Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results and Discussion
Conclusion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(16):4305-4320. doi:10.7150/thno.26506 This issue Cite
Research Paper
Cholangiocarcinoma therapy with nanoparticles that combine downregulation of MicroRNA-210 with inhibition of cancer cell invasiveness
Ying Xie1, Yazhe Wang1, Jing Li1,2, Yu Hang1, Lee Jaramillo1,2, Cody J. Wehrkamp3, Mary Anne Phillippi3, Ashley M. Mohr3, Yi Chen1, Geoffrey A. Talmon4, Justin L. Mott3, David Oupický1,2 ![]()
1. Center for Drug Delivery and Nanomedicine, Department of Pharmaceutical Sciences, University of Nebraska Medical Center, Omaha, NE 68198, USA
2. Bohemica Pharmaceuticals, LLC, La Vista, NE 68128, USA
3. Department of Biochemistry and Molecular Biology, University of Nebraska Medical Center, Omaha, NE 68198, USA
4. Department of Pathology and Microbiology, University of Nebraska Medical Center, Omaha, NE 68198, USA
Received 2018-4-5; Accepted 2018-7-4; Published 2018-7-30
Abstract

Cholangiocarcinoma (CCA) is the second most common primary liver malignancy with extremely poor therapeutic outcome due to high drug resistance, widespread metastasis and lack of effective treatment options. CCA progression and metastasis are regulated by multiple biological factors including multiple miRNAs and chemokine receptor CXCR4. The goal of this study was to test if nanotherapeutic blockade of CXCR4 by polymeric CXCR4 antagonist (PCX) combined with inhibition of hypoxia-inducible miR-210 cooperatively enhances therapeutic efficacy in CCA through reducing invasiveness, inducing cell killing, and reversing drug resistance.
Methods: We first tested the activity of PCX to inhibit migration of CCA cells. We then prepared PCX/anti-miRNA nanoparticles and analyzed their miRNA delivery efficacy and anticancer activity in vitro. Finally, in vivo biodistribution assay and anticancer activity study were performed in CCA tumor-bearing mice.
Results: Our results show that PCX had a broad inhibitory effect on cell migration, effectively delivered anti-miR-210, and downregulated miR-210 expression in CCA cells. Combination PCX/anti-miR-210 nanoparticles showed cytotoxic activity towards CCA cells and reduced the number of cancer stem-like cells. The nanoparticles reversed hypoxia-induced drug resistance and sensitized CCA cells to standard gemcitabine and cisplatin combination treatment. Systemic intravenous treatment with the nanoparticles in a CCA xenograft model resulted in prominent combined antitumor activity.
Conclusion: Our findings support PCX-based nanoparticles as a promising delivery platform of therapeutic miRNA in combination CCA therapies.
Keywords: nanoparticles, miRNA, hypoxia, drug resistance, cholangiocarcinoma
Introduction
Cholangiocarcinoma (CCA) is an aggressive cancer of the biliary duct system. Intrahepatic CCA arises from the epithelial cells of the intrahepatic bile duct and is the second most common primary liver cancer [1]. CCA is a deadly disease with dismal prognosis as evident from its low 10% five-year survival rate. Surgery is the only curative option for CCA. However, fewer than 30% of CCA patients are eligible for surgical resection at diagnosis [2]. The worldwide incidence and mortality of CCA have increased over the past several decades. Advanced CCA is currently treated with a combination of gemcitabine (GEM) and cisplatin (CDDP). Unfortunately, the therapeutic outcome of systemic GEM/CDDP treatment is poor due to drug resistance [3, 4]. The high mortality of CCA is also attributed to early invasion and widespread metastasis [5, 6]. No effective treatment options exist for metastatic CCA. These therapeutic challenges highlight the urgent need to develop new therapeutics to reverse drug resistance and inhibit metastasis in CCA.
The C-X-C receptor type 4 (CXCR4) and its ligand (CXCL12) are two key factors in tumor growth, metastasis, angiogenesis, and cancer cell-microenvironment interactions, which make them potential targets for CCA therapy [7, 8]. CXCR4 overexpression has been found in more than 20 major human cancer types, including CCA. CXCR4 was upregulated in ~65% of all CCA samples but was undetectable in normal biliary epithelium [9, 10]. The upregulation of CXCR4 is highly dependent on multiple transcription factors, growth factors, and hypoxia-inducible factors. The hypoxic tumor microenvironment can induce CXCR4 based on activation of the hypoxia-inducible factor-1α (HIF1α) and transcript stabilization [11]. The binding of CXCL12 to CXCR4 activates intracellular signaling to promote migration and invasion of cancer cells. CXCR4 then facilitates metastasis of the primary tumor cells to those sites where CXCL12 is highly expressed [12, 13]. CXCL12 is preferentially expressed in the liver, lymph nodes, lung, and bone marrow, which overlaps the metastatic profile of CCA [14]. Inhibition of the signaling pathway with CXCR4 antagonists decreases the motility and invasion of CCA cells [15-17]. Inhibition of CXCR4 can also reverse drug resistance and improve chemotherapy [18, 19], as documented by the chemosensitizing effect of a CXCR4 antagonist AMD3100 to GEM in human CCA cells [20]. Several studies reported that CXCR4 inhibition reduced the stemness of cancer cells to overcome drug resistance [21, 22]. Current evidence strongly supports the potential of CXCR4 inhibition to reverse drug resistance and inhibit metastasis in CCA.
MicroRNAs (miRNAs) are small noncoding RNAs that post-transcriptionally regulate gene expression [23]. In cancer, miRNAs can act as tumor suppressors or oncogenes and play an important role in tumorigenesis, tumor growth, angiogenesis, and metastasis [24-26]. Therapies that target miRNAs exhibit great promise for cancer because single miRNA typically targets multiple genes simultaneously, enabling regulation of multiple signaling pathways involved in various cancers [27]. A growing number of studies confirm the important role of miR-25, miR-34a, and let-7c in CCA formation and progression [28-31]. Recent evidence already suggests that miRNA therapy can inhibit CCA growth, metastasis, and improve survival in animal models [32-34]. Hypoxia in CCA induces the upregulation of miR-210, which helps the cancer cells to adapt to the hypoxic microenvironment through multiple biological pathways [35-37]. Oncogenic activity of miR-210 is responsible for cell proliferation, apoptosis, metastasis, DNA repair, cell metabolism, and antitumor drug resistance [38-41]. Hence, inhibition of miR-210 provides a promising target for the treatment of CCA.
Despite the great potential of miRNAs in metastatic cancer, their clinical translation has been limited by a lack of efficient and safe systemic delivery systems [42-45]. Nanosized polyelectrolyte complexes of miRNA with polycations (polyplexes) have been among the most investigated delivery platforms, which effectively promote miRNA delivery by protecting them from degradation and overcoming multiple biological barriers [46, 47]. Traditional polycations used in the preparation of polyplexes are pharmacologically inert with no inherent anticancer activity. Recently, alternative strategies from our lab and others have suggested development of pharmacologically active polymers as a novel approach for combination therapies of cancer [48-51]. These delivery systems present several advantages, including simple formulation and high content of active agents.
We have reported a series of polymeric CXCR4 antagonists (PCX) capable of delivering miRNA, siRNA, and DNA [52-56]. PCX simultaneously inhibit cancer cell invasion by CXCR4 axis blockade and deliver nucleic acids into various cancer cells for improved anticancer effect. PCX were synthesized from AMD3100 and modified with cholesterol to improve the stability of their nanoparticles for better systemic delivery [57, 58]. Due to the key roles of CXCR4 and miR-210 in CCA, we hypothesized that combining inhibition of CXCR4 by PCX and inhibition of hypoxia-inducible miR-210 would cooperatively enhance the therapy of CCA through reducing invasiveness, inducing cell killing and overcoming drug resistance (Scheme 1). To test this hypothesis, we used PCX modified with cholesterol to prepare PCX/anti-miR-210 nanoparticles and evaluated their delivery activity and anticancer efficacy in CCA both in vitro and in vivo.
Proposed mechanism of action of PCX/anti-miR-210 nanoparticles.
Methods
Materials
Cholesterol-modified polymeric CXCR4 inhibitors PCX (Mw = 16.7 kDa, Mw/Mn = 1.9, cholesterol wt% = 16.8%) were synthesized and characterized as previously described [58]. Succinimidyl ester of Alexa Fluor® 647 carboxylic acid was from Life Technologies (Eugene, OR). AlexaFluor 647-labeled PCX polymers (AF647-PCX) were produced according to the manufacturer's instructions and purified by dialysis to remove unreacted dye. AMD3100 was from Biochempartner (Shanghai, China). Dulbecco's modified Eagle medium (DMEM), Dulbecco's phosphate buffered saline (PBS), and fetal bovine serum (FBS) were from Thermo Scientific (Waltham, MA). Hsa-miR-210-3p-Hairpin Inhibitor (anti-miR-210, mature miRNA sequence: 5′-CUGUGCGUGUGACAGCGGCUGA-3′), negative control miR-NC inhibitor (anti-miR-NC, mature miRNA sequence: 5′-UCACAACCUCCUAGAAAGAGUAGA-3′), and carboxyfluorescein (FAM) labeled FAM-anti-miRNA were purchased from Dharmacon (Lafayette, CO). Cell culture inserts (for 24-well plates, 8.0 μm pores, Translucent PET Membrane, cat# 353097) were purchased from BD Biosciences (Billerica, MA). Real-time (RT)-PCR primers were purchased from Invitrogen (Carlsbad, CA). All other reagents were from Fisher Scientific and used as received unless otherwise noted.
Cell culture
Human malignant cholangiocarcinoma Mz-ChA-1 cell line was kindly provided by Dr. Gregory Gores, Mayo Clinic, Rochester, MN. Mz-ChA-1 cells were grown in high-glucose DMEM supplemented with 10% FBS, penicillin (100 U/mL), streptomycin (100 μg/mL), G418 (50 μg/mL), and insulin (0.5 μg/mL) at 37 °C with 5% CO2 in a humidified chamber. To induce hypoxia, cells were incubated in an atmosphere of 2% O2, 5% CO2, and 93% N2 at 37 °C.
Surface expression of CXCR4
Mz-ChA-1 cells were detached with enzyme-free Cell Dissociation Buffer (Thermo Scientific) and suspended in a staining buffer. Cells were stained live with allophycocyanin (APC)-conjugated anti- CXCR4 antibody (Abcam, USA) for 1 h at 4 °C. Isotype-matched negative control was used in the panel of mAb to assess background fluorescence intensity. Samples were analyzed on a BD FACS Calibur flow cytometer (BD Bioscience, Bedford, MA). The results were processed using FlowJo software (Tree Star Inc., Ashland, OR).
Transwell migration
Mz-ChA-1 cells (2 × 105) were seeded into 6-well plates and cultured in complete DMEM medium. The cultured cells were subsequently treated with AMD3100 (300 nM) or PCX (3 μg/mL). After 48 h of incubation, the cells were trypsinized and suspended in medium without serum. Subsequently, 3 × 104 cells were seeded in the top chambers in 300 μL of serum-free medium and 500 μL of complete medium containing 10% FBS was added to the lower transwell chambers. After 12 h, the nonmigrated cells in the top chamber were removed with a cotton swab. The migrated cells were fixed in 100% methanol and stained with 0.2% Crystal Violet solution for 10 min at room temperature. The images were taken by EVOS xl microscope. Three 20× visual fields were randomly selected for each insert, and each group was conducted in triplicate.
Preparation and characterization of nanoparticles
The ability of PCX to condense anti-miRNA was determined by electrophoresis in a 2% agarose gel containing 0.5 μg/mL ethidium bromide (EtBr). PCX/anti-miRNA nanoparticles were prepared by adding a predetermined volume of PCX to an anti-miRNA solution (20 μM in 10 mM HEPES pH 7.4) to achieve the desired w/w ratio and vigorously vortexed for 10 s. Nanoparticles were then incubated at room temperature for 20 min before further use. Nanoparticles formed at various polycation-to-anti-miRNA weight ratios were loaded (20 μL of the sample containing 0.5 μg of microRNA) and run for 15 min at 100 V in 0.5 × Tris/Borate/EDTA buffer. The gels were visualized under UV illumination with a KODAK Gel Logic 100 imaging system. Hydrodynamic diameter and zeta potential of the nanoparticles were determined by dynamic light scattering (DLS) using a ZEN3600 Zetasizer Nano-ZS (Malvern Instruments Ltd., Massachusetts, USA). Morphology was observed under transmission electron microscopy (TEM, Tecnai G2 Spirit, FEI Company, USA) using NanoVan® negative staining (Nanoprobes, USA). Anti-miRNA release from nanoparticles was analyzed by heparin displacement assay. The nanoparticles (w/w = 2) were incubated with increasing concentrations of heparin for 30 min at room temperature. The samples (20 μL of the sample containing 0.5 μg of anti-miRNA) were then analyzed by agarose gel electrophoresis. For the serum stability test, free anti-miRNA solution and nanoparticles (w/w = 2) solution were incubated with FBS (50% v/v) at 37 °C, respectively. Each sample was collected at a specified time interval (0.5, 1, 2, 4, 8, and 24 h) and analyzed by gel electrophoresis. Prior to gel electrophoresis, nanoparticles solution was pre-treated with heparin to replace anti-miRNA from the nanoparticles.
Cellular uptake and intracellular trafficking of nanoparticles
Flow cytometry analysis was performed to study the cellular uptake of the nanoparticles. Mz-ChA-1 cells (5 × 104) were seeded in 24-well plates. After 24 h growth, the cells were incubated for 4 h with nanoparticles prepared with AF647-PCX and FAM-anti-miRNA (w/w =2, 100 nM FAM-anti-miRNA). The cells were then trypsinized, washed with cold PBS, and subjected to analysis using a BD FACS Calibur flow cytometer (BD Bioscience, Bedford, MA). The results were processed using FlowJo software. Intracellular localization of AF647-PCX/FAM-anti-miRNA was also observed by confocal laser scanning microscopy. Cells were cultured on a 20 mm glass-bottom cell culture dish (Nest) using 1 × 105 cells per dish. After 24 h, the medium was replaced with fresh medium and AF647-PCX/FAM-anti-miRNA nanoparticles were added (100 nM FAM-anti-miRNA). After incubation for 4 h, the cells were washed twice with PBS, stained with Hoechst 33258 for 10 min. The cells were visualized using a LSM 800 Laser Scanning Microscope (Zeiss, Jena, Germany). Moreover, to follow the endosomal release of nanoparticles, AF647-PCX/anti-miRNA nanoparticles were incubated with cells for 4 h. Then, the cells were washed twice with PBS and stained with LysoTracker Red DND-99 (Life Technology, USA) for 30 min. Finally, the cells were rinsed three times with PBS and visualized with a confocal microscope.
Quantitative real-time polymerase chain reaction (qRT-PCR)
Cells were seeded in 12-well plates at 1 × 105 cells per well 24 h prior to treatment. Then, the cells were incubated with the nanoparticles containing anti-miRNA (100 nM) in 1 mL of medium for 48 h. The expression levels of miR-210 were evaluated by TaqMan qRT-PCR. The mirVana miRNA Isolation Kit (Ambion, USA) was used for total RNA extraction from cultured cells. 10 ng of total RNA was converted into cDNA using specific primers for miR-210 (or the internal control Z30 (Applied Biosystems, Foster City, CA)) and the TaqMan microRNA reverse transcription kit (Applied Biosystems). qRT-PCR was performed using TaqMan Universal Master Mix II, No AmpErase UNG (2×) and specific primers for miR-210 or Z30 (Applied Biosystems, Foster City, CA) on a Rotor-Gene Q instrument (QIAGEN) according to the manufacturer's instructions. MiRNA expression levels were expressed relative to the internal control according to the comparative threshold cycle (Ct) method.
Apoptosis
Apoptosis was analyzed using the Annexin V-FITC/PI Apoptosis Detection Kit (BioLegend, USA). Briefly, cells were seeded in 12-well plates at a density of 1 × 105 cells per well 24 h prior to treatment. Then, the cells were incubated with the nanoparticles containing anti-miRNA (100 nM) in 1 mL of medium for 48 h. The Annexin V-FITC apoptosis detection was performed using flow cytometry in accordance with the manufacturer's protocol, and the data were processed using FlowJo. Moreover, apoptosis percentage was also quantified by nuclear morphology and visualized by treatment with the fluorescent DNA-binding dye, DAPI (4′, 6-diamidine-2′-phenylindole dihydrochloride). Briefly, cells were stained with 2 μg/mL of DAPI for 30 min at 37 °C. Apoptotic nuclei (condensed, fragmented) were counted and presented as a percent of total nuclei. At least 100 cells were counted per well and experiments were performed in triplicate. Caspase 3/7 activity was measured by enzymatic fluorophore release (Apo-One) according to the manufacturer's protocol (Promega).
Cytotoxicity
Toxicity of the nanoparticles carrying anti-miRNA was evaluated by Cell Titer Blue assay. Briefly, cells were plated in 96-well microplates at a density of 5000 cells/well. After 24 h, cells were incubated with the nanoparticles containing miRNA (100 nM) for another 48 h prior to measuring cell viability. The medium was then removed and replaced with a mixture of 100 μL serum-free media and 20 μL of CellTiter-Blue reagent (CellTiter-Blue Cell Viability Assay, Promega). After a 2 h incubation, fluorescence (560/590 nm) was measured on a SpectraMax iD3 Multi-Mode Microplate Reader (Molecular Devices, CA). The relative cell viability (%) was calculated as [I]treated/[I]untreated × 100%.
Colony formation
Cells were transfected with nanoparticles containing miRNA (100 nM) for 4 h. Then, transfected cells were reseeded in 6-well plates (200 cells per plate). Cells were allowed to grow for 14 days. Cells were fixed in 100% methanol and stained with 0.2% Crystal Violet solution for 10 min. Finally, plates were washed with distilled water and photographed. The number of colonies in each well was quantified.
Aldefluor activity
Cells were transfected with nanoparticles containing miRNA (100 nM) for 4 h and then incubated for 48 h in fresh medium. Then, Aldefluor kit (Stem Cell Technologies, Durham, NC, USA) was used to analyze ALDH enzymatic activity according to the manufacturer's instructions. Cells were suspended in Aldefluor assay buffer containing ALDH substrate (BODIPY-aminoacetaldehyde) and incubated for 30 min at 37 °C. As the negative control, cells were treated with 50 mM diethylaminobenzaldehyde (DEAB) inhibitor before adding ALDH substrate. The cells were subjected to analysis using a BD FACSCalibur flow cytometer (BD Bioscience, Bedford, MA).
Tumor spheroid formation
Cells were transfected with nanoparticles containing miRNA (100 nM) for 4 h and then cells were dissociated and plated in ultralow attachment 6-well plates (Corning, New York) at a density of 5000 viable cells per well. Cells were cultured in serum-free DMEM/F12 medium containing 5 μg/mL insulin (Sigma-Aldrich, St. Louis, MO, USA), 0.4% bovine serum albumin (Sigma-Aldrich, St. Louis, MO, USA), 10 ng/mL basic fibroblast growth factor (Invitrogen, Waltham, MA, USA), and 20 ng/mL human recombinant epidermal growth factor (Invitrogen, Waltham, MA, USA) and observed after 14 days.
Combinational cell killing assay
First, cell viability assay was performed to study the activity of nanoparticles to sensitize cells to chemotherapy. Cells were seeded in 96-well microplates at a density of 5000 cells/well. After 24 h, cells were incubated with indicated concentrations of gemcitabine/cisplatin (GEM/CDDP, w/w=10), PCX/anti-miR-NC (100 nM) plus GEM/CDDP and PCX/anti-miR-210 (100 nM) plus GEM/CDDP, respectively. After incubation for another 48 h, CellTiter-Blue assay was performed to measure cell viability. The IC50 were calculated in GraphPad Prism using a built-in dose-response analysis as the total drugs (gemcitabine plus cisplatin) concentration that achieves 50% growth inhibition relative to untreated cells (n = 3). Moreover, synergy calculations were performed to study the combinational effects between PCX/anti-miR-210 and GEM/CDDP. Briefly, cells were seeded in 96-well microplates at a density of 5000 cells/well. After 24 h, cells were incubated with indicated concentrations of gemcitabine/cisplatin (GEM/CDDP, w/w=10), PCX/anti-miR-210 and PCX/anti-miR-210 plus GEM/CDDP, respectively. After incubation for another 48 h, CellTiter-Blue assay was performed to measure cell viability. The data from cell viability assays were analyzed using Combenefit software (Cancer Research UK Cambridge Institute). The "Mapped Surface" views (Bliss model) were selected as graphical outputs for the synergy distribution.
Xenograft tumor model
All animal experiments followed a protocol approved by the University of Nebraska Medical Center Institutional Animal Care and Use Committee. Animals were placed in a facility accredited by the Association for Assessment and Accreditation of Laboratory Animal Care upon arrival. Male athymic nu/nu mice (6 weeks) were purchased from Charles River Laboratories. The xenograft tumor model was generated by subcutaneous injection of 5 × 106 Mz-ChA-1 cells in 1:1 mixture of PBS/Matrigel (100 µL) into the flank region of the mouse.
Biodistribution
Xenograft tumor mice (~300 mm3) were injected with AF647-PCX/FAM-miRNA nanoparticles (w/w=2, 2.4 mg/kg AF647-PCX, 1.2 mg/kg FAM-miRNA) through tail vein. The mice were sacrificed 24 h post administration, and the tumors and major organs were harvested and subjected to ex vivo fluorescence imaging using Xenogen IVIS 200 (Ex = 640 nm, Em = 680 nm). The fluorescence from each organ was analyzed by the instrument software. Then, the isolated tumors were embedded in an OCT compound, cut into 10 μm sections, stained with DAPI, and imaged with a confocal microscope.
In vivo therapeutic effect
When the tumor sizes reached about 150-200 mm3 at day 10 post-tumor inoculation, mice were randomly assigned to 5 groups (n = 5) and injected with PBS, GEM/CDDP, PCX/anti-miR-NC, PCX/anti-miR-210 and PCX/anti-miR-210 + GEM/CDDP, respectively. PCX/anti-miRNA (w/w = 2, 2.4 mg/kg PCX, 1.2 mg/kg anti-miRNA) were injected at days 11, 13, 15 and 17 through tail vein. GEM/CDDP (15 mg/kg GEM, 1.5 mg/kg CDDP) were given at days 12 and 16 by intraperitoneal injection. Tumor volume was monitored by measuring the perpendicular size of the tumors using digital calipers. The estimated volume was calculated according to the following formula: tumor volume (mm3) = 0.5 × length × width2. Body weight of the mice was also recorded. On days 22, mice were sacrificed, and all tumor tissues and major organs were harvested, fixed in 4% paraformaldehyde, sectioned, and stained with H&E. The apoptosis of tumor cells was determined using Caspase 3 immunohistochemical staining according to the manufacturer's instructions. Blinded histological analysis of the tissues was conducted by a trained pathologist at the UNMC core facility.
Statistical analysis
Data are presented as the mean ± SD. Statistical significance was determined using ANOVA followed by Bonferroni post hoc correction with p < 0.05 as the minimal level of significance.
Results and Discussion
Inhibition of cancer cell migration
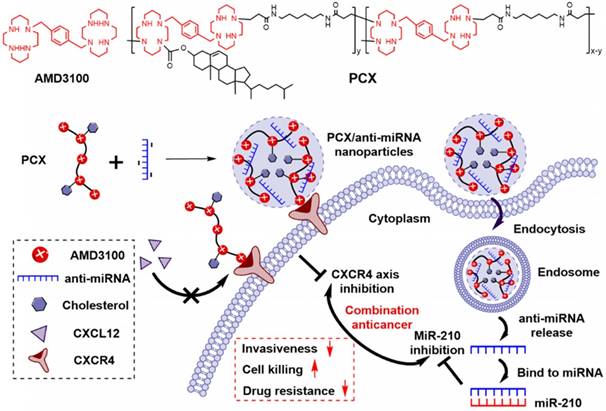
We analyzed the expression of CXCR4 in human CCA cells Mz-ChA-1 in normoxic (20% oxygen) and hypoxic (2% oxygen) conditions. The expression of CXCR4 mRNA was quantified with qRT-PCR. Hypoxia increased the CXCR4 expression 5.8-fold when compared with normoxic cells (Figure 1A). Surface expression of the CXCR4 receptor was then analyzed by flow cytometry (Figure 1B). The percentage of CXCR4-positive cells increased from 31% under normoxia to 91% in hypoxia. These results confirmed that hypoxia induces CXCR4 expression in CCA cells. High CXCR4 expression typically leads to increased cell motility, migration and invasion, which leads to enhanced metastasis [59].
(A) CXCR4 mRNA expression in Mz-ChA-1 cells quantified by qRT-PCR. (B) CXCR4 expression on Mz-ChA-1 cell surface measured by flow cytometry. The percent of CXCR4-positive cells and mean fluorescence intensity (MFI) were analyzed using FlowJo software. (C) Inhibition of FBS-induced cancer cell migration in normoxia and hypoxia. Mz-ChA-1 cells were treated with AMD3100 (300 nM) or PCX (3 µg/mL) for 48 h and then allowed to migrate through a transwell membrane insert (3 × 104 cells per insert) upon stimulation with 10% FBS for 12 h. Three 20× imaging areas were randomly selected for each insert and each group was conducted in triplicate. Scale bar 200 µm. Data are shown as mean ± SD (n = 3). **P < 0.01, ***P < 0.001.
A migration assay was performed to test if hypoxia enhanced cell migration and if the migration could be inhibited by CXCR4 antagonism of PCX. To avoid the effect of PCX cytotoxicity on migration activity, we carefully selected a safe concentration of PCX (Figure S1). PCX at the used concentration of 3 µg/mL showed no cytotoxicity in either normoxia or hypoxia. AMD3100 is a highly specific and potent (nM) small molecule inhibitor of CXCR4. Here, 300 nM of AMD3100 was selected as an effective and safe positive control. To confirm that PCX inhibits CXCR4 in CCA cells, we evaluated the downstream extracellular-signal-regulated kinase (Erk) signaling pathway by Western blot analysis. Erk is a main downstream signaling pathway of CXCR4/CXCL12 axis, which regulates cell survival, migration and invasion [8]. As shown in Figure S2, AMD3100 inhibited SDF-1-induced phosphorylation of Erk in Mz-Cha-1 cells. PCX showed similar activity to inhibit pErk as AMD3100, which confirmed PCX as a CXCR4 inhibitor. Then, the anti-migration activity of PCX was evaluated and compared with AMD3100. As shown in Figure 1C, hypoxia increased the FBS-induced migration of CCA cells ~3-fold when compared with normoxic conditions. AMD3100 effectively inhibited migration under both normoxia (57% inhibition) and hypoxia (63% inhibition). Interestingly, PCX showed better anti-migration activity and almost completely inhibited migration of CCA cells in both conditions (95% inhibition in normoxia, 97% inhibition in hypoxia). To understand why PCX presented such a superior activity, we hypothesized that PCX affected additional signaling pathways involved in cell motility. The bioactive phospholipid lysophosphatidic acid (LPA) and its G-protein-coupled receptors play an important role in cancer migration, invasion and metastasis through regulating GTPases Ras, Rho, and Rac pathways. These LPA-regulated GTPases are normally overexpressed in human CCA and are involved in the invasion and metastasis process. Inhibition of LPA signaling represents an effective approach to inhibit CCA cells migration [60-64]. Here, we performed a migration assay using LPA as the chemoattractive agent. As shown in Figure S3, LPA treatment induced migration of CCA cells. Hypoxia also increased LPA-induced migration when compared with normoxic conditions. As expected, AMD3100 failed to inhibit migration under both normoxia and hypoxia. However, PCX effectively inhibited LPA-induced migration, with 59% inhibition observed in normoxia and 65% inhibition found in hypoxia. AMD3100 reduced CCA cells invasiveness through CXCR4 pathway. However, PCX inhibited invasiveness not only through CXCR4 pathway but also LPA signaling. These findings support the utility of PCX as a potential antimetastatic therapy due to more efficient prevention of CCA migration through multiple pathways than conventional CXCR4 inhibitor.
Preparation and characterization of nanoparticles
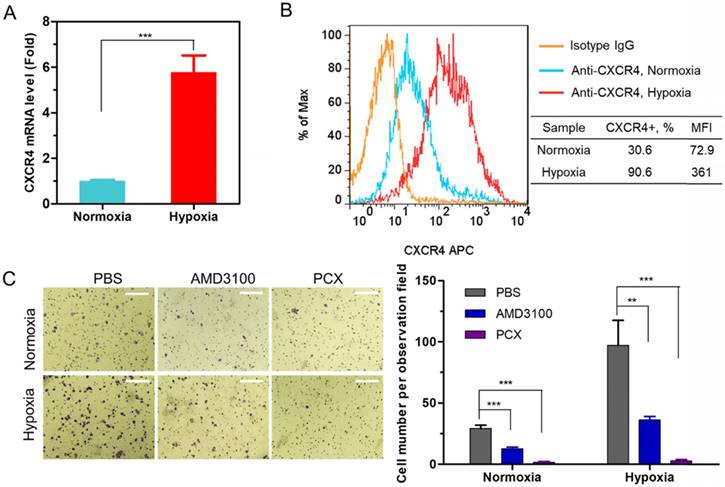
Besides inhibiting CXCR4, PCX are polycations that form polyplexes with nucleic acids. The ability of PCX to form nanoparticles with miRNA was first evaluated using agarose gel electrophoresis. Nanoparticles were prepared by adding PCX to anti-miRNA solution at increasing polycation/anti-miRNA ratios (Figure 2A). PCX fully condensed the miRNA at and above PCX/miRNA (w/w) ratios of 2. Partial condensation was observed at lower w/w ratios (0.5-1) as indicated by the smear of the RNA in the gel.
Physicochemical characterization of PCX/anti-miRNA nanoparticles. (A) Anti-miRNA condensation by PCX using agarose gel electrophoresis. (B) Hydrodynamic size distribution of PCX/anti-miRNA nanoparticles (w/w=2). (C) ζ-potential of PCX/anti-miRNA nanoparticles (w/w=2). (D) TEM image of PCX/anti-miRNA nanoparticles (w/w=2). (E) Heparin induced anti-miRNA release from the PCX/anti-miRNA nanoparticles (w/w=2) with increasing concentration of heparin. (F) Serum stability assays of naked anti-miRNA and PCX/anti-miRNA nanoparticles. Agarose gel electrophoresis of samples after treatment with serum.
Hydrodynamic size and ζ-potential of PCX/anti-miRNA nanoparticles (w/w = 2) were measured by dynamic light scattering. As shown in Figure 2B, nanoparticles exhibited sizes of 67.5 ± 0.3 nm with low polydispersity index (0.10 ± 0.05). The nanoparticles presented a positive surface charge with ζ potential of 21.5 ± 1.3 mV (Figure 2C). The shape and morphology of the nanoparticles were analyzed by transmission electron microscopy (TEM) (Figure 2D) and they were found to be uniform particles with a mostly spherical morphology.
Release of the miRNA from the particles was analyzed by heparin displacement assay. The nanoparticles completely released miRNA above 160 μg/mL heparin (Figure 2E). Poor stability against degradation by serum nucleases hinders the in vivo application of miRNA. We studied the serum stability of PCX/anti-miRNA nanoparticles in 50% FBS at 37 °C using gel electrophoresis (Figure 2F). Naked anti-miRNA was rapidly degraded within 2 h of serum incubation. The PCX/anti-miR-210 nanoparticles protected the anti-miRNA for at least 8 h. Intact anti-miRNA was observed even after serum incubation for 24 h. This result confirmed the improved serum stability provided by the PCX particles.
In vitro delivery of anti-miRNA
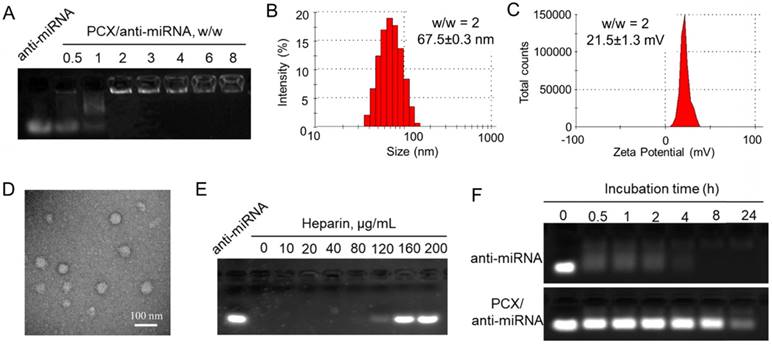
After preparation and characterization of the nanoparticles, we examined their delivery to CCA cells. We prepared PCX nanoparticles using fluorescently labeled PCX (AF647-PCX) and anti-miRNA (FAM-anti-miRNA). Mz-ChA-1 cells were incubated in normoxic conditions with the AF647-PCX/FAM-anti-miRNA (w/w = 2) for 4 h and observed under a confocal microscope. As shown in Figure 3A, both AF647-PCX (red) and FAM-anti-miRNA (green) were observed in the Mz-ChA-1 cells after 4 h of incubation, indicating effective uptake and internalization of the nanoparticles. Partial colocalization of the PCX and miRNA fluorescence (yellow) suggested that at least part of the particles remained assembled in the cells. Disassembly of a significant fraction of the nanoparticles was evident from the separation of the PCX and miRNA fluorescence signals. The disassembly of the nanoparticles is important for delivery of bioavailable miRNA inhibitors. Cell uptake was also quantified by flow cytometry (Figure 3B). We found that ~96% CCA cells have internalized both PCX and anti-miRNA. We also quantified the cellular uptake of nanoparticles in hypoxia and found no difference in the uptake percentage or mean fluorescence intensity when compared with the above results in normoxia (Figure S4). The lack of difference despite a large increase in CXCR4 expression in hypoxic cells suggests that CXCR4 was not directly involved in the cell uptake of the particles. This conclusion was further supported by a competition assay in which pretreatment of cells with AMD3100 failed to inhibit the internalization of the nanoparticles (Figure S4).
Effective delivery of anti-miRNA to CCA cells. (A) Confocal microscopy images of Mz-ChA-1 cells incubated with AF647-PCX/FAM-anti-miRNA for 4 h. Scale bar 20 µm. (B) Flow cytometry analysis of cells treated with AF647-PCX/FAM-anti-miRNA for 4 h. (C) Intracellular trafficking of AF647-PCX/anti-miRNA in cells after 4 h of incubation. Scale bar 20 µm. (D) MiR-210 expression in normoxic and hypoxic conditions measured by TaqMan qRT-PCR in Mz-ChA-1 cells after treatment with PCX/anti-miRNA for 48 h. Data are shown as mean ± SD (n = 3). **P < 0.01, ***P < 0.001.
Endosomal escape is an important step in delivery of cytoplasmically active nucleic acids like miRNA. Intracellular distribution and trafficking of the PCX nanoparticles were studied using confocal microscopy. Mz-ChA-1 cells were incubated with PCX/FAM-anti-miRNA nanoparticles for 4 h and lysosomes were stained with LysoTracker Red (Figure 3C). The fluorescence of the FAM-miRNA (green) was localized predominantly in the cytoplasm with only a limited extent of co-localization with the LysoTracker signal (red). This result confirmed endosomal escape of the nanoparticles.
After reaching cytoplasm, free anti-miRNA can inhibit targeted mature miRNA and result in therapeutic effect. Transfection activity was evaluated using nanoparticles prepared with anti-miR-210 and its negative control (anti-miR-NC). CCA cells were treated with PCX/ miRNA under normoxia or hypoxia and miR-210 expression was measured using qRT-PCR (Figure 3D). As expected, hypoxia induced a 6-fold upregulation of miR-210 expression. Incubation with PCX/anti-miR-210 significantly downregulated the miR-210 levels under both normoxia (~57% decrease) and hypoxia (~85% decrease). PCX/anti-miR-NC had no effect on miR-210 expression. These results confirmed effective delivery of functional anti-miR-210 into CCA cells and inhibition of the targeted miRNA by the PCX nanoparticle.
Anticancer activity in vitro
The CCA cell killing activity of PCX/anti-miR-210 was first studied by determining apoptosis in the treated cells using Annexin V assay (Figure 4A). PCX/anti-miR-210 treatment increased the number of apoptotic cells (~32%) and necrotic cells (∼13%) compared with the control groups. This pro-apoptotic effect of the nanoparticles was further corroborated by staining with DAPI and evaluation based on nuclear morphology. Upon treatment with PCX/anti-miR-210, Mz-ChA-1 cells demonstrated at least a 26% increase in apoptosis compared to cells treated with PCX/anti-miR-NC (Figure 4B and Figure S5). The PCX/anti-miR-210 nanoparticles also increased caspase 3/7 activity (∼3.2-fold) in the CCA cells (Figure 4C), validating that the nanoparticles induced caspase activation, apoptotic nuclear morphology, and externalization of phosphatidylserine. Next, we studied the contribution of PCX/anti-miR-210 nanoparticles to the overall cell killing activity in Mz-ChA-1 cells using cell viability assay (Figure 4D). PCX/anti-miR-210 exhibited significantly higher cell killing activity (∼28%) than the untreated and miR-NC controls. Colony formation assay was also performed to study the effect of the nanoparticles on the tumorigenic potential of CCA cells (Figure 4E). Treatment with PCX/anti-miR-210 reduced colony formation by 92% compared to PBS. As expected, CXCR4 inhibition by PCX alone had no direct cell killing effect in the CCA cells. Overall, the combined results from the anticancer activity assays confirmed the promising activity of PCX/anti-miR-210 nanoparticles in CCA cells due to their effect on apoptosis and colony formation.
Therapeutic effect of nanoparticles in vitro. (A) Mz-ChA-1 cells were treated with PCX/anti-miRNA nanoparticles (100 nM anti-miRNA) for 48 h. Flow cytometry analysis of apoptosis was performed using Annexin V-FITC/PI staining. (B) Apoptotic nuclei were counted after DAPI staining and expressed as a percent of total nuclei. (C) Quantitation of caspase 3/7 activity in cells. (D) Cell viability measured by CellTiterBlue assay after treatment for 48 h. Results are normalized to the viability of PBS-treated cells. (E) Quantification and representative images of colonies from the colony formation assay at 14 days. (F) ALDH activity in the CCA cells by flow cytometry after treatment with nanoparticles. Upper panels: representative plot showing the percentage of ALDH-positive population. Lower panels: negative control after addition of ALDH inhibitor DEAB. (G) Tumor spheroid formation in CCA cells treated with the nanoparticles and then cultured for 14 days. Data are shown as mean ± SD (n = 3). **P < 0.01, ***P < 0.001.
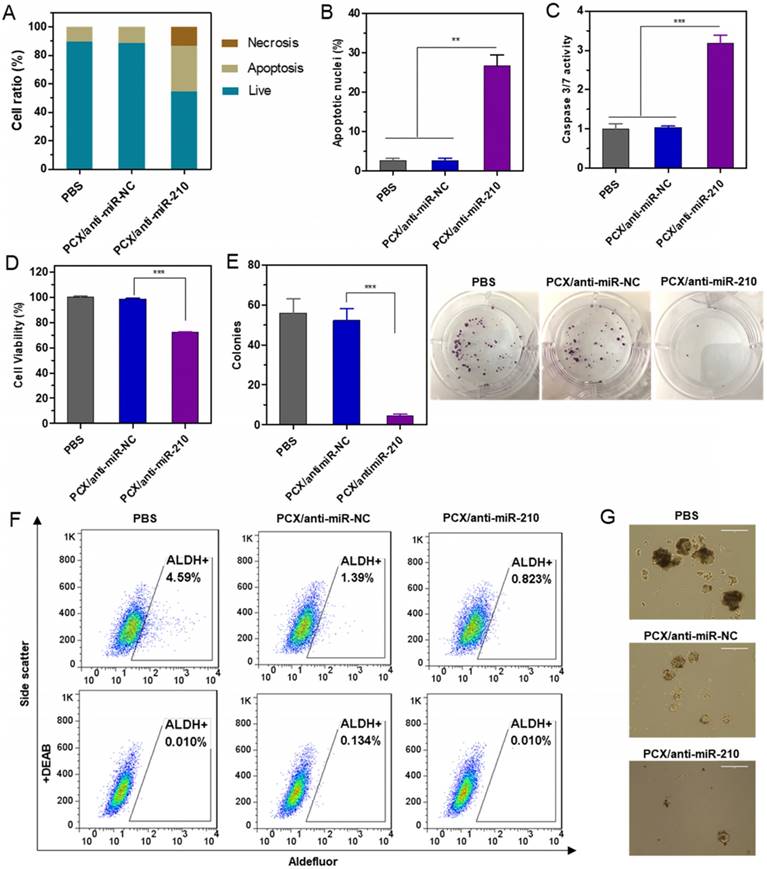
Cancer stem cells play important roles in growth, recurrence, and metastasis. CXCR4/CXCL12 axis is involved in modulating the cancer stem cell niche. Inhibition of CXCR4/CXCL12 can decrease cancer stem cells through reducing phosphorylation of ERK and STAT3 [65, 66]. Knockdown of miR-210 previously reduced stemness of cancer cells by rescuing the expression of Myc antagonist protein [67]. The combined inhibition of CXCR4 and miR-210 was thus expected to cooperatively reduce stemness of the CCA cells. Aldehyde dehydrogenase (ALDH) is a reliable marker for cancer stem cells that exhibit high ALDH enzymatic activity. The Mz-ChA-1 cells were treated with the particles, incubated with Aldefluor fluorescent reagent, and ALDH activity was analyzed by flow cytometry (Figure 4F). In the untreated cells, the ALDH-positive subpopulation of CCA cells was 4.6%. Treatment with PCX/anti-miR-NC reduced the ALDH+ population to 1.4%. Further decrease to 0.8% was observed in cells treated with the combined PCX/anti-miR-210 nanoparticles. Tumor spheroid formation assay was then used to validate the effect of the nanoparticles on cancer stem cells. Compared with the untreated (PBS) control group with large and abundant tumor spheroids, PCX/anti-miR-NC reduced the formation of large spheres. PCX/anti-miR-210 treatment further reduced the activity of cancer cells to form tumor spheroids as indicated by the formation of small and fragmentary spheroids (Figure 4G). Based on the combined evidence from the ALDH and tumor spheroid assays, we confirmed that both CXCR4 antagonism by PCX and miR-210 inhibition by anti-miR-210 contributed to the reduction of stemness in CCA cancer cells.
Chemosensitizing effect of the nanoparticles
Chemotherapy using GEM and CDDP has limited therapeutic effect in CCA patients due to drug resistance. Cancer stem cells have been shown to be involved in drug resistance through multiple cellular and molecular mechanisms. Decrease of the cancer stem cell population thus emerged as a promising approach to overcome therapy resistance and improve efficacy of cancer therapy [68]. After confirming the ability of the nanoparticles to decrease stemness of CCA cells, we aimed to study whether PCX/anti-miRNA nanoparticles could reverse drug resistance and sensitize CCA cancer cells to the chemotherapy. Mz-ChA-1 cells were treated with GEM/CDDP or GEM/CDDP+PCX/anti-miRNA under both normoxia and hypoxia. Then, cell viability was measured and half-maximal inhibition concentrations (IC50) of the GEM/CDDP combination were calculated (Figure 5A). Treatment with PCX/anti-miR-NC (anti-miRNA 100 nM) in normoxia had no direct cell killing effect (Figure 4D) but the CXCR4 inhibition by the particles sensitized the cells to the effects of GEM/CDDP as indicated by the decrease of IC50 from 1.48 µg/mL to 0.49 µg/mL (Figure 5A). Combining the CXCR4 inhibition with miR-210 inhibition further enhanced the chemosensitizing effect of the particles (IC50 ~0.14 µg/mL). Incubation of the CCA cells in hypoxic conditions greatly increased resistance to GEM/CDDP treatment as shown by the more than 3-fold increase of IC50 to >5 µg/mL. Both PCX/anti-miR-NC and PCX/anti-miR-210 nanoparticles sensitized the cancer cells to GEM/CDDP therapy in hypoxic conditions.
Nanoparticles sensitize CCA cells to chemotherapy. (A) Cell viability assay of Mz-ChA-1 cells after treatment with gemcitabine/cisplatin (GEM/CDDP, w/w=10), GEM/CDDP + PCX/anti-miR-NC, and GEM/CDDP + PCX/anti-miR-210 (anti-miRNA100 nM) for 48 h under both normoxia and hypoxia. IC50 values in combined GEM/CDDP concentrations calculated from the dose-response curves. Data are shown as mean ± SD (n = 3). (B) Synergy calculations of the combination effects between PCX/anti-miR-210 and GEM/CDDP treatments. Cells were treated with indicated concentrations of GEM/CDDP (w/w=10) and PCX/anti-miR-210 plus GEM/CDDP for 48 h. The data were analyzed using Combenefit software (Cancer Research UK Cambridge Institute).
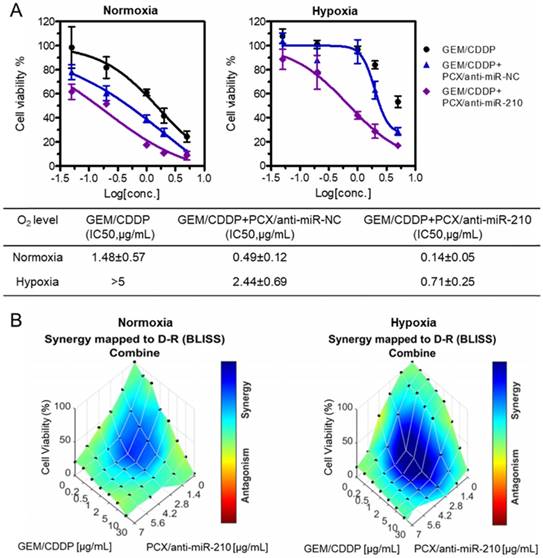
The chemosensitizing effect of the PCX/anti-miR-210 nanoparticles was further investigated using synergy analysis. Cell viability was measured in CCA cells treated with different doses and ratios of PCX/anti-miR-210 and GEM/CDDP. Combenefit software was then used to calculate synergy scores for each combination. A positive score (0-100) indicates synergy, a score of 0 is additive, and a negative score indicates antagonism. Higher score indicates stronger synergistic effect. The mapped surface of synergy/antagonism with the Bliss model is shown in Figure 5B. In normoxia, PCX/anti-miR-210 and GEM/CDDP showed synergistic cell killing effect with a score of ~30. Much stronger synergy of PCX/anti-miR-210 and GEM/CDDP was achieved under hypoxia with a score of ~60. These results confirmed that PCX/anti-miR-210 sensitized the CCA cells to chemotherapy in a synergistic manner and reversed hypoxia-induced drug resistance.
Biodistribution
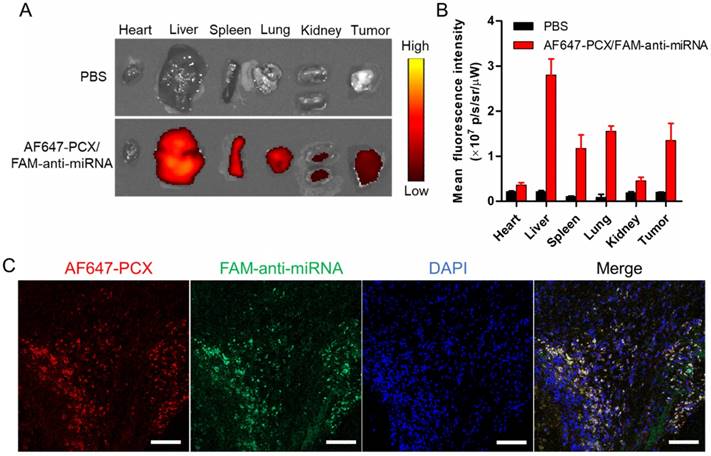
Following the promising in vitro findings, we explored the delivery efficacy of the nanoparticles to tumors in vivo. Fluorescently labeled nanoparticles (AF647-PCX/FAM-anti-miRNA) were administered intravenously into mice bearing xenograft Mz-ChA-1 tumors. Following animal sacrifice 24 h post injection, we measured ex vivo fluorescence (AF647) in the excised tissues to analyze the biodistribution (Figure 6A-B). We observed significant tumor accumulation of the nanoparticles. As expected, the nanoparticles were also found in the liver, spleen and lung. To evaluate the ability of the nanoparticles to deliver both PCX and anti-miRNA into the tumors, frozen tumor sections were observed under a confocal microscope (Figure 6C). Both AF647-PCX (red) and FAM-anti-miRNA (green) fluorescence were clearly present in the tumor. The high colocalization (yellow) of AF647-PCX and FAM-anti-miRNA also indicated good stability of the nanoparticles during systemic delivery.
Biodistribution of the nanoparticles in xenograft MZ-ChA-1 tumor after intravenous injection. (A) Ex vivo images of the tumors and other tissues 24 h postinjection of AF647-PCX/FAM-anti-miRNA nanoparticles (Ex = 640 nm, Em = 680 nm). (B) Semiquantitative analysis of the nanoparticle biodistribution 24 h postinjection. Results are expressed as mean fluorescence intensity ± SD (n = 3). (C) Confocal images of frozen tumor sections. PCX is shown in red (AF647), anti-miRNA in green (FAM) and the nucleus in blue (DAPI). Scale bar 100 µm.
In vivo therapeutic effect
Therapeutic efficacy of the nanoparticles was tested in a xenograft CCA model in nude mice. We confirmed the presence of the hypoxic tumor microenvironment needed for the mechanism of action of our PCX/anti-miR-210 nanoparticles using pimonidazole staining in frozen tumor sections (Figure S6). Systemic intravenous treatment with PCX/anti-miR-210 nanoparticles effectively inhibited tumor growth (51%) when compared with PBS (Figure 7A). Treatment with control PCX/anti-miR-NC nanoparticles or with GEM/CDDP chemotherapy showed negligible effect on tumor growth. These results suggested that CXCR4 inhibition alone has no effect on the growth of CCA tumors. In contrast, combined downregulation of miR-210 and inhibition of CXCR4 by PCX/anti-miR-210 nanoparticles cooperatively achieved antitumor activity. As expected, the CCA tumor model was resistant to GEM/CDDP chemotherapy. We used a suboptimal dose of GEM/CDDP (GEM 15 mg/kg, CDDP 1.5 mg/kg) to better demonstrate the benefits of the nanoparticles. When we treated the animals with the combination of PCX/anti-miR-210 and GEM/CDDP, we observed greatly enhanced tumor growth inhibition (~79%), indicating PCX/anti-miR-210 nanoparticles sensitized the tumors to chemotherapy. At the end of the study, tumors were photographed (Figure 7B-C) and weighed (Figure 7D). The mean tumor weight in the PCX/anti-miR-210 group (0.34 g) was significantly lower than the weight in the PCX/anti-miR-NC group (0.68 g) and the untreated group (~0.81 g). Combination PCX/anti-miR-210 with GEM/CDDP resulted in the lowest tumor weight (~0.15 g). These findings confirmed the strong antitumor and chemosensitizing effect of PCX/anti-miR-210 nanoparticles in CCA.
Antitumor efficacy in a CCA xenograft model. (A) Mz-ChA-1 tumor growth after intravenous injection of PCX/anti-miRNA (2.4 mg/kg PCX, 1.2 mg/kg anti-miRNA) and intraperitoneal injection of GEM/CDDP (15 mg/kg GEM, 1.5 mg/kg CDDP). Data are shown as mean ± SD (n = 5). (B) Representative images of the mice on day 22. (C) Tumor tissues resected from mice on day 22. (D) Weights of tumors collected from the sacrificed mice. Data are shown as mean ± SD (n = 5). (E) Body weight during the treatment. Data are shown as mean ± SD (n = 5). (F) Caspase-3 immunohistochemistry analysis (magnification 40×, scale bar 100 µm.) and H&E staining (magnification 20×, scale bar 200 µm.) of tumor tissues after various treatments. The percentage of caspase-3-positive cells in tumors. Data are shown as mean ± SD (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001.
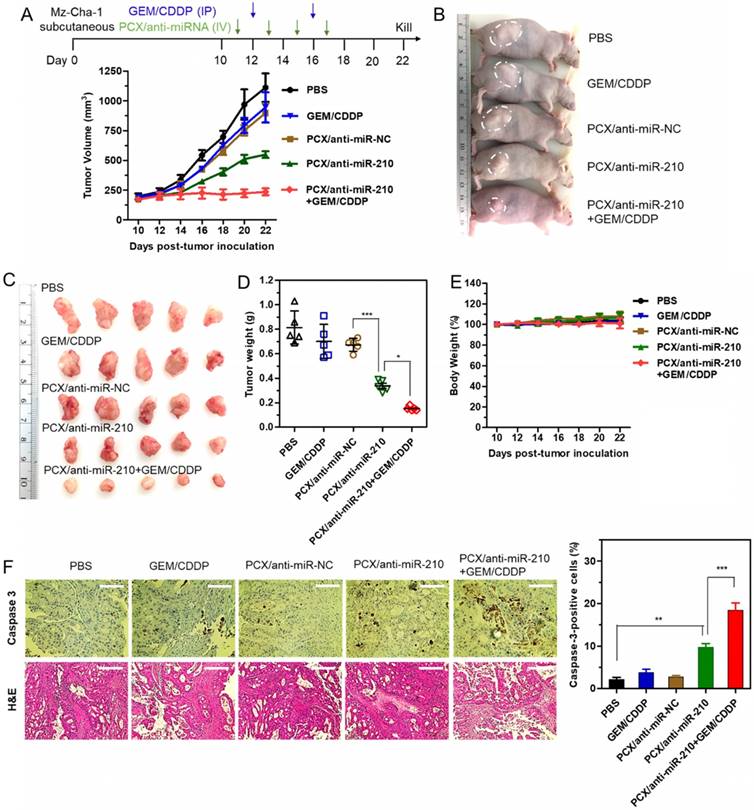
As shown by the unchanged body weight during the treatment, PCX/anti-miR-210 had no apparent signs of gross toxicity (Figure 7E). H&E staining of major organs demonstrated no obvious tissue damage when compared to the PBS group (Figure S7). Due to the low dose of the chemotherapy drugs in this study, we observed no reduction in body weight or presence of tissue injury caused by GEM/CDDP. Immunohistochemical staining of caspase 3 in the tumors was used to validate the proapoptotic activity of the nanoparticles (Figure 7F). When compared with the PBS group (2% caspase-3-positive cells), the PCX/anti-miR-210 treatment increased the fraction of apoptotic caspase-3-positive cells to ~10%. Combination of PCX/anti-miR-210 and GEM/CDDP showed even higher proapoptotic activity, with 19% of caspase-3-positive cells. Analysis of the H&E tumor sections also suggested considerably enhanced necrosis in the PCX/anti-miR-210 and GEM/CDDP combination group when compared with the control groups.
Conclusion
In this study, we developed an innovative combination nanoparticle treatment approach through simultaneous inhibition of CXCR4 and miR-210. The results showed that CXCR4-inhibiting polycation PCX could efficiently block the hypoxia-induced migration of CCA cells apparently through CXCR4 and LPA pathways. PCX/anti-miRNA nanoparticles delivered functional anti-miRNA to CCA cells and downregulated miR-210 expression, which resulted in significant cell killing through induction of apoptosis. PCX/anti-miR-210 nanoparticles sensitized CCA cells to GEM/CDDP chemotherapy by reducing stemness and synergistically reversed hypoxia-induced drug resistance. The combination nanoparticles achieved effective systemic delivery to CCA xenograft tumors and enhanced antitumor therapy in vivo through direct tumor growth inhibition and chemotherapy sensitization. The nanoparticles represent a promising dual-function delivery platform for miRNA delivery and provide safe and effective nanomedicines for systemic CCA therapy. Future studies will focus on the evaluation of the nanoparticles in orthotopic and metastatic CCA animal models to facilitate their clinical translation. We will further optimize the formulation of the nanoparticles to improve their systemic delivery to tumors. For example, the high positive charge of the nanoparticles is likely to reduce their circulation time due to increased binding affinity with polyanion components in the blood and opsonization. Optimized PEGylation or zwitterionic modifications of PCX is thus expected to improve their in vivo pharmacokinetic performance.
Abbreviations
CCA: cholangiocarcinoma; CDDP: cisplatin; CXCR4: C-X-C receptor type 4; DLS: dynamic light scattering; FBS: fetal bovine serum; GEM: gemcitabine; IC50: half-maximal inhibition concentrations; LPA: lysophosphatidic acid; miRNA: microRNA; PBS: phosphate buffered solution; PCX: polymeric CXCR4 antagonist.
Supplementary Material
Supplementary figures.
Acknowledgements
This work was supported by the National Institutes of Health (R41-TR001312), Nebraska Department of Economic Development (16-01-298), and the University of Nebraska Medical Center. China Scholarship Council support for Y. X. and Y. H. is gratefully acknowledged.
Author contribution
YX and YW performed experiments, analyzed data and wrote the manuscripts. JL supervised the animal study. YH and LJ synthesized the polymers. CW, MP and AM assisted with the animal studies. GT performed the pathology analysis. JM supervised the animal study and edited the manuscript. DO supervised the study, designed experiments, and edited the manuscript text.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Razumilava N, Gores GJ. Cholangiocarcinoma. Lancet. 2014;383:2168-2179
2. Bridgewater J, Galle PR, Khan SA, Llovet JM, Park J-W, Patel T. et al. Guidelines for the diagnosis and management of intrahepatic cholangiocarcinoma. J Hepatol. 2014;60:1268-1289
3. Valle J, Wasan H, Palmer DH, Cunningham D, Anthoney A, Maraveyas A. et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N Engl J Med. 2010;362:1273-1281
4. Valle JW, Furuse J, Jitlal M, Beare S, Mizuno N, Wasan H. et al. Cisplatin and gemcitabine for advanced biliary tract cancer: a meta-analysis of two randomised trials. Ann Oncol. 2013;25:391-398
5. Valle JW, Wasan H, Johnson P, Jones E, Dixon L, Swindell R, Baka S. et al. Gemcitabine alone or in combination with cisplatin in patients with advanced or metastatic cholangiocarcinomas or other biliary tract tumours: a multicentre randomised phase II study-The UK ABC-01 Study. Br J Cancer. 2009;101:621-627
6. Sia D, Tovar V, Moeini A, Llovet J. Intrahepatic cholangiocarcinoma: pathogenesis and rationale for molecular therapies. Oncogene. 2013;32:4861-4870
7. Guo F, Wang Y, Liu J, Mok S, Xue F, Zhang W. CXCL12/CXCR4: a symbiotic bridge linking cancer cells and their stromal neighbors in oncogenic communication networks. Oncogene. 2016;35:816-826
8. Wang Y, Xie Y, Oupický D. Potential of CXCR4/CXCL12 chemokine axis in cancer drug delivery. Curr Pharmacol Rep. 2016;2:1-10
9. Domanska UM, Kruizinga RC, Nagengast WB, Timmer-Bosscha H, Huls G, de Vries EG. et al. A review on CXCR4/CXCL12 axis in oncology: no place to hide. Eur J Cancer. 2013;49:219-230
10. Tan X-Y, Chang S, Liu W, Tang H-H. Silencing of CXCR4 inhibits tumor cell proliferation and neural invasion in human hilar cholangiocarcinoma. Gut Liver. 2014;8:196-204
11. Schioppa T, Uranchimeg B, Saccani A, Biswas SK, Doni A, Rapisarda A. et al. Regulation of the chemokine receptor CXCR4 by hypoxia. J Exp Med. 2003;198:1391-1402
12. Balkwill F. Cancer and the chemokine network. Nat Rev Cancer. 2004;4:540-550
13. Sleightholm RL, Neilsen BK, Li J, Steele MM, Singh RK, Hollingsworth MA. et al. Emerging roles of the CXCL12/CXCR4 axis in pancreatic cancer progression and therapy. Pharmacol Ther. 2017;179:158-170
14. Müller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME. et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50-56
15. Leelawat K, Keeratichamroen S, Leelawat S, Tohtong R. CD24 induces the invasion of cholangiocarcinoma cells by upregulating CXCR4 and increasing the phosphorylation of ERK1/2. Oncol Lett. 2013;6:1439-1446
16. Ohira S, Sasaki M, Harada K, Sato Y, Zen Y, Isse K. et al. Possible regulation of migration of intrahepatic cholangiocarcinoma cells by interaction of CXCR4 expressed in carcinoma cells with tumor necrosis factor-α and stromal-derived factor-1 released in stroma. Am J Pathol. 2006;168:1155-1168
17. Sung Y-C, Liu Y-C, Chao P-H, Chang C-C, Jin P-R, Lin T-T. et al. Combined delivery of sorafenib and a MEK inhibitor using CXCR4-targeted nanoparticles reduces hepatic fibrosis and prevents tumor development. Theranostics. 2018;8:894-905
18. Burger J, Peled A. CXCR4 antagonists: targeting the microenvironment in leukemia and other cancers. Leukemia. 2009;23:43-52
19. Chen Y, Ramjiawan RR, Reiberger T, Ng MR, Hato T, Huang Y. et al. CXCR4 inhibition in tumor microenvironment facilitates anti-programmed death receptor-1 immunotherapy in sorafenib-treated hepatocellular carcinoma in mice. Hepatology. 2015;61:1591-1602
20. Mayr C, Neureiter D, Pichler M, Berr F, Wagner A, Kiesslich T. et al. Cytotoxic effects of chemokine receptor 4 inhibition by AMD3100 in biliary tract cancer cells: Potential drug synergism with gemcitabine. Mol Med Rep. 2015;12:2247-2252
21. Singh AP, Arora S, Bhardwaj A, Srivastava SK, Kadakia MP, Wang B. et al. CXCL12/CXCR4 protein signaling axis induces sonic hedgehog expression in pancreatic cancer cells via extracellular regulated kinase-and Akt kinase-mediated activation of nuclear factor κB. J Biol Chem. 2012;287:39115-39124
22. Jung M, Rho J, Kim Y, Jung J, Jin Y, Ko Y. et al. Upregulation of CXCR4 is functionally crucial for maintenance of stemness in drug-resistant non-small cell lung cancer cells. Oncogene. 2013;32:209-221
23. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281-297
24. Croce CM. Causes and consequences of microRNA dysregulation in cancer. Nature Rev Genet. 2009;10:704-714
25. Mott JL, Mohr AM. Overview of microRNA biology. Semin Liver Dis. 2015;35:3-11
26. Yang T, Zhao P, Rong Z, Li B, Xue H, You J. et al. Anti-tumor efficiency of lipid-coated cisplatin nanoparticles co-loaded with microRNA-375. Theranostics. 2016;6:142-154
27. Rupaimoole R, Calin GA, Lopez-Berestein G, Sood AK. miRNA deregulation in cancer cells and the tumor microenvironment. Cancer Discov. 2016;6:235-246
28. Mott JL. MicroRNAs involved in tumor suppressor and oncogene pathways: implications for hepatobiliary neoplasia. Hepatology. 2009;50:630-637
29. Razumilava N, Bronk SF, Smoot RL, Fingas CD, Werneburg NW, Roberts LR. et al. miR-25 targets TNF-related apoptosis inducing ligand (TRAIL) death receptor-4 and promotes apoptosis resistance in cholangiocarcinoma. Hepatology. 2012;55:465-475
30. Natarajan SK, Stringham BA, Mohr AM, Wehrkamp CJ, Lu S, Phillippi MA. et al. FoxO3 increases miR-34a to cause palmitate-induced cholangiocyte lipoapoptosis. J Lipid Res. 2017;58:866-875
31. Xie Y, Zhang H, Guo X-J, Feng Y-C, He R-Z, Li X. et al. Let-7c inhibits cholangiocarcinoma growth but promotes tumor cell invasion and growth at extrahepatic sites. Cell Death Dis. 2018;9:249
32. Li L, Piontek K, Ishida M, Fausther M, Dranoff JA, Fu R. et al. Extracellular vesicles carry microRNA-195 to intrahepatic cholangiocarcinoma and improve survival in a rat model. Hepatology. 2017;65:501-514
33. Kwon H, Song K, Han C, Zhang J, Lu L, Chen W. et al. Epigenetic silencing of miRNA-34a in human cholangiocarcinoma via EZH2 and DNA methylation: impact on regulation of Notch pathway. Am J Pathol. 2017;187:2288-2299
34. Li H, Zhou ZQ, Yang ZR, Tong DN, Guan J, Shi BJ. et al. MicroRNA-191 acts as a tumor promoter by modulating the TET1-p53 pathway in intrahepatic cholangiocarcinoma. Hepatology. 2017;66:136-151
35. Pulkkinen K, Malm T, Turunen M, Koistinaho J, Ylä-Herttuala S. Hypoxia induces microRNA miR-210 in vitro and in vivo. FEBS Lett. 2008;582:2397-2401
36. Huang X, Le Q-T, Giaccia AJ. MiR-210-micromanager of the hypoxia pathway. Trends Mol. Med. 2010;16:230-237
37. Morine Y, Shimada M, Utsunomiya T, Imura S, Ikemoto T, Mori H, Hanaoka J. et al. Hypoxia inducible factor expression in intrahepatic cholangiocarcinoma. Hepatogastroenterology. 2011;58:1439-1444
38. Ying Q, Liang L, Guo W, Zha R, Tian Q, Huang S. et al. Hypoxia-inducible MicroRNA-210 augments the metastatic potential of tumor cells by targeting vacuole membrane protein 1 in hepatocellular carcinoma. Hepatology. 2011;54:2064-2075
39. Chan SY, Loscalzo J. MicroRNA-210: a unique and pleiotropic hypoxamir. Cell Cycle. 2010;9:1072-1083
40. Chan YC, Banerjee J, Choi SY, Sen CK. miR-210: The master hypoxamir. Microcirculation. 2012;19:215-223
41. Lee D, Sun S, Zhang XQ, De Zhang P, Ho AS, Kiang KM. et al. MicroRNA-210 and endoplasmic reticulum chaperones in the regulation of chemoresistance in glioblastoma. J Cancer. 2015;6:227-232
42. Sun S, Wang Y, Zhou R, Deng Z, Han Y, Han X. et al. Targeting and regulating of an oncogene via nanovector delivery of MicroRNA using patient-derived xenografts. Theranostics. 2017;7:677-693
43. Lin L, Fan Y, Gao F, Jin L, Li D, Sun W. et al. UTMD-promoted co-delivery of gemcitabine and miR-21 inhibitor by dendrimer-entrapped gold nanoparticles for pancreatic cancer therapy. Theranostics. 2018;8:1923-1939
44. Choi KY, Silvestre OF, Huang X, Hida N, Liu G, Ho DN. et al. A nanoparticle formula for delivering siRNA or miRNAs to tumor cells in cell culture and in vivo. Nature Protoc. 2014;9:1900-1915
45. Choi KY, Silvestre OF, Huang X, Min KH, Howard GP, Hida N. et al. Versatile RNA interference nanoplatform for systemic delivery of RNAs. ACS Nano. 2014;8:4559-4570
46. Chen Y, Gao D-Y, Huang L. In vivo delivery of miRNAs for cancer therapy: challenges and strategies. Adv Drug Deliv Rev. 2015;81:128-141
47. Lächelt U, Wagner E. Nucleic acid therapeutics using polyplexes: a journey of 50 years (and beyond). Chem Rev. 2015;115:11043-11078
48. Kwak G, Kim D, Nam GH, Wang SY, Kim IS, Kim SH. et al. Programmed cell death protein ligand-1 silencing with polyethylenimine-dermatan sulfate complex for dual inhibition of melanoma growth. ACS Nano. 2017;11:10135-10146
49. Li J, Yu F, Chen Y, Oupický D. Polymeric drugs: Advances in the development of pharmacologically active polymers. J Control Release. 2015;219:369-382
50. Xie Y, Murray-Stewart T, Wang Y, Yu F, Li J, Marton LJ. et al. Self-immolative nanoparticles for simultaneous delivery of microRNA and targeting of polyamine metabolism in combination cancer therapy. J Control Release. 2017;246:110-119
51. Xie Y, Yu F, Tang W, Alade BO, Peng ZH, Wang Y. et al. Synthesis and evaluation of chloroquine-containing DMAEMA copolymers as efficient anti-miRNA delivery vectors with improved endosomal escape and antimigratory activity in cancer cells. Macromol Biosci. 2018;18:1700194
52. Li J, Zhu Y, Hazeldine ST, Li C, Oupický D. Dual-function CXCR4 antagonist polyplexes to deliver gene therapy and inhibit cancer cell invasion. Angew Chem Int Ed. 2012;51:8740-8743
53. Wang Y, Hazeldine ST, Li J, Oupický D. Development of functional poly (amido amine) CXCR4 antagonists with the ability to mobilize leukocytes and deliver nucleic acids. Adv Healthc Mater. 2015;4:729-738
54. Li J, Oupický D. Effect of biodegradability on CXCR4 antagonism, transfection efficacy and antimetastatic activity of polymeric Plerixafor. Biomaterials. 2014;35:5572-5579
55. Xie Y, Wehrkamp CJ, Li J, Wang Y, Wang Y, Mott JL. et al. Delivery of miR-200c mimic with poly (amido amine) CXCR4 antagonists for combined inhibition of cholangiocarcinoma cell invasiveness. Mol Pharm. 2016;13:1073-1080
56. Peng Z-H, Xie Y, Wang Y, Li J, Oupický D. Dual-function polymeric HPMA prodrugs for the delivery of miRNA. Mol Pharm. 2017;14:1395-1404
57. Wang Y, Kumar S, Rachagani S, Sajja BR, Xie Y, Hang Y. et al. Polyplex-mediated inhibition of chemokine receptor CXCR4 and chromatin-remodeling enzyme NCOA3 impedes pancreatic cancer progression and metastasis. Biomaterials. 2016;101:108-120
58. Wang Y, Li J, Chen Y, Oupický D. Balancing polymer hydrophobicity for ligand presentation and siRNA delivery in dual function CXCR4 inhibiting polyplexes. Biomater. Sci. 2015;3:1114-1123
59. Su L, Zhang J, Xu H, Wang Y, Chu Y, Liu R. et al. Differential expression of CXCR4 is associated with the metastatic potential of human non-small cell lung cancer cells. Clin Cancer Res. 2005;11:8273-8280
60. Mills GB, Moolenaar WH. The emerging role of lysophosphatidic acid in cancer. Nat Rev Cancer. 2003;3:582-591
61. Liu S, Umezu-Goto M, Murph M, Lu Y, Liu W, Zhang F. et al. Expression of autotaxin and lysophosphatidic acid receptors increases mammary tumorigenesis, invasion, and metastases. Cancer Cell. 2009;15:539-550
62. Cadamuro M, Nardo G, Indraccolo S, Dall'Olmo L, Sambado L, Moserle L. et al. Platelet-derived growth factor-D and Rho GTPases regulate recruitment of cancer-associated fibroblasts in cholangiocarcinoma. Hepatology. 2013;58:1042-1053
63. Wang Q, Tang H, Yin S, Dong C. Downregulation of microRNA-138 enhances the proliferation, migration and invasion of cholangiocarcinoma cells through the upregulation of RhoC/p-ERK/MMP-2/MMP-9. Oncol. Rep. 2013;29:2046-2052
64. Shi Z, Chen M-L, He Q-L, Zeng J-H. Antisense RhoC gene suppresses proliferation and invasion capacity of human QBC939 cholangiocarcinoma cells. Hepatobiliary Pancreat Dis Int. 2007;6:516-520
65. Balic A, Sørensen MD, Trabulo SM, Sainz B, Cioffi M, Vieira CR. et al. Chloroquine targets pancreatic cancer stem cells via inhibition of CXCR4 and hedgehog signaling. Mol Cancer Ther. 2014;13:1758-1771
66. Trautmann F, Cojoc M, Kurth I, Melin N, Bouchez LC, Dubrovska A. et al. CXCR4 as biomarker for radioresistant cancer stem cells. Int J Radiat Biol. 2014;90:687-699
67. Yang W, Wei J, Guo T, Shen Y, Liu F. Knockdown of miR-210 decreases hypoxic glioma stem cells stemness and radioresistance. Exp. Cell Res. 2014;326:22-35
68. Batlle E, Clevers H. Cancer stem cells revisited. Nat Med. 2017;23:1124-1134
Author contact
![]() Corresponding author: E-mail: david.oupickyedu
Corresponding author: E-mail: david.oupickyedu