Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Methods
Results
Discussion
Abbreviations
Acknowledgements
Supplementary Material
References
Introduction
Methods
Results
Discussion
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(13):3571-3583. doi:10.7150/thno.25194 This issue Cite
Research Paper
Transcription factor AP-4 promotes tumorigenic capability and activates the Wnt/β-catenin pathway in hepatocellular carcinoma
Junwei Song1,2,*, Chan Xie3,*, Lili Jiang4,*, Geyan Wu2, Jinrong Zhu2, Shuxia Zhang2, Miaoling Tang5, Libing Song5, Jun Li1,2, ![]()
1. Guangdong Provincial Key Laboratory of Liver Disease, The Third Affiliated Hospital of Sun Yat-Sen University, Guangzhou 510630, China
2. Guangdong Province Key Laboratory of Brain Function and Disease, Department of Biochemistry, Zhongshan School of Medicine, Sun Yat-sen University, Guangzhou 510080, China
3. Department of Infectious Diseases, Third Affiliated Hospital, Sun Yat-sen University Guangzhou 510630, China
4. Key Laboratory of Protein Modification and Degradation, School of Basic Medical Sciences; Affiliated Cancer Hospital &Institute of Guangzhou Medical University, Guangzhou Medical University, Guangzhou 510030, China
5. State Key Laboratory of Oncology in Southern China, Department of Experimental Research, Sun Yat-sen University Cancer Center, Guangzhou 510060, China
* These authors contributed equally to this work.
Received 2018-1-28; Accepted 2018-4-19; Published 2018-6-7
Citation:
Song J, Xie C, Jiang L, Wu G, Zhu J, Zhang S, Tang M, Song L, Li J. Transcription factor AP-4 promotes tumorigenic capability and activates the Wnt/β-catenin pathway in hepatocellular carcinoma. Theranostics 2018; 8(13):3571-3583. doi:10.7150/thno.25194. https://www.thno.org/v08p3571.htm
Other stylesAbstract

It has been reported that the transcription factor activating enhancer-binding protein 4 (TFAP4) is upregulated and associated with an aggressive phenotype in several cancers. However, the precise mechanisms underlying the oncogenic role of TFAP4 remain largely unknown.
Methods: TFAP4 expression levels in hepatocellular carcinoma (HCC) cells and tissues were detected by quantitative real-time PCR (qPCR) and immunohistochemistry (IHC). In vitro and in vivo assays were performed to investigate the oncogenic function of TFAP4 in the tumor-initiating cell (TIC)-like phenotype and the tumorigenic capability of HCC cells. Luciferase reporter and chromatin immunoprecipitation (ChIP)-qPCR assays were performed to determine the underlying mechanism of TFAP4-mediated HCC aggressiveness.
Results: TFAP4 was markedly upregulated in human HCC, and was associated with significantly poorer overall and relapse-free survival in patients with HCC. Furthermore, we found that overexpression of TFAP4 significantly enhanced, whereas silencing TFAP4 inhibited, the tumor sphere formation ability and proportion of side-population cells in HCC cells in vitro, and ectopic TFAP4 enhanced the tumorigenicity of HCC cells in vivo. Mechanistically, we demonstrated that TFAP4 played an important role in activating Wnt/β-catenin signaling by directly binding to the promoters of DVL1 (dishevelled segment polarity protein 1) and LEF1 (lymphoid enhancer binding factor 1).
Conclusions: Our results provide new insight into the mechanisms underlying hyperactivation of the Wnt/β-catenin pathway in HCC, as well the oncogenic ability of TFAP4 to enhance the tumor-forming ability of HCC cells.
Keywords: TFAP4, hepatocellular carcinoma, tumorigenicity, Wnt/β-catenin signaling
Introduction
Hepatocellular carcinoma (HCC) is the third most common cause of cancer-related deaths; the 5-year survival rate for patients who do not receive treatment is less than 5% [1]. In China, patients with HCC account for nearly 50% of all new cases worldwide [2]. Unfortunately, the post-resection recurrence rate at 2 years is 50% [3] and the 5-year recurrence rate for patients with intermediate or advanced HCC is as high as 74% [4]. Recurrence is mainly attributed to the high tumorigenic potential and drug resistance of HCC cells [5]. Interestingly, these features can be detected in a small subpopulation of cells in HCC tissues, termed tumor-initiating cells (TICs) [6-8]. TICs have high self-renewal capability and are believed to give rise to tumor heterogeneity [9, 10]. An increasing number of studies support the notion that liver TICs are responsible for tumorigenesis and local recurrence in subgroups of HCC [6, 11]. Therefore, targeting such liver TIC-like traits may be a potential strategy to reduce tumor recurrence.
Aberrant activation of Wnt/β-catenin signaling is essential for the self-renewal capacity and drug-resistant properties of human colorectal cancer stem cells, and is associated with poor survival [12, 13]. Approximately 30% of cases of HCC exhibit hyperactivation of the Wnt/β-catenin signaling pathway [14-16]. It has been reported that aberrant Wnt/β-catenin pathway activation in HCC promotes proliferation and sorafenib resistance [17], whereas Wnt/β-catenin pathway inhibition weakens tumorigenic potential [18], suggesting hyperactivation of the Wnt/β-catenin signaling pathway contributes to tumorigenic properties and the TIC-like phenotype in HCC. Hence, exploration of the molecular mechanisms underlying Wnt/β-catenin signaling hyperactivation in HCC may reveal valuable therapeutic targets to prevent recurrence.
Transcription factor activating enhancer-binding protein 4 (TFAP4), a ubiquitously expressed transcription factor, is upregulated in multiple human malignancies, such as colorectal and gastric carcinoma [19-23], suggesting TFAP4 plays an oncogenic role in cancer pathogenesis and progression. Importantly, Jung et al. demonstrated that overexpressing TFAP4 directly repressed p21 transcription induced by transforming growth factor (TGF)-β in human keratinocytes and reduced p21 expression during myelomonoblast differentiation. They also found that TFAP4 is specifically expressed in colonic progenitor cells and colorectal carcinoma cells and that it can maintain cells in progenitor-like state. These results suggest that TFAP4 may be involved in maintaining cancer cells in a progenitor-like state [24, 25]. However, the precise mechanism underlying the oncogenic function of TFAP4 remains largely unknown.
Herein, we report that TFAP4 overexpression promoted Wnt/β-catenin pathway activation by directly binding with the promoters of DVL1 (dishevelled segment polarity protein 1) and LEF1 (lymphoid enhancer binding factor 1) to enhance the tumorigenicity and TIC-like phenotype of HCC cells in vitro and in vivo. These results provide new insight into TFAP4-mediated tumorigenicity and the mechanisms by which the Wnt/β-catenin pathway is hyperactivated in HCC.
Methods
Cell culture
Immortalized LO2 human liver cells were maintained in RPMI-1640 medium (Gibco, Grand Island, NY, USA, Cat. No. 61870044) supplemented with 10% fetal bovine serum (FBS; HyClone, Logan, UT, USA), 100 U/mL penicillin and 100 mg/mL streptomycin in a 5% CO2 atmosphere at 37 °C. HCC cell lines, including HepG2, Hep3B, PLC/PRF-5, MHCC-97H, HCC-LM3, MHCC-97L and QGY-7701 cells, were cultured in DMEM supplemented with 10% FBS.
Patients and tissue specimens
This study was conducted on a total of 197 primary paraffin-embedded, archived HCC specimens that had been histopathologically and clinically diagnosed at the Sun Yat-Sen University Cancer Center from 2003 to 2010. Clinical and clinicopathological classification and staging were determined according to the American Joint Committee on Cancer criteria [26]. Detailed clinical information on the 197 patients is summarized in Table S1. In addition, four freshly non-relapse HCC tissues and four relapse HCC tissues were collected. Percentage tumor purity in sections adjacent to regions used for RNA extraction was estimated during routine histopathologic analysis. Prior Patients' Consents and Approval were obtained from the Institutional Research Ethics Committee (IREC) for the clinical materials used in this study.
Western blotting analysis
Western blotting was performed according to a standard protocol, as described previously [27]. The following primary antibodies were used: anti-TFAP4 (Sigma, St. Louis, MO, USA, Cat. No. HP001912), anti-β-catenin (Pharmingen/BD Biosciences, Bedford, MA, USA, Cat. No. 610254), anti-DVL1 (Abcam, Cambridge, MA, USA, Cat. No. ab106844), and anti-LEF1 (Abcam, Cat. No. ab137872). To confirm equal sample loading, the membranes were stripped and re-probed using anti-α-tubulin antibody (Sigma-Aldrich, Cat. No. T6199), anti-β-actin antibody (Abcam, Cat. No. ab6276), or anti-p84 antibody (Abcam, Cat. No. ab487).
Immunohistochemistry
Immunohistochemistry (IHC) was performed in 197 HCC specimens, which were assessed using an anti-TFAP4 antibody (Sigma-Aldrich, Cat. No. HP001912), anti-active- β-catenin antibody (EMD Millipore, Temecula, CA, USA, Cat. No. 05-665-AF555), anti-DVL1 antibody (Proteintech, Rosemont, IL, USA, Cat. No. 27384-1-AP), or anti-LEF1 antibody (Abcam, Cat. No. ab22884) according to a standard protocol as described previously [28, 29]. The degree of immunostaining, scored separately by two independent pathologists who were blinded to the histopathological features and patient data of the samples, was determined by the staining index (SI). The SI was calculated as the product of the grade of tumor cell proportions and the staining intensity score. The tumor cell proportions were graded as follows: 0, no positive tumor cells; 1, < 2% positive tumor cells; 2, 2-8% positive tumor cells; 3, 8-20% positive tumor cells; and 4, > 20% positive tumor cells. Staining intensity was scored as: 1, no staining; 2, weak staining (light yellow); 3, moderate staining (yellow-brown); and 4, strong staining (brown). Accordingly, the protein expression as evaluated by SI has a possible score of 0, 1, 2, 3, 4, 6, 8, 9, 12, or 16. Samples with a SI ≥ 8 were determined as high expression, and those with a SI < 8 were determined as low expression. Cutoff values were determined based on a measure of heterogeneity using the log-rank test with respect to overall survival. β-Catenin activation was scored as nuclear positivity if > 5% of cells were stained.
IHC staining for protein expression was also analyzed quantitatively using an Olympus BX51 image analysis system along with Olympus cellSens Dimension 1.5 Imaging software (Olympus, Tokyo, Japan). The stained sections were assessed at 200× magnification, and 10 representative staining fields of view in each section were analyzed to determine mean optical density (MOD), which represents the strength of the staining signals measured as the proportion of positive pixels. MOD values for different groups of tissues were compared using the Student's t-test. Cut-off values were determined based on a measure of heterogeneity using the log-rank test with respect to survival. P < 0.05 was considered significant.
Plasmids, retroviral infection, and transfection
The human TFAP4 gene was PCR-amplified from cDNA and cloned into a pSin-EF2 lentiviral vector. To silence TFAP4, two human TFAP4-targeting short hairpin RNA (shRNA) sequences were cloned into a SUPER.retro.puro vector (OligoEngine, Washington, USA) to generate the respective pSUPER.retro.TFAP4-RNAi(s). HepG2 or LM3 cells were plated at 2×106 cells per p100 plate and transfected with 10 µg of the indicated plasmids. Stable cell lines expressing TFAP4 or TFAP4 shRNA were generated via retroviral infection using HEK293T cells as previously described [30]. The stable cell lines were selected for 10 days with 0.5 mg/mL puromycin. The human DVL1 promoter region spanning nucleotides -348 to -193 and the human LEF1 promoter region spanning nucleotides -321 to -173 (relative to the transcription initiation site) generated by PCR-amplification from HepG2 cells were cloned, respectively, into the NheI/BglII sites of pGL3-basic luciferase reporter plasmid (Promega, Madison, WI, USA) to generate DVL1 and LEF1 luciferase reporters. The primers are listed in the Supplemental Methods section on Plasmids, Retroviral Infection and Transfection.
Immunofluorescence assay
Cells were seeded on coverslips and cultured routinely. Once the cells had reached 70% confluence, they were fixed for 15 min in 4% paraformaldehyde (Beyotime, Shanghai, China). After washing with phosphate-buffered saline (PBS) three times, the cells were incubated with 0.1 mL Triton X-100 (0.5%) for 20 min and blocked for 20 min with 10% bovine serum albumin (BSA, Sigma-Aldrich, Cat. No. A1933-1G). Then, the cells were incubated with primary antibodies (anti-β-catenin, Pharmingen/BD Biosciences, Cat. No. 610254) at 4 °C for 8-10 h, washed with PBS, and incubated with fluorescein-conjugated goat anti-rabbit secondary antibodies (GeneCopoeia, Rockville, MD, USA) for 1 h. Nuclei were stained using 4,6-diamidino-2-phenylindole (DAPI, GeneCopoeia). The MOD method was used to quantify the nuclear β-catenin staining signals in the HepG2 cells and LM3 cells. Briefly, the stained slides were evaluated at 400× magnification using the FV10-ASW 4.2 computerized image analysis system (Olympus, Tokyo, Japan) assisted by ImageJ software (Version 1.48, https://imagej.nih.gov/ij/); 10 randomly picked fields per slide were analyzed to determine the nuclear β-catenin MOD, which represents the strength of staining signals as measured per positive pixels. The MOD data were statistically analyzed using the t-test to compare the MOD differences between groups; P < 0.05 was considered statistically significant.
Tumor sphere formation assay
The cells (5×102) were seeded in 6-well ultra-low cluster plates for 10-12 days. The tumor spheres were cultured in DMEM/F12 serum-free medium (Invitrogen, Carlsbad, CA, USA, Cat. No. 88215) supplemented with 2% B27 (Invitrogen, Cat. No. 12587010), 20 ng/mL epidermal growth factor (EGF, PeproTech, Rocky Hill, USA, Cat. No. 37000015), 20 ng/mL basic fibroblast growth factor (bFGF, PeproTech, Cat. No. 100-18B), 5 μg/mL insulin (PeproTech, Cat. No. 100-11), and 0.4% BSA (Sigma-Aldrich, Cat. No. A1933-1G). After 10-12 days, the tumor spheres (tight, spherical, non-adherent masses > 50 µm in diameter) were counted, and their images were captured under an inversed microscope. Sphere formation efficiency was calculated as colonies/input cells×100%.
Flow cytometric analysis
HCC cells were digested with trypsin and resuspended at 1×106 cells per mL in DMEM containing 2% FBS and then pre-incubated at 37 °C for 30 min with or without 100 μM verapamil (Sigma-Aldrich, Cat. No. 1711202) to inhibit ATP-binding cassette (ABC) transporters. The cells were then labeled with 5 μg/mL Hoechst 33342 (Sigma-Aldrich, Cat. No. B2261), and incubated for 90 min at 37 °C and were swirled every 10 min. After 90 min, the cells were washed with ice-cold PBS. After centrifugation using a desk centrifuge (Heraeus, Hanau, Germany), the cells were washed with PBS and processed for flow cytometry analysis. The cells were counterstained with 2 μg/mL propidium iodide (PI; Sigma-Aldrich, Cat. No. P4170) to identify dead cells. Then, 1×106 viable cells were analyzed and sorted in an EPICS ALTRA flow cytometer (Beckman Coulter, Brea, CA, USA). The data were analyzed by Summit5.2 software (Beckman Coulter). All experiments were performed in triplicate. The results were consistent and considered statistically significant for all tests.
Luciferase reporter assay
Luciferase reporter assay was performed according to a standard protocol as described previously [28]. Briefly, HCC cells (3×104 cells/well) were seeded in 24-well plates in triplicate and allowed to settle for 24 h. The indicated plasmids and 1.5 ng pRL-TK Renilla plasmid were transfected using Lipofectamine 3000 Reagent (Thermo Fisher Scientific, Waltham, MA, USA, Cat. No. L3000008). At 48 h post-transfection, luciferase and Renilla signals were determined by a Dual Luciferase Reporter Assay Kit (Promega, Cat. No. E1980) according to the manufacturer's instructions as previously described [31].
Preparation of nuclear extracts
Confluent HepG2 and LM3 cells in T75 flasks were washed with 5 mL PBS/phosphatase inhibitors, the supernatant aspirated, and 3 mL ice-cold PBS/phosphatase inhibitors added. The cells were removed by gently scraping with a cell lifter and transferred to a prechilled 15-mL conical tube. The cell suspension was centrifuged for 5 min at 200 rcf in a centrifuge precooled at 4 °C, and the supernatant was discarded. A Nuclear Extract Kit (Active Motif, Rixensart, Belgium, Cat. No. 40010) was used according to the manufacturer's instructions for isolating nuclear extracts from cell pellets.
Chromatin immunoprecipitation-qPCR (ChIP-qPCR)
Chromatin immunoprecipitation (ChIP) was performed as described previously [32]. Briefly, crosslinking was performed with 1% formalin, and the cells were lysed in SDS buffer and sonication was used to fragment the DNA. ChIP for TFAP4 was performed using a Flag antibody (Sigma, SAB4301135). Eluted DNA fragments were analyzed by qPCR. The primers are listed in section 'Chromatin immunoprecipitation-qPCR' in Supplementary Material.
Tumor xenograft
Male BALB/c nude mice (5 to 6-weeks-old, 16-18 g) were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China) and were housed in a barrier facility on a 12-h light/dark cycle. Based on a previously described standard protocol [28], the mice were randomly divided into four groups (n = 10 per group). HepG2 cells (1×106, 1×105, 1×104, 1×103, or 1×102 cells), stably transfected with TFAP4-RNAi#1, RNAi-vector, TFAP4 or vector, with Matrigel (final concentration, 25%) were inoculated into the inguinal folds of mice. Tumor volumes were measured with an external caliper, and calculated using the equation (L × W2)/2. At 36 days after inoculation, the mice were sacrificed, and the tumors were excised and subjected to pathologic examination.
Bioinformatics analysis
Gene set enrichment analysis (GSEA, http://software.broadinstitute.org/gsea/msigdb/index.jsp) software programs were used to analyze The Cancer Genome Atlas (TCGA) HCC dataset.
Statistical analysis
All statistical analyses were carried out using SPSS version 21.0 statistical software (SPSS Inc., New York, NY, USA). The two-tailed paired Student's t-test was used to assess the comparisons between groups. The relationships between TFAP4 expression and clinicopathological characteristics were determined using the χ2 test. Survival curves were plotted using the Kaplan-Meier method and compared using the log-rank test. Survival data were evaluated using univariate and multivariate Cox regression analyses. Bivariate correlations between variables were calculated using Spearman's rank correlation coefficients. P-values of < 0.05 were considered statistically significant.
Results
Elevated expression of TFAP4 is associated with recurrence in human HCC
To explore the role of TFAP4 in HCC and the mechanisms underlying the oncogenic function of TFAP4, we initially analyzed TFAP4 mRNA expression between HCC specimens and normal controls in a publicly available HCC dataset (The Cancer Genome Atlas, TCGA). As shown in Figure 1A, TFAP4 mRNA expression was significantly elevated in HCC tissues compared to tumor-adjacent liver tissues. Importantly, TFAP4 mRNA expression was negatively associated with relapse-free survival (P = 0.008, Figure 1B) and overall survival (P = 0.031, Figure S1A), suggesting that TFAP4 gene expression might be an indicator of the risk of recurrence in HCC. Consistently, both TFAP4 protein and mRNA expression were significantly higher in the HCC cell lines than in the immortalized LO2 human liver cell line (Figure 1C-D) and in the HCC relapse tissues compared with the non-relapse HCC tissues (Figure 1E-F).
Figure 1
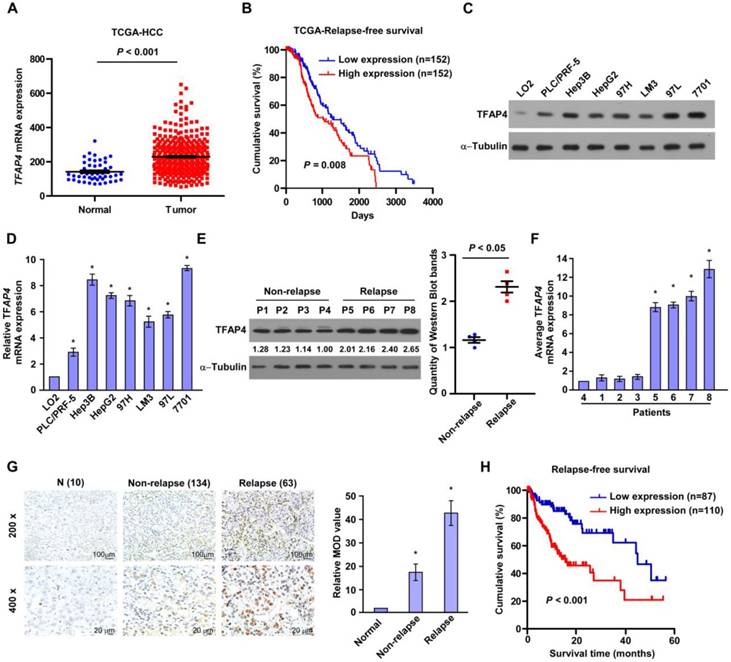
Elevated TFAP4 expression is associated with recurrence in human HCC. (A) TFAP4 mRNA levels in the HCC TCGA dataset. (B) Relapse-free survival of patients in TCGA HCC dataset with low versus high levels of TFAP4 mRNA. (C, D) Western blot analysis (C) and qRT-PCR analysis (D) of TFAP4 expression in LO2 cells and HCC cell lines. (E) Western blot analysis (left) and quantification (right) of TFAP4 expression in four HCC relapse tissues and four non-relapse HCC tissues. TFAP4 protein levels were normalized to α-tubulin. (F) qRT-PCR analysis of TFAP4 expression in four HCC relapse tissues and four non-relapse HCC tissues. TFAP4 mRNA levels were normalized to that of GAPDH. (G) Representative images of IHC staining of 134 primary HCC specimens versus 63 primary HCC specimens that relapsed later after treatment (top, 200× magnification; bottom, 400× magnification) (left). Statistical analysis of the average MOD of TFAP4 staining in normal liver tissues and HCC specimens (right). Bars represent the mean ± SD of three independent experiments; *P < 0.05. (H) Relapse-free survival of patients with HCC with low versus high TFAP4 expression. HCC: hepatocellular carcinoma; IHC: immunohistochemistry; MOD: mean optical density; qRT-PCR: quantitative real-time PCR; TCGA: the cancer genome atlas; TFAP4: transcription factor AP-4.
To evaluate the relationship between TFAP4 expression and the clinicopathological features of HCC, 197 primary HCC tissues (134 primary HCC specimens without relapse and 63 primary HCC specimens that relapsed after treatment) were subjected to IHC staining using a human TFAP4 antibody. TFAP4 expression was significantly higher in the HCC relapse tissues compared to the non-relapse tissues (Figure 1G). Furthermore, statistical analyses of the 197 specimens revealed that TFAP4 protein expression was associated with clinical stage (P < 0.001), tumor size (P = 0.001), histological differentiation (P = 0.038) and recurrence (P = 0.007), but not with age, gender, or alpha-fetoprotein (AFP), and hepatitis B surface antigen (HBsAg) (Table S2 and Table S3). Moreover, Kaplan-Meier analysis and log-rank testing revealed that TFAP4 protein expression was negatively associated with relapse-free survival (P < 0.001; n = 197; Figure 1H) and overall survival (P < 0.001; Figure S1B). Moreover, univariate and multivariate analysis revealed that TFAP4 expression was an independent prognostic factor of overall survival of HCC (P < 0.001; Table S4). Taken together, these data indicate that upregulation of TFAP4 contributes to recurrence and is associated with a poorer prognosis in HCC.
Table 1
Effect of Transcription factor AP-4 on the tumorigenicity of hepatocellular carcinoma cells in vivo (n = 10/group).
| Cell type | Cell number | Tumor incidence | TIC Frequency (95% CI) | P value |
|---|---|---|---|---|
| HepG2-Vector | 1000000 | 10/10 | 1/40,070 (1/79,901-1/20,095) | |
| 100000 | 7/10 | |||
| 10000 | 5/10 | |||
| 1000 | 3/10 | |||
| 100 | 0/10 | < 0.01 | ||
| HepG2-TFAP4 | 1000000 | 10/10 | 1/12025 (1/26,124-1/5,535) | |
| 100000 | 9/10 | |||
| 10000 | 7/10 | |||
| 1000 | 5/10 | |||
| 100 | 2/10 | |||
| HepG2-RNAi-vector | 1000000 | 10/10 | 1/28,214 (1/5,8953-1/13,503) | |
| 100000 | 8/10 | |||
| 10000 | 6/10 | |||
| 1000 | 3/10 | |||
| 100 | 0/10 | < 0.05 | ||
| HepG2-TFAP4-RNAi#1 | 1000000 | 10/10 | 1/65,094 (1/129,823-1/3,2638) | |
| 100000 | 6/10 | |||
| 10000 | 4/10 | |||
| 1000 | 1/10 | |||
| 100 | 0/10 |
Overexpression of TFAP4 in HCC cells promotes a TIC-like phenotype in vitro
TFAP4 expression was inversely correlated with tumor differentiation status (Table S2), which suggested that TFAP4 may be involved in the regulation of stemness. Consistently, GSEA of a publicly available HCC dataset (TCGA) revealed that TFAP4 expression was positively associated with stemness signatures (Figure 2A). Furthermore, real-time PCR revealed that the mRNA levels of the stemness-related markers, including CD133, NANOG, SOX2, ABCG2, BMI1, CD44, EpCAM and ALDH1A1 were significantly upregulated in TFAP4-transduced HepG2 and HCC-LM3 cells, and conversely downregulated in TFAP4-silenced HCC cells (Figure 2B-C). Next, we conducted the tumor sphere formation assay to examine the effect of TFAP4 on the self-renewal ability of spherogenic HCC cells. Notably, TFAP4-transduced cells formed more spheres with higher cell numbers than the vector control cells (Figure 2D), whereas TFAP4-silenced cells formed fewer spheres with lower cell numbers than the vector control cells (Figure 2E). Taken together, our results suggest that TFAP4 overexpression promotes HCC cell tumorigenicity in vitro.
The side population (SP) is a subpopulation of cells that may exhibit a TIC-like phenotype [33]. Consistently, overexpression of TFAP4 increased the proportion of HepG2 SP+ cells from 0.99% to 2.39%, and HCC-LM3 SP+ cells from 0.60% to 2.27% (Figure 2F-G). Conversely, silencing TFAP4 decreased the proportion of HepG2 SP+ cells from 1.00% to 0.15% and HCC-LM3 SP+ cells from 0.58% to 0.12% (Figure 2F-G). Collectively, these results indicate that overexpression of TFAP4 enhances the TIC subpopulation and promotes a TIC-like phenotype in HCC cells in vitro.
Overexpression of TFAP4 enhances the tumorigenicity of HCC cells in vivo
To explore the oncogenic role of TFAP4 in the promotion of the TIC-like phenotype in HCC cells in vivo, we subcutaneously inoculated different numbers of HCC cells mixed with Matrigel into the inguinal folds of BALB/c nude mice. The tumors formed by TFAP4-transduced HepG2 cells were significantly larger than those formed by vector control cells after implantation of 1×106, 1×105, 1×104, 1×103, or 1×102 cells (Figure 3A-D and Table 1). Conversely, TFAP4-silenced cells formed significantly smaller tumors and had lower tumorigenicity (Figure 3A-D and Table 1). Importantly, only TFAP4/HepG2 cells formed tumors when 1×102 cells were implanted (Figure 3A-D and Table 1). These results suggest that TFAP4 overexpression promotes the tumorigenicity of HCC cells in vivo.
Wnt/β-catenin signaling contributes to TFAP4-mediated TIC-like phenotype in HCC cells
To explore the mechanism related to the TFAP4-mediated TIC-like phenotype in HCC, we analyzed the expression of TFAP4 and the genes regulated by various signaling signatures using GSEA of TCGA HCC datasets. TFAP4 mRNA expression correlated positively with Wnt-activated gene signatures (Figure 4A) and negatively with Wnt-suppressed gene signatures (Figure S2A), indicating that TFAP4 may activate the Wnt/β-catenin signaling pathway. Furthermore, TOP/FOP flash assays revealed that overexpression of TFAP4 markedly increased whereas TFAP4 knockdown attenuated the transcriptional activation of TCF/LEF in the HepG2 and HCC-LM3 cells (Figure 4B). Additionally, nuclear extract and immunofluorescence assays showed that TFAP4 overexpression substantially increased the β-catenin nuclear signals, whereas knockdown of TFAP4 reduced them (Figure 4C-D). Meanwhile, the mRNA levels of the downstream targets of Wnt/β-catenin signaling, including CCND1, CD44, JUN, and TCF1, were increased in TFAP4-transduced cells but were decreased in TFAP4-silenced cells (Figure 4E). Collectively, these data suggest that TFAP4 overexpression activates the Wnt/β-catenin signaling pathway.
Figure 2
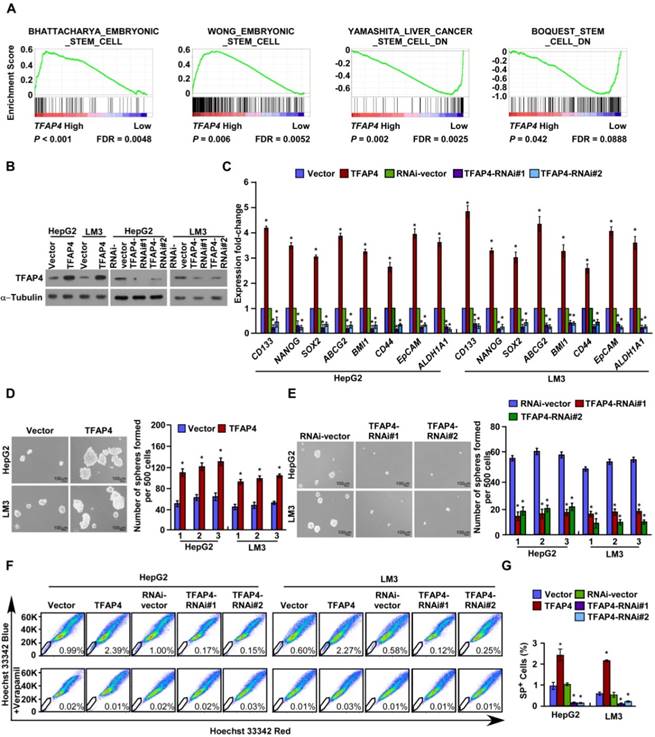
TFAP4 overexpression promotes a TIC-like phenotype in HCC cells in vitro. (A) GSEA indicating significant correlations between TFAP4 mRNA expression and stemness-related gene signatures (BHATTACHARYA_ EMBRYONIC_ STEM_CELL, WONG_EMBRYONIC_STEM_CELL, YAMASHITA_LIVER_ CANCER_ STEM_CELL_DN, BOQUEST_STEM_CELL_DN). (B) Western blot analysis of TFAP4 in the indicated TFAP4-transduced, TFAP4-silenced, or vector control cells. α-Tubulin was used as the loading control. (C) Real-time PCR analysis of stemness-related markers in the indicated cells. (D) Representative micrographs (left) and quantification (right) of tumor sphere formation by TFAP4-overexpressing cells or vector control cells. Bars represent the mean ± SD of three independent experiments; *P < 0.05. (E) Representative micrographs (left) and quantification (right) of tumor sphere formation by TFAP4-silenced cells or vector control cells. Bars represent the mean ± SD of three independent experiments; *P < 0.05. (F) Hoechst 33342 dye exclusion assay of the effect of TFAP4 on the proportion of SP+ cells in the indicated cells. (G) Statistical analysis of the proportion of SP+ cells. Bars represent the mean ± SD of three independent experiments; *P < 0.05. GSEA: gene set enrichment analysis; TIC: tumor-initiating cell.
Figure 3
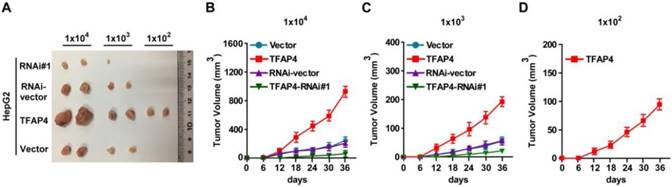
Overexpression of TFAP4 enhances the tumorigenicity of HCC cells in vivo. (A) Tumors formed by TFAP4-transduced HCC cells were larger than the vector control tumors. Conversely, the tumors formed by TFAP4-silenced cells were smaller than those formed by the RNAi-vector cells. Cells (1×106, 1×105, 1×104, 1×103 or 1×102) were implanted into BALB/c nude mice. When 1×102 cells were injected, only the TFAP4-transduced cells formed tumors. Representative images of the tumors are shown. (B-D) Tumor formation growth curves following implantation of 1×104 (B), 1×103 (C), or 1×102 (D) cells.
Figure 4
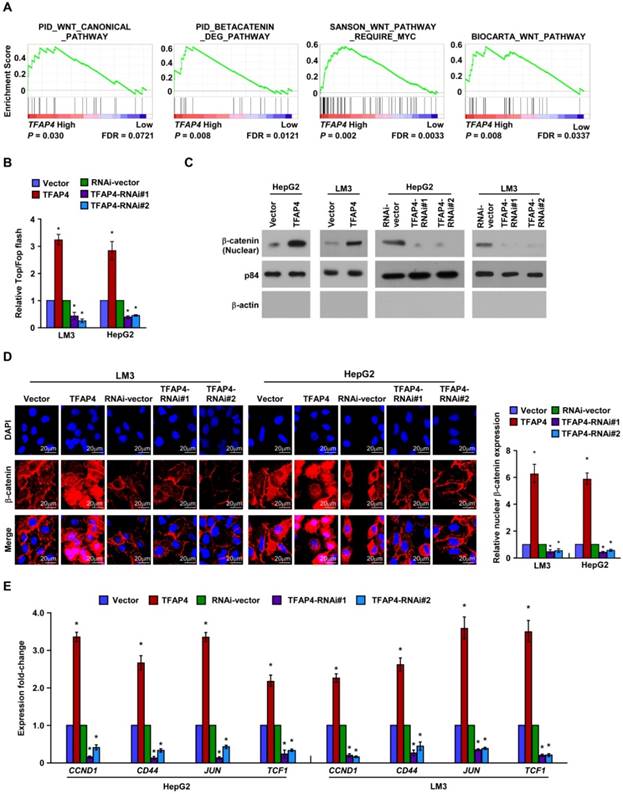
Overexpression of TFAP4 activates the Wnt/β-catenin signaling. (A) GSEA showing the positive correlations between TFAP4 mRNA levels and Wnt signaling based on publicly available HCC profiles (PID_WNT_CANONICAL _PATHWAY, PID_BETACATENIN_DEG_PATHWAY, SANSON_WNT_PATHWAY_ REQUIRE _MYC, BIOCARTA_WNT_PATHWAY). (B) Luciferase-reporter assays of TOP/FOP transcriptional activity in the indicated cells. (C) Western blotting analysis of β-catenin in the nuclear fractions of the indicated cells. (D) Immunofluorescence staining (left) and quantification (right) of nuclear β-catenin expression in the indicated cells. (E) Real-time PCR of the mRNA expression of Wnt/β-catenin downstream genes. Bars indicate the mean ± SD of three independent experiments; *P < 0.05. GSEA: gene set enrichment analysis.
Figure 5
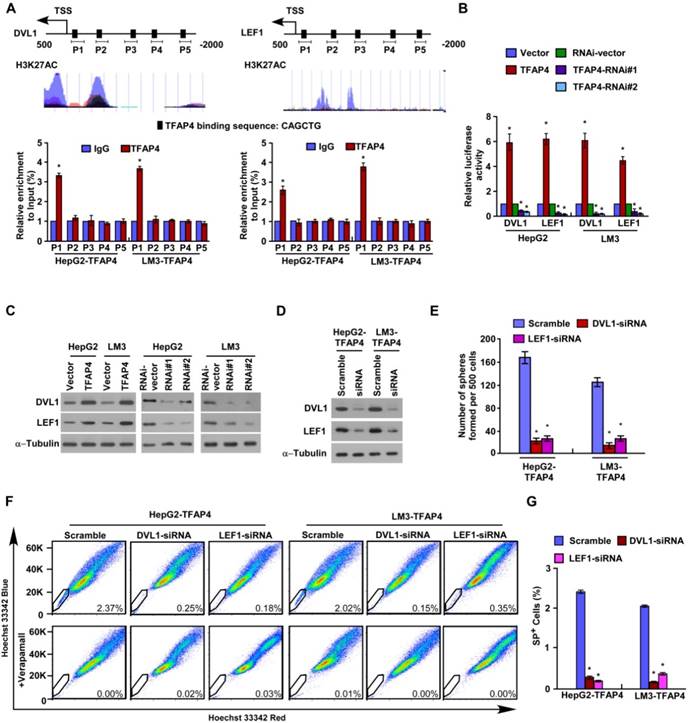
TFAP4 directly upregulates DVL1 and LEF1 in HCC cells. (A) Prediction and validation of possible TFAP4-target genes. (B) Luciferase reporter assay of DVL1 and LEF1 transcriptional activity. (C) DVL1 and LEF1 protein levels in the cells. α-Tubulin was used as the loading control. (D) Protein levels of DVL1 and LEF1 in TFAP4-transduced HCC cells transfected with DVL1 or LEF1 siRNAs. (E) Quantification of tumor sphere formation in TFAP4-transduced cells transfected with DVL1 or LEF1 siRNA. Bars represent the mean ± SD of three independent experiments; *P < 0.05. (F) Hoechst 33342 dye exclusion assay of the effect of knocking down DVL1 or LEF1 on the proportion of SP+ cells. (G) Statistical analysis of the proportion of SP+ cells in the cells. Bars represent the mean ± SD of three independent experiments; *P < 0.05. DVL1: dishevelled segment polarity protein 1; LEF1: lymphoid enhancer binding factor 1.
To further validate the downstream targets of TFAP4 in the Wnt/β-catenin signaling pathway, we performed JASPAR database analysis and predicted that the promoters of the DVL1, LEF1 and TCF4 genes contain TFAP4 binding sites (Figure 5A and Figure S2B). Chromatin immunoprecipitation (ChIP) assay confirmed that TFAP4 can directly associate with the DVL1 and LEF1 promoters but does not bind to the TCF4 promoter (Figure 5A and Figure S2B). Furthermore, TFAP4 overexpression significantly increased DVL1/LEF1 promoter-driven reporter activity and DVL1 and LEF1 protein expression levels in HCC cells, whereas silencing TFAP4 had the opposite effects (Figure 5B-C), confirming TFAP4 directly targets and positively regulates DVL1 and LEF1. Consistently, overexpressing TFAP4 increased the LEF1 and DVL1 mRNA levels in HCC cells, but silencing TFAP4 decreased them (Figure S2C). However, no obvious alteration of TCF4 mRNA was observed in TFAP4-dysregulated cells and vector control cells, which was consistent with the ChIP experiment results (Figure S2B). Next, we investigated whether DVL1 or LEF1 activation was required for the ability of TFAP4 to enhance the TIC-like phenotype in HCC cells. Silencing DVL1 or LEF1 significantly reduced the sphere-forming ability of TFAP4-overexpressing cells and the expression of the stemness-related markers, including CD133, NANOG, SOX2, ABCG2, BMI1, CD44, EpCAM, and ALDH1A1, as well as the expression of the downstream targets of Wnt/β-catenin signaling (Figure 5D-E and Figure S3A-B). The increased proportion of SP+ cells induced by overexpression of TFAP4 was also dramatically abrogated by the DVL1 or LEF1 small interfering RNA (siRNA) (Figure 5F-G). These results suggest that activation of DVL1 or LEF1 is required for the ability of TFAP4 to promote a TIC-like phenotype in HCC cells. Taken together, these data indicate that TFAP4 enhances the tumorigenicity of HCC cells by directly targeting and positively regulating DVL1 and LEF1 to activate the Wnt/β-catenin signaling pathway.
Figure 6
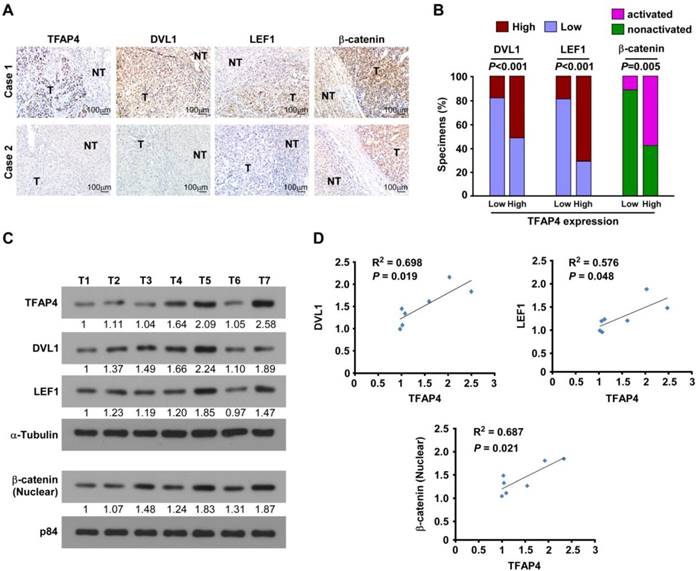
TFAP4 expression correlates with Wnt/β-catenin pathway activation in human HCC. (A) DVL1, LEF1, and activated β-catenin expression levels were associated with TFAP4 expression in the 197 primary human HCC specimens; two representative cases are shown. Scale bar, 100 μm. (B) Percentages of the 197 primary human HCC specimens showing low or high DVL1, LEF1 and activated β-catenin expression relative to low/high TFAP4 expression. IHC quantification was performed using the staining intensity (SI). (C, D) Western blotting analysis (C) and correlation analyses (D) of TFAP4 expression with expression of DVL1, LEF1 and activated β-catenin in seven freshly collected human HCC samples; α-tubulin was used as a loading control. DVL1: dishevelled segment polarity protein 1; LEF1: lymphoid enhancer binding factor 1.
TFAP4 expression correlates with Wnt/β-catenin signaling activation in human HCC
Next, we examined whether the Wnt/TFAP4/DVL1-LEF1 axis identified in HCC cells were clinically relevant using IHC analysis. TFAP4 protein expression was correlated positively with the levels of DVL1, LEF1, and activated β-catenin (P < 0.05; n = 87) (Figure 6A-B). These observations were validated in seven freshly collected HCC tissue specimens (Figure 6C), in which TFAP4 expression correlated positively with the expression of DVL1 (P = 0.019, R2 = 0.698), LEF1 (P = 0.048, R2 = 0.576), and activated β-catenin (P = 0.021, R2 = 0.687; Figure 6D). Taken together, our results indicate that upregulation of TFAP4 in HCC correlates positively with expression DVL1 and LEF1, which in turn activates the Wnt/β-catenin signaling pathway to enhance the tumorigenicity of HCC cells.
Discussion
The key findings of the present report are that TFAP4 promotes tumor sphere formation, increases the proportion of SP+ cells, enhances tumorigenicity and activates the Wnt/β-catenin signaling pathway in HCC. Meanwhile, TFAP4 was overexpressed in human HCC and high TFAP4 expression was associated with poor overall survival and relapse-free survival, indicating TFAP4 may represent a valuable prognostic factor for recurrence in HCC.
TFAP4 belongs to the basic helix-loop-helix-leucine zipper (bHLH-LZ) subgroup of proteins and recognizes the symmetrical DNA core sequence CAGCTG [34, 35]. Overexpression of TFAP4 promotes neuroblastoma cell growth [36] and induces the epithelial-mesenchymal transition (EMT) and metastasis in colorectal cancer [32]. Importantly, it is believed that TFAP4 is involved in maintaining cancer cells in a progenitor-like state [25], which confers a degree of tumor stemness. TICs, which have self-renewal, multiple differentiation and tumorigenic properties, are critical determinants of the capacity for malignant proliferation, invasion, metastasis, tumor recurrence and drug resistance [37, 38]. Several studies have shown that TICs most likely give rise to HCC and a progenitor cell phenotype is detected in a substantial proportion of HCC cases [39, 40]. In fact, patients with HCC whose tumors express stem cell markers such as CD133, CD44 and CD90 have a significantly poorer prognosis and higher rate of recurrence [41]. We found that overexpressing TFAP4 in HCC cells promoted tumor sphere formation and increased the proportion of SP+ cells (these features are associated with poor overall survival and relapse-free survival in patients with HCC), whereas silencing TFAP4 inhibited sphere formation and reduced the proportion of SP+ cells (which is associated with improved survival). These results suggest that upregulation of TFAP4 enhances tumorigenicity and promotes a TIC-like phenotype in HCC cells, which may hold promise for development of novel therapeutic treatments to prevent recurrence.
The Wnt/β-catenin pathway, also termed the canonical Wnt pathway, is crucial to embryonic development and adult tissue self-renewal [42, 43]. Aberrant activation of this pathway can cause uncontrolled cell growth and malignant transformation [42, 43]. Malanchi et al. demonstrated that periostin interacts with Wnt1 and Wnt3a, inducing Wnt signaling and sustaining a TIC phenotype in breast tumor cells [44]. MMP3 secreted by mammary epithelial cells stimulated Wnt/β-catenin signaling in mammary stem cells [45]. Importantly, Zhu et al. found that β-catenin methylation suppressed β-catenin ubiquitination and stability, leading to Wnt/β-catenin signaling activation and sustaining tumorigenicity of HCC cell lines [46]. The Wnt/β-catenin pathway also participates in TIC self-renewal and proliferation in HCC cells [47-49]. Consistently, we found that Wnt/β-catenin pathway was hyperactivated in human HCC. Herein, we demonstrate that TFAP4 directly targets and positively regulates DVL1 and LEF1, which results in Wnt/β-catenin pathway activation and upregulation of multiple downstream genes. These results reveal a novel molecular mechanism of Wnt/β-catenin pathway hyperactivation in HCC. Therefore, these results suggest that TFAP4 may not only contribute to Wnt/β-catenin activation but may also selectively regulate the downstream targets of β-catenin, which would be worthy of further investigation.
Jung and colleagues reported that the oncoprotein c-MYC could upregulate TFAP4 transcriptionally by directly binding to a region within the first intron of the AP4 gene, which contains four canonical c-MYC binding motifs. They further demonstrated that TFAP4 directly represses p21 by occupying four CAGCTG motifs in the p21 promoter via its basic region. Importantly, TFAP4 overexpression could block p53/p21-induced cell cycle arrest in cells with DNA damage and antagonize p21 induction mediated by TGF-β and 12-O-tetradecanoylphor-bol-13-acetate (TPA) during myelomonoblast differentiation. Therefore, this finding not only describes a novel mechanism by which c-MYC mediates p21 repression but also suggests that TFAP4 may be involved in maintaining cancer cells in a progenitor-like state [24, 25]. Moreover, c-MYC-mediated TFAP4 induction is required for c-MYC overexpression-induced cell cycle progression, metastasis, senescence suppression, and transformation, as well as CD8+ T cell activation and sustained expansion of germinal center B cells [50-54]. Interestingly, c-MYC is a downstream target of Wnt/β-catenin signaling [55], which is constitutively activated in HCC and in other tumor types [56, 57]. Herein, we demonstrate that TFAP4 overexpression activates the Wnt/β-catenin pathway in HCC by directly binding to the DVL1 and LEF1 promoters, thereby enhancing tumorigenicity. Accordingly, these results suggest that TFAP4-mediated c-MYC upregulation might take place be through the activation of Wnt/β-catenin signaling. Therefore, c-MYC and TFAP4 might form a positive feedback loop through Wnt/β-catenin signaling in HCC cells, which would contribute to HCC initiation and progression.
In summary, we demonstrate that TFAP4 overexpression in HCC directly upregulates DVL1 and LEF1 and activates the Wnt/β-catenin pathway, thereby enhancing tumorigenicity, promoting a TIC-like phenotype and conferring a poor prognosis. Further exploration of the biological function of TFAP4 in HCC would increase our knowledge of the mechanisms underlying the high rate of recurrence in HCC, and establish whether TFAP4 represents a prognostic factor or potential therapeutic target for recurrence in HCC.
Abbreviations
ChIP: chromatin immunoprecipitation; GSEA: gene set enrichment analysis; HCC: hepatocellular carcinoma; IHC: immunohistochemistry; MOD: mean optical density; qRT-PCR: quantitative real-time PCR; SI: staining index; TFAP4: transcription factor AP4; TICs: tumor-initiating cells; TCGA: the cancer genome atlas.
Acknowledgements
This work was supported by the Natural Science Foundation of China [No. 91740119, 91740118, 81530082, 91529301, 81472259, 81672874, 81621004]; the Natural Science Foundation of Guangdong Province [No. 2015A030313468, 2015A020212007, 2016A030308002]; the Guangzhou Science and Technology Plan Projects [No. 201510010292, 201803010098]; the Distinguished Young Scholar of Guangdong Province [No. 2015A030306033]; the Young Scholar of Science and Technology of Guangdong Province [No. 2016TQ03R801]; and the Innovative Academic Team of Guangzhou Education System [No. 1201610014]; the Fundamental Research Funds for the Central Universities [No. 17ykjc02].
Supplementary Material
Supplementary materials and methods, figures and tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11-30
2. Ferlay J, Soerjomataram I, Dikshit R. et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359-86
3. Maluccio M, Covey A. Recent progress in understanding, diagnosing, and treating hepatocellular carcinoma. CA Cancer J Clin. 2012;62:394-9
4. Zhong JH, Ke Y, Gong WF. et al. Hepatic resection associated with good survival for selected patients with intermediate and advanced-stage hepatocellular carcinoma. Ann Surg. 2014;260:329-40
5. Yamashita T, Wang XW. Cancer stem cells in the development of liver cancer. J Clin Invest. 2013;123:1911-8
6. Chen ZZ, Huang L, Wu YH. et al. LncSox4 promotes the self-renewal of liver tumour-initiating cells through Stat3-mediated Sox4 expression. Nat Commun. 2016;7:12598
7. Tang Y, Kitisin K, Jogunoori W. et al. Progenitor/stem cells give rise to liver cancer due to aberrant TGF-beta and IL-6 signaling. Proc Natl Acad Sci U S A. 2008;105:2445-50
8. Oishi N, Yamashita T, Kaneko S. Molecular biology of liver cancer stem cells. Liver Cancer. 2014;3:71-84
9. Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8:755-68
10. Jordan CT, Guzman ML, Noble M. Cancer stem cells. N Engl J Med. 2006;355:1253-61
11. Han H, Du Y, Zhao W. et al. PBX3 is targeted by multiple miRNAs and is essential for liver tumour-initiating cells. Nat Commun. 2015;6:8271
12. Li J, Yu B, Deng P. et al. KDM3 epigenetically controls tumorigenic potentials of human colorectal cancer stem cells through Wnt/β-catenin signalling. Nat Commun. 2017;8:15146
13. van de Wetering M, Sancho E, Verweij C. et al. The beta-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell. 2002;111:241-50
14. Lachenmayer A, Alsinet C, Savic R. et al. Wnt-pathway activation in two molecular classes of hepatocellular carcinoma and experimental modulation by sorafenib. Clin Cancer Res. 2012;18:4997-5007
15. Guichard C, Amaddeo G, Imbeaud S. et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet. 2012;44:694-8
16. Kan Z, Zheng H, Liu X. et al. Whole-genome sequencing identifies recurrent mutations in hepatocellular carcinoma. Genome Res. 2013;23:1422-33
17. Liu Y, Ye X, Zhang JB. et al. PROX1 promotes hepatocellular carcinoma proliferation and sorafenib resistance by enhancing beta-catenin expression and nuclear translocation. Ongene. 2015;34:5524-35
18. Yamashita T, Ji J, Budhu A. et al. EpCAM-positive hepatocellular carcinoma cells are tumor-initiating cells with stem/progenitor cell features. Gastroenterology. 2009;136:1012-24
19. Lau WY, Lai EC. Hepatocellular carcinoma: current management and recent advances. Hepatobiliary Pancreat Dis Int. 2008;7:237-57
20. Koorey D. Hepatocellular carcinoma: prevention, detection and treatment.. in the real world. Intern Med J. 2007;37:513-5
21. Pleguezuelo M, Marelli L, Misseri M. et al. TACE versus TAE as therapy for hepatocellular carcinoma. Expert Rev Anticancer Ther. 2008;8:1623-41
22. Massari ME, Murre C. Helix-loop-helix proteins: regulators of transcription in eucaryotic organisms. Mol Cell Biol. 2000;20:429-40
23. Edge SB, Compton CC. The American Joint Committee on Cancer: the 7th edition of the AJCC cancer staging manual and the future of TNM. Ann Surg Oncol. 2010;17:1471-4
24. Jung P, Hermeking H. The c-MYC-AP4-p21 cascade. Cell Cycle. 2009;8:982-9
25. Jung P, Menssen A, Mayr D. et al. AP4 encodes a c-MYC-inducible repressor of p21. Proc Natl Acad Sci U S A. 2008;105:15046-51
26. Edge SB, Compton CC. The American Joint Committee on Cancer: the 7th edition of the AJCC cancer staging manual and the future of TNM. Ann Surg Oncol. 2010;17:1471-4
27. Li J, Zhang N, Song LB. et al. Astrocyte elevated gene-1 is a novel prognostic marker for breast cancer progression and overall patient survival. Clin Cancer Res. 2008;14:3319-26
28. Gong L, Song J, Lin X. et al. Serine-arginine protein kinase 1 promotes a cancer stem cell-like phenotype through activation of Wnt/beta-catenin signalling in NSCLC. J Pathol. 2016;240:184-96
29. Lee TK, Castilho A, Cheung VC. et al. CD24(+) liver tumor-initiating cells drive self-renewal and tumor initiation through STAT3-mediated NANOG regulation. Cell Stem Cell. 2011;9:50-63
30. Li J, Gong LY, Song LB. et al. Oncoprotein Bmi-1 renders apoptotic resistance to glioma cells through activation of the IKK-nuclear factor-kappaB Pathway. Am J Pathol. 2010;176:699-709
31. Hahn WC, Dessain SK, Brooks MW. et al. Enumeration of the simian virus 40 early region elements necessary for human cell transformation. Mol Cellu Biol. 2002;22:2111-23
32. Bernt KM, Zhu N, Sinha AU. et al. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell. 2011;20:66-78
33. Ho MM, Ng AV, Lam S. et al. Side population in human lung cancer cell lines and tumors is enriched with stem-like cancer cells. Cancer Res. 2007;67:4827-33
34. Mermod N, Williams TJ, Tjian R. Enhancer binding factors AP-4 and AP-1 act in concert to activate SV40 late transcription in vitro. Nature. 1988;332:557-61
35. Hu YF, Luscher B, Admon A. et al. Transcription factor AP-4 contains multiple dimerization domains that regulate dimer specificity. Genes Dev. 1990;4:1741-52
36. Xue C, Yu DM, Gherardi S. et al. MYCN promotes neuroblastoma malignancy by establishing a regulatory circuit with transcription factor AP4. Oncotarget. 2016;7:54937-51
37. Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010;29:4741-51
38. Li S, Li Q. Cancer stem cells and tumor metastasis (Review). Int J Oncol. 2014;44:1806-12
39. Fausto N. Liver regeneration and repair: hepatocytes, progenitor cells, and stem cells. Hepatology. 2004;39:1477-87
40. Michalopoulos GK, DeFrances MC. Liver regeneration. Science. 1997;276:60-6
41. Roskams T. Liver stem cells and their implication in hepatocellular and cholangiocarcinoma. Oncogene. 2006;25:3818-22
42. Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Ann Rev Cell Dev Biol. 2004;20:781-810
43. Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell. 2012;149:1192-205
44. Malanchi I, Santamaria-Martinez A, Susanto E. et al. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature. 2011;481:85-9
45. Kessenbrock K, Dijkgraaf GJ, Lawson DA. et al. A role for matrix metalloproteinases in regulating mammary stem cell function via the Wnt signaling pathway. Cell Stem Cell. 2013;13:300-13
46. Zhu P, Wang Y, Huang G. et al. lnc-beta-Catm elicits EZH2-dependent beta-catenin stabilization and sustains liver CSC self-renewal. Nat Struct Mol Biol. 2016;23:631-9
47. Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature. 2005;434:843-50
48. Reya T, Morrison SJ, Clarke MF. et al. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105-11
49. Tsai RY. A molecular view of stem cell and cancer cell self-renewal. Int J Biochem Cell Biol. 2004;36:684-94
50. Jackstadt R, Hermeking H. AP4 is required for mitogen- and c-MYC-induced cell cycle progression. Oncotarget. 2014;5:7316-27
51. Jackstadt R, Jung P, Hermeking H. AP4 directly downregulates p16 and p21 to suppress senescence and mediate transformation. Cell Death Dis. 2013;4:e775
52. Jackstadt R, Roh S, Neumann J. et al. AP4 is a mediator of epithelial-mesenchymal transition and metastasis in colorectal cancer. J Exp Med. 2013;210:1331-50
53. Chou C, Pinto AK, Curtis JD. et al. c-Myc-induced transcription factor AP4 is required for host protection mediated by CD8+ T cells. Nat Immunol. 2014;15:884-93
54. Chou C, Verbaro DJ, Tonc E. et al. The Transcription Factor AP4 Mediates Resolution of Chronic Viral Infection through Amplification of Germinal Center B Cell Responses. Immunity. 2016;45:570-82
55. He TC, Sparks AB, Rago C. et al. Identification of c-MYC as a target of the APC pathway. Science. 1998;281:1509-12
56. Clevers H. Axin and hepatocellular carcinomas. Nat Genet. 2000;24:206-8
57. Polakis P. Wnt signaling and cancer. Genes Dev. 2000;14:1837-51
Author contact
![]() Corresponding author: Jun Li, Ph.D. Guangdong Provincial Key Laboratory of Liver Disease, The Third Affiliated Hospital of Sun Yat-Sen University, Guangzhou, Guangdong 510630, China; Phone: +86(20)87335828; Fax: +86(20)87335828; E-mail: lijun37sysu.edu.cn
Corresponding author: Jun Li, Ph.D. Guangdong Provincial Key Laboratory of Liver Disease, The Third Affiliated Hospital of Sun Yat-Sen University, Guangzhou, Guangdong 510630, China; Phone: +86(20)87335828; Fax: +86(20)87335828; E-mail: lijun37sysu.edu.cn
Citation styles
APA
Song, J., Xie, C., Jiang, L., Wu, G., Zhu, J., Zhang, S., Tang, M., Song, L., Li, J. (2018). Transcription factor AP-4 promotes tumorigenic capability and activates the Wnt/β-catenin pathway in hepatocellular carcinoma. Theranostics, 8(13), 3571-3583. https://doi.org/10.7150/thno.25194.
ACS
Song, J.; Xie, C.; Jiang, L.; Wu, G.; Zhu, J.; Zhang, S.; Tang, M.; Song, L.; Li, J. Transcription factor AP-4 promotes tumorigenic capability and activates the Wnt/β-catenin pathway in hepatocellular carcinoma. Theranostics 2018, 8 (13), 3571-3583. DOI: 10.7150/thno.25194.
NLM
Song J, Xie C, Jiang L, Wu G, Zhu J, Zhang S, Tang M, Song L, Li J. Transcription factor AP-4 promotes tumorigenic capability and activates the Wnt/β-catenin pathway in hepatocellular carcinoma. Theranostics 2018; 8(13):3571-3583. doi:10.7150/thno.25194. https://www.thno.org/v08p3571.htm
CSE
Song J, Xie C, Jiang L, Wu G, Zhu J, Zhang S, Tang M, Song L, Li J. 2018. Transcription factor AP-4 promotes tumorigenic capability and activates the Wnt/β-catenin pathway in hepatocellular carcinoma. Theranostics. 8(13):3571-3583.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.