Theranostics
13.3
Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2018; 8(17):4664-4678. doi:10.7150/thno.26619 This issue Cite
Research Paper
Pepducin-mediated cardioprotection via β-arrestin-biased β2-adrenergic receptor-specific signaling
Laurel A. Grisanti1, Toby P. Thomas2, Rhonda L. Carter2, Claudio de Lucia2, Erhe Gao2, Walter J. Koch2, Jeffrey L. Benovic3, Douglas G. Tilley2 ![]()
1. Department of Biomedical Sciences, College of Veterinary Medicine, University of Missouri, Columbia, MO, USA
2. Center for Translational Medicine, Lewis Katz School of Medicine, Temple University, Philadelphia, PA, USA
3. Department of Biochemistry and Molecular Biology, Thomas Jefferson University, Philadelphia, PA, USA
Received 2018-4-11; Accepted 2018-8-21; Published 2018-9-9
Citation:
Grisanti LA, Thomas TP, Carter RL, de Lucia C, Gao E, Koch WJ, Benovic JL, Tilley DG. Pepducin-mediated cardioprotection via β-arrestin-biased β2-adrenergic receptor-specific signaling. Theranostics 2018; 8(17):4664-4678. doi:10.7150/thno.26619. https://www.thno.org/v08p4664.htm
Other stylesAbstract

Reperfusion as a therapeutic intervention for acute myocardial infarction-induced cardiac injury itself induces further cardiomyocyte death. β-arrestin (βarr)-biased β-adrenergic receptor (βAR) activation promotes survival signaling responses in vitro; thus, we hypothesize that this pathway can mitigate cardiomyocyte death at the time of reperfusion to better preserve function. However, a lack of efficacious βarr-biased orthosteric small molecules has prevented investigation into whether this pathway relays protection against ischemic injury in vivo. We recently demonstrated that the pepducin ICL1-9, a small lipidated peptide fragment designed from the first intracellular loop of β2AR, allosterically engaged pro-survival signaling cascades in a βarr-dependent manner in vitro. Thus, in this study we tested whether ICL1-9 relays cardioprotection against ischemia/reperfusion (I/R)-induced injury in vivo.
Methods: Wild-type (WT) C57BL/6, β2AR knockout (KO), βarr1KO and βarr2KO mice received intracardiac injections of either ICL1-9 or a scrambled control pepducin (Scr) at the time of ischemia (30 min) followed by reperfusion for either 24 h, to assess infarct size and cardiomyocyte death, or 4 weeks, to monitor the impact of ICL1-9 on long-term cardiac structure and function. Neonatal rat ventricular myocytes (NRVM) were used to assess the impact of ICL1-9 versus Scr pepducin on cardiomyocyte survival and mitochondrial superoxide formation in response to either serum deprivation or hypoxia/reoxygenation (H/R) in vitro and to investigate the associated mechanism(s).
Results: Intramyocardial injection of ICL1-9 at the time of I/R reduced infarct size, cardiomyocyte death and improved cardiac function in a β2AR- and βarr-dependent manner, which led to improved contractile function early and less fibrotic remodeling over time. Mechanistically, ICL1-9 attenuated mitochondrial superoxide production and promoted cardiomyocyte survival in a RhoA/ROCK-dependent manner. RhoA activation could be detected in cardiomyocytes and whole heart up to 24 h post-treatment, demonstrating the stability of ICL1-9 effects on βarr-dependent β2AR signaling.
Conclusion: Pepducin-based allosteric modulation of βarr-dependent β2AR signaling represents a novel therapeutic approach to reduce reperfusion-induced cardiac injury and relay long-term cardiac remodeling benefits.
Keywords: Pepducin, β-arrestin, β2-adrenergic receptor, cardiac ischemia/reperfusion, cardiomyocyte
Introduction
Reperfusion has substantially reduced patient mortality following acute cardiac ischemic events, such as myocardial infarction (MI), but still induces oxidative damage and further cardiomyocyte death, contributing to ongoing risk of recurrent MI, sudden death, heart failure, and stroke [1-3]. Thus, current drug discovery efforts focus on mitigating cardiomyocyte death via engagement of pro-survival signaling pathways or inhibition of cardiotoxic signaling events at the time of reperfusion. G protein-coupled receptors (GPCRs) constitute the largest class of drug targets, are expressed abundantly in the heart and have been mainstays of therapeutic investigation for the last two decades. β-adrenergic receptors (βARs) in particular play critical roles in the regulation of cardiac function and remodeling in response to acute ischemic injury, classically via G protein-dependent signaling. Both β1AR and β2AR are expressed in cardiomyocytes and although βAR stimulation has been shown capable of engaging pro-survival signaling pathways in cardiomyocytes, including epidermal growth factor receptor (EGFR), extracellular-regulated kinase 1/2 (ERK1/2) and Akt activation [4, 5], β1AR has also been shown to relay cardiomyocyte death in response to ischemic injury through either protein kinase A (PKA) or Ca2+/calmodulin-dependent protein kinase II (CAMKII) signaling [6, 7]. While β-blockers are used as a standard therapy post-infarction to reduce recurrent MI or sudden death [3], the development of pharmacologic tools that can differentially engage proximal βAR isoform-specific signaling pathways at the time of injury to promote cardioprotection and improve cardiac remodeling outcomes has been at the forefront of recent drug discovery efforts.
It has been postulated for some time that proximal engagement of β-arrestin (βarr)-dependent βAR signaling in the absence of G protein activation has the capacity to impart survival benefits in cardiomyocytes [4, 5], but the ability to selectively engage this pathway with small molecules to mitigate I/R-induced myocyte death in vivo has been challenging. Pepducins, small lipidated peptide fragments designed from the amino acid sequences of the intracellular loop (ICL) domains of GPCRs, were originally shown to selectively engage proximal signaling events via allosteric modulation of protease-activated receptors [8]. Since their discovery, pepducins have been demonstrated capable of regulating an increasing cohort of GPCRs in a biased manner and have even begun to be tested in vivo [9]. Recently, we reported the development of β2AR-specific pepducins that selectively engage either G protein- or βarr-dependent pathways [10]. In particular, a cohort of pepducins based on the ICL1 region of β2AR were shown in HEK 293 cells to induce βarr signaling, in the absence of G protein activation, the most potent of which was ICL1-9 [11].
Subsequent analysis of ICL1-9 revealed that it increased the activation of EGFR and ERK1/2 in HEK 293 cells and enhanced isolated adult cardiomyocyte contractility, independent of changes in canonical G protein-dependent signaling [11]. These results highlighted the ability of ICL1-9 to exert functional responses in cardiomyocytes and suggested that it may be capable of promoting cardiomyocyte survival signaling. Therefore, we sought to determine whether ICL1-9 confers cardioprotection against acute ischemic injury and identify the mechanism(s) by which such protection is mediated. Here, we show that intramyocardial injection of ICL1-9 administered at the time of injury reduces I/R-mediated infarct size, cardiomyocyte death and improves cardiac function acutely and long-term fibrotic remodeling. Further, we demonstrate that although the pro-survival effect of ICL1-9 in I/R-treated hearts is dependent upon β2AR and βarr1 or βarr2, the downstream survival signaling is mechanistically not relayed via EGFR, ERK1/2 or Akt, but is dependent instead upon the activation of RhoA. This is the first study to demonstrate the ability of a βarr-biased β2AR-selective allosteric modulator to promote cardiomyocyte survival signaling and impart structural and functional benefits following I/R, offering a novel therapeutic approach to reduce reperfusion-induced cardiac injury.
Methods
Experimental animals
WT C57BL/6 mice as well as β2ARKO, βarr1KO and βarr2KO mice, all backcrossed onto a C57BL/6 background, were used in this study (equal numbers male and female, randomly assigned to treatment/surgery groups, 8-12 weeks of age). Numbers of animals used per experimental condition are listed in figure legends. Animal procedures were performed in accordance with the Institutional Animal Care and Use Committee at Temple University and in accordance to the NIH Guidelines on the Use of Laboratory Animals.
Intracardiac injection and ischemia/reperfusion (I/R) surgery
Three evenly spaced injections (10 μL each at a concentration of 1 µg/µL) were administered into the left ventricle (LV) with either ICL1-9 (Palm-TAIAKFERLQTVTNYFIT-NH2, Peptide 2.0 Inc.) or scrambled (Scr) pepducin control (Palm-TTKYFQTINVLFAIRATE-NH2, Peptide 2.0 Inc.). Pepducins were injected during needle retraction to enable their administration throughout the LV wall in both the remote zone and area at risk (AAR) of the LV. Injections occurred ~1 min prior to left coronary artery ligation. I/R injury was induced as previously described [12] with the surgeon blinded to the treatment groups. Briefly, mice were anesthetized via 2% isoflurane inhalation, a small skin incision was made and the pectoral muscles retracted to expose the fourth intercostal space where a small hole was formed to pop out the heart. The left coronary artery was sutured ~2-3 mm from its origin with a slipknot and the heart was placed back into the intrathoracic space followed by closure of muscle and skin. Sham animals received the same surgical procedures without tying of the suture. After 30 min of ischemia, the slipknot was released to allow reperfusion of the myocardium. Animals received a single dose (0.3 mg/kg) of buprenorphine immediately following surgery.
LV area at risk, infarct size and immunohistochemistry
I/R-induced injury was detected by Evans blue/triphenyltetrazolium chloride (TTC) double staining. Hearts were excised after 24 h reperfusion and the ligature around the LCA was retied. Aortas were injected with 0.2 mL of 2% Evans blue dye and hearts were extracted and sliced into five 1.0 mm-thick sections. The sections were incubated with 1% TTC (Sigma) in phosphate buffered saline (PBS; pH 7.4) for 5 min at 37 °C. Slices were immediately imaged for infarct size using NIS Elements software for recording images and image analysis. Myocardial infarcts were imaged using NIS Elements software to measure the unstained portion (area at risk; AAR), the Evans blue-stained portion (area not at risk; ANAR), the TTC-stained area (red) and TTC-negative stained area (white; infarcted myocardium), expressed as a % of AAR. For Masson trichrome and Wheat germ agglutinin (WGA) staining, excised hearts were fixed in 4% paraformaldehyde, paraffin embedded and sectioned at 5 μm thickness. Deparaffinized sections were stained for Masson trichrome (Sigma-Aldrich) or wheat germ agglutinin (WGA, Vector Labs). An In Situ Cell Death Detection Kit, TMR Red (Roche Diagnostics; Mannheim, Germany) was used to measure cell death via the TUNEL (TdT-mediated dUTP-X nick end labeling) method in deparaffinized heart sections. Deparaffinized sections were incubated with proteinase K and DNA strand breaks were labeled according to the manufacturer's instructions using tetra-methyl-rhodamine-dUTP. Hearts were counterstained with troponin I (TNNI, 1:100; Cell Signaling; Danvers, MA) to identify cardiomyocytes. Cells were visualized at 20X magnification using a Nikon Eclipse microscope and TUNEL-positive cardiomyocytes were calculated in relation to the number of DAPI-stained cardiomyocyte nuclei from 10 random fields in the ANAR and AAR.
Echocardiography
Cardiac function was performed at baseline, 1, 3, 7, 14, 21 and 28 days post-I/R via transthoracic two-dimensional echocardiography using a VisualSonics Vevo 2100 System with a 12 MHz probe on mice anesthetized with isoflurane (1.5%) and body temperature was maintained on a heated table with embedded ECG leads to monitor heart rate, temperature of the animal, electrocardiogram and breathing. The parasternal short-axis views of the heart were obtained in B-mode by placing the transducer in the parasternal long-axis and rotating the transducer 90° in a clockwise fashion to find the parasternal short-axis. For measurements, M-mode echocardiography was performed in the parasternal short-axis view at the level of the papillary muscle to assess several cardiac parameters including left ventricular (LV) volumes and internal diameters (ID) in systole (s) and diastole (d), wall thickness, LV fractional shortening and ejection fraction. Percent fractional shortening was calculated using the equation (LVIDd - LVIDs) / LVIDd × 100%. Percent ejection fraction was calculated using the equation (LVvold - LVvols) / LVvold × 100%. The assessor was blinded to the groups and the data was compiled into treatment and surgical groups only after the analyses were completed.
Primary neonatal rat ventricular myocyte (NRVM) isolation
Primary neonatal cardiomyocytes were prepared from 1 to 2 day old Sprague Dawley rat pups (Harlan Laboratories; Indianapolis, IN) by manual and enzymatic digestion. Hearts were excised and placed in sterile ADS solution (116 mM NaCl, 20 mM HEPES, 80 μM Na2HPO4, 56 mM glucose, 5.4 mM KCl, 80 mM MgSO4; pH 7.4). Blood and connective tissue were removed and the ventricles were minced and subjected to five 15 min enzymatic digestions using collagenase II (Worthington; Lakewood, NJ) and pancreatin. Myocytes and fibroblasts were separated via pre-plating for 2 h. NRVM were cultured overnight in F-10 media containing 10% horse serum, 5% fetal bovine serum (FBS) and 1% PSF at 37 °C in a humidified incubator with 5% CO2. After 24 h, the media was replaced with F-10 media containing 5% FBS. At least 3 independent cell preparations were used per experimental condition.
Radioligand binding
The density of βAR on cardiac membranes, prepared as previously described [13], or NRVM were determined by saturation binding experiments. Cardiac membranes (25 μg of protein) or single cell suspensions (1×105 cells) of NRVM were incubated with [125I]-cyanopindolol ([125I]-CYP; 200 pM; PerkinElmer) in binding buffer (75 mM Tris, pH 7.4, 2 mM EDTA, 12.5 mM MgCl2, 1 μg/mL aprotinin, 1 μg/mL leupeptin). Incubations were performed in the presence or absence of propranolol (10 μM) to determine non-specific binding of CGP20712A and ICI 118,551 (100 nM) to determine βAR subtype expression. Reactions were performed in a 250 μL volume and allowed to equilibrate at 37 °C for 1 h before filtering through a glass fiber filter (Whatman GF/C; Brandel). Each filter was washed five times with 5 mL of ice-cold wash buffer (10 mM Tris, pH 7.4, 10 mM EDTA) to remove unbound drug. The amount of total and nonspecific radiolabel bound to cells was determined on a gamma counter and receptor density was normalized to cell number (NRVM) or protein amount (cardiac membranes). All assays were performed in duplicate.
Caspase 3/7 activity assay and TUNEL measurements
Caspase 3/7 activity was measured using a Caspase-Glo® 3/7 Assay according to the manufacturer's instructions (Promega; Madison, WI). In brief, NRVM were plated at 10,000 cells/well in white-walled 96-well plates. Following stabilization in culture, cells underwent 3 h serum starvation followed by inhibitor pre-treatment: PD98059 (10 μM for 10 min), LY294002 (10 μM for 10 min), AG1478 (10 μM for 10 min) or rhosin (10 μM for 1 h). NRVM were subsequently treated with Scr or ICL1-9 pepducin (10 μM each) for 24 h and 100 μL Caspase-Glo® 3/7 Reagent was added to each well. Plates were incubated for 1 h prior to reading luminescence. For TUNEL assays, NRVM were plated on fibronectin-coated glass-bottom dishes and underwent serum deprivation ± Scr or ICL1-9 pepducin treatment with or without rhosin (10 µM each) for 24 h, following which they were fixed with 4% paraformaldehyde and underwent staining with TUNEL (red) and DAPI (blue) as described above as well as α-sarcomeric actin (αSrA, green; 1:500 overnight incubation (Sigma) followed by goat anti-mouse Alexa Fluor-488 1:1000 for 1 h (ThermoFisher)). Images were captured using a Zeiss Axio Observer Z1 fluorescent microscope with a 40X oil immersion lens. TUNEL-positive α-SrA-positive cardiomyocytes were calculated as a % of the total α-SrA-positive cardiomyocytes from 5 images per dish.
Immunoblot analysis
Heart samples or primary cardiomyocytes were homogenized in RIPA buffer containing 1X HALT protease inhibitor cocktail (78437; Thermo Scientific; Rockford, IL) and phosphatase inhibitor cocktail set IV (524628; Calbiochem, USA). Equal amounts of lysates were resolved by SDS-polyacrylamide gel electrophoresis (10% gels) and transferred to Immobilon-PSQ polyvinylidene fluoride 0.2 μm pore size membranes (Millipore; Billerica, MA). Odyssey Blocking Buffer (LI-COR Biosciences; Lincoln, NE) was used to prevent non-specific binding. Immunoblotting was performed overnight at 4 °C with diluted antibodies against phospho-ERK1/2 (Cell Signaling), total ERK1/2 (1:5000; Cell signaling), phospho-Akt (1:1000; Cell Signaling), total-Akt (1:1000; Cell Signaling), RhoA (1:500; Cytoskeleton, Inc) or GAPDH (1:1000; Cell Signaling). After washing with TBS-T, membranes were incubated at room temperature for 60 min with the appropriate diluted secondary antibody (IRDye680 Donkey anti-rabbit IgG (H + L) at 1:20,000; IRDye800CW Goat anti-mouse IgG (H + L) at 1:15,000; LI-COR Biosciences; IRDye680 Donkey anti-goat IgG (H+L) at 1:20,000). Bound antibody was detected using the LI-COR Biosciences Odyssey System (LI-COR Biosciences).
RhoA activity assay
Activated RhoA was determined using a RhoA Pull-Down Activation Assay Biochem Kit (Cytoskeleton, Inc) according to the manufacturer's instructions. In brief, NRVM were washed with ice-cold PBS prior to lysis or hearts were homogenized in lysis buffer and centrifuged at 10,000 ×g for 1 min to clarify samples. Equal amounts of protein (1000 μg for NRVM and 3000 μg for hearts) were added to 15 μg GST-Rho binding domain (RBD) beads and incubated for 1 h at 4 °C. The beads were washed and boiled for 2 min at 95 °C in 2X Laemmli Buffer and samples separated by SDS-PAGE as described above. Precipitated GTP-bound RhoA was normalized to total RhoA and GAPDH in the whole cell lysate (100 µg loaded).
In vitro hypoxia/reoxygenation (H/R)
NRVM were pre-treated with ICI 118,551 (100 nM for 10 min), Y-27632 (10 μM for 10 min) or rhosin (10 μM for 1 h) prior to treatment with Scr or ICL1-9 pepducin (10 μM each). Cells were subjected to hypoxia in Hanks Balanced Salt Solution supplemented with 1 mM MgCl2, 2 mM CaCl2 and 10 mM HEPES under 95% N2 and 5% CO2 at 37 °C for 4 h in the presence of inhibitors and pepducins. Cells were then changed to fresh culture medium (F-10 media containing 5% FBS and 1% PSF) and cultured under normoxic conditions at 37 °C with 1% O2 and 5% CO2 for 20 h.
Mitochondrial superoxide production
Mitochondrial superoxide production was measured using MitoSOX™ in NRVM. Cells were loaded for 10 min with 5 μM MitoSox™ in PBS with Ca2+ and Mg2+ at 37 °C. Cells were washed three times and counterstained with DAPI. Cells were imaged in warm PBS with Ca2+ and Mg2+ at 100X magnification on a Nikon Eclipse microscope and fluorescence was calculated on a plate reader with excitation/emission of 510/580 nm. MitoSOX fluorescence was normalized to DAPI.
Statistical analysis
Data presented are expressed as mean ± standard error of the mean (SEM). Statistical analyses were performed using a two-tailed unpaired Student's t-test, a one-way ANOVA with Tukey's multiple comparison test or a two-way repeated measures ANOVA with Bonferroni post test as appropriate using Prism 5.0 software (GraphPad Software; San Diego, CA) with n and p values indicated in figure legends. No statistical differences were observed between male and female animals, thus all data is a combination of both sexes within treatment/surgical groups.
Results
ICL1-9 reduces cardiac injury after I/R
To determine if direct cardiac administration of a pepducin results in sustained localization within the myocardium in vivo, we administered TMR-labeled ICL1-9 (ICL1-9TMR) via intracardiac injection and monitored its localization over time. Within 3 h of injection, ICL1-9TMR localized within or just proximal to the injection track; however, more diffuse localization distal to the injection site was observed at 6 and 24 h (Figure 1A), both within cardiomyocytes and interstitially, but was largely undetectable by 72 h post-injection (not shown). We previously observed that ICL1-9 induces β2AR internalization over time in a HEK 293 cell receptor overexpression model [11]; therefore, we performed radioligand binding analysis to determine whether downregulation of β2AR from cardiac membranes occurs in response to the long-lasting presence of ICL1-9 in the heart. Neither β1AR nor β2AR densities were altered by ICL1-9 compared to scrambled pepducin control (Scr) 24 h after injection (Figure 1B). Together, these data suggest that ICL1-9 may be capable of imparting effects in the heart that last hours to days, well within the timeframe in which cardiac damage occurs in response to acute ischemic injury. Thus, to determine whether ICL1-9 administration confers protection against acute ischemic injury, wild-type C57Bl6/J (WT) mice were given intramyocardial injections of either Scr or ICL1-9 pepducin followed by 30 min ischemia and 24 h reperfusion (I/R), after which the hearts were excised and infarct size assessed (Figure 1C). Although there was no difference in the area at risk (AAR) between the Scr and ICL1-9 groups (Figure 1D), ICL1-9-treated mice had a decreased infarct size within the AAR (Figure 1E), correspondingly decreased cardiomyocyte death within the AAR (Figure 1F) and reciprocally enhanced cardiac contractility (Figure 1G).
ICL1-9-mediated cardioprotection requires β2AR and β-arrestins
Since we previously demonstrated that ICL1-9-induced changes in isolated adult cardiomyocyte function were dependent upon β2AR and βarr expression [11], we tested their requirement for relaying the protective effect of ICL1-9 against I/R in vivo. Cardiac AAR and infarct area values in response to I/R were not statistically different between the various KO mice and their WT controls treated with Scr pepducin. However, in contrast to WT mice, intracardiac injection of ICL1-9 was unable to reduce infarct size or improve cardiomyocyte survival within the AAR in β2ARKO mice (Figure 2A-E). Similarly, ICL1-9 was unable to impart protection against I/R in either βarr1KO or βarr2KO mice as compared to WT control mice (Figure 2F-J). Therefore, direct intracardiac administration of ICL1-9 imparts protection against I/R-induced cardiomyocyte death to attenuate the acute loss of contractile function via βarr-dependent β2AR signaling.
ICL1-9 relays improvements in cardiac remodeling and function after I/R
We next sought to determine whether the acute cardioprotection offered by ICL1-9 translated to improvements in cardiac structure and function following I/R. WT mice that received intracardiac injection of Scr of ICL1-9 pepducin at the time of either sham or I/R surgery were monitored via echocardiography for up to 4 weeks (Figure S1, Table 1 and Table 2). Analysis of LV internal diameter and volume indicated that ICL1-9-treated hearts displayed progressively less post-I/R dilation than Scr-treated hearts over time (Table 1). However, ICL1-9-mediated improvements in both fractional shortening and ejection fraction occurred only at early post-I/R timepoints, whereas Scr pepducin-treated hearts were able to recover contractile function by the end of 4 weeks. This long-term functional recovery to I/R is similar to that observed in previous studies using the same conditions of I/R [12, 14, 15]. Notably, wall thicknesses were unchanged between Scr- and ICL1-9 pepducin-treated groups (Table 2). Indeed, gravimetric analysis showed similar changes in size of the Scr- and ICL1-9-treated hearts at 4 weeks post-I/R (Figure 3A), and wheat germ agglutinin staining demonstrated similar cardiomyocyte cross sectional area changes in response to I/R regardless of pepducin treatment (Figure 3B-C). Although ICL1-9 treatment did not influence cardiac hypertrophy, it conveyed a dramatic impact on fibrosis. Masson Trichrome staining revealed a significantly reduced fibrotic infarct scar area in ICL1-9-treated hearts as compared to the Scr pepducin control at 4 weeks post-I/R (Figure 3D-E), which likely reflects the decreased cardiomyocyte death imparted by ICL1-9 immediately following I/R. These results demonstrate that the acute cardioprotective effect of even a single administration of ICL1-9 at the time of I/R is sufficient to relay long-term benefits on cardiac remodeling.
Figure 1
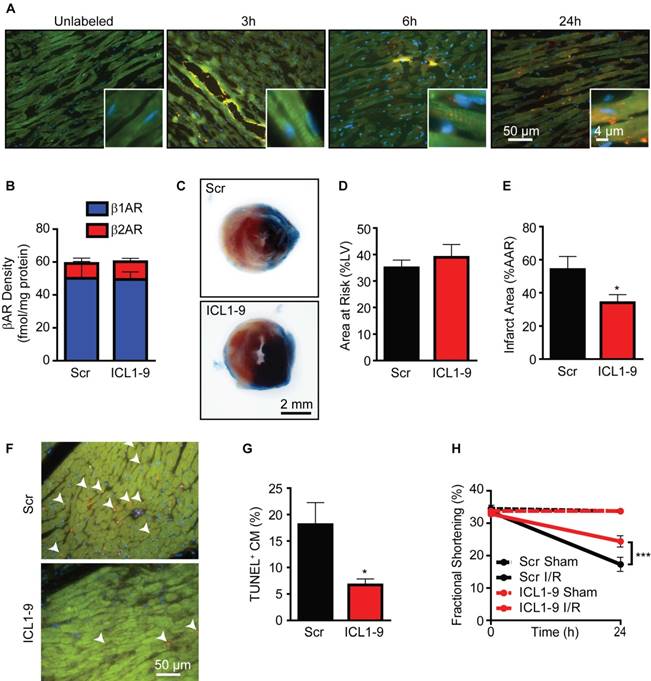
ICL1-9 attenuates cardiac injury after I/R. (A) WT mice received an intracardiac injection of unlabeled ICL1-9 or TMR-conjugated ICL1-9 (red). Representative heart sections from at least 4 independent mice per timepoint. Sections were stained with troponin I (TNNI, green) to identify cardiomyocytes and DAPI (blue) to label nuclei. n=4 ICL1-9 (Unlabeled), n=5 ICL1-9TMR at each timepoint. (B) Radioligand binding analysis of βAR isoform expression in the LV of WT mice 24 h after intracardiac injection of Scr or ICL1-9 pepducin. n=5 Scr, n=6 ICL1-9, no significant differences observed, two-tailed unpaired t-test for each isoform. (C) Representative Evan's Blue/TTC staining of hearts from mice receiving Scr or ICL1-9 pepducin injection followed by 30 min ischemia/24 h reperfusion (I/R). Quantification of (D) the area at risk (AAR) expressed as a percentage of the total LV area and (E) infarct size expressed as a percentage of the area at risk. n=9 each, * p < 0.05, two-tailed unpaired t-test. (F) Representative TUNEL staining (red) from the AAR of hearts injected with ICL1-9 or Scr control pepducin prior to I/R. Sections were counterstained with troponin I (TNNI, green) to identify cardiomyocytes and DAPI (blue) to label nuclei and are quantified as a percentage of the TUNEL-positive cardiomyocytes in (G). n=9 each, * p < 0.05, two-tailed unpaired t-test. (H) LV fractional shortening (FS) was measured at the short axis from M-mode using VisualSonics Analysis software. n=6 Scr Sham, n=6 ICL1-9 Sham, n=9 Scr I/R, n=10 ICL1-9 I/R, *** p < 0.001, two-way repeated measures ANOVA with Bonferroni post test.
ICL1-9 protects against cardiomyocyte death in vitro
Beyond a requirement for β2AR and either βarr1 or βarr2, the mechanism by which ICL1-9 promotes cardiomyocyte survival is unknown. We previously demonstrated that ICL1-9 increases the phosphorylation of EGFR and ERK1/2 in HEK 293 cells [11] and that βAR-mediated transactivation of EGFR and downstream engagement of ERK1/2 and Akt provides protection against cardiomyocyte death in vitro [4]; thus, we hypothesized that this pathway would be responsible for relaying the protective effects of ICL1-9. In order to investigate the mechanism responsible for the protective effects of ICL1-9, we employed primary neonatal rat ventricular myocytes (NRVM), which we initially treated with ICL1-9 for increasing timepoints, to determine whether it impacts βAR membrane density in cardiomyocytes in vitro. Treatment of NRVM for either 1 h (Figure 4A) or 24 h (Figure 4B) with ICL1-9 did not reduce membrane density of either β1AR or β2AR, although decreased β1AR density in response to ISO was detected at both timepoints. These data recapitulate our in vivo data, suggesting that ICL1-9 may be capable of imparting long-lasting effects via regulation of membrane-localized β2AR in cardiomyocytes.
Figure 2
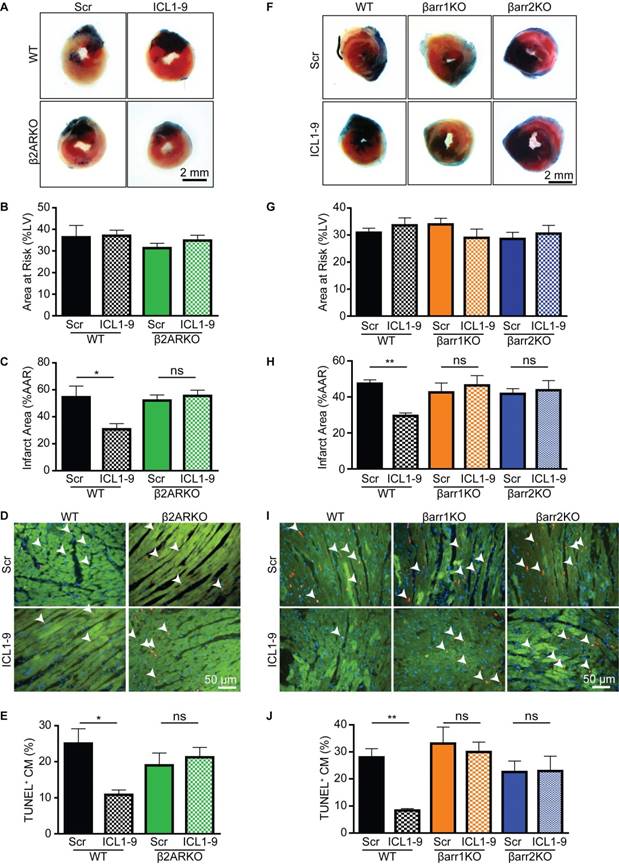
ICL1-9-mediated protection against I/R-induced injury is β2AR- and β-arrestin-dependent. Representative images of hearts from WT controls versus β2ARKO (A) or βarr1KO and βarr2KO (F) mice that received Scr or ICL1-9 pepducin at the time of I/R, stained with Evan's Blue/TTC 24 h following I/R injury. Quantification of (B, G) the area at risk (AAR) expressed as percentage of the total LV area and (C, H) infarct size expressed as a percentage of the AAR for WT controls versus β2ARKO (B-C) or βarr1KO and βarr2KO (G-H) mouse hearts. Representative TUNEL staining (red) within the AAR of WT controls versus β2ARKO (D) or βarr1KO and βarr2KO (I) mouse hearts. Hearts were counterstained for troponin I (TNNI, green) to label cardiomyocytes and DAPI (blue) to identify nuclei. Quantification of TUNEL+ cardiomyocytes expressed as a percentage of the total number of cardiomyocytes for WT versus β2ARKO (E) or βarr1KO and βarr2KO (J) mouse hearts. For (A-E): n=7 Scr WT, n=7 ICL1-9 WT, n=10 Scr β2ARKO, n=11 ICL1-9 β2ARKO. For (F-J): n=18 Scr WT, n=10 ICL1-9 WT, n=10 Scr βarr1KO, n=10 ICL1-9 βarr1KO, n=11 Scr βarr2KO, n=10 ICL1-9 βarr2KO. * p < 0.05, ** p < 0.01, one-way ANOVA with Tukey's multiple comparison test.
Figure 3
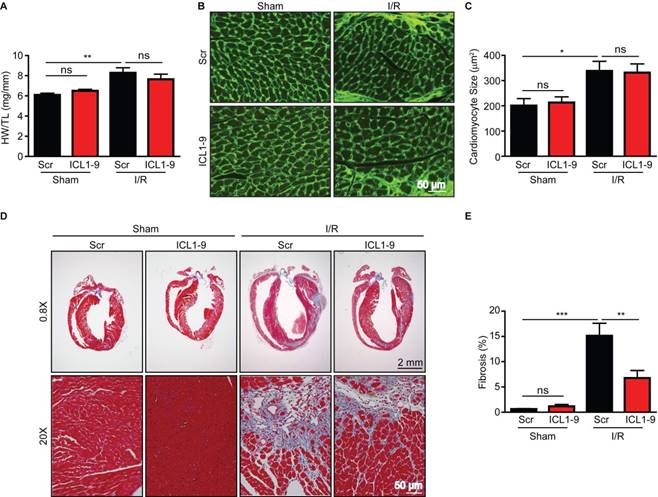
ICL1-9 reduces fibrotic remodeling following I/R. Cardiac remodeling responses to Scr or ICL1-9 pepducin were assessed via gravimetric and immunohistological analyses 4 weeks post-I/R. (A) Quantification of gravimetric analysis of heart weight normalized to tibia length. n=8 Sham Scr, n=8 Sham ICL1-9, n=12 I/R Scr, n=11 I/R ICL1-9, ns = not significant, ** p < 0.01, one-way ANOVA with Tukey's multiple comparison test. (B) Representative wheat germ agglutinin (WGA) staining of hearts to assess cardiomyocyte cross sectional area. (C) Quantification of cardiomyocyte size from WGA-stained hearts. n=7 Scr Sham, n=8 ICL1-9 Sham, n=12 Scr I/R, n=11 ICL1-9 I/R, ns = not significant, * p < 0.05, one-way ANOVA with Tukey's multiple comparison test. (D) Representative Masson trichrome staining of Scr or ICL1-9 pepducin-injected hearts following I/R injury. (E) Quantification of fibrosis from Masson trichrome-stained hearts, expressed as a percentage of the total LV area. n=8 Sham Scr, n=8 Sham ICL1-9, n=10 I/R Scr, n=10 I/R ICL1-9, ns = not significant, ** p < 0.01, *** p < 0.001, one-way ANOVA with Tukey's multiple comparison test.
Table 1
Echocardiographic contractile measurements
| LV Volume-systolic (μL) | LV Volume-diastolic (μL) | LVID-systolic (mm) | LVID-diastolic (mm) | Ejection Fraction (%) | Fractional Shortening (%) | Cardiac Output (mL/min) | Stroke Volume (µL) | Heart Rate (BPM) | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Scr Sham | Baseline | 18.7±1.9 | 50.7±2.7 | 2.39±0.11 | 3.49±0.08 | 65.1±1.9 | 33.8±1.2 | 15.2±0.9 | 33.4±1.7 | 460±9 |
| n=16 | 1 d | 17.7±1.8 | 45.4±2.3 | 2.33±0.11 | 3.32±0.10 | 60.2±1.9 | 33.6±1.0 | 16.3±1.5 | 26.9±1.2 | 527±15 |
| 3 d | 17.2±1.4 | 49.3±1.6 | 2.35±0.11 | 3.47±0.03 | 63.2±2.4 | 33.5±1.1 | 14.7±1.6 | 32.0±1.1 | 455±34 | |
| 7 d | 18.2±1.1 | 50.7±2.3 | 2.34±0.05 | 3.41±0.07 | 64.1±1.2 | 34.2±0.8 | 16.6±1.0 | 32.9±1.6 | 506±15 | |
| 14 d | 20.6±1.2 | 54.8±2.4 | 2.46±0.06 | 3.58±0.06 | 62.6±1.0 | 33.2±0.7 | 16.9±0.8 | 34.1±1.4 | 493±8 | |
| 21 d | 20.3±1.4 | 57.5±2.6 | 2.49±0.07 | 3.58±0.07 | 65.0±1.3 | 34.9±1.0 | 18.9±0.8 | 37.3±1.5 | 507±9 | |
| 28 d | 20.7±2.0 | 56.5±2.9 | 2.50±0.09 | 3.57±0.07 | 63.2±1.6 | 31.6±1.8 | 18.5±0.7 | 35.8±1.1 | 514±9 | |
| Scr I/R | Baseline | 21.4±1.2 | 56.1±2.1 | 2.47±0.07 | 3.56±0.07 | 63.2±1.2 | 33.7±1.0 | 15.2±0.8 | 34.2±1.5 | 443±10 |
| n=19 | 1 d | 31.1±2.7 | 58.0±3.4 | 2.96±0.10 | 3.70±0.10 | 40.4±1.7 | 19.5±0.9 | 12.0±0.6 | 24.5±1.1 | 485±11 |
| 3 d | 24.8±3.6 | 49.4±8.1 | 2.74±0.18 | 3.43±0.25 | 41.2±3.1 | 19.2±1.4 | 11.8±1.4 | 23.5±2.8 | 501±7 | |
| 7 d | 40.6±3.5 | 73.8±4.7 | 3.20±0.13 | 3.99±0.11 | 44.1±2.5 | 22.0±1.5 | 15.4±0.9 | 31.4±2.1 | 489±12 | |
| 14 d | 37.8±4.7 | 73.6±5.5 | 3.04±0.15 | 3.95±0.14 | 50.8±2.1 | 25.2±1.2 | 16.9±0.9 | 34.9±2.1 | 507±10 | |
| 21 d | 55.0±0.5 | 90.4±11.5 | 3.68±0.29 | 4.13±0.34 | 46.4±4.6 | 24.6±2.6 | 18.3±0.9 | 35.4±2.0 | 518±9 | |
| 28 d | 59.5±3.3 | 105.7±14.7 | 3.76±0.33 | 4.31±0.23 | 51.1±4.2 | 24.3±2.5 | 21.3±1.8 | 40.9±2.8 | 512±12 | |
| ICL1-9 Sham | Baseline | 20.2±1.9 | 53.9±2.2 | 2.60±0.12 | 3.75±0.06 | 64.9±2.4 | 34.8±1.1 | 16.0±0.7 | 35.3±1.4 | 451±9 |
| n=16 | 1 d | 20.4±1.7 | 49.6±3.0 | 2.64±0.20 | 3.47±0.12 | 59.0±2.4 | 32.4±1.0 | 15.5±2.3 | 28.9±2.1 | 477±16 |
| 3 d | 16.1±1.2 | 45.2±2.6 | 2.25±0.05 | 3.37±0.09 | 63.7±0.8 | 33.7±0.4 | 13.2±0.7 | 29.1±1.6 | 456±2 | |
| 7 d | 23.5±1.2 | 58.9±1.1 | 2.67±0.08 | 3.71±0.05 | 62.0±1.2 | 32.9±0.8 | 19.2±0.8 | 38.5±0.9 | 498±15 | |
| 14 d | 22.9±1.2 | 57.8±1.9 | 2.53±0.06 | 3.67±0.07 | 63.3±1.0 | 34.0±0.7 | 19.6±1.2 | 38.0±1.4 | 498±15 | |
| 21 d | 24.9±3.5 | 60.3±4.5 | 2.69±0.12 | 3.67±0.10 | 58.9±2.8 | 31.0±1.4 | 18.5±1.1 | 35.3±2.0 | 525±16 | |
| 28 d | 23.4±1.6 | 62.3±2.8 | 2.60±0.07 | 3.73±0.07 | 60.3±1.5 | 32.9±0.9 | 19.8±0.6 | 38.3±1.6 | 520±10 | |
| ICL1-9 I/R | Baseline | 19.4±1.3 | 50.8±1.9 | 2.64±0.09 | 3.46±0.07 | 64.4±1.8 | 33.7±1.1 | 14.6±0.5 | 33.6±0.9 | 438±9 |
| n=17 | 1 d | 30.1±3.1 | 51.7±3.5 | 2.89±0.12 | 3.45±0.11 | 48.4±2.1* | 23.6±1.3 | 12.4±0.8 | 24.4±1.1 | 478±12 |
| 3 d | 27.8±3.3 | 56.1±4.4 | 2.72±0.10 | 3.54±0.14 | 49.0±1.8* | 24.3±1.1* | 14.0±1.0 | 28.2±2.2 | 499±13 | |
| 7 d | 27.7±2.0* | 64.0±3.2 | 2.68±0.07* | 3.80±0.08 | 55.3±1.4*** | 28.5±0.9*** | 17.6±0.5 | 34.9±1.3 | 515±5 | |
| 14 d | 28.3±1.7 | 63.7±2.6 | 2.80±0.08 | 3.80±0.07 | 55.4±1.5 | 28.5±1.0 | 17.7±1.0 | 37.1±2.0 | 491±10 | |
| 21 d | 29.1±4.2*** | 69.9±6.4* | 2.94±0.10** | 3.96±0.16 | 54.7±2.4 | 30.6±3.4* | 19.6±2.0 | 40.8±6.0 | 491±29 | |
| 28 d | 37.5±3.2*** | 81.3±5.5** | 3.14±0.12* | 4.19±0.12 | 53.4±1.8 | 27.8±1.3 | 22.2±1.6 | 43.8±3.5 | 512±12 |
* p < 0.05, ** p < 0.01, *** p < 0.001 versus Scr I/R at same timepoint, two-way repeated measures ANOVA with Bonferroni post test.
Table 2
Echocardiographic wall thickness measurements
| LVAW-systolic (mm) | LVAW-diastolic (mm) | LVPW-systolic (mm) | LVPW-diastolic (mm) | ||
|---|---|---|---|---|---|
| Scr Sham | Baseline | 1.28±0.04 | 0.94±0.04 | 1.20±0.04 | 0.76±0.02 |
| n=16 | 1 d | 1.52±0.08 | 1.11±0.07 | 1.33±0.12 | 0.92±0.07 |
| 3 d | 1.64±0.08 | 1.11±0.07 | 1.25±0.19 | 0.92±0.16 | |
| 7 d | 1.27±0.05 | 0.91±0.04 | 1.23±0.04 | 0.84±0.03 | |
| 14 d | 1.32±0.07 | 0.95±0.05 | 1.27±0.04 | 0.87±0.04 | |
| 21 d | 1.29±0.06 | 0.95±0.05 | 1.18±0.06 | 0.89±0.05 | |
| 28 d | 1.28±0.04 | 0.95±0.03 | 1.10±0.06 | 0.81±0.03 | |
| Scr I/R | Baseline | 1.34±0.03 | 0.96±0.03 | 1.21±0.04 | 0.87±0.03 |
| n=19 | 1 d | 1.24±0.07 | 1.00±0.05 | 1.15±0.04 | 0.96±0.04 |
| 3 d | 1.57±0.12 | 1.28±0.16 | 1.21±0.06 | 1.05±0.11 | |
| 7 d | 1.20±0.07 | 0.91±0.04 | 1.07±0.05 | 0.85±0.03 | |
| 14 d | 1.28±0.07 | 0.96±0.05 | 1.15±0.06 | 0.86±0.03 | |
| 21 d | 1.15±0.06 | 0.82±0.05 | 1.05±0.05 | 0.86±0.04 | |
| 28 d | 1.19±0.07 | 0.92±0.05 | 1.10±0.06 | 0.81±0.04 | |
| ICL1-9 Sham | Baseline | 1.29±0.05 | 0.91±0.04 | 1.14±0.04 | 0.77±0.03 |
| n=16 | 1 d | 1.40±0.06 | 1.01±0.05 | 1.18±0.05 | 0.90±0.05 |
| 3 d | 1.57±0.18 | 1.10±0.23 | 1.43±0.15 | 0.93±0.14 | |
| 7 d | 1.51±0.06 | 1.15±0.05 | 1.30±0.043 | 0.90±0.03 | |
| 14 d | 1.33±0.06 | 0.95±0.04 | 1.24±0.04 | 0.86±0.02 | |
| 21 d | 1.30±0.07 | 0.99±0.07 | 1.12±0.03 | 0.86±0.04 | |
| 28 d | 1.19±0.03 | 0.87±0.03 | 1.21±0.05 | 0.88±0.03 | |
| ICL1-9 I/R | Baseline | 1.33±0.04 | 1.00±0.04 | 1.16±0.04 | 0.84±0.04 |
| n=17 | 1 d | 1.43±0.07 | 1.13±0.07 | 1.25±0.06 | 1.04±0.06 |
| 3 d | 1.43±0.16 | 1.17±0.12 | 1.41±0.08 | 1.05±0.07 | |
| 7 d | 1.41±0.06 | 1.02±0.04 | 1.16±0.04 | 0.86±0.04 | |
| 14 d | 1.34±0.08 | 0.94±0.04 | 1.09±0.05 | 0.84±0.03 | |
| 21 d | 1.14±0.09 | 0.78±0.02 | 1.11±0.07 | 0.89±0.09 | |
| 28 d | 1.29±0.06 | 1.01±0.05 | 1.12±0.05 | 0.78±0.05 |
To investigate the impact of ICL1-9 on cardiomyocyte survival, we employed a 24 h serum-deprivation model of stress we previously used to induce NRVM death [4]. Serum deprivation of NRVM for 24 h increased Caspase 3/7 activity, which was not impacted by treatment with Scr pepducin, but was ablated by ICL1-9 treatment (Figure 4C). To determine if ICL1-9 promotes survival via EGFR, ERK1/2 and/or Akt signaling, serum-deprived NRVM were co-incubated with inhibitors of each pathway, including PD98059 (upstream MEK1/2 inhibitor), LY294002 (upstream PI3K inhibitor) and AG1478 (EGFR inhibitor) and in each case, ICL1-9-mediated protection of NRVM was not blocked (Figure 4D). Further, we could not detect a significant increase in ERK1/2 or Akt phosphorylation at any timepoint measured after stimulation of either NRVM or mouse hearts with ICL1-9 (Figure S2), revealing that ICL1-9 does not engage EGFR, ERK1/2 or Akt signaling to mediate its cardioprotective effects in cardiomyocytes in vitro or in whole heart in vivo.
Figure 4
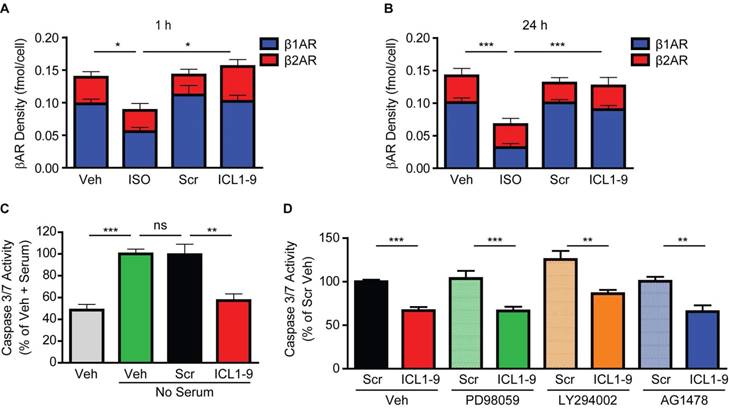
ICL1-9 protects against cardiomyocyte death in vitro. 125I-cyanopindolol binding analysis of NRVM treated for 1 h (A) or 24 h (B) with Veh, ISO, Scr or ICL1-9 pepducin (10 μM each). n=4 Veh, n=4 ISO, n=5 Scr, n=5 ICL1-9. *p < 0.05, *** p < 0.001 for β1AR density, one-way ANOVA with Tukey's multiple comparison test. (C) Caspase 3/7 activity was measured in NRVM subjected to 24 h with or without serum in the presence of Scr or ICL1-9 pepducin (10 µM each) with values represented as a percentage of no serum Veh control. n=7 Veh, n=7 Veh no serum, n=10 Scr no serum, n=10 ICL1-9 no serum. ns = not significant, ** p < 0.01, *** p < 0.001, one-way ANOVA with Tukey's multiple comparison test. (D) Caspase 3/7 activity was measured in NRVM subjected to 24 h without serum in the presence of Scr or ICL1-9 pepducin (10 µM each) and pretreated with PD98059, LY294002 or AG1478 (10 µM each) with values represented as a percentage of Veh Scr. n=18 Veh Scr, n=20 Veh ICL1-9, n=10 PD98059 Scr and PD98059 ICL1-9, n=8 for all other treatment groups. ns = not significant, ** p < 0.01, *** p < 0.001, one-way ANOVA with Tukey's multiple comparison test.
ICL1-9 relays cardiomyocyte survival via RhoA activation
Previous work by others demonstrated that GPCR-mediated pro-survival effects in cardiomyocytes can be relayed via activation of RhoA [16-19]; thus, we tested whether ICL1-9 activates RhoA in cardiomyocytes. Activated RhoA (GTP-bound RhoA) immunoprecipitations were performed using NRVM treated with Scr or ICL1-9 pepducin for either 5 min or 24 h, revealing significantly increased RhoA activation in response to ICL1-9, an effect completely prevented by incubation of the cells with the β2AR-selective inverse agonist ICI 118,551 (Figures 5A-B). Further, RhoA activation was increased in WT mouse hearts 24 h following intracardiac injection with ICL1-9, but not Scr pepducin, an effect that was absent in β2ARKO mouse hearts (Figure 5C). To determine whether RhoA inhibition would prevent ICL1-9-mediated protection of cardiomyocytes from serum deprivation-induced cell death, we assessed the percentage of TUNEL-positive cardiomyocytes treated with ICL1-9 or Scr pepducin in the presence or absence of the small molecule RhoA inhibitor Rhosin. Indeed, while ICL1-9 decreased serum deprivation-induced cardiomyocyte TUNEL staining, incubation with Rhosin completely blocked this effect (Figures 6A-B), as well as prevented the ICL1-9-mediated reduction in caspase 3/7 activation (Figure 6C).
Figure 5
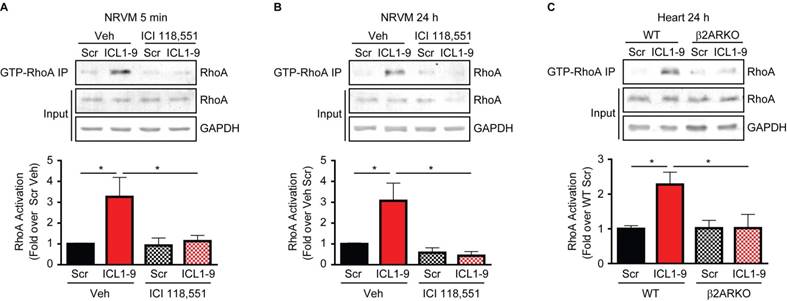
ICL1-9 activates RhoA in cardiomyocytes and whole heart. RhoA activation was assessed via anti-GTP-bound RhoA pulldown in NRVM pretreated with Veh or 100 nM ICI 118,551 and stimulated with Scr or ICL1-9 pepducin for 5 min (A) or 24 h (B), or in left ventricular lysates of WT or β2ARKO mice 24 h after intracardiac injection (C). Activated RhoA (IP) was normalized to total RhoA/GAPDH (input) and expressed in histograms as fold change from Veh Scr (A-B) or WT Scr (C). For (A): n=7 Veh Scr, n=6 Veh ICL1-9, n=5 ICI Scr and n=6 ICI ICL1-9; for (B): n=9 Veh Scr and Veh ICL1-9, n=4 ICI Scr and ICI ICL1-9; for (C): n=14 WT Scr, n=16 WT ICL1-9, n=13 β2ARKO Scr, n=14 β2ARKO ICL1-9. * p < 0.05, one-way ANOVA with Tukey's multiple comparison test.
Figure 6
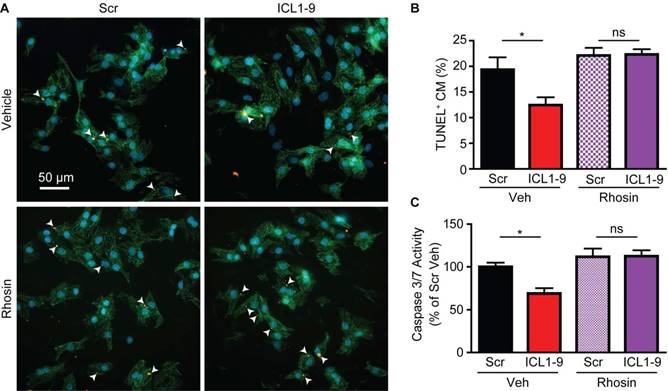
RhoA inhibition prevents ICL1-9-mediated cardiomyocyte survival. NRVM underwent serum deprivation for 24 h in the presence or absence of ICL1-9 or Scr pepducin with or without Rhosin (10 µM each). Cells were fixed, stained for TUNEL, DAPI and αSrA (A) and TUNEL-positive cardiomyocytes (arrowheads) were quantified as a percentage of total cardiomyocytes (B). n=27 Veh+Scr, Veh+ICL1-9, n=24 Rhosin+Scr, n=25 Rhosin+ICL1-9 (number of fields of view assessed from 5 plates per treatment and 3 independent primary NRVM isolations). ns = not significant, * p < 0.05, one-way ANOVA with Tukey's multiple comparison test. (C) Caspase 3/7 activity was measured in NRVM subjected to 24 h without serum in the presence of Scr or ICL1-9 pepducin (10 µM each) and pretreated with Rhosin (10 µM) with values represented as a percentage of Veh Scr. n=9 Veh Scr, n=8 for all other groups. ns = not significant, * p < 0.05, one-way ANOVA with Tukey's multiple comparison test.
ICL1-9 reduces hypoxia/reoxygenation-induced caspase activation and mitochondrial superoxide formation via RhoA/ROCK activation in cardiomyocytes
Since acute ischemic injury results in cardiomyocyte death due to enhanced mitochondrial oxidative stress [20], we sought to determine whether ICL1-9 acts to reduce this response. Thus, we used an in vitro model of hypoxia-reoxygenation (H/R, [19]) to mimic in vivo I/R. H/R-exposed NRVM displayed increased Caspase 3/7 activity (Figure 7A) and mitochondrial superoxide formation (Figure 7C) that were each significantly reduced by ICL1-9 but not Scr pepducin. The ICL1-9-mediated protection against both Caspase 3/7 activity (Figure 7B) and mitochondrial superoxide formation (Figure 7D-E) was blocked by co-incubation with either ICI 118,551 or Rhosin. Further, since the downstream RhoA effector ROCK was shown previously to relay RhoA-dependent survival signaling [16, 19], we tested whether inhibition of ROCK with its small molecule inhibitor Y-27632 would prevent ICL1-9-mediated pro-survival effects. Indeed, the ICL1-9-mediated protection against both Caspase 3/7 activity (Figure 7B) and mitochondrial superoxide formation (Figures 7D-E) was blocked by co-incubation with Y-27632. Altogether, our study shows for the first time that direct administration of a β-arrestin-biased β2AR-selective pepducin promotes cardiomyocyte survival via RhoA/ROCK activation to prevent adverse cardiac remodeling in response to acute ischemic injury.
Figure 7
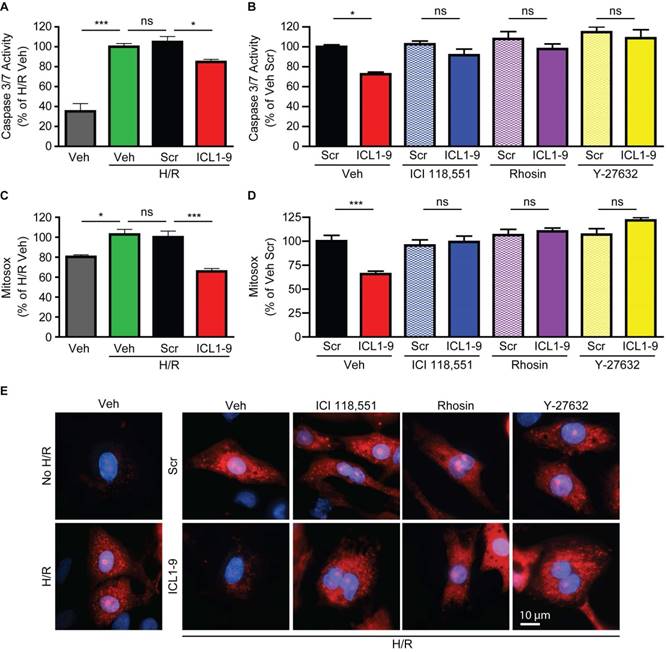
ICL1-9 decreases H/R-induced caspase 3/7 activity and superoxide formation in cardiomyocytes via RhoA/ROCK activation. NRVM underwent hypoxia for 4 h followed by reoxygenation for 20 h in the presence of Veh, Scr or ICL1-9 pepducin (10 µM each) with or without pretreatment with ICI 118,551 (100 nM), Y-27632 (10µM) or Rhosin (10 µM). Caspase 3/7 activity (A-B) was assessed as in Figure 5 and superoxide formation (C-D) was measured using mitosox red. Values are expressed as a percentage of H/R Veh (A, C) or Veh Scr (B, D). For (A): n=9 for all treatment groups, for (B): n=12 for all treatment groups, for (C): n=8 Veh and H/R Veh, n=14 H/R Scr and H/R ICL1-9, for (C): n=14 for Veh Scr and Veh ICL1-9, n=8 for all other treatment groups. ns = not significant, * p < 0.05, *** p < 0.001, one-way ANOVA with Tukey's multiple test. (E) Representative images of mitoSOX staining (red) in NRVM with or without H/R (left panels) or H/R plus Veh, ICI 118,551 (100 nM), Y-27632 (10µM) or Rhosin (10 µM) and Scr or ICL1-9 pepducin (10 µM each, right panels). Cells were counterstained with DAPI (blue) to detect nuclei. n=5 for Veh No H/R and Veh H/R, n=6 for Veh H/R Scr and Veh H/R ICL1-9, n=4 for all other H/R treatment groups.
Discussion
Targeting GPCRs to treat I/R-induced cardiac injury has been a mainstay of therapeutic research for decades. The activation or inhibition of numerous cardiac GPCRs, including adenosine, adrenergic, apelin, formyl peptide, opioid, relaxin, sphingosine 1 phosphate and urocortin receptors, have been investigated for their potential cardioprotective effects following I/R-induced injury [17, 21-29]. Recently, research efforts have focused more precisely on harnessing the potential of biased signaling, wherein distinct proximal signaling branches of GPCRs, such as G protein- versus βarr-dependent pathways, may be engaged to promote cardioprotection. For instance, recent work described the impact of a formyl peptide receptor (FPR) ligand that acts in a biased manner to promote ERK1/2- and Akt-associated survival signaling with reduced pro-death Ca2+ signaling, which conferred greater improvement in post-I/R outcomes in vivo compared to a conventional FPR ligand [23]. With regard to βarr-dependent signaling, it has been known for some time that βAR stimulation relays pro-survival effects via engagement of βarr [5, 30] and, therefore, may represent a novel therapeutic pathway to mitigate I/R-induced myocyte death; however, the ability to promote this pathway with orthosteric ligands to test this theory in vivo has been challenging. Here, we demonstrate for the first time that pepducin-mediated allosteric modulation of β2AR engages βarr-dependent cardioprotection against acute I/R-induced injury in vivo to relay structural and functional benefits. Thus, this signaling paradigm represents a bona fide therapeutic strategy to promote cardiac survival signaling at the time of I/R injury.
We previously demonstrated that ICL1-9 enhanced the activation of known survival signaling pathways in a βarr1- and βarr2-sensitive manner in a HEK 293 cell model with heterologous overexpression of β2AR, and that ICL1-9 could increase isolated adult cardiomyocyte contractility, which was dependent upon endogenous expression of β2AR and sensitive to the deletion of either βarr1 or βarr2 [11]. These results suggested that ICL1-9 may be capable of engaging pro-survival signaling in vivo to relay protection against I/R-induced myocyte death via an endogenous βarr-dependent β2AR pathway. Our current study confirms this paradigm as ICL1-9 was unable to reduce infarct size and cardiomyocyte death in response to I/R in mice lacking expression of β2AR, βarr1 or βarr2. With regard to these genetic mouse models, we observed that β2ARKO mice are as susceptible to I/R-induced injury as WT mice, which aligns with previous studies demonstrating that β2ARKO mice are not protected, or are even more susceptible to injury and death, from either permanent coronary ligation-, catecholamine- or anthracycline-induced cardiac toxicity [6, 31, 32]. However, in contrast to a recent study in which βarr2KO mouse hearts experienced less damage following I/R than WT hearts [33], in our study neither βarr1KO nor βarr2KO mice exhibited a statistically significant difference in I/R-induced injury compared to WT mice. Notably, the authors of the previous study modeled I/R using an ex vivo Langendorff perfusion system in which they could assess readouts of cardiac damage only at early timepoints (minutes to hours), while our in vivo assessment occurred 24 h post-I/R in vivo. In addition, others have reported that βarr1KO mice exhibit less cardiac cell death 24 h post ischemic injury [34], though infarct sizes and function were not assessed at this timepoint and the study employed permanent coronary ligation, making comparisons between the models difficult. Regardless of these disparate outcomes with regard to the potential impact of isoform-selective deletion of βarr on the response to acute ischemic injury, we definitively show here that the ability of ICL1-9 to relay acute cardioprotection against I/R-induced injury in vivo occurs via βarr-dependent β2AR signaling since deletion of any one of β2AR, βarr1 or βarr2 was sufficient to abolish the effect.
Previous work, including our own, has shown that βarr-dependent βAR signaling mediates cardiomyocyte survival via transactivation of EGFR with subsequent downstream activation of ERK1/2 and Akt [4, 5, 35]. Since we had demonstrated that ICL1-9 activated EGFR and increased ERK1/2 phosphorylation for hours in a HEK 293 cell model [11], we postulated that its cardioprotective effect against I/R would be relayed via this mechanism. However, we were unable to detect a significant increase in phosphorylation of either ERK1/2 or Akt at multiple timepoints following treatment of primary cardiomyocytes or injection of mouse hearts with ICL1-9. Further, antagonists of EGFR, ERK1/2 and Akt signaling pathways were completely unable to prevent ICL1-9-mediated protection against pro-death caspase 3/7 activation in NRVM undergoing serum deprivation. However, GPCR signaling paradigms characterized in HEK 293 cell models do not necessarily translate to those used in cardiomyocytes, possibly due to factors including distinct expression and localization patterns of the GPCRs themselves or their downstream signaling components [4, 36].
Therefore, an unrecognized βarr-dependent molecular pathway was responsible for promoting cardiomyocyte survival downstream of β2AR, which we postulated to be mediated via RhoA activation based on the following lines of evidence: 1) previous work by Brown and colleagues detailed the ability of RhoA to promote cardiomyocyte survival, either via cardiomyocyte-specific low-level expression of constitutively active RhoA or in response to sphingosine-1-phosphate (S1P) stimulation of the S1P receptor 3 [17-19]; 2) several studies, albeit in HEK 293 cells, have shown that angiotensin type 1A receptor-mediated regulation of RhoA activity is βarr-sensitive [37-39]; and 3) β2AR stimulation was shown to induce βarr2-dependent membrane translocation of p115RhoGEF and subsequent activation of RhoA, also in HEK 293 cells [40]. Thus, we tested whether ICL1-9-mediated pro-survival effects in cardiomyocytes were sensitive to inhibition of RhoA. Indeed, treatment with ICL1-9 induced activation of RhoA in cardiomyocytes and in whole heart, and, although we had not detected an ICL1-9-mediated increase in ERK1/2 or Akt phosphorylation at any timepoint, RhoA activation occurred rapidly and was sustained for at least 24 h both in vitro and in vivo. This activation was essential to the promotion of cardiomyocyte survival since inhibition or deletion of β2AR prevented ICL1-9-mediated RhoA activation and protection from ischemia in response to H/R in vitro or I/R in vivo. Further, although the exact biochemical nature by which pepducins induce signaling at their receptors remains elusive [41], Rhosin-mediated inhibition of RhoA activation by ICL1-9 is consistent with βarr-dependent regulation of a RhoGEF since Rhosin directly targets the RhoGEF binding domain of RhoA [42], though the RhoGEF in question remains to be identified in future studies. We additionally observed that inhibition of the RhoA effector ROCK also prevented ICL1-9-mediated pro-survival effects in cardiomyocytes, consistent with other reports [16, 19], though the molecular determinants downstream of ROCK remain to be identified in future studies. In all, our data supports a mechanism by which βarr-biased β2AR stimulation leads to prolonged activation of RhoA to confer cardioprotection against ischemic injury.
An additional unexpected observation in our study was the lack of β2AR downregulation induced by ICL1-9 treatment. We had previously shown in HEK 293 cells that ICL1-9 caused β2AR internalization [11]; however, in both isolated cardiomyocytes and in whole heart, ICL1-9 did not alter β2AR levels, even though the canonical non-selective βAR agonist ISO was able to reduce the level of β1AR after stimulation. Since ICL1-9 acted as a partial agonist for internalization in the HEK 293 cell model with β2AR overexpression [11], this result could be due to an undetectable or negligible loss of endogenous levels of cardiac β2AR, which others have recently shown to be expressed at low or sporadic levels in adult cardiomyocytes [43]. Alternatively, the allosteric modulation of β2AR by ICL1-9 in cardiomyocytes may result in the engagement of a conformation of βarr that preferentially promotes pro-survival signaling over time at the expense of appreciable receptor loss. This notion would fit with current interpretations of GPCR-βarr interaction wherein specific intracellular phosphorylation residues on the receptor are differentially induced by distinct ligands/modulators, which in turn promote unique βarr-assembled scaffolds [44, 45]. Thus, ICL1-9 likely induces a preference for a distinct β2AR-dependent βarr signaling module in cardiomyocytes that includes a RhoGEF, as discussed above, rather than components of endocytic machinery. Further, ICL1-9 localization within the heart and cardiomyocytes for at least 24 h after treatment, corresponding with sustained activation of RhoA and decreased cell death, offers an explanation as to how a single intramyocardial injection of ICL1-9 at the time of I/R led to both immediate improvements in cardiac function and decreased fibrotic remodeling over time. Thus, ICL1-9 acts primarily to maintain cardiomyocyte survival through reperfusion/reoxygenation, thereby leading secondarily to improvements in cardiac function and fibrotic remodeling. Future studies should address whether single or repeated administrations of ICL1-9 after injury offers further functional benefit, or if the 24 h post-I/R window is the only timeframe in which it can relay its benefits via acute cardiomyocyte retention.
Until now the in vivo, and even early clinical, use of pepducins has been focused on those targeting GPCRs expressed in easily accessible cells and compartments, including platelets and the liver, in which intravenous administration of pepducins results in sufficient uptake into the desired tissues [8, 46, 47]. As such, a potential limitation of the use of ICL1-9 in particular for prevention of cardiac injury is its method of delivery. In this study we used direct intramyocardial injections to allow the greatest accumulation of the pepducin within the myocardium, which diffused away from the injection site over time, ultimately enhancing cardiomyocyte survival in response to I/R. However, the ability of ICL1-9 to diffuse from the bloodstream through the endothelial cell layer and into the cardiac muscle following intracoronary delivery at the time of reperfusion, and its resulting efficacy in comparison to direct intramyocardial injection, is as yet untested. Thus, development of small molecules to allosterically promote βarr-dependent β2AR signaling in a similar fashion to ICL1-9 could offer an improved approach for a more generalized systemic delivery. Regardless, this study shows for the first time that allosteric engagement of βarr-biased β2AR signaling via pepducin administration at the time of ischemic injury in vivo promotes cardiomyocyte survival, thereby relaying structural and functional benefits, and represents a desirable and specific therapeutic target for cardioprotection.
Supplementary Material
Supplementary figures.
Acknowledgements
Author Contributions
L.A.G. and D.G.T. wrote the manuscript and designed the experiments. L.A.G., T.P.T., R.L.C. and C.d.L. conducted the experiments and analyzed the data. E.G. performed intracardiac injections and the sham and I/R surgeries. W.J.K., J.L.B. and D.G.T. provided intellectual guidance and manuscript revision.
Sources of Funding
This work was supported by NIH grants R01 HL105414 (D.G.T.), R01 HL136219 (D.G.T. and J.L.B.), R01 HL139522 (D.G.T.) and P01 HL091799 (W.J.K.) and by American Heart Association Scientific Development Grant 17SDG33400114 (L.A.G.) and Postdoctoral Fellowship 17POST33660942 (C.d.L.).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Hausenloy DJ, Yellon DM. Myocardial ischemia-reperfusion injury: a neglected therapeutic target. J Clin Invest. 2013;123:92-100
2. Neri M, Riezzo I, Pascale N, Pomara C, Turillazzi E. Ischemia/reperfusion injury following acute myocardial infarction: a critical issue for clinicians and forensic pathologists. Mediators Inflamm. 2017;2017:7018393
3. Jneid H, Addison D, Bhatt DL, Fonarow GC, Gokak S, Grady KL. et al. 2017 AHA/ACC clinical performance and quality measures for adults with ST-elevation and non-ST-elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Performance Measures. J Am Coll Cardiol. 2017;70:2048-90
4. Grisanti LA, Talarico JA, Carter RL, Yu JE, Repas AA, Radcliffe SW. et al. beta-Adrenergic receptor-mediated transactivation of epidermal growth factor receptor decreases cardiomyocyte apoptosis through differential subcellular activation of ERK1/2 and Akt. J Mol Cell Cardiol. 2014;72:39-51
5. Noma T, Lemaire A, Naga Prasad SV, Barki-Harrington L, Tilley DG, Chen J. et al. Beta-arrestin-mediated beta1-adrenergic receptor transactivation of the EGFR confers cardioprotection. J Clin Invest. 2007;117:2445-58
6. Yoo B, Lemaire A, Mangmool S, Wolf MJ, Curcio A, Mao L. et al. Beta1-adrenergic receptors stimulate cardiac contractility and CaMKII activation in vivo and enhance cardiac dysfunction following myocardial infarction. Am J Physiol Heart Circ Physiol. 2009;297:H1377-86
7. Zhang X, Szeto C, Gao E, Tang M, Jin J, Fu Q. et al. Cardiotoxic and cardioprotective features of chronic beta-adrenergic signaling. Circ Res. 2013;112:498-509
8. Covic L, Gresser AL, Talavera J, Swift S, Kuliopulos A. Activation and inhibition of G protein-coupled receptors by cell-penetrating membrane-tethered peptides. Proc Natl Acad Sci U S A. 2002;99:643-8
9. Zhang P, Covic L, Kuliopulos A. Pepducins and other lipidated peptides as mechanistic probes and therapeutics. Methods Mol Biol. 2015;1324:191-203
10. Carr R 3rd, Du Y, Quoyer J, Panettieri RA Jr, Janz JM, Bouvier M. et al. Development and characterization of pepducins as Gs-biased allosteric agonists. J Biol Chem. 2014;289:35668-84
11. Carr R 3rd, Schilling J, Song J, Carter RL, Du Y, Yoo SM. et al. beta-arrestin-biased signaling through the beta2-adrenergic receptor promotes cardiomyocyte contraction. Proc Natl Acad Sci U S A. 2016;113:E4107-16
12. Gao E, Lei YH, Shang X, Huang ZM, Zuo L, Boucher M. et al. A novel and efficient model of coronary artery ligation and myocardial infarction in the mouse. Circ Res. 2010;107:1445-53
13. Wasilewski MA, Grisanti LA, Song J, Carter RL, Repas AA, Myers VD. et al. Vasopressin type 1A receptor deletion enhances cardiac contractility, beta-adrenergic receptor sensitivity and acute cardiac injury-induced dysfunction. Clin Sci (Lond). 2016 Sep 2. pii: CS20160363
14. Fan Q, Huang ZM, Boucher M, Shang X, Zuo L, Brinks H. et al. Inhibition of Fas-associated death domain-containing protein (FADD) protects against myocardial ischemia/reperfusion injury in a heart failure mouse model. PLoS One. 2013;8:e73537
15. Li X, Mikhalkova D, Gao E, Zhang J, Myers V, Zincarelli C. et al. Myocardial injury after ischemia-reperfusion in mice deficient in Akt2 is associated with increased cardiac macrophage density. Am J Physiol Heart Circ Physiol. 2011;301:H1932-40
16. Del Re DP, Miyamoto S, Brown JH. Focal adhesion kinase as a RhoA-activable signaling scaffold mediating Akt activation and cardiomyocyte protection. J Biol Chem. 2008;283:35622-9
17. Xiang SY, Ouyang K, Yung BS, Miyamoto S, Smrcka AV, Chen J. et al. PLCepsilon, PKD1, and SSH1L transduce RhoA signaling to protect mitochondria from oxidative stress in the heart. Sci Signal. 2013;6:ra108
18. Xiang SY, Vanhoutte D, Del Re DP, Purcell NH, Ling H, Banerjee I. et al. RhoA protects the mouse heart against ischemia/reperfusion injury. J Clin Invest. 2011;121:3269-76
19. Zhao X, Ding EY, Yu OM, Xiang SY, Tan-Sah VP, Yung BS. et al. Induction of the matricellular protein CCN1 through RhoA and MRTF-A contributes to ischemic cardioprotection. J Mol Cell Cardiol. 2014;75:152-61
20. Chouchani ET, Pell VR, James AM, Work LM, Saeb-Parsy K, Frezza C. et al. A unifying mechanism for mitochondrial superoxide production during ischemia-reperfusion injury. Cell Metab. 2016;23:254-63
21. Valle Raleigh J, Mauro AG, Devarakonda T, Marchetti C, He J, Kim E. et al. Reperfusion therapy with recombinant human relaxin-2 (Serelaxin) attenuates myocardial infarct size and NLRP3 inflammasome following ischemia/reperfusion injury via eNOS-dependent mechanism. Cardiovasc Res. 2017;113:609-19
22. Shao Q, Casin KM, Mackowski N, Murphy E, Steenbergen C, Kohr MJ. Adenosine A1 receptor activation increases myocardial protein S-nitrosothiols and elicits protection from ischemia-reperfusion injury in male and female hearts. PLoS One. 2017;12:e0177315
23. Qin CX, May LT, Li R, Cao N, Rosli S, Deo M. et al. Small-molecule-biased formyl peptide receptor agonist compound 17b protects against myocardial ischaemia-reperfusion injury in mice. Nat Commun. 2017;8:14232
24. Qin C, Yang YH, May L, Gao X, Stewart AG, Tu Y. et al. Cardioprotective potential of annexin-A1 mimetics in myocardial infarction. Pharmacol Ther. 2015;148:47-65
25. Headrick JP, See Hoe LE, Du Toit EF, Peart JN. Opioid receptors and cardioprotection - 'opioidergic conditioning' of the heart. Br J Pharmacol. 2015;172:2026-50
26. Walczewska J, Dzieza-Grudnik A, Siga O, Grodzicki T. The role of urocortins in the cardiovascular system. J Physiol Pharmacol. 2014;65:753-66
27. Folino A, Accomasso L, Giachino C, Montarolo PG, Losano G, Pagliaro P. et al. Apelin-induced cardioprotection against ischaemia/reperfusion injury: roles of epidermal growth factor and Src. Acta Physiol (Oxf). 2018:222 (2), e12924
28. Yung BS, Brand CS, Xiang SY, Gray CB, Means CK, Rosen H. et al. Selective coupling of the S1P3 receptor subtype to S1P-mediated RhoA activation and cardioprotection. J Mol Cell Cardiol. 2017;103:1-10
29. Bhushan S, Kondo K, Predmore BL, Zlatopolsky M, King AL, Pearce C. et al. Selective beta2-adrenoreceptor stimulation attenuates myocardial cell death and preserves cardiac function after ischemia-reperfusion injury. Arterioscler Thromb Vasc Biol. 2012;32:1865-74
30. Maudsley S, Pierce KL, Zamah AM, Miller WE, Ahn S, Daaka Y. et al. The beta(2)-adrenergic receptor mediates extracellular signal-regulated kinase activation via assembly of a multi-receptor complex with the epidermal growth factor receptor. J Biol Chem. 2000;275:9572-80
31. Bernstein D, Fajardo G, Zhao M, Urashima T, Powers J, Berry G. et al. Differential cardioprotective/cardiotoxic effects mediated by beta-adrenergic receptor subtypes. Am J Physiol Heart Circ Physiol. 2005;289:H2441-9
32. Patterson AJ, Zhu W, Chow A, Agrawal R, Kosek J, Xiao RP. et al. Protecting the myocardium: a role for the beta2 adrenergic receptor in the heart. Crit Care Med. 2004;32:1041-8
33. Wang Y, Jin L, Song Y, Zhang M, Shan D, Liu Y. et al. beta-arrestin 2 mediates cardiac ischemia-reperfusion injury via inhibiting GPCR-independent cell survival signalling. Cardiovasc Res. 2017;113:1615-26
34. Bathgate-Siryk A, Dabul S, Pandya K, Walklett K, Rengo G, Cannavo A. et al. Negative impact of beta-arrestin-1 on post-myocardial infarction heart failure via cardiac and adrenal-dependent neurohormonal mechanisms. Hypertension. 2014;63:404-12
35. Chen H, Ma N, Xia J, Liu J, Xu Z. beta2-Adrenergic receptor-induced transactivation of epidermal growth factor receptor and platelet-derived growth factor receptor via Src kinase promotes rat cardiomyocyte survival. Cell Biol Int. 2012;36:237-44
36. Xiang YK. Compartmentalization of beta-adrenergic signals in cardiomyocytes. Circ Res. 2011;109:231-44
37. Anthony DF, Sin YY, Vadrevu S, Advant N, Day JP, Byrne AM. et al. beta-Arrestin 1 inhibits the GTPase-activating protein function of ARHGAP21, promoting activation of RhoA following angiotensin II type 1A receptor stimulation. Mol Cell Biol. 2011;31:1066-75
38. Barnes WG, Reiter E, Violin JD, Ren XR, Milligan G, Lefkowitz RJ. beta-Arrestin 1 and Galphaq/11 coordinately activate RhoA and stress fiber formation following receptor stimulation. J Biol Chem. 2005;280:8041-50
39. Godin CM, Ferguson SS. The angiotensin II type 1 receptor induces membrane blebbing by coupling to Rho A, Rho kinase, and myosin light chain kinase. Mol Pharmacol. 2010;77:903-11
40. Ma X, Zhao Y, Daaka Y, Nie Z. Acute activation of beta2-adrenergic receptor regulates focal adhesions through betaArrestin2- and p115RhoGEF protein-mediated activation of RhoA. J Biol Chem. 2012;287:18925-36
41. Carr R 3rd, Benovic JL. From biased signalling to polypharmacology: unlocking unique intracellular signalling using pepducins. Biochem Soc Trans. 2016;44:555-61
42. Shang X, Marchioni F, Sipes N, Evelyn CR, Jerabek-Willemsen M, Duhr S. et al. Rational design of small molecule inhibitors targeting RhoA subfamily Rho GTPases. Chem Biol. 2012;19:699-710
43. Myagmar BE, Flynn JM, Cowley PM, Swigart PM, Montgomery MD, Thai K. et al. Adrenergic receptors in individual ventricular myocytes: the beta-1 and alpha-1B are in all cells, the alpha-1A Is in a subpopulation, and the beta-2 and beta-3 are mostly absent. Circ Res. 2017;120:1103-15
44. Yang Z, Yang F, Zhang D, Liu Z, Lin A, Liu C. et al. Phosphorylation of G protein-coupled receptors: from the barcode hypothesis to the flute model. Mol Pharmacol. 2017;92:201-10
45. Nobles KN, Xiao K, Ahn S, Shukla AK, Lam CM, Rajagopal S. et al. Distinct phosphorylation sites on the beta(2)-adrenergic receptor establish a barcode that encodes differential functions of beta-arrestin. Sci Signal. 2011;4:ra51
46. Shearer AM, Rana R, Austin K, Baleja JD, Nguyen N, Bohm A. et al. Targeting liver fibrosis with a cell-penetrating protease-activated receptor-2 (PAR2) pepducin. J Biol Chem. 2016;291:23188-98
47. Gurbel PA, Bliden KP, Turner SE, Tantry US, Gesheff MG, Barr TP. et al. Cell-penetrating pepducin therapy targeting PAR1 in subjects with coronary artery disease. Arterioscler Thromb Vasc Biol. 2016;36:189-97
Author contact
![]() Corresponding author: Douglas G. Tilley, PhD, Room 945A MERB, Center for Translational Medicine, Lewis Katz School of Medicine, Temple University, 3500 N. Broad St., Philadelphia, PA 19140. Tel.: 215-707-9758; Fax: 215-707-9890; E-mail: douglas.tilleyedu
Corresponding author: Douglas G. Tilley, PhD, Room 945A MERB, Center for Translational Medicine, Lewis Katz School of Medicine, Temple University, 3500 N. Broad St., Philadelphia, PA 19140. Tel.: 215-707-9758; Fax: 215-707-9890; E-mail: douglas.tilleyedu
Citation styles
APA
Grisanti, L.A., Thomas, T.P., Carter, R.L., de Lucia, C., Gao, E., Koch, W.J., Benovic, J.L., Tilley, D.G. (2018). Pepducin-mediated cardioprotection via β-arrestin-biased β2-adrenergic receptor-specific signaling. Theranostics, 8(17), 4664-4678. https://doi.org/10.7150/thno.26619.
ACS
Grisanti, L.A.; Thomas, T.P.; Carter, R.L.; de Lucia, C.; Gao, E.; Koch, W.J.; Benovic, J.L.; Tilley, D.G. Pepducin-mediated cardioprotection via β-arrestin-biased β2-adrenergic receptor-specific signaling. Theranostics 2018, 8 (17), 4664-4678. DOI: 10.7150/thno.26619.
NLM
Grisanti LA, Thomas TP, Carter RL, de Lucia C, Gao E, Koch WJ, Benovic JL, Tilley DG. Pepducin-mediated cardioprotection via β-arrestin-biased β2-adrenergic receptor-specific signaling. Theranostics 2018; 8(17):4664-4678. doi:10.7150/thno.26619. https://www.thno.org/v08p4664.htm
CSE
Grisanti LA, Thomas TP, Carter RL, de Lucia C, Gao E, Koch WJ, Benovic JL, Tilley DG. 2018. Pepducin-mediated cardioprotection via β-arrestin-biased β2-adrenergic receptor-specific signaling. Theranostics. 8(17):4664-4678.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.