaging of saline (a), 0.5 mg/mL Pt(IV) NP-cRGD (b), 1.0 mg/mL Pt(IV) NP-cRGD (c) and 1.5 mg/mL Pt(IV) NP-cRGD (d) at 1 min. (e-h) US imaging of saline (e) and 1.5 mg/mL Pt(IV) NP-cRGD after different US exposure times (f-h separately represent saline after US treatment at 5 min, 10 min and 20 min). B-mode (left) and contrast-enhanced ultrasound (CEUS) (right) image of Pt(IV) NP-cRGD. (B, C) Grayscale values of the Pt(IV) NP-cRGD changed along with the changes in concentration and US exposure time. The data are shown as the mean ± SD of three independent experiments. (D) Ultrasound images of tumors injected with saline, Pt(IV) NP-cRGD (1.5 mg/mL) and Sonovue (SF6 = 8 μL/mL). The upper panel shows the preinjection images of the tumors, and the lower panel shows the postinjection images of the tumors. B-mode (left) and CEUS (right).
Integrin-dependent activity of Pt(IV) NP-cRGD
As shown in Figure 3A, the introduction of the cRGD ligands increased the in vitro cytotoxicity of the Pt(IV) NPs. The data obtained after 48 h of incubation showed that the 50% growth inhibitory concentration (IC50) values were lower with 1% and 2% cRGD. These results demonstrated that ligand density is essential for increasing the cytotoxicity of the nanoparticles, and the ligand density of 1% cRGD displayed the highest cytotoxicity in vitro (Table 1). It should be noted that the cytotoxicity of cRGD itself is very low. In our system, the concentrations of cRGD did not reach a toxic range (Figure S8A).
To confirm the recognition of integrin by the cRGD ligand, we analyzed cellular uptake in αvβ3- and αvβ5-positive SKOV3 human ovarian tumor cells and αvβ3- and αvβ5-negative A2780 human ovarian tumor cells (Figure S8C-D). Compared with Rhodamine B-labeled Pt(IV) NPs (RB NPs), the RB-labeled Pt(IV) NP-cRGD (RB NP-cRGD) were internalized more rapidly into the SKOV3 cells after 4 h (Figure 3B-C). However, the RB NPs and RB NP-cRGD did not exhibit a significant difference in mean fluorescence intensity after incubating with the A2780 cells (Figure 3B, D). The data indicated that the introduction of the cRGD ligands improved cellular uptake by ligand-receptor recognition. In addition, we established αv integrin-downregulated SKOV3 cells by RNA silencing (Figure S8B). Flow cytometric analysis confirmed that the SKOV3 cells expressed αvβ3 (red line) and αvβ5 (blue line) integrins, whereas the αv integrin-downregulated SKOV3 cells expressed almost no αvβ3 or αvβ5 integrin (Figure 3E). Subsequently, the αv integrin-downregulated SKOV3 cells were used in the in vitro cytotoxicity assays. As shown in Figure 3F, the IC50 of Pt(IV) NP-cRGD incubated with the αv integrin-downregulated SKOV3 cells became similar to that of the Pt(IV) NPs, thus indicating a crucial role for combining the cRGD ligands with the αvβ3 and αvβ5 integrins in enhancing the cytotoxicity of Pt(IV) NP-cRGD against the SKOV3 cells. In conclusion, these results emphasized the significance of the cRGD ligand, particularly when the surface payload of the peptide ligand molecules on the nanoparticles reached 1%, which enhanced cytotoxicity through improving cellular uptake by receptor-mediated endocytosis [39].
Figure 3
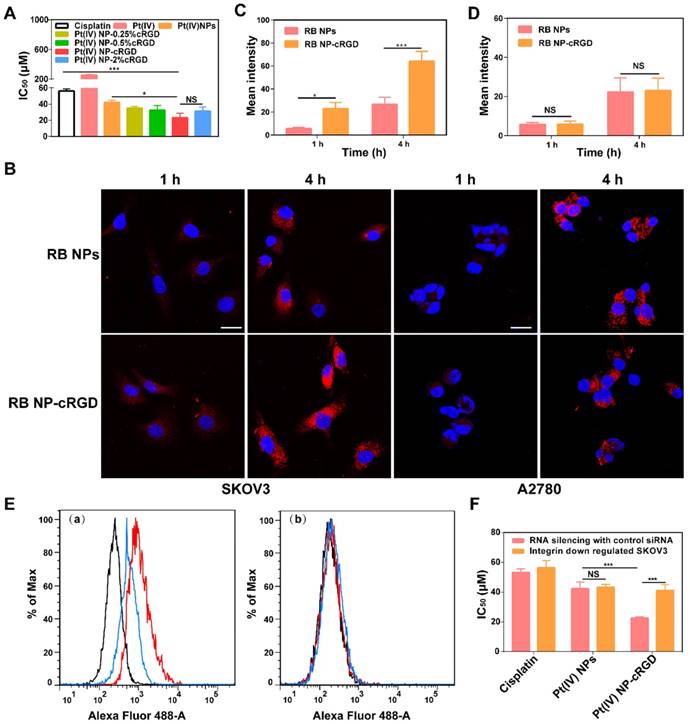
Integrin-dependent activity of Pt(IV) NP-cRGD. (A) In vitro cytotoxicities of cisplatin, Pt(IV) or Pt(IV) NPs with different densities of the cRGD ligand in the SKOV3 cells, IC50: 50% growth inhibitory concentration. (B) In vitro confocal laser scanning microscopy (CLSM) images of RB-labeled Pt(IV) NPs (RB NPs) and RB-labeled Pt(IV) NP-cRGD (RB NP-cRGD) uptake in SKOV3 and A2780 cells, RB: Rhodamine B. The cells were incubated with the respective nanoparticle solutions (20 μg/mL RB) for 1 h and 4 h at 37 °C. All pictures show merged images, which include the nuclei (blue) and nanoparticles (red). Scale bar 20 μm. (C, D) Quantitative analysis of the internalized nanoparticles in the SKOV3 (C) and A2780 cells (D) based on the CLSM images. (E) Flow cytometric analysis of integrin expression. (a) SKOV3 cells and (b) αv integrin-downregulated SKOV3 cells. Alexa Flour 488-labeled mouse IgG: control, black; Alexa Flour 488-labeled anti-αvβ3 integrin antibody: red; and Alexa Flour 488-labeled anti-αvβ5 integrin antibody: blue. (F) In vitro cytotoxicities of cisplatin, Pt(IV) NPs and Pt(IV) NP-cRGD in αv integrin-downregulated SKOV3 cells. The data are presented as the mean ± SD of three independent experiments. Statistical significance in (A) was calculated by one-way ANOVA with Tukey's post hoc test. Statistical significance in (C), (D) and (F) was calculated by two-way ANOVA with Tukey's post hoc test. *P < 0.05, **P < 0.01, ***P < 0.005, NS indicates P > 0.05.
Antitumor effects of Pt(IV) NP-cRGD under US exposure in vitro
The MTT assay was used for measuring the cytotoxicity of the nanocarrier. The viability of the SKOV3 cells did not exhibit a significant change at 24 h and 48 h, even at concentrations of up to 1500 μg/mL, which implied that the PFH and the hybrid lipid-polymer shell material had relatively low cytotoxicities and, together, can be a good drug nanovector (Figure S9A-B). In addition, to determine the effect of US exposure, we tested whether power and duration time can influent SKOV3 proliferation. US exposure showed apparent power-dependent and time-dependent cytotoxicity when the frequency was 1 MHz (Figure S9C-D). In addition, 1 MHz US at 2 W/cm2 for 1 min had little effect on the viability of the SKOV3 cells and thus was adopted in the following study.
The antitumor efficiency of the Pt(IV) NP-cRGD was higher than that of the Pt(IV) NPs because the cRGD ligand improved cellular uptake (Figure 4A and Figure S9E-F). In addition, compared to the equivalent dose of Pt(IV) NP-cRGD, the cytotoxicity of Pt(IV) NP-cRGD with US increased clearly, whose cell viability value was calculated as 28.49 ± 5.09% at the concentration of 25 μM.
Figure 4
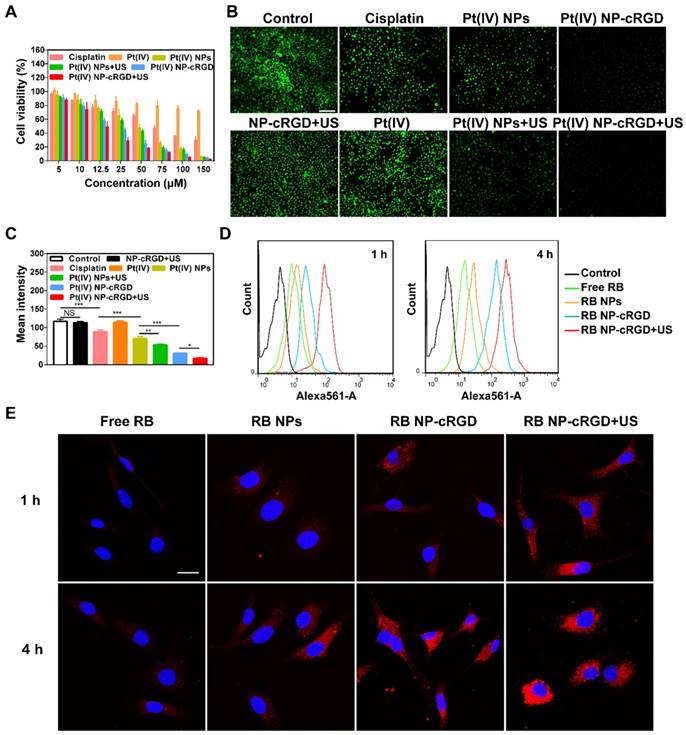
Antitumor effects of Pt(IV) NP-cRGD under US exposure in vitro. (A) Cell viability of the SKOV3 cells treated with various concentrations of cisplatin, Pt(IV), Pt(IV) NPs, Pt(IV) NPs+US, Pt(IV) NP-cRGD and Pt(IV) NP-cRGD+US for 48 h. (B) Fluorescence images and (C) quantitative fluorescence intensity of Calcein-AM in the SKOV3 cells treated with NP-cRGD+US, cisplatin, Pt(IV), Pt(IV) NPs, Pt(IV) NPs+US, Pt(IV) NP-cRGD and Pt(IV) NP-cRGD+US (containing an equal amount of 30 μM platinum) for 48 h. Original magnification, 100×; Scale bar 200 μm. (D) Flow cytometric analysis and (E) CLSM images of SKOV3 cells treated with free RB, RB NPs, RB NP-cRGD and RB NP-cRGD+US for 1 h and 4 h, RB: Rhodamine B. The cells were incubated with the respective nanoparticle solutions (20 μg/mL RB) for 1 h and 4 h. All pictures show merged images, which include the nuclei (blue) and nanoparticles (red). Scale bar 20 μm. The data are presented as the mean ± SD of three independent experiments. Statistical significance in (C) was calculated by one-way ANOVA with Tukey's post hoc test. *P < 0.05, **P < 0.01, ***P < 0.005, NS indicates P > 0.05.
According to Figure 3A and 4A, the different nanoparticle formulations contained an equal amount of platinum, 30 μM, in the following experiments unless otherwise stated. The calcein-AM assay results also confirmed that US enhanced the cytotoxicity of Pt(IV) NP-cRGD (Figure 4B-C). The previous results indicated that drug release depended on GSH concentration and that US promoted drug release due to US cavitation. In addition, the cellular uptake experiments showed that the Pt(IV) NP-cRGD+US group had higher intracellular fluorescence intensity after incubating for 4 h than without US (Figure 4D-E). These results confirmed that US not only promoted drug release but also accelerated intracellular uptake to enhance cytotoxicity.
Figure 5
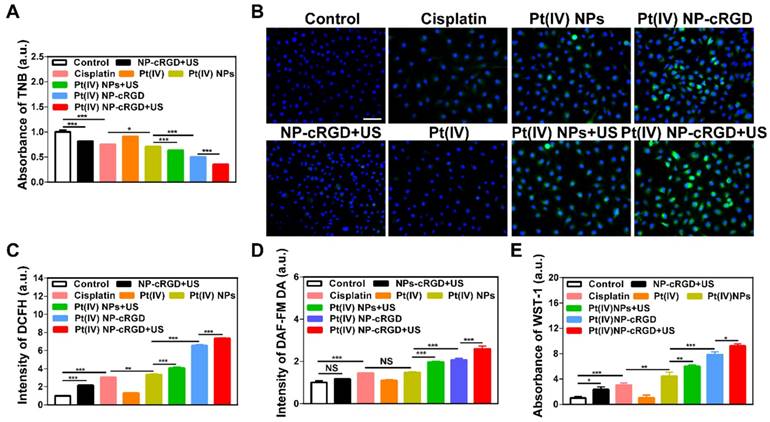
The consumption of GSH and overproduction of ROS. (A) The concentration of GSH in cells treated with NP-cRGD+US, cisplatin, Pt(IV), Pt(IV) NPs, Pt(IV) NPs+US, Pt(IV) NP-cRGD and Pt(IV) NP-cRGD+US (30 μM eq.) for 48 h, as tested by the absorbance of TNB. (B, C) The ROS level using the DCFH-DA dye after treatment with NP-cRGD+US, cisplatin, Pt(IV), Pt(IV) NPs, Pt(IV) NPs+US, Pt(IV) NP-cRGD and Pt(IV) NP-cRGD+US (30 μM eq.) for 48 h, as detected by fluorescence microscope (B) and flow cytometry (C). All pictures show merged images, which include the nuclei (blue) and DCFH (green). Original magnification, 200×; Scale bar 100 μm. (D) Concentration of nitric oxide in the cells, as tested by the intensity of DAF-FM DA. (E) Concentration of oxygen superoxides in the cells, as tested by the absorbance of WST-1. The data are shown as the mean ± SD of three independent experiments. Statistical significance was calculated by one-way ANOVA with Tukey's post hoc test. *P < 0.05, **P < 0.01, ***P < 0.005, NS indicates P > 0.05.
Previous work has indicated that there are two main pathways of drug release and intracellular uptake: 1) prodrugs can be taken up by passive diffusion and sonoporation after ultrasound exposure and interact favorably with and adsorb onto the cancer cell membrane; 2) nanoparticles can be uptaken by receptor-mediated endocytosis and escaped from the lysosomes, activated by GSH to release the drug inside the cell [40]. These data demonstrated that combining US with Pt(IV) NP-cRGD led to higher drug release and cellular uptake, which resulted in higher antitumor activity.
Mitochondrial apoptosis signaling pathways
Both GSH and ROS play an important role in maintaining the redox balance of cells. The transformation of the Pt(IV) prodrug into Pt(II) complexes requires a large consumption of GSH during drug release. If the concentration of GSH decreases, the redox balance is destroyed, which results in an overproduction of ROS [41]. In addition, studies have confirmed that US exposure can induce the production of ROS [42, 43]. Thus, the combination of Pt(IV) NP-cRGD with US can consume GSH and overproduce ROS in tumor cells. The concentration of GSH in the SKOV3 cells was measured to determine the GSH consumption (Figure 5A). The SKOV3 cells incubated with Pt(IV) NP-cRGD+US showed the lowest concentration of GSH of all the groups, which indicated the high consumption of GSH. The SKOV3 cells incubated with NP-cRGD+US exhibited significant increased fluorescence intensity of DCFH, which demonstrated that US exposure induced ROS overproduction (Figure 5B-C and Figure S10). The SKOV3 cells incubated with Pt(IV) NP-cRGD or Pt(IV) NP-cRGD+US exhibited significantly higher fluorescence intensities than those incubated with the other drug formulations, suggesting a stronger ability to induce high levels of ROS in the tumor cells. The quantitative data of ROS production indicated that Pt(IV) NP-cRGD and Pt(IV) NP-cRGD+US resulted in much higher fluorescence intensity, in good agreement with the above results (Figure 5C). Moreover, compared to the Pt(IV) NP-cRGD group, the Pt(IV) NP-cRGD+US group showed even stronger fluorescence signals. These results demonstrated that Pt(IV) NP-cRGD combined with US can induce high levels of ROS in cancer cells. Considering that ROS is a generic term of various chemically active oxygen-containing molecules, we further studied whether the Pt(IV) NP-cRGD+US induced a single kind of ROS or all kinds of ROS. As shown in Figure 5D-E, the Pt(IV) NP-cRGD+US group exhibited higher fluorescence intensity than any other groups in both tests, indicating a higher ability of these species to be induced by Pt(IV) NP-cRGD+US. These results confirmed that Pt(IV) NP-cRGD with US induced the overproduction of different kinds of ROS instead of a specific one. The above results confirmed that Pt(IV) NP-cRGD with US consumed GSH and overproduced ROS.
Several research studies have revealed that the high levels of ROS are relative to mitochondrial damage and can decrease mitochondrial membrane potential (Δψm) [44-46]. The reduction of Δψm is usually considered an early event in mitochondria-mediated apoptosis. To further elucidate the antitumor mechanism, various apoptotic markers were measured in the SKOV3 cells after incubating with various drug formulations. The mitochondrial membrane potential was measured by a fluorescent probe, JC-1, which can be used as an indicator of mitochondrial depolarization and is revealed by an increase in the green (monomer)/red (aggregate) fluorescence intensity ratio. Flow cytometry results in Figure 6A and Figure S11 shows that after applying NP-cRGD+US or cisplatin, the Δψm of the SKOV3 cells did not decrease significantly.
Figure 6
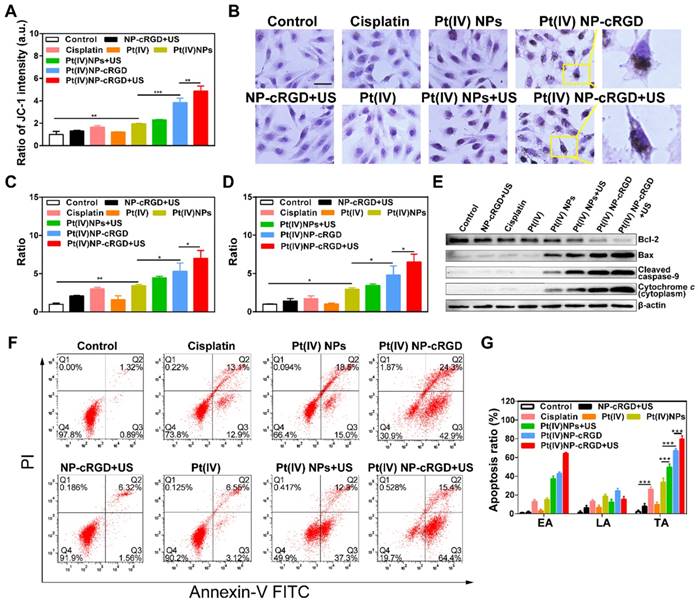
Mitochondrial apoptosis signaling pathways. (A) Mitochondrial membrane potential (Δψm) in cells with the treatment of NP-cRGD+US, cisplatin, Pt(IV), Pt(IV) NPs, Pt(IV) NPs+US, Pt(IV) NP-cRGD and Pt(IV) NP-cRGD+US (30 μM eq.) for 48 h, as detected by the ratio of fluorescent intensity ([green]/[red]) of JC-1. (B) Immunohistochemically stained images of cytochrome c translocated from the mitochondria to the cytosol. Original magnification, 200×; Scale bar, 100 μm. (C, D) Activities of caspase 9 and caspase 3 as determined by a microplate reader. (E) Western blotting analyses of Bcl-2, Bax, Cleaved caspase 9 and Cytochrome c after different drug formulations incubations. (F, G) Cell apoptosis of the SKOV3 cells treated with NP-cRGD+US, cisplatin, Pt(IV), Pt(IV) NPs, Pt(IV) NPs+US, Pt(IV) NP-cRGD and Pt(IV) NP-cRGD+US (30 μM eq.) for 48 h, using the Annexin V and PI kit to detect the apoptosis ratio through the flow cytometry analysis, along with the statistics of cell apoptosis ratio, EA: early apoptosis; LA: late apoptosis; TA: total apoptosis. The data are presented as the mean ± SD of three independent experiments. Statistical significance in (A), (C) and (D) was calculated by one-way ANOVA with Tukey's post hoc test. Statistical significance in (G) was calculated by two-way ANOVA with Tukey's post hoc test. *P < 0.05, **P < 0.01, ***P < 0.005, NS indicates P >0.05.
Although the above two groups raised the levels of ROS, the increase might not have been enough to decrease the mitochondrial membrane potential. The fluorescence intensity ratio of green to red increased significantly after treatment with Pt(IV) NP-cRGD+US, which was 4.87-fold that of control. It indicated that the Δψm decreased dramatically. After the mitochondrial membrane is permeabilized, cytochrome c translocates from the mitochondria to the cytosol, which is detected as a marker of cell apoptosis. Figure 6B depicts the immunocytochemically stained images of cytochrome c in the SKOV3 cells after being treated with different groups, as evidenced by the brown staining. After incubating with the culture medium, NP-cRGD+US, cisplatin and Pt(IV), no cytochrome c was seen in the cytosol of the SKOV3 cells. Cytochrome c release was more clearly observed in the cytosol of the SKOV3 cells incubated with Pt(IV) NP-cRGD+US compared with all other formulations. Cytochrome c further activates a cascade of caspase 9 and caspase 3 reactions. After the Pt(IV) NP-cRGD+US treatment, the caspase 9 activity ratios in the SKOV3 cells were 7.00-fold and caspase 3 activity ratios approximately 6.51-fold higher than those of the controls (Figure 6C-D). These data demonstrated that Pt(IV) NP-cRGD+US had the most distinct ability for releasing cytochrome c and activating caspase 9 and caspase 3 of SKOV3 cells. In addition, the western blotting results in Figure 6E further confirmed our finding. The expression of the pro-apoptotic protein Bax, cleaved caspase 9 and cytochrome c in the SKOV3 cells treated with Pt(IV) NP-cRGD+US increased significantly, while the expression of the anti-apoptotic protein Bcl-2 decreased as compared to the other groups (Figure 6E). Moreover, apoptotic cell death induced by various treatments was determined by counting the apoptotic cells in early and late periods. As shown in Figure 6F-G, the percentages of apoptosis induced were 26.0 ± 2.9%. 33.5 ± 4.6%, 49.6 ± 3.7%, 67.2 ± 2.6% and 79.8 ± 3.4%, respectively, when the cells were incubated with cisplatin, Pt(IV) NPs, Pt(IV) NPs+US, Pt(IV) NP-cRGD and Pt(IV) NP-cRGD+US, indicating that Pt(IV) NP-cRGD with US significantly enhanced the apoptosis in SKOV3 cells. In addition, the percentage of apoptosis in the NP-cRGD+US group was 7.88 ± 3.21%, similar to that in the controls (2.21 ± 1.48%), suggesting that NP-cRGD with US had no direct effect on mitochondria-mediated apoptosis.
Overall, the above results demonstrated a route of ROS-induced mitochondria-mediated apoptosis. Pt(IV) NP-cRGD with US promoted tumor cell apoptosis, notably by consuming GSH and highly enhancing the levels of ROS, further inducing Δψm decreasing and releasing cytochrome c into the cytosol, activating caspase 9 and caspase 3 and finally activating tumor cell apoptosis.
Antitumor efficiency of Pt(IV) NP-cRGD under US exposure in vivo
The higher expression of integrin receptors in the endothelial cells of tumor angiogenic vessels and SKOV3 cells, when compared to that of normal cells, can contribute to Pt(IV) NP-cRGD targeting of tumor tissues. The tumor-targeting ability of Pt(IV) NP-cRGD in vivo was investigated by using SKOV3 tumor-xenografted nude mice as the model. A time-dependent biodistribution was observed with a NightOWL II LB 983 in vivo imaging system, as shown in Figure 7A. The fluorescence signal of free Cy7 was primarily distributed in the liver, and very little was found in the tumor at 24 h. However, the fluorescence signals of the Cy7 NPs and Cy7 NP-cRGD groups were mainly located at the liver and tumor and increased in the tumor as time increased. These data demonstrated that both Pt(IV) NPs and Pt(IV) NP-cRGD could passively accumulate at the tumor sites via the EPR effect. In addition, concentration of platinum in blood at different time after drug injection was tested by ICP-MS, as shown in Figure S12. Pt(IV) NPs-cRGD exhibited long circulation half-life and an increase of area under the curve (AUC). The prolonged circulation time properties of Pt(IV) NPs-cRGD may exhibit protection from hepatic phagocytosis and benefit for passive tumor targeting via EPR effect. Compared with that of Cy7 NPs, Cy7 NP-cRGD showed significantly better tumor selectivity (Figure 7B-C), indicating that cRGD ligand modification improved the accumulation of the nanoparticles in the tumor tissue through active targeting.
Antitumor efficiency was determined by tumor volume changes over 30 days in SKOV3 cell-grafted tumor-bearing nude mice. As shown in Figure 7D and E, Pt(IV) NP-cRGD with US had the most effective inhibition on tumor growth compared with saline and other drug formulations with an equivalent dose. In addition, the body weight of each treatment group exhibited no significant change to a certain extent, which was similar to that of the saline-treated group (Figure S13). The H&E assay in Figure 7F shows the pathological characteristics of the tumor tissue in six groups. The tumor obtained from the Pt(IV) NP-cRGD+US-treated mice showed larger area of dead cells than those from the other five groups. These results demonstrated that Pt(IV) NP-cRGD+US exhibited higher antitumor efficiency. Subsequently, we further confirmed whether the antitumor efficiency of Pt(IV) NP-cRGD+US was induced by mitochondrial apoptosis in vivo. Compared with that of any other group, the expression of procaspase 9 (brown) in the tumor treated with Pt(IV) NP-cRGD+US was decreased significantly, and the expression of cleaved-caspase 3 (brown) was enhanced. Next, the Ki67 staining and TUNEL assay were conducted to measure the cell proliferation and apoptosis of the tumor in vivo. Ki67 immunohistochemical analysis exhibited a dramatic decrease in cell proliferation in the tumors of Pt(IV) NP-cRGD+US-treated mice. In addition, the TUNEL assay indicated that an obvious increase in apoptotic cells was observed in the tumors of Pt(IV) NP-cRGD+US-treated mice. Generally, cisplatin therapy is often accompanied by dose-limiting nephrotoxicity, which results in increased serum levels of creatinine (CREA) and UREA. According to Figure S14, serum CREA and UREA levels were significantly increased in the cisplatin-treated mice. And H&E staining of kidney sections obtained from the cisplatin-treated group exhibited glomerular atrophy. In contrast, the serum CREA and UREA levels of Pt(IV) NP-cRGD+US-treated mice did not significantly increase. And H&E staining showed no obvious pathological signs of nephrotoxicity. These results further demonstrated that Pt(IV) NP-cRGD with US maximized the therapeutic effect while reducing the toxicity of chemotherapy.
Figure 7
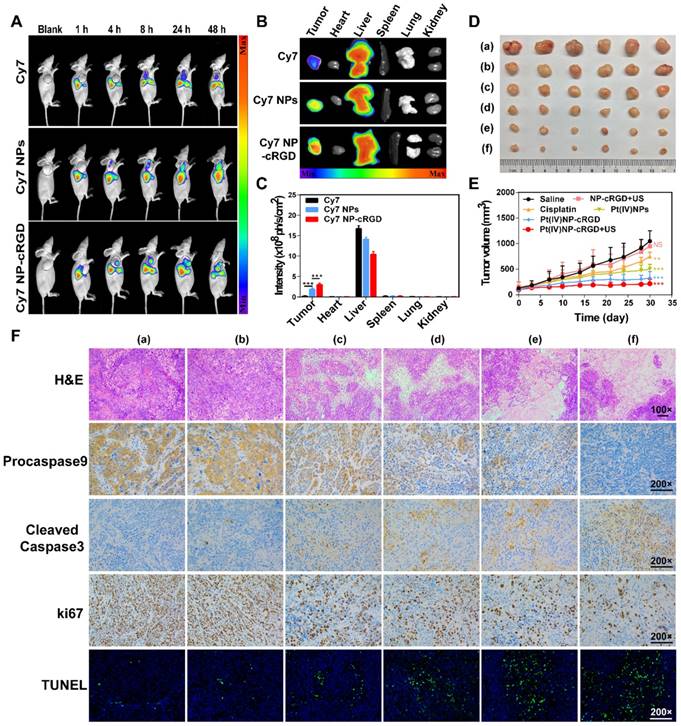
Antitumor efficiency of Pt(IV) NP-cRGD with US in vivo. (A) In vivo distribution of free Cy7, Cy7 NPs and Cy7 NP-cRGD (at a Cy7 dose of 1 μg/mL) in SKOV3 tumor-bearing mice after intravenous injection for 1, 4, 8, 24 and 48 h (n = 3). (B) Fluorescence images of excised tumors and organs 48 h after the intravenous injection. (C) Quantitative analysis of the mean fluorescence intensity of Cy7 in excised tumors and organs 48 h after the intravenous injection. (D) Sizes of the tumors collected from different groups of mice 3 days after the last treatment. (a) saline, (b) NP-cRGD+US, (c) cisplatin, (d) Pt(IV) NPs, (e) Pt(IV) NP-cRGD and (f) Pt(IV) NP-cRGD+US (n = 6). (E) Tumor growth curves of tumor-bearing mice after treatment with different formulations of 2.0 mg/kg. (F) The tumor sections were stained with H&E, procaspase 9, cleaved caspase 3, Ki67 and TUNEL. The upper panels (H&E) were magnified 100-fold, and the lower panels were magnified 200-fold, Scale bar, 100 μm. (a) saline, (b) NP-cRGD+US, (c) cisplatin, (d) Pt(IV) NPs, (e) Pt(IV) NP-cRGD and (f) Pt(IV) NP-cRGD+US. The data are presented as the mean ± SD. Statistical significance in (C) was calculated by two-way ANOVA with Tukey's post hoc test. Statistical significance in (E) was calculated by two-way ANOVA with Sidak's post hoc test. *P < 0.05, **P < 0.01, ***P < 0.005, NS indicates P > 0.05.
Figure 8
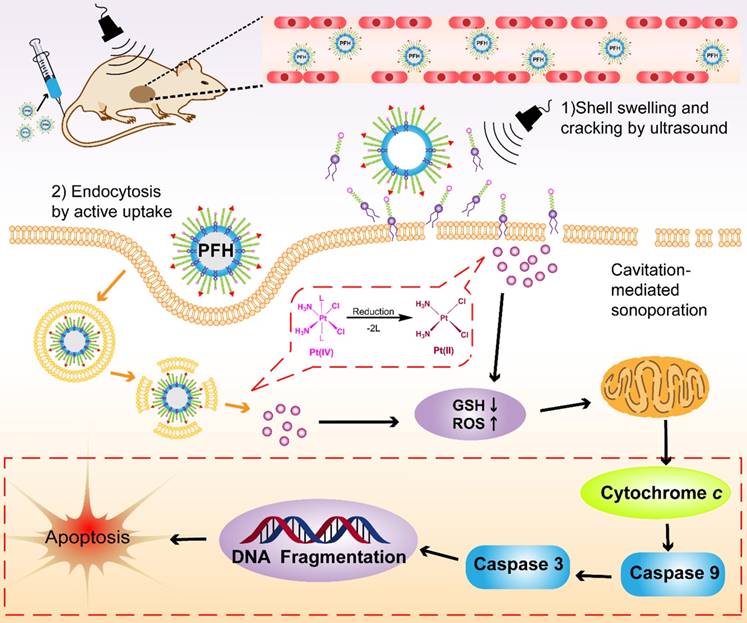
The schematic illustration of Pt(IV) NP-cRGD assisted by US for tumor treatment. The Pt(IV) NP-cRGD are rapidly accumulated at the tumor sites via the EPR effect and active targeting and then uptaken by tumor cells through two main pathways: (1) Prodrugs are taken up by passive diffusion and sonoporation after ultrasound exposure and interact favorably with and adsorb onto the cancer cell membrane; (2) Pt(IV) NP-cRGD are selectively taken up via receptor-mediated endocytosis. Next, the prodrug and nanoparticles that escape from the endosome respond to endogenous GSH and release the drug. Finally, Pt(IV) NP-cRGD with US induce ROS overproduction and cause cell apoptosis to maximize the antitumor efficiency.
Several factors contribute to the higher antitumor efficiency induced by Pt(IV) NP-cRGD with US. As shown in Figure 8, the excellent properties of Pt(IV) NP-cRGD (for example, size, geometry, surface potential, etc) were beneficial for passive tumor targeting via EPR effect. And Pt(IV) NP-cRGD further accumulated at the tumor sites through cRGD-mediated active targeting [39]. Additionally, Pt(IV) NP-cRGD avoided the leakage of the drug in the blood circulation and rapidly released drug in the tumor tissue and cells because of GSH-sensitive and US-triggered drug release [11]. Then, the nanoparticles under US exposure were taken up rapidly by the tumor cells through two main pathways [40]: (1) The prodrugs are taken up by passive diffusion and sonoporation after ultrasound exposure. In addition, the prodrug interacts favorably with and adsorb onto the cancer cell membrane due to the amphiphilicity of DSPE [12]; (2) Pt(IV) NP-cRGD are selectively taken up via receptor-mediated endocytosis. Next, the prodrug and nanoparticles that escape from the endosome respond to the endogenous GSH and release the drug. Finally, Pt(IV) NP-cRGD with US induces ROS overproduction and further causes mitochondria-mediated apoptosis to maximize the overall antitumor efficiency in ovarian cancer.
Conclusion
In summary, we successfully constructed a GSH-sensitive and US-triggered drug delivery system using multifunctional nanosized UCAs for US imaging and ovarian carcinoma therapy. The nanosized UCAs were composed of a PFH liquid core, a hybrid lipid-polymer shell with PLGA12k-PEG2k and DSPE-PEG1k, and an active ligand-targeting cRGD peptide. Pt(IV) was covalently attached to the DSPE-PEG1k to form the prodrug, which fine-tuned lipophilicity and improved cellular uptake. In addition, the active pharmaceutical ingredient release of DSPE-PEG1k-Pt(IV) prodrug was sensitive to GSH levels and US stimulation. The nanosized UCAs based on the Pt(IV) prodrug (Pt(IV) NP-cRGD) exhibited several advantages, such as the phase-transition behavior of Pt(IV) NP-cRGD for passive tumor-targeting and US imaging, a hybrid lipid-polymer shell for improved US imaging, proper cRGD ligand peptide density of Pt(IV) NP-cRGD for active tumor-targeting and enhanced accumulation, GSH-sensitive and US-triggered drug release of Pt(IV) NP-cRGD for sufficient drug concentration to treat the tumor. Both the in vitro and in vivo experiments demonstrated that Pt(IV) NP-cRGD with US exhibited excellent echogenic signals and brilliant antitumor therapeutic efficiency. The mechanistic studies confirmed that they can consume GSH and enhance ROS levels, which further decreased the mitochondrial membrane potential, released cytochrome c into the cytoplasm, induced a cascade of caspase 9 and caspase 3 reactions and activated tumor cell apoptosis. Thus, the nanosized UCAs in this work may provide valuable insight into the development of various multifunctional UCAs for precise theranostics against various cancers.
Experimental Section
Materials: cis-Diamineplatinum-(II) dichloride (cisplatin), mPEG2k-NH2 (Mw = 2000 Da), PLGA12k-COOH (Mw = 12000 Da), N-hydroxysuccinimide (NHS), 1-ethyl-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC·HCl), 3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), and Rhodamine B (RB) were purchased from Sigma-Aldrich Co., Ltd. (Shanghai, China). DSPE-PEG1k-NH2, DSPE-PEG1k-RB, DSPE-PEG1k-Cy7, and PLGA12k-PEG2k-Mal were purchased from Ponsure Biotechnology Company (Shanghai, China). Poly(vinyl alcohol) (PVA, 99%, Mw = 22000 Da) was obtained from Sigma-Aldrich Co., Ltd. (Shanghai, China). c(RGDfKC) (cRGD, 99.4% purity, Mw = 706.83, sequence: cyclo[Arg-Gly-Asp-(D-Phe)-Lys(Cys)]) was synthesized by ChinaPeptides Co., Ltd. (Shanghai, China). All other chemicals and organic solvents were of analytical grade purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China).
Preparation and characterization of DSPE-PEG1k-Pt(IV): c,c,t-[Pt(NH3)2Cl2(OH)2] and c,c,t-[Pt(NH3)2Cl2(OOCCH2CH2COOH)(OH)] (Pt(IV)) were synthesized by following the procedure described by S Dhar et al [47]. Briefly, to a solution of c,c,t-[Pt(NH3)2Cl2(OH)2] (50 mg, 0.15 mmol) in DMSO (3 mL) was added succinic anhydride (15 mg, 0.15 mmol), and the reaction mixture was stirred at room temperature for 24 h. The solution was lyophilized, and 10 mL acetone was added to precipitate a light yellow solid, which was washed several times with acetone and diethyl ether and then dried. The chemical structures of c,c,t-[Pt(NH3)2Cl2(OH)2] and Pt(IV) in deuterated dimethyl sulfoxide (DMSO-d6) were confirmed by nuclear magnetic resonance (1H NMR) spectra using a Bruker Avance 500 (500 MHz) spectrometer (Beijing, China). Mass Spectroscopy (ESI-MS) measurements were performed on a Quattro Premier XE system (Waters) equipped with an electrospray interface (ESI).
The amphiphilic polymer, 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino (polyethylene glycol)-1000] (ammonium salt, DSPE-PEG1k-NH2), was functionalized with Pt(IV) via an amide reaction between the amino groups of DSPE-PEG1k-NH2 and the carboxyl groups of Pt(IV) in the presence of EDC/NHS [48]. Briefly, Pt(IV) (108 mg, 0.25 mmol), EDC·HCl (191 mg, 1 mmol) and NHS (115 mg, 1 mmol) were dissolved in DMSO (3 mL). After 30 min, a DMSO solution of DSPE-PEG1k-NH2 (435 mg, 0.25 mmol, 2 mL) was added to the solution containing the activated Pt(IV) complex. The reaction mixture was kept stirring at room temperature for 72 h and then dialyzed in a Slide-A-Lyzer dialysis cassette (Thermo Scientific, Shanghai, China) (MWCO: 1000 Da) against water. The dialysate containing the pure product was lyophilized, and the residue was dried in vacuo over P2O5. The chemical structure of DSPE-PEG1k-Pt(IV) in deuterated chloroform (CDCl3) was measured by 1H NMR spectra using a Bruker Avance 500 (500 MHz) spectrometer (Beijing, China).
Preparation and characterization of PLGA12k-mPEG2k: mPEG2k-NH2 was conjugated to the PLGA12k-COOH polymer using the EDC/NHS method. Briefly, PLGA12k-COOH (250 mg, 20 μmol), EDC·HCl (15 mg, 80 μmol) and NHS (12 mg, 80 μmol) were dissolved in DMSO (5 mL). After 30 min, a DMSO solution of mPEG2k-NH2 (40 mg, 20 μmol, 5 mL) was added to the solution containing the activated PLGA12k-COOH. The reaction mixture was kept stirring at room temperature for 4 h and then dialyzed in a Slide-A-Lyzer dialysis cassette (Thermo Scientific, Shanghai, China) (MWCO: 3500 Da) against water. The dialysate containing the pure product was lyophilized, and the residue was dried in vacuo over P2O5. The 1H NMR (500 MHz) spectrum of the PLGA12k-mPEG2k mixture was measured in DMSO-d6.
Preparation and characterization of Pt(IV) NP-cRGD: 1%cRGD-functionalized Pt(IV)-loaded nanoparticles (Pt(IV) NP-cRGD) was formulated using a newly modified emulsion evaporation process [49, 50]. In a typical procedure, 24 mg of DSPE-PEG1k-Pt(IV) was added to a solution of 1.3 mL of CHCl3 and 36 mg of PLGA12k (PLGA12k-mPEG2k:PLGA12k-PEG2k-Mal = 2:1, (w/w)). Subsequently, 40 μL of liquid PFH was added to the mixture, which was then oscillated slightly. The solution was poured into 8 mL of cold PVA solution (1% w/v) and emulsified in an ice bath using an ultrasonic probe (YJ92-II ultrasonic cell pulverizer, Shanghai, China) at 85% energy for 5 min. The final emulsion was then mechanically mixed at room temperature for 4 h to extract CHCl3. Then, the mixture was centrifuged at 7500 rpm for 5 min, the precipitate was discarded, and the supernatant yielded the maleimide-functionalized Pt(IV) NPs (Pt(IV) NP-Mal). The centrifugation was repeated three times. c(RGDfKC) (1.2 mg, 1.7 μmol, 2.0 eq. vs the maleimide group) was added to the Pt(IV) NP-Mal solution (8.0 mL) and mechanically mixed at room temperature for 4 h. Subsequently the nanoparticles solution was purified by ultrafiltration (MWCO: 100000 Da, Thermo Scientific, Shanghai, China) to yield Pt(IV) NP-cRGD. Finally, the nanoparticles were collected and stored at 4°C for further use. The 1H NMR (500 MHz) spectrum of the polymer mixture was measured in DMSO-d6. The preparations of Pt(IV) NP-0.25%cRGD, Pt(IV) NP-0.5%cRGD and Pt(IV) NP-2%cRGD were performed in the same manner as described above, with a slight exception: the mass ratios of the PLGA12k-PEG2k-Mal/nanoparticles were changed from 20/100 to 100/0 for Pt(IV) NPs, 5/100 for Pt(IV) NP-0.25%cRGD, 10/100 for Pt(IV) NP-0.5%cRGD and 40/100 for Pt(IV) NP-2%cRGD.
Transmission electron microscopy (TEM) was performed by a Talos F200X transmission electron microscope (Shanghai, China) with a field emission gun operating at 200 kV. The Pt(IV) drug loading capacity was measured by an inductively coupled plasma mass spectrometry (ICP-MS, Leeman Prodigy, USA). The dynamic light scattering (DLS) and zeta potential were determined by a Malvern Zetasizer Nano ZS (Malvern Instruments, Malvern, UK).
Storage stability assay: Pt(IV) NPs and Pt(IV) NP-cRGD were stored at 4 °C, 25 °C and 37 °C in closed EP tubes without any other precautions and were periodically removed for routine analysis. The average size of Pt(IV) NPs and Pt(IV) NP-cRGD were determined in PBS (1×, pH = 7.4) by DLS.
Serum-induced aggregation assay: Aggregation was monitored by measuring turbidity [51, 52]. Briefly, Pt(IV) NPs and Pt(IV) NP-cRGD were mixed with 30% FBS (v/v) and incubated at 37 °C. The absorbance of the complexes in the absence and presence of 30% (v/v) serum was measured at 500 nm, and a corresponding amount of serum alone was used as a reference. The absorbance was measured at 1 min, 30 min, 1 h, 4 h, 8 h, 12 h, 24 h, 30 h, 48 h, 72 h, 96 h, 120 h, 144 h and 168 h. A relative turbidity value of 1 indicated that the turbidity of the serum-incubated complexes was equal to the turbidity of a buffer-incubated sample.
Drug release of Pt(IV) NP-cRGD: The release profile of the nanoparticles in vitro was assessed by using the dialysis method [53]. A mixture of 0.9 mL of Pt(IV) NP-cRGD and 0.1 mL of 200 mM GSH was first placed into a dialysis bag (MWCO: 3500 Da, Thermo Scientific, Shanghai, China) and then sonicated using a 1 MHz transducer at 2 W/cm2 for 1 min (NewAge Pocket Sonovit, Hongkong, China). Next, the dialysis bags were placed in a reservoir of a phosphate-buffered saline solution of GSH (20 mM, 1×PBS, pH = 7.4, 20 mL) with stirring at 80 rpm at 37 °C. At appropriate intervals, 1 mL samples were removed from the 20 mL release medium, and the same volume of PBS was added at the same temperature. Furthermore, because the linkage between DSPE-PEG1k and Pt(IV) is affected by reducing agents such as GSH, different concentrations of GSH were added to the above dissolved medium. The amounts of platinum released from Pt(IV) NP-cRGD were determined by ICP-MS (Leeman Prodigy, USA). The weight ratio of the accumulated released drug to the total drug payload in the nanoparticle was measured as a function of release time. Each assay was repeated in triplicate.
In vitro and in vivo US imaging: In vitro US imaging of Pt(IV) NP-cRGD was carried out in a latex tube (with an inner diameter of ~5 mm) using an Acuson S2000 Ultrasound System unit with a 9 L4 transducer (Siemens Healthcare, Shanghai, China). The Pt(IV) NP-cRGD were dispersed in 0.9% saline at different concentrations and injected into the latex tube. Saline was used as the negative control. The US imaging was performed using the transducer in both contrast-enhanced ultrasound (CEUS) mode (mechanical index, MI = 0.065) and conventional B-mode at the same time.
The in vivo US imaging of Pt(IV) NP-cRGD was measured in tumor-bearing nude mice using a VisualSonics 2100 imaging system with a MS-201 transducer (VisualSonics Inc., Canada) (Figure S 15). The anesthetized tumor-bearing nude mice were randomly divided into three groups (3 mice per group) and were separately injected intratumorally with 0.2 mL of saline, Pt(IV) NP-cRGD (1.5 mg/mL) and Sonovue (SF6 = 8 μL/mL). Saline was used as the negative control, and Sonovue was used as the positive control. The postinjection images of the tumors were captured ten seconds after the injection by ultrasonography (12.5 MHz in frequency).
Cell culture: The SKOV3 human ovarian tumor cell line was obtained from the Cell Resource Center, Shanghai Institutes for Biological Sciences (SIBS, Shanghai, China). The cells were grown in Dulbecco's modified Eagle medium (DMEM) (Gibco, Grand Island, USA) supplemented with 10% fetal bovine serum (Gibco, Grand Island, USA) and 1% penicillin/streptomycin (Gibco, Grand Island, USA) at 37 °C in a 5% CO2-humidified atmosphere. The A2780 human ovarian tumor cell line was purchased from KeyGEN Biotech Co., Ltd. (Nanjing, Jiangsu). The cells were grown in DMEM (Gibco, Grand Island, USA) supplemented with 10% newborn calf serum (Gibco, Grand Island, USA) and 1% penicillin/streptomycin (Gibco, Grand Island, USA) at 37 °C in a 5% CO2-humidified atmosphere.
Cytotoxicity Assays: Cytotoxicity toward the SKOV-3 cells was determined quantitatively in vitro by the MTT colorimetric assay. Briefly, the SKOV-3 cells were distributed in a 96-well culture plate at a density of 5000 cells/well and incubated for 24 h. Different drug formulations were added in different concentrations for different incubation times. The blank culture medium was treated as control. Cell viability was then determined. Data were computed as the mean ± SD of triplicates. The statistical analysis was conducted using an analysis of variance. Each assay was repeated in triplicate.
Ultrasound in vitro cell: A Pocket Sonovit therapeutic ultrasound apparatus from NewAge (Hongkong, China), with 1 MHz in frequency, 2 W/cm2 in intensity, 1 min in exposure time and 25 mm2 cross sectional area of the probe, was applied for in vitro cell experiments. The facilities were fixed in a supporter with a hole on the upper surface. The tranducer was placed on it and then 1-3 cm thick couplants were coated to form a conductive pathway of ultrasound waves. Finally, the cell culture plate was put onto the surface of the transducer. The ultrasound parameters were adjusted in the range of 1/3 MHz of US frequency, 1.0∼3.0 W/cm2 of US intensity, 1∼60 min of US exposure time, respectively.
Cell uptake: The SKOV-3 and A2780 cells were seeded in a 6-well culture plate at a density of 1×104 cells/well overnight and then exposed to RB NPs (20 μg/mL) and RB NP-cRGD (20 μg/mL). After they were incubated in triplicate for 1 h and 4 h, the cells were washed three times with PBS (1×, pH = 7.4), and the nucleus was labeled with Hoechst 33342 (5 μg/mL) and imaged using confocal laser scanning microscopy (FV1000, Olympus, Japan). To prepare the samples for flow cytometry, the SKOV3 cells were seeded in a 6-well culture plate at a density of 2×105 cells/well overnight and incubated with 20 μg/mL nanoparticles at different formulations for 1 h and 4 h. The blank culture medium was treated as control. After incubation, the cells were washed three times with PBS. Then, the cells were detached with trypsin, washed three times with PBS, resuspended in PBS, and analyzed by flow cytometry (Becton Dickinson, USA). Each assay was repeated in triplicate.
Integrin alpha V (ITGAV) gene knockdown: siRNAs against ITGAV (siITGAVs) and the control siRNA were purchased from Guangzhou RiboBio Co., Ltd. (Guangzhou, China). The three siRNA target sequences against ITGAV were as follows: siITGAV-1, GAA CAT GTC CTC CTT ATA C; siITGAV-2, GAA TAT CGG TTG GAT TAT A; siITGAV-3, GAT GGA TAC TTC TGT GAA A. The siRNAs were transfected into the cells using Lipofectamine™ 2000 (Life Technologies Corporation, Shanghai, China) at 50 nM for 24 h, 48 h, or 72 h according to the manufacturer's instructions.
RNA extraction and quantitative real-time PCR analysis: After incubation, the cells were collected for RNA extraction. The total RNA was extracted according to the instruction for the TRIzol reagent (Invitrogen, Shanghai, China), and the concentration of each RNA sample was measured by a spectrophotometer (NanoDrop 2000, Thermo Scientifc, USA). The cDNA was synthesized according to the manufacturer's instructions (Takara, Dalian, China). For the quantitative real-time PCR (qRT-PCR) assay, the primers for the ITGAV genes were synthesized by Sangon Biotech Co., Ltd. (Shanghai, China). The qRT-PCR reactions were carried out using SYBR Premix Ex Taq (Ribo, Guangzhou, China). The relative amount of the gene transcripts was normalized to actin. The relative expression levels of ITGAV were calculated by the 2-ΔΔCt method. Each assay was repeated in triplicate.
Detection of αvβ3 and αvβ5 integrin receptors: After detachment with trypsin, the cells were washed two times with PBS and resuspended in 100 mL of growth media. Alexa Fluor 488-labeled antibodies against αvβ3 and αvβ5 integrins (5 μg/106 cells, R&D systems, USA) were added to the cell suspension and incubated on ice for 1 h in the dark. The cells were washed three times with cold medium, resuspended in PBS, and analyzed by flow cytometry (Becton Dickinson, USA). Each assay was repeated in triplicate.
Calcein-AM Assay: The cytotoxicity of different formulations of nanoparticles was further confirmed through the Calcein-AM Assay. The SKOV3 cells were seeded in a 6-well culture plate at a density of 1×105 cells/well overnight and treated with 2 mL of NP-cRGD+US, cisplatin, Pt(IV), Pt(IV) NPs, Pt(IV) NPs+US, Pt(IV) NP-cRGD and Pt(IV) NP-cRGD+US for 48 h. The concentration of platinum was 30 μM. Nontreated tumor cells were used as control. After washing with PBS three times, 1 mL of 2.0 μM Calcein-AM (Yeasen, Shanghai, China) was added into each well, and the calcein fluorescence was immediately measured using fluorescence microscopy (CFM-500, Zeiss, Germany). Each assay was repeated in triplicate.
ROS determination: The total induction of ROS in the SKOV3 cells caused by different drugs was determined by fluorescence microscopy and flow cytometry. The SKOV3 cells were seeded in a 6-well culture plate at a density of 2×104 cells/well overnight and treated with 2 mL of NP-cRGD+US, cisplatin, Pt(IV), Pt(IV) NPs, Pt(IV) NPs+US, Pt(IV) NP-cRGD, Pt(IV) NP-cRGD+US at an equivalent concentration of 30 μM for 48 h. Nontreated tumor cells were used as control. The supernatants were removed, and dichlorofluorescein diacetate (DCFH-DA, 5 mg/mL) in DMEM was added. After incubating at 37 °C for 30 min, the cells were washed three times with PBS. Then, the cells were incubated with 4% paraformaldehyde (1 mL per dish) for 5 min and stained with Hoechst 33342 (5 μg/mL) for 5 min. The cells were washed with PBS three times and then observed on a fluorescence microscope (CFM-500, Zeiss, Germany) using Ex/Em of 488/525 nm and 350/460 nm. To prepare the samples for flow cytometry, the SKOV3 cells were seeded in a 6-well culture plate at a density of 2×105 cells/well overnight and incubated with different formulations of nanoparticles at 30 μM for 48 h. The blank culture medium was treated as control. The supernatants were removed, and DCFH-DA (5 mg/mL) in DMEM was added. After incubating for 30 min, the cells were washed three times with PBS. Then, the cells were detached with trypsin, washed three times with PBS, resuspended in PBS, and analyzed by flow cytometry (Becton Dickinson, USA).
The intracellular concentrations of oxygen superoxide and nitric oxide were detected with similar methods. Oxygen superoxide was monitored by a Superoxide Assay Kit (Beyotime Biotechnology Company, Jiangsu, China). Nitric oxide was determined by a DAF-FM DA (3-amino, 4-aminomethyl-2',7'-difluorescein diacetate) assay kit (Beyotime Biotechnology Company, Jiangsu, China) according to the manufacturer's instructions. Each assay was repeated in triplicate.
Intracellular glutathione assay: The SKOV3 cells were cultured in 96-well plates at a density of 5000 cells/well overnight and treated with 100 μL of NP-cRGD+US, cisplatin, Pt(IV), Pt(IV) NPs, Pt(IV) NPs+US, Pt(IV) NP-cRGD and Pt(IV) NP-cRGD+US at an equivalent concentration of 30 μM for 48 h, and the total intracellular glutathione level was determined with an assay kit (Beyotime Biotechnology Company, Jiangsu, China). The procedure was similar to the manufacturer's instructions. Each assay was repeated in triplicate.
Cell apoptosis: The apoptosis analysis of the SKOV3 cells was measured using the Annexin V-FITC Apoptosis Detection kit (Beyotime Biotechnology Company, Jiangsu, China) and analyzed by flow cytometry. Briefly, the SKOV3 cells were seeded in a 6-well culture plate at a density of 2×105 cells/well overnight and treated with 2 mL of NP-cRGD+US, cisplatin, Pt(IV), Pt(IV) NPs, Pt(IV) NPs+US, Pt(IV) NP-cRGD and Pt(IV) NP-cRGD+US for 48 h. The concentration of platinum was 30 μM. The control experiments were performed by adding only culture medium. The collected cell pellets were washed twice with cold PBS, and the cells were stained with annexin V-FITC and propidium iodide (PI, provided in kit) and analyzed by flow cytometry (Becton Dickinson, USA). Each assay was repeated in triplicate.
Mitochondrial membrane potential (Δψm): Mitochondrial membrane potential (Δψm) change was determined using the JC-1 (5,5',6,6'-tetrachloro-1,1',3,3'-tetraethylbenzimidazolyl carbocyanine iodide) assay kit (Beyotime Biotechnology Company, Jiangsu, China). Briefly, the SKOV3 cells were seeded in a 6-well culture plate at a density of 2×105 cells/well overnight and treated with 2 mL of NP-cRGD+US, cisplatin, Pt(IV), Pt(IV) NPs, Pt(IV) NPs+US, Pt(IV) NP-cRGD and Pt(IV) NP-cRGD+US for 48 h. The concentration of platinum was 30 μM. Nontreated tumor cells were used as control. The collected cell pellets were washed twice with cold PBS. Then, the cells were incubated with JC-1 (5 mg/mL) at 37 °C for 20 min in the dark, washed twice with cold PBS and analyzed by flow cytometry (Becton Dickinson, USA). Each assay was repeated in triplicate.
Western Blotting: The western blotting was performed according to standard procedures. The primary antibody against Bcl-2, cytochrome c, cleaved caspase 9 was obtained from Abcam plc (Cambridge, MA, UK). The primary antibody against Bax and β-actin was purchased from Cell Signaling Technology (Danvers, MA, USA). All the secondary antibodies were obtained from Cell Signaling Technology. In order to detect the protein expression of cytochrome c in cytoplasm, the isolation of mitochondrial and cytosolic proteins was performed by the Mitochondria/cytosol Fractionation Kit (Beyotime Biotechnology Company, Jiangsu, China) according to the manufacturer's instructions. Each assay was repeated in triplicate.
Release of cytochrome c from mitochondria: The cytochrome c content released from the mitochondria of the SKOV3 cells to the cytoplasm was detected using a streptavidin-peroxidase immunohistochemical kit (Zhongshan Goldenbridge Biotechnology, Co., Ltd, Beijing, China). Briefly, the SKOV3 cells were cultured in a 6-well culture plate (2×104 cells/well) overnight. Then, the cells were treated with 2 mL of NP-cRGD+US, cisplatin, Pt(IV), Pt(IV) NPs, Pt(IV) NPs+US, Pt(IV) NP-cRGD and Pt(IV) NP-cRGD+US for 48 h. The concentration of platinum was 30 μM. The control experiments were performed by adding culture medium. Subsequently, the cells were fixed in 4% paraformaldehyde for 15 min and then incubated with 3% H2O2 (v/v), blocking buffer (provided in the kit), primary anti-cytochrome c antibody (Abcam, Cambridge, MA, UK), enhanced secondary antibody (provided in the kit) and enhanced streptavidin HRP conjugate (provided in the kit). The samples were visualized using a diaminobenzidine (DAB) kit and observed under a light microscope. Each assay was repeated in triplicate.
Caspase 9 and Caspase 3 activities: Caspase 9 and Caspase 3 activities were measured using colorimetric assay kits (Beyotime Biotechnology Company, Jiangsu, China). Briefly, the SKOV3 cells were cultured in a 6-well culture plate (2×105 cells/well) overnight. Then, the cells were treated with 2 mL of NP-cRGD+US, cisplatin, Pt(IV), Pt(IV) NPs, Pt(IV) NPs+US, Pt(IV) NP-cRGD and Pt(IV) NP-cRGD+US for 48 h. The concentration of platinum was 30 μM. Control experiments were performed by adding the culture medium. The collected cell pellets were washed with PBS and suspended in 100 μL of the supplied lysis buffer for 30 min in an ice bath. The obtained cell lysates were centrifuged at 16000g for 15 min at 4 °C. The supernatant was collected and incubated with the caspase 9 or caspase 3 substrates at 37 °C for 4 h in the dark. The activities of caspase 9 and caspase 3 were then detected by measuring the absorbance at 405 nm using a microplate reader (ELX800, BioTek, USA). Each assay was repeated in triplicate.
Animal Experiments: All the animal experimental procedures were performed according to the protocols approved by the Animal Care and Use Committee of the Shanghai Cancer Institute. The BALB/c nude female mice were obtained from Shanghai Cancer Institute and raised in the SPF animal facility.
Tumor Targeting in vivo: To confirm the targeting delivery of Pt(IV) NP-cRGD in vivo. the drug in the Pt(IV) NP-cRGD was replaced with a lipophilic carbocyanine dye, Cy7, due to its strong fluorescence emission in the NIR region (Ex/Em: 748/780). The biodistribution of the nanoparticles in vivo was then examined using SKOV3 tumor-bearing female BALB/c nude mice. The SKOV3 cells (2×106) were subcutaneously injected into the lower right backs of 4- to 5-week-old female nude mice. Once the tumors reached a volume of 200 mm3, the mice were intravenously administered with 100 μL of Cy7, Cy7 NPs or Cy7 NP-RGD at a Cy7 dose of 1 μg/mL. Whole body fluorescence images were obtained at 1 h, 4 h, 8 h, 24 h, and 48 h after injection using a NightOWL II LB 983 in vivo imaging system (Berthold Technologies, Germany). The mice were sacrificed 48 h after the injection to examine the nanoparticle distribution in the major organs.
Assessment of circulation half-life: Eighteen SD rat were divided into three groups, which were injected intravenously with cisplatin, Pt(IV) NPs and Pt(IV) NPs-cRGD (2 mg/kg), respectively. Blood samples were collected after 10 min, 1 h, 2 h, 4 h, 8 h, 10 h, 12 h, 24 h, 48 h and 72 h. The concentration of platinum was measured with ICP-MS (Leeman Prodigy, USA).
Antitumor Efficacy in vivo: The SKOV3 cells (2×106) were subcutaneously injected into the lower right backs of 4- to 5-week-old female nude mice. Once the tumors reached a volume of 100 mm3, 6 mice/group were randomly allocated to different treatments. The treatments were administered every 3 days for 30 days via i.v. (tail vein) injection of 0.9% NaCl (saline), NP-cRGD+US, cisplatin, Pt(IV) NPs, Pt(IV) NP-cRGD and Pt(IV) NP-cRGD+US at a dose of 2 mg/kg on a Pt basis. To quantify the antitumor efficacy, the tumor volumes were measured according to the following equation: Volume = (length × width2)/2. Three days after the final injection, the mice were sacrificed. The tumors tissues harvested from the different groups were fixed in 4% paraformaldehyde and sectioned into 5-μm slices for hematoxylin and eosin (H&E) staining. In addition, the tumor sections were further stained with procaspase 9 and cleaved caspase 3 antibodies to confirm whether the antitumor effect of Pt(IV) NP-cRGD+US was due to the induction of mitochondrial apoptosis involved with ROS. In addition, the cell proliferation ability of the tumor was measured using Ki67 staining. Finally, the TUNEL assay was conducted to detect the apoptotic cells in the tumor tissue sections.
Statistical analysis: All results were expressed as the mean value ± standard deviation (SD) of three independent measurements. Analysis of variance (ANOVA) was performed when more than two groups were compared, and when the result was significant (P < 0.05), multiple comparisons were performed using Tukey's post hoc test or Sidak's post hoc test. All statistical analyses were performed with GraphPad Prism (7.0). *P < 0.05, **P < 0.01, ***P < 0.005.
Abbreviations
CEUS: contrast-enhanced ultrasound; cRGD: cyclic Arg-Gly-Asp; DAB: diaminobenzidine; DAF-FM DA: 3-amino, 4-aminomethyl-2',7'-difluorescein diacetate; DCFH-DA: dichlorofluorescein diacetate; DLS: dynamic light scattering; DSPE: 1,2-distearoyl-sn-glycero-3-phosphoethanolamine; EDC·HCl: 1-ethyl-(3-dimethylaminopropyl) carbodiimide hydrochloride; FBS: fetal bovine serum; GSH: glutathione; H&E: hematoxylin and eosin; 1H NMR: nuclear magnetic resonance; ICP-MS: Inductively coupled plasma mass spectrometry; MI: mechanical index; MTT: 3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyltetrazolium bromide; NHS: N-hydroxysuccinimide; PBS: phosphate-buffered saline; PEG: Polyethylene glycol; PFH: perfluorohexane; PLGA: poly (lactic-co-glycolic acid); PVA: Poly(vinyl alcohol); qRT-PCR: quantitative real-time PCR; RB: Rhodamine B; RES: reticuloendothelial systems; ROS: reactive oxygen species; siITGAVs : siRNAs against ITGAV; TEM: Transmission electron microscopy; UCAs: ultrasound contrast agents; Δψm: Mitochondrial membrane potential.
Supplementary Material
Supplementary figures.
Acknowledgements
This research was supported by the National Natural Science Foundation of China (No. 81572999, No. 81771839, No. 81773272 and No. 21807071), the State Key Laboratory of Oncogenes and Related Genes (No. 91-17-20).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7-30
2. Reid BM, Permuth JB, Sellers TA. Epidemiology of ovarian cancer: a review. Cancer Biol Med. 2017;14:9-32
3. White JD, Haley MM, DeRose VJ. Multifunctional Pt(II) reagents: covalent modifications of Pt complexes enable diverse structural variation and in-cell detection. Acc Chem Res. 2016;49:56-66
4. Oberoi HS, Nukolova NV, Kabanov AV, Bronich TK. Nanocarriers for delivery of platinum anticancer drugs. Adv Drug Del Rev. 2013;65:1667-85
5. Qi R, Wang Y, Bruno PM, Xiao H, Yu Y, Li T. et al. Nanoparticle conjugates of a highly potent toxin enhance safety and circumvent platinum resistance in ovarian cancer. Nat Commun. 2017;8:2166-77
6. Cascales Campos P, Gil J, Parrilla P. Morbidity and mortality outcomes of cytoreductive surgery and hyperthermic intraperitoneal chemotherapy in patients with primary and recurrent advanced ovarian cancer. Eur J Surg Oncol. 2014;40:970-5
7. Yu C, Ding B, Zhang X, Deng X, Deng K, Cheng Z. et al. Targeted iron nanoparticles with platinum-(IV) prodrugs and anti-EZH2 siRNA show great synergy in combating drug resistance in vitro and in vivo. Biomaterials. 2018;155:112-23
8. He S, Li C, Zhang Q, Ding J, Liang XJ, Chen X. et al. Tailoring platinum(IV) amphiphiles for self-targeting all-in-one assemblies as precise multimodal theranostic nanomedicine. ACS Nano. 2018;12:7272-81
9. Cong YW, Xiao HH, Xiong HJ, Wang ZG, Ding JX, Li C. et al. Dual drug backboned shattering polymeric theranostic nanomedicine for synergistic eradication of patient-derived lung cancer. Adv Mater. 2018;30:1706220-30
10. Johnstone TC, Suntharalingam K, Lippard SJ. The next generation of platinum drugs: targeted Pt(II) agents, nanoparticle delivery, and Pt(IV) prodrugs. Chem Rev. 2016;116:3436-86
11. Huang C, Sun Y, Shen M, Zhang XY, Gao P, Duan YR. Altered cell cycle arrest by multifunctional drug-loaded enzymatically-triggered nanoparticles. ACS Appl Mater Interfaces. 2016;8:1360-70
12. Misra SK, Ghoshal G, Gartia MR, Wu Z, De AK, Ye M. et al. Trimodal therapy: combining hyperthermia with repurposed bexarotene and ultrasound for treating liver cancer. ACS Nano. 2015;9:10695-718
13. Wexselblatt E, Gibson D. What do we know about the reduction of Pt(IV) pro-drugs? J Inorg Biochem. 2012;117:220-9
14. Chen QL, Yang YY, Lin X, Ma W, Chen G, Li WL. et al. Platinum(iv) prodrugs with long lipid chains for drug delivery and overcoming cisplatin resistance. Chem Commun. 2018;54:5369-72
15. Il Yoon Y, Tang W, Chen XY. Ultrasound-mediated diagnosis and therapy based on ultrasound contrast agents. Small Methods. 2017;1:1700173-85
16. Zhou Y, Gu H, Xu Y, Li F, Kuang S, Wang Z. et al. Targeted antiangiogenesis gene therapy using targeted cationic microbubbles conjugated with CD105 antibody compared with untargeted cationic and neutral microbubbles. Theranostics. 2015;5:399-417
17. Sirsi SR, Borden MA. State-of-the-art materials for ultrasound-triggered drug delivery. Adv Drug Del Rev. 2014;72:3-14
18. Wu Q, Zhang MQ, Luo H, Yi T. Self-assembled honokiol-loaded microbubbles in the treatment of ovarian cancer by ultrasound irradiation. J Biomed Nanotechnol. 2018;14:1796-805
19. Wang LV, Hu S. Photoacoustic tomography: in vivo imaging from organelles to organs. Science. 2012;335:1458-62
20. Chen Y, Gao Y, Chen H, Zeng D, Li Y, Zheng Y. et al. Engineering inorganic nanoemulsions/nanoliposomes by fluoride-silica chemistry for efficient delivery/co-delivery of hydrophobic agents. Adv Funct Mater. 2012;22:1586-97
21. Chen Q, Ke HT, Dai ZF, Liu Z. Nanoscale theranostics for physical stimulus-responsive cancer therapies. Biomaterials. 2015;73:214-30
22. Huynh E, Rajora MA, Zheng G. Multimodal micro, nano, and size conversion ultrasound agents for imaging and therapy. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2016;8:796-813
23. Wang R, Zhou Y, Zhang P, Chen Y, Gao W, Xu J. et al. Phase-transitional Fe3O4/perfluorohexane microspheres for magnetic droplet vaporization. Theranostics. 2017;7:846-54
24. Cao Y, Chen Y, Yu T, Guo Y, Liu F, Yao Y. et al. Drug release from phase-changeable nanodroplets triggered by low-intensity focused ultrasound. Theranostics. 2018;8:1327-39
25. Wilson K, Homan K, Emelianov S. Biomedical photoacoustics beyond thermal expansion using triggered nanodroplet vaporization for contrast-enhanced imaging. Nat Commun. 2012;3:618-27
26. Fan X, Guo Y, Wang L, Xiong X, Zhu L, Fang K. Diagnosis of prostate cancer using anti-PSMA aptamer A10-3.2-oriented lipid nanobubbles. Int J Nanomed. 2016;11:3939-50
27. Tian J, Yang F, Cui H, Zhou Y, Ruan X, Gu N. A novel approach to making the gas-filled liposome real: based on the interaction of lipid with free nanobubble within the solution. ACS Appl Mater Interfaces. 2015;7:26579-84
28. Fan X, Wang L, Guo Y, Xiong X, Zhu L, Fang K. Inhibition of prostate cancer growth using doxorubicin assisted by ultrasound-targeted nanobubble destruction. Int J Nanomed. 2016;11:3585-96
29. Huang HY, Hu SH, Hung SY, Chiang CS, Liu HL, Chiu TL. et al. SPIO nanoparticle-stabilized PAA-F127 thermosensitive nanobubbles with MR/US dual-modality imaging and HIFU-triggered drug release for magnetically guided in vivo tumor therapy. J Control Release. 2013;172:118-27
30. Deng LM, Cai XJ, Sheng DL, Yang Y, Strohm EM, Wang ZG. et al. A laser-activated biocompatible theranostic nanoagent for targeted multimodal imaging and photothermal therapy. Theranostics. 2017;7:4410-23
31. Huang HY, Liu HL, Hsu PH, Chiang CS, Tsai CH, Chi HS. et al. A multitheragnostic nanobubble system to induce blood-brain barrier disruption with magnetically guided focused ultrasound. Adv Mater. 2015;27:655-61
32. Huang YK, Su CH, Chen JJ, Chang CT, Tsai YH, Syu SF. et al. Fabrication of silica-coated hollow carbon nanospheres encapsulating Fe3O4 cluster for magnetical and MR imaging guided NIR light triggering hyperthermia and ultrasound imaging. ACS Appl Mater Interfaces. 2016;8:14470-80
33. Hamidi H, Ivaska J. Every step of the way: integrins in cancer progression and metastasis. Nat Rev Cancer. 2018;18:533-48
34. Xu X, Wu J, Liu Y, Yu M, Zhao L, Zhu X. et al. Ultra-pH-responsive and tumor-penetrating nanoplatform for targeted siRNA delivery with robust anti-cancer efficacy. Angew Chem Int Ed Engl. 2016;55:7091-4
35. Yang YY, Wang XF, Liao GC, Liu XQ, Chen QL, Li HM. et al. iRGD-decorated red shift emissive carbon nanodots for tumor targeting fluorescence imaging. J Colloid Interface Sci. 2018;509:515-21
36. Zhou Y, Wang ZG, Chen Y, Shen HX, Luo ZC, Li A. et al. Microbubbles from gas-generating perfluorohexane nanoemulsions for targeted temperature-sensitive ultrasonography and synergistic HIFU ablation of tumors. Adv Mater. 2013;25:4123-30
37. Li W, Hou W, Guo X, Luo L, Li Q, Zhu C. et al. Temperature-controlled, phase-transition ultrasound imaging-guided photothermal-chemotherapy triggered by NIR light. Theranostics. 2018;8:3059-73
38. Guo XS, Cheng Y, Zhao XT, Luo YL, Chen JJ, Yuan WE. Advances in redox-responsive drug delivery systems of tumor microenvironment. J Nanobiotechnol. 2018;16:74-83
39. Miura Y, Takenaka T, Toh K, Wu SR, Nishihara H, Kano MR. et al. Cyclic RGD-linked polymeric micelles for targeted delivery of platinum anticancer drugs to glioblastoma through the blood-brain tumor barrier. ACS Nano. 2013;7:8583-92
40. Lee JY, Carugo D, Crake C, Owen J, de St Victor M, Seth A. et al. Nanoparticle-loaded protein-polymer nanodroplets for improved stability and conversion efficiency in ultrasound imaging and drug delivery. Adv Mater. 2015;27:5484-92
41. Andrisic L, Dudzik D, Barbas C, Milkovic L, Grune T, Zarkovic N. Short overview on metabolomics approach to study pathophysiology of oxidative stress in cancer. Redox Biol. 2018;14:47-58
42. Feng QH, Li YZ, Yang XM, Zhang WX, Hao YW, Zhang HL. et al. Hypoxia-specific therapeutic agents delivery nanotheranostics: A sequential strategy for ultrasound mediated on-demand tritherapies and imaging of cancer. J Control Release. 2018;275:192-200
43. Shi JJ, Chen ZY, Wang BH, Wang L, Lu TT, Zhang ZZ. Reactive oxygen species-manipulated drug release from a smart envelope-type mesoporous titanium nanovehicle for tumor sonodynamic-chemotherapy. ACS Appl Mater Interfaces. 2015;7:28554-65
44. Tan YA, Zhu Y, Zhao Y, Wen LJ, Meng TT, Liu X. et al. Mitochondrial alkaline pH-responsive drug release mediated by Celastrol loaded glycolipid-like micelles for cancer therapy. Biomaterials. 2018;154:169-81
45. Li TY, Xiang WT, Li F, Xu HP. Self-assembly regulated anticancer activity of platinum coordinated selenomethionine. Biomaterials. 2018;157:17-25
46. Ma P, Xiao H, Yu C, Liu J, Cheng Z, Song H. et al. Enhanced cisplatin chemotherapy by iron oxide nanocarrier-mediated generation of highly toxic reactive oxygen species. Nano Lett. 2017;17:928-37
47. Yang Y, Xu LG, Zhu WJ, Feng LZ, Liu JJ, Chen Q. et al. One-pot synthesis of pH-responsive charge-switchable PEGylated nanoscale coordination polymers for improved cancer therapy. Biomaterials. 2018;156:121-33
48. Ruggiero E, Hernandez-Gil J, Mareque-Rivas JC, Salassa L. Near infrared activation of an anticancer Pt-IV complex by Tm-doped upconversion nanoparticles. Chem Commun. 2015;51:2091-4
49. Zhao YJ, Song WX, Wang D, Ran HT, Wang RH, Yao YZ. et al. Phase-shifted PFH@PLGA/Fe3O4 nanocapsules for MRI/US imaging and photothermal therapy with near-infrared irradiation. ACS Appl Mater Interfaces. 2015;7:14231-42
50. Zhu X, Wu J, Shan W, Zhou Z, Liu M, Huang Y. Sub-50 nm nanoparticles with biomimetic surfaces to sequentially overcome the mucosal diffusion barrier and the epithelial absorption barrier. Adv Funct Mater. 2016;26:2728-38
51. Chen J, Gao P, Yuan S, Li R, Ni A, Chu L. et al. Oncolytic adenovirus complexes coated with lipids and calcium phosphate for cancer gene therapy. ACS Nano. 2016;10:11548-60
52. Zhang Y, Anchordoquy TJ. The role of lipid charge density in the serum stability of cationic lipid/DNA complexes. Biochim Biophys Acta. 2004;1663:143-57
53. Wang Q, Zhang XY, Liao HZ, Sun Y, Ding L, Teng YW. et al. Multifunctional shell-core nanoparticles for treatment of multidrug resistance hepatocellular carcinoma. Adv Funct Mater. 2018;28:1706124-40
Author contact
![]() Corresponding authors: yrduanorg; ysunorg
Corresponding authors: yrduanorg; ysunorg
Citation styles
APA
Huang, H., Dong, Y., Zhang, Y., Ru, D., Wu, Z., Zhang, J., Shen, M., Duan, Y., Sun, Y. (2019). GSH-sensitive Pt(IV) prodrug-loaded phase-transitional nanoparticles with a hybrid lipid-polymer shell for precise theranostics against ovarian cancer. Theranostics, 9(4), 1047-1065. https://doi.org/10.7150/thno.29820.
ACS
Huang, H.; Dong, Y.; Zhang, Y.; Ru, D.; Wu, Z.; Zhang, J.; Shen, M.; Duan, Y.; Sun, Y. GSH-sensitive Pt(IV) prodrug-loaded phase-transitional nanoparticles with a hybrid lipid-polymer shell for precise theranostics against ovarian cancer. Theranostics 2019, 9 (4), 1047-1065. DOI: 10.7150/thno.29820.
NLM
Huang H, Dong Y, Zhang Y, Ru D, Wu Z, Zhang J, Shen M, Duan Y, Sun Y. GSH-sensitive Pt(IV) prodrug-loaded phase-transitional nanoparticles with a hybrid lipid-polymer shell for precise theranostics against ovarian cancer. Theranostics 2019; 9(4):1047-1065. doi:10.7150/thno.29820. https://www.thno.org/v09p1047.htm
CSE
Huang H, Dong Y, Zhang Y, Ru D, Wu Z, Zhang J, Shen M, Duan Y, Sun Y. 2019. GSH-sensitive Pt(IV) prodrug-loaded phase-transitional nanoparticles with a hybrid lipid-polymer shell for precise theranostics against ovarian cancer. Theranostics. 9(4):1047-1065.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.