Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(12):3224-3236. doi:10.7150/thno.23259 This issue Cite
Research Paper
Oncogenic TRIM31 confers gemcitabine resistance in pancreatic cancer via activating the NF-κB signaling pathway
Chao Yu*, Shiyu Chen*, Yuntao Guo, Chengyi Sun ![]()
Department of Hepatobiliary Surgery, Affiliated Hospital of Guizhou Medical, University, Guiyang, Guizhou 550004, China.
*These authors contributed equally to this work
Received 2017-10-10; Accepted 2018-3-31; Published 2018-5-11
Citation:
Yu C, Chen S, Guo Y, Sun C. Oncogenic TRIM31 confers gemcitabine resistance in pancreatic cancer via activating the NF-κB signaling pathway. Theranostics 2018; 8(12):3224-3236. doi:10.7150/thno.23259. https://www.thno.org/v08p3224.htm
Other stylesAbstract

Background: Drug resistance is well known as a major obstacle for cancer recurrence and treatment failure, leading to poor survival in pancreatic cancer, which is a highly aggressive tumor. Identifying effective strategies to overcome drug resistance would have a significant clinical impact for patients with pancreatic cancer.
Methods: The protein and mRNA expression of TRIM31 in pancreatic cancer cell lines and patient tissues were determined using Real-time PCR and Western blot, respectively. 89 human pancreatic cancer tissue samples were analyzed by IHC to investigate the association between TRIM31 expression and the clinicopathological characteristics of pancreatic cancer patients. Functional assays, such as MTT, FACS, and Tunel assay used to determine the oncogenic role of TRIM31 in human pancreatic cancer progression. Furthermore, western blotting and luciferase assay were used to determine the mechanism of TRIM31 promotes chemoresistance in pancreatic cancer cells.
Results: The expression of TRIM31was markedly upregulated in pancreatic cancer cell lines and tissues, and high TRIM31 expression was associated with an aggressive phenotype and poor prognosis with pancreatic cancer patients. TRIM31 overexpression confers gemcitabine resistance on pancreatic cancer cells; however, inhibition of TRIM31 sensitized pancreatic cancer cell lines to gemcitabine cytotoxicity both in vitro and in vivo. Additionally, TRIM31 upregulated the levels of nuclear p65 by promoting K63-linked polyubiquitination of tumor necrosis factor receptor-associated factor 2 (TRAF2) and sustained the activation of nuclear transcription factor kappa B (NF-κB) in pancreatic cancer cells.
Conclusions: Our findings provided evidence that TRIM31 is a potential therapeutic target for patients with pancreatic cancer. Targeting TRIM31 signaling may be a promising strategy to enhance gemcitabine response during pancreatic cancer chemo-resistance.
Keywords: TRIM31, chemoresistance, pancreatic cancer, NF-κB signaling
Introduction
Pancreatic cancer is the fourth leading cause of cancer-related mortality worldwide, as manifested by a 5-year survival probability of just 7% for all patients [1-3]. The major challenge responsible for this poor prognosis is the extreme chemoresistance phenotype of the tumor. The currently available treatment for pancreatic cancer is cytoreductive surgery, followed by gemcitabine-based chemotherapy, which leads to a slight improvement in overall survival [4-6]. However, most patients with pancreatic cancer still remain refractory and undergo tumor relapse, ultimately succumbing to this malignant tumor [7]. Therefore, a better understanding the molecular basis of chemoresistance in pancreatic cancer would provide novel therapeutic opportunities for patients with pancreatic cancer.
The nuclear transcription factor kappa B (NF-κB) pathway, which is constitutively activated in various cancers, not only orchestrates immune and inflammatory responses, but also plays a crucial role in cancer development [8-10]. During the past decade, ubiquitin modification has become an important factor in the regulation of NF-κB signaling [11-13]. In response to tumor necrosis factor-α (TNF-α) or interleukin 1β (IL-1β), signaling intermediaries, such as TNF receptor associated factors (TRAFs) and receptor interacting protein (RIP), are rapidly modified with K63-linked polyubiquitin chains, facilitating recruitment and activation of TGF-β-activated kinase1 (TAK1) and I kappa B kinase (IKK) complexes by binding to TGFβ-activated kinase 1 (TAK1)-binding proteins (TABs) and NF-kappa-B essential modulator (NEMO), respectively [14-16]. The activated IKK complex phosphorylates IκBs, resulting in phosphorylation and K48-ubiquitination/degradation of IκBs, consequently leading to NF-κB activation [17, 18]. Collectively, these findings indicated that ubiquitin modification plays important roles in virtually every step of NF-κB signaling cascades.
Most pancreatic tumors show high levels of constitutive activated NF-κB, which mediates survival signaling and confers chemo-resistance to conventional therapeutics [19, 20]. Increased expression of NF-kappa B subunits and enhanced NF-κB activity were found in pancreatic cancer cells, which correlated with a worse prognosis in pancreatic cancer. Zhang et al. demonstrated that overexpression and activation of the epidermal growth factor (EGF) receptor contributes to tumor progression of pancreatic tumors by permanent activation of NF-κB [21]. More importantly, blockade of NF-κB expression by delivery of a p65-specific short interfering RNA (siRNA) to pancreatic cancer cells in vivo could effectively inhibit apoptosis, indicating that inhibition of NF-κB activity by siRNAs might have therapeutic potential [22]. Despite these promising findings, the technology requires significant improvement, including the discovery of novel molecular(s) that can regulate the aberrant activation of the NF-κB pathway.
Tripartite motif containing 31 (TRIM31) is a newly identified protein comprising a RING finger, B-box, and coiled-coil domains. TRIM31 functions as an E3 ubiquitin-protein ligase. Recently, several reports have shown that TRIM31 attenuates NLR family pyrin domain containing 3 (NLRP3) inflammasome activation by promoting proteasomal degradation of NLRP3 and the aggregation and activation of the signaling adaptor mitochondrial antiviral-signaling protein (MAVS) through Lys63-linked polyubiquitination [23, 24]. These studies demonstrated that TRIM31 plays a vital role in inflammatory responses and viral infection. Importantly, TRIM31 showed rapid kinetics of induction during retinoid-induced growth arrest of breast carcinoma cells and is upregulated in gastric adenocarcinoma, suggesting that TRIM31 is involved in carcinogenesis [25, 26]. However, the role and the molecular mechanism of TRIM31 in the aggression and treatment failure of pancreatic cancer remain ambiguous.
Methods
Cell lines and cell culture
Primary cultures of normal human pancreatic duct epithelial cells (HPDECs) were established from fresh specimens of the adjacent non-tumor pancreatic tissue and maintained in bronchial epithelial basal medium (BEBM; Lonza Walkersville, Walkersville, MD, USA) containing 10% FBS and supplemented with BEGM Single-Quots (Lonza Walkersville), according to a previous report. The human pancreatic cell lines PANC-1, CFPAC-1, BxPC-3, AsPC-1, Capan-1, Capan-2, MIA PaCa-2, Hs 766T, and MIN6 were cultured as described in the ATCC protocol.
Patient information and tissue specimens
A total of 89 paraffin-embedded pancreatic adenocarcinoma samples, which were histopathologically and clinically diagnosed, were obtained from Affiliated Hospital of Guizhou Medical University. Clinical information on the samples is summarized in Table S1. Prior patient consent and approval from the Institutional Research Ethics Committee were obtained.
Vectors, retroviral infection, and transfection
A TRIM31 expression construct was generated by subcloning the PCR-amplified full-length human TRIM31 cDNA into the pMSCV retrovirus plasmid, and human TRIM31-targeting short hairpin RNA (shRNA) oligonucleotides sequences were cloned into pSuper-retro-puro to generate pSuper-retro-TRIM31-RNAi(s). The shRNA sequences were: RNAi#1, TTCCCGTC AAAGGAAGTTTGG; RNAi#2, TATGATGGACTCATGCCTTGC (synthesized by RIBO). pNF-κB-luc and control plasmids (Clontech) were used to examine NF-κB activity. pBabe-Puro-IκBα-mut (plasmid#15291) expressing IκBα dominant-negative mutant (IκBα-mut) was purchased from Addgene (Cambridge, MA, USA). Transfection of siRNA or plasmids was performed using the Lipofectamine 3000 reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instruction. Stable cell lines expressing TRIM31 or TRIM31/RNAi were selected for 10 days with 0.5 μg/mL puromycin 48 h after infection.
Western blotting analysis
Western blotting was performed using anti-TRIM31, anti-cleaved caspase 3, anti-PARP, anti-p65, anti-IκBα, anti-IKK-β, anti-BCL2, anti-XIAP, anti-SURVIVIN, and anti-TRAF2 antibodies, (Cell Signaling, Danvers, MA, USA). The membranes were stripped and re-probed with an anti-α-tubulin antibody or anti-α-tubulin antibody (Sigma, St Louis, MO, USA) as a loading control.
Xenografted tumor model
In the subcutaneous tumor model, BALB/c nude mice were randomly divided into four groups (n = 6/group). Four groups of mice were inoculated subcutaneously with 2×106 PANC-1/Vector, PANC-1/TRIM31, PANC-1/shRNA-Vector, PANC-1/TRIM31-shRNA#1 cells, respectively, in the left dorsal flank per mouse. After the xenografts reached 0.5 cm in diameter, gemcitabine (100 mg/kg) was given intraperitoneally twice a week for 43 days. Tumor growth was monitored by measurements of the length and width and the tumor volume was calculated using the equation (L × W2)/2. Tumors were detected by an IVIS imaging system, and animals were euthanized, tumors were excised, weighed and paraffin-embedded. A TUNEL assay was performed on paraffin-embedded tissue section according to the manufacturer's instructions (Promega). All experimental procedures were approved by the Institutional Animal Care and Use Committee of Affiliated Hospital of Guizhou Medical University.
Cytotoxicity assay
The sensitivity to gemcitabine of pancreatic cancer cells was determined using the MTT assay. Briefly, 2×103 cells were seeded onto 96-well plates and incubated at 37 °C overnight. Cells were then transfected with different concentrations of gemcitabine (50 µM). After incubation for 48 h, 50 μL of the MTT solution (0.15%) was added to each well, and the plates were further incubated for 2 h. One hundred microliters of DMSO was added to solubilize the MTT formazan product. Absorbance at 540 nm was measured using a Falcon microplate reader (BD-Labware). Dose-response curves were plotted on a semilog scale as the percentage of the control cell number, which was obtained from the sample with no drug exposure. The IC50 was determined by the intersection of the gemcitabine concentration and the midpoint of the 540-nm reading.
Apoptosis assay
To evaluate apoptosis, a PE Annexin V Apoptosis Detection Kit I (BD Pharmingen) was used. Briefly, 1×106 pancreatic cancer cells were plated in 10-cm plates and incubated for 24 h. Treatment was started with gemcitabine for the indicated time. Cell morphology was assessed using phase-contrast microscopy. The cells were then removed from the plate using trypsin-EDTA, washed twice with phosphate-buffered saline (PBS), and re-suspended with binding buffer at 106 cells/mL. FITC Annexin V and propidium iodide were added (each at 5 μL /105 cells). Cells were incubated for 15 min at room temperature in the dark. Percentage of apoptosis was analyzed using an EPICS XL flow cytometer (Beckman-Coulter). Each sample was analyzed in triplicate.
Luciferase assay
Cells (1×104) were seeded in triplicate in 48-well plates and allowed to settle for 24 h. One hundred nanograms of luciferase reporter plasmids or the control plasmid, plus 1 ng of pRL-TK renilla plasmid (Promega), were transfected into cells using the Lipofectamine 3000 reagent (Invitrogen) according to the manufacturer's instructions. Luciferase and renilla signals were measured using the Dual Luciferase Reporter Assay Kit (Promega) according to a protocol provided by the manufacturer.
Statistical analysis
Statistical tests for data analysis included Fisher's exact test, log-rank test, Chi-square test, and Student's 2-tailed t test. Multivariate statistical analysis was performed using a Cox regression model. Statistical analyses were performed using the SPSS 11.0 statistical software package. Data represent mean ± standard deviation (SD). P < 0.05 was considered statistically significant.
Results
TRIM31 overexpression correlates with progression and poor prognosis in human pancreatic cancer
By analyzing the published mRNA expression profiles (GSE62452; GSE60980; GSE28735) obtained from NCBI (https://www.ncbi.nlm.nih.gov/geo/), we found that TRIM31 mRNA was significantly upregulated in pancreatic cancer tissues compared with non-tumor tissues (Figure 1A). This upregulation of TRIM31 was confirmed by The Cancer Genome Atlas (TCGA; http://cancergenome.nih.gov/) dataset (Figure 1B). Furthermore, analysis of TCGA datasets showed that patients with pancreatic cancer with higher TRIM31 expression had a shorter survival time and demonstrated an earlier relapse disease-free survival time (P < 0.05; Figure 1C). Consistently, real-time PCR and western blotting analyses revealed that TRIM31 was upregulated at both the protein and mRNA levels in all nine pancreatic cancer cell lines and in human pancreatic cancer tissues, compared with two primary normal human pancreatic duct epithelial cells (HPDEC) or adjacent non-tumor tissues (Figure 1D-E and Figure S1A-B), suggesting that TRIM31 is upregulated in human pancreatic cancer.
Figure 1
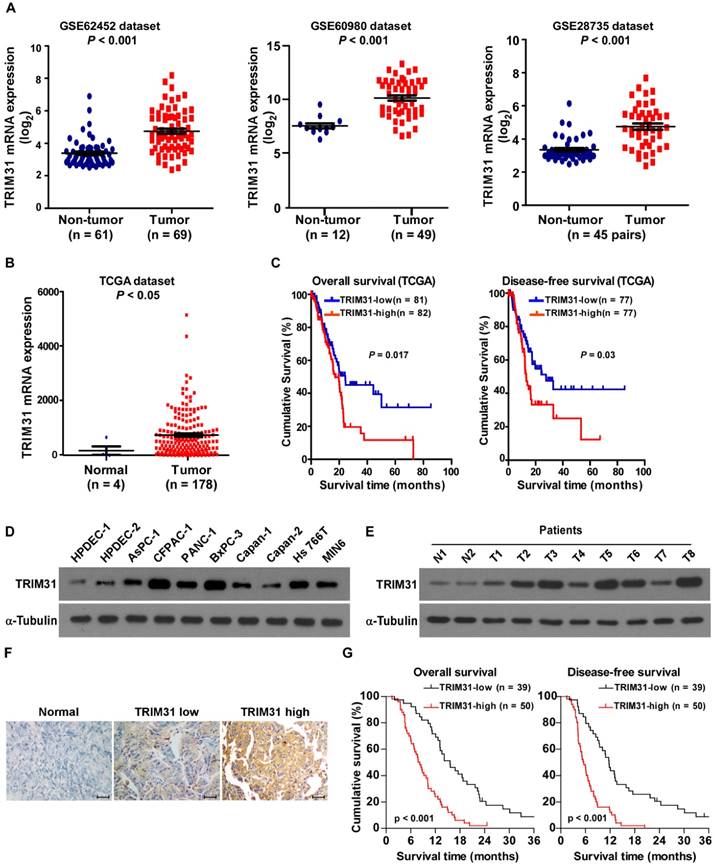
TRIM31 overexpression correlates with progression and poor prognosis in human pancreatic cancer. (A) Expression profiling of mRNAs showing that TRIM31 is upregulated in pancreatic cancer tissues (T) compared with non-tumor tissues (GSE62452; GSE60980; GSE28735). (B) Expression profiling of mRNAs showing that TRIM31 is upregulated in pancreatic cancer tissues (T) compared with normal tissues (n =182; TCGA dataset, p < 0.05). (C) Kaplan-Meier analysis of overall (left) or disease-free (right) survival curves from the TCGA dataset of patients with pancreatic cancer with low vs. high TRIM31 expression, p < 0.05. (D-E) Western blotting analysis of TRIM31 expression in two primary normal HPDEC lines, nine cultured pancreatic cancer cell lines (D), and in nine primary pancreatic cancer tissues (T) and their matched adjacent non-tumor tissues (ANT) (E); α-Tubulin was used as protein loading control. (F) IHC staining indicating the TRIM31 protein levels in human pancreatic cancer compared with pancreatic non-tumor tissue, (Scale bars: 100μm). (G) The Kaplan-Meier overall (left) or disease-free (right) survival curves comparing pancreatic cancer patients with low and high TRIM31 expression levels (n = 89; P < 0.05). HPDEC, human pancreatic duct epithelial cells; IHC, immunohistochemistry; TCGA, The Cancer Genome Atlas.
Furthermore, TRIM31 expression was examined in 89 paraffin-embedded, archived pancreatic cancer tissues and in 20 paraffin-embedded, archived adjacent non-tumor tissues by an immunohistochemistry (IHC) assay. As shown in Figure 1F, TRIM31 was markedly upregulated in pancreatic cancer tissues, but was only marginally detectable in adjacent non-tumor tissues. Importantly, statistical analysis showed that patients with pancreatic cancer with high TRIM31 expression had worse overall and disease-free survival than those with low TRIM31 expression (Figure 1G). These results suggested that TRIM31 has potential clinical value as a predictive biomarker for disease outcome in pancreatic cancer.
Figure 2
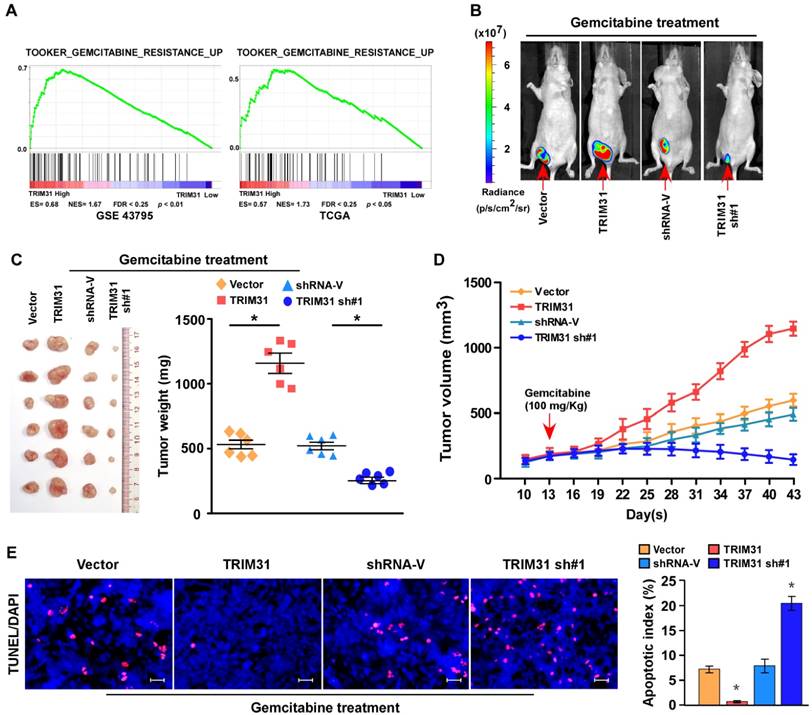
Upregulation of TRIM31 conferred gemcitabine resistance on pancreatic cancer in vivo. (A) GSEA plot indicating a significant correlation between the mRNA levels of TRIM31 in pancreatic cancer and the gemcitabine resistance-related gene signatures in published datasets. (B) Representative images of tumor-bearing mice in the indicated cells treated with gemcitabine (100 mg/kg). (C) Tumors from all the mice in the indicate cell (left) together with the mean tumor weights (right). (D) Tumor volumes were measured on the indicated days. (E) IHC staining demonstrating the expression of TUNEL-positive cells in the indicated tissues (Scale bars: 100μm). Each bar represents the mean ± SD of three independent experiments. * P <0.05. GSEA, gene set enrichment analysis; IHC, immunohistochemistry; TUNEL, terminal deoxynucleotidyl transferase (TdT) dUTP nick-end labeling.
Upregulation of TRIM31 conferred gemcitabine resistance on pancreatic cancer in vivo
Cancer relapse mainly stems from resistance to chemotherapy. Interestingly, gene set enrichment analysis (GSEA) revealed that TRIM31 overexpression strongly correlated with gene signatures associated with gemcitabine-based chemotherapy, suggesting that TRIM31 overexpression might contribute to gemcitabine-resistance in pancreatic cancer (Figure 2A).
To explore the function of TRIM31 in pancreatic cancer chemoresistance, we used nude mouse models to assess the anti-tumor effect of TRIM31 in pancreatic cancer. Nude mice were subcutaneously inoculated with PANC-1/Vector and PANC-1-TRIM31, PANC-1/Vector-shRNA and PANC-1/TRIM31-shRNA respectively, and then treated with gemcitabine twice per week as soon as the tumor became palpable. As shown in Figure 2B-D and Table S2, treatment with the TRIM31-shRNA plus gemcitabine resulted in a significant reduction in tumor growth; however, overexpression of TRIM31 resulted in a significant increase compared with that in the control group. Furthermore, TRIM31 overexpression conferred marked resistance to chemotherapy-induced apoptosis, as determined by the decrease in the proportion of TUNEL+-cells compared with that in the control group (Figure 2E). However, silencing TRIM31 using TRIM31-shRNA enhanced the cytotoxic effect of gemcitabine on pancreatic cancer cells, which resulted in increased TUNEL+-cells compared with those in the control group (Figure 2E). However, we did not observe significant alterations between the dysregulated-TRIM31 group and the vector control group under vehicle treatment in the in vivo experiments (Figure S2C-E). Therefore, these results demonstrated that TRIM31 overexpression contributes to pancreatic cancer chemoresistance.
Upregulation of TRIM31 conferred gemcitabine resistance on pancreatic cancer in vitro
Abnormal regulation of apoptosis is one of the important mechanisms of drug resistance. To investigate the anti-apoptosis role of TRIM31 in pancreatic cancer progression, PANA-1 and AsPC-1 cell lines, which stably express TRIM31, were established (Figure S2A-B). Interestingly, the protein level of cleaved caspase3 and PARP were significantly decreased in TRIM31 overexpressing pancreatic cancer cells but increased in TRIM31 downregulated cells (Figure 3A). The percentage of apoptotic cells in TRIM31-overexpressing pancreatic cancer cells treated with gemcitabine was much lower than that in the control cells, but much higher in the TRIM31-silenced cells (Figure 3B). Moreover, upregulation of TRIM31 drastically decreased the gemcitabine-induced apoptosis rates and induced the colony-forming capacity of PANA-1 and AsPC-1 cells (Figure 3C). In contrast, the IC50 value for gemcitabine was significantly decreased in TRIM31-overexpression pancreatic cancer cells, but increased in TRIM31-silenced cells (Figure 3D), suggesting that TRIM31 enhanced the resistance of pancreatic cancer cells to gemcitabine in vitro. Furthermore, we found that overexpressing TRIM31 or silencing TRIM31 only resulted in a slight change in the apoptosis rate of pancreatic cancer cells without gemcitabine treatment (Figure S2F, D). Taken together, these results indicated that deregulation of TRIM31 is involved in gemcitabine resistance of pancreatic cancer cells.
Upregulation of TRIM31 activates the NF-κB signaling pathway in pancreatic cancer
Through analysis of TRIM31 mRNA expression levels and NF-κB-regulated gene signatures from two collections of published pancreatic cancer profiles (NCBI/GEO/GSE43795 and GSE35141), we found that TRIM31 Gene ontology (GO) enrichment analysis showed that NF-κB signaling pathway was enriched in both datasets (Figure 4A). By analyzing TRIM31 mRNA expression levels and NF-κB-regulated gene signatures from published profiles of patients with pancreatic cancer, we found that TRIM31 expression correlated positively with NF-κB signaling gene signatures (Figure 4B). In addition, the overexpression of TRIM31 significantly enhanced the activity of an NF-κB luciferase reporter, whereas silencing of TRIM31 significantly reduced the reporter activity (Figure 4B). Moreover, western blotting revealed that the levels of nuclear p65 were dramatically upregulated in TRIM31-overexpressing cells, but were downregulated in TRIM31-silenced cells (Figure 4C). Consistently, we also observed that upregulation of TRIM31 substantially increased, whereas downregulation of TRIM31 decreased, the protein levels of phosphorylated-IKK-β and-IκBα (Figure 4D). Furthermore, the mRNA and protein levels of numerous well characterized anti-apoptotic factors, as well as NF-κB downstream genes (BCL2, XIAP, and SURVIVIN) increased in TRIM31 overexpressing cells, but were lower in TRIM31-silenced cells (Figure 4E and Figure S4). These results suggested that TRIM31 upregulates anti-apoptosis genes in pancreatic cancer by activating the NF-κB signaling pathway.
The NF-κB signaling pathway is required for TRIM31-induced chemoresistance
To further validate that TRIM31-mediates pancreatic cancer chemoresistance through NF-κB activation, we blocked the NF-κB pathway in TRIM31-overexpressing cells by overexpressing an IκBα dominant negative mutant (IκBα-mut) or by treatment with an NF-κB inhibitor. As expected, the stimulatory effect of TRIM31 overexpression on NF-κB activation was inhibited by IκBα-mut and the NF-κB inhibitor (Figure S3A). Strikingly, IκBα-mut or treatment with an NF-κB inhibitor significantly enhanced the effects of gemcitabine in vitro, as determined by an IC50 assay and Annexin V-FITC assay, but decreased colony formation by the cells compared with that in the control group (Figure S3B-D). We then investigated whether TRIM31-mediated pancreatic cancer progression occurs via NF-κB activation in vivo. Strikingly, treatment with an NF-κB inhibitor significantly enhanced the effects of gemcitabine in vivo, as determined by quantification of bioluminescence signal, tumor volume, and proportion of TUNEL+-cells compared with that in the control group (Figure 5A-C). Taken together, these results indicated that activation of the NF-κB signaling pathway mediates the functional effects of TRIM31 on pancreatic cancer drug resistance.
Figure 3
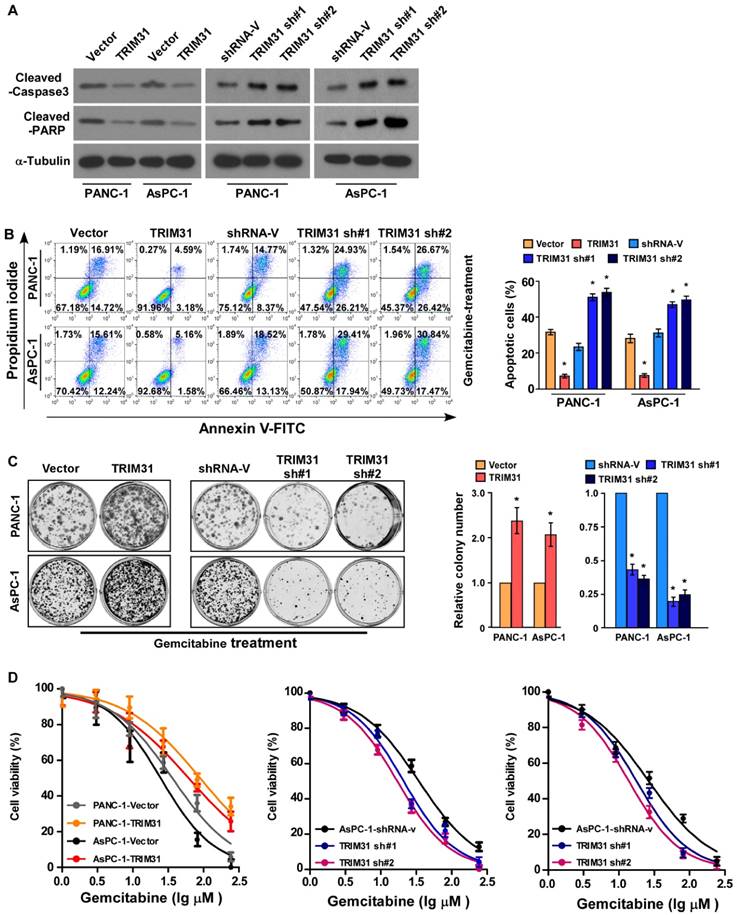
Upregulation of TRIM31 conferred gemcitabine resistance on pancreatic cancer in vitro. (A) Western blotting analysis of cleaved caspase3 and PARP in the indicated cells; α-tubulin was used as a loading control. (B) Annexin V-FITC and PI staining of the indicated cells treated with gemcitabine (50 μM) for 48 h. Each bar represents the mean ± SD of three independent experiments. * P <0.05. (C) Representative images (left panel) and quantification (right panel) of the indicated cells after crystal violet staining. Each bar represents the mean ± SD of three independent experiments. * P <0.05. (D) IC50 of gemcitabine in the indicated cells. Each bar represents the mean ± SD of three independent experiments. * P <0.05. FITC, fluorescein isothiocyanate; PI, propidium iodide.
Figure 4
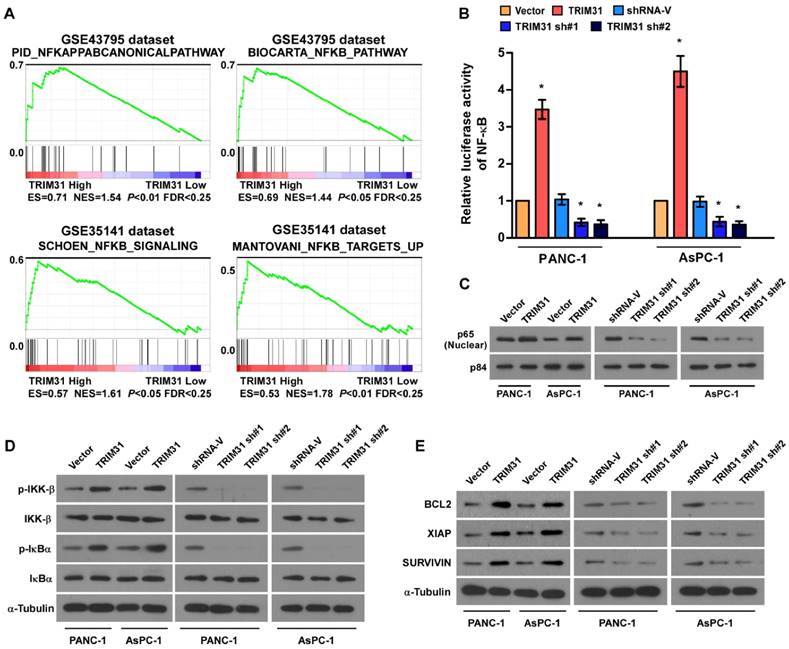
Upregulation of TRIM31 activates the NF-κB signaling pathway in pancreatic cancer. (A) GSEA plot indicating a significant correlation between the mRNA levels of TRIM31 in pancreatic cancer and the NF-κB-activated gene signatures in published datasets. (B) Analysis of luciferase reporter activity in the indicated cells after transfection with 100 ng of pNF-κB-luc plasmids or control-luciferase plasmid. Each bar represents the mean ± SD of three independent experiments. * P <0.05. (C) Western blotting analysis of the protein levels of P65 in the indicated cells; P84 was used as a loading control, (Scale bars: 100μm). (D) Western blotting analysis of the levels of the indicated proteins in the indicated cells; α-tubulin was used as a loading control. (E) Western blotting analysis of the levels of BCL2, XIAP, and Survivin in the indicated cells; α-tubulin was used as a loading control. GSEA, gene set enrichment analysis.
TRIM31 sustains NF-κB signaling pathway activation
Previous studies have confirmed that TNF stimulation rapidly induces the recruitment and ubiquitination of tumor necrosis factor receptor-associated factor 2 (TRAF2), RIP, and NEMO in the NF-κB receptor complex, and this process is crucial for TNF-induced NF-κB activation. Interestingly, coimmunoprecipitation and western blotting assays demonstrated that TRIM31 could form a complex with TRAF2, suggesting that TRIM31 might be involved in regulation of TNF-induced NF-κB activation (Figure 5D). This interaction between TRIM31 and TRAF2 was further confirmed by checking their endogenous levels (Figure 5E). As shown in Figure 5F, the K63-polyubiquitination levels of TRAF2 were higher in TRIM31-transduced cells and lower in TRIM31-silenced cells than in the corresponding control cells, suggesting that TRIM31 promotes ubiquitin conjugation in NF-κB signaling (Figure 5F). Moreover, we found that the decreased levels of IκB after TNFα treatment were significantly prolonged in TRIM31-overexpressing cells and reduced in TRIM31-silenced cells (Figure 5G). Taken together, these results indicated that TRIM31 promotes ubiquitin conjugation of NF-κB signaling-related proteins and sustains NF-κB activation.
Clinical relevance of TRIM31 induced NF-κB activation in pancreatic cancer
The clinical relevance of TRIM31 expression and NF-κB activation were further characterized in human pancreatic cancer. As shown in Figure 6A and Table S1, TRIM31 levels in ten freshly collected clinical pancreatic cancer samples correlated positively with nuclear p65 signals (r = 0.65, P = 0.042) and the mRNA expression of anti-apoptotic downstream genes: XIAP (r = 0.65, P = 0.043), Bcl-xL (r = 0.74, P = 0.012) and SURVIVIN (r = 0.66, P = 0.039). The correlation of TRIM31 expression and nuclear p65, as an indicator of NF-κB signaling activation, was further confirmed in a cohort of clinical pancreatic cancer tissues using IHC analysis (P < 0.01; Figure 6B). Importantly, statistical analysis showed that patients with pancreatic cancer with high p65 expression had worse overall and disease-free survival than those with low p65 expression (Figure 6C). These data further supported the notion that TRIM31 upregulation promotes pancreatic cancer chemoresistance by activating the NF-κB signaling pathway, which might lead to tumor relapse and poor clinical outcome for patients with pancreatic cancer.
Figure 5
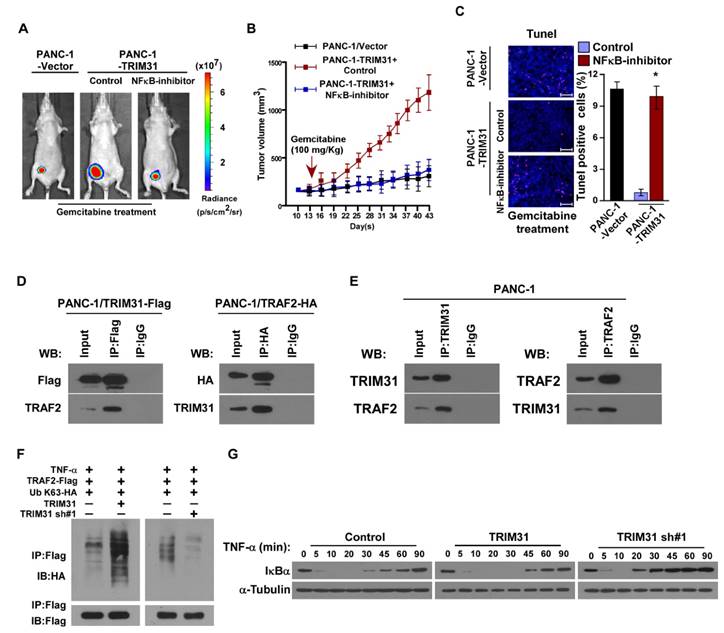
The NF-κB signaling pathway is required for TRIM31-induced chemoresistance. (A) Representative images of tumor-bearing mice treated with gemcitabine (100 mg/kg) and treated with the NF-κB inhibitor (JSH-23). (B) Tumor volumes were measured on the indicated days. (C) IHC staining demonstrating the expression of TUNEL-positive cells in the indicated tissues (Scale bars: 100μm). Each bar represents the mean ± SD of three independent experiments. * P <0.05. (D) Immunoprecipitation assay indicating that TRIM31 interacts with TRAF2 in the indicated cells (exogenous). (E) Immunoprecipitation assay indicating that TRIM31 interacts with TRAF2 in the indicated cells (endogenous). (F) Western blotting analysis of the K63-linked polyubiquitination levels of TRAF2 in the indicated cells treated with TNFα (10 ng/mL). (G) Western blotting analysis of IκBα levels in the indicated cells treated with TNFα (10 ng/mL); α-tubulin was used as a loading control. TUNEL, terminal deoxynucleotidyl transferase (TdT) dUTP nick-end labeling.
Discussion
In this study, we presented the first demonstration that TRIM31 overexpression conferred gemcitabine resistance on pancreatic cancer, both in vitro and in vivo. Mechanically, TRIM31 overexpression promotes K63-linked polyubiquitination of TRAF2 and sustains the activation of NF-κB, which subsequently activates multiple anti-apoptosis downstream genes. More importantly, our study demonstrated that inhibition of the NF-κB signaling pathway significantly enhanced the sensitivity of tumor cells to gemcitabine chemotherapy, resulting in suppressed tumor growth. Hence, these findings uncover a novel mechanism to activate NF-κB signaling in pancreatic cancer, and suggest a promising strategy for TRIM31 to enhance the gemcitabine response during pancreatic cancer chemo-resistance.
Figure 6
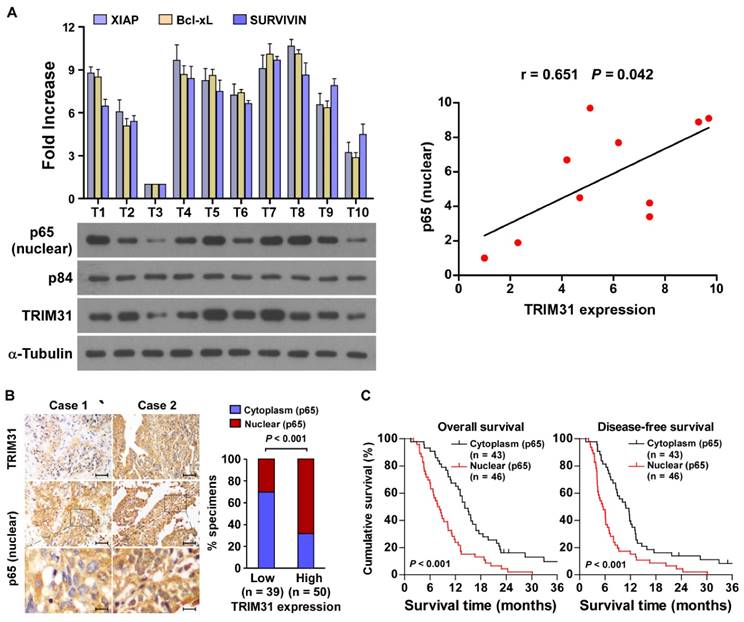
Clinical relevance of TRIM31 induced NF-κB activation in pancreatic cancer. (A) Expression analysis (left) and correlation (right) of TRIM31 and nuclear p65 expression in 10 freshly collected human pancreatic cancer tissue samples (T); α-Tubulin and nuclear protein p84 were used as loading controls. Each bar represents the mean ± SD of three independent experiments. (B) The nuclear expression levels of p65 were associated with the expression of TRIM31 in 89 primary human pancreatic specimens, (Scale bars: 100μm, insets, 20 μm). (C) The Kaplan-Meier overall (left) or disease-free (right) survival curves comparing patients with pancreatic cancer with low and high p65 expression levels (n = 89; P < 0.05).
Activated NF-κB signaling is associated with the development and progression of pancreatic cancer [27-29]. Over-activation of NF-κB pathway is observed in 70% of pancreatic cancer cell lines, and in most cases, canonical NF-κB activity is significantly increased in pancreatic cancer tissues. Nomura et al. reported that over-activation of NF-κB signaling contributed to the modulation of epithelial-mesenchymal transition induction, lymphovascular invasion, and neural invasion in pancreatic cancer [30]. Recently, it was shown that overexpression of G protein-coupled receptor (GPR87) promotes proliferation and angiogenesis, and increased resistance to gemcitabine-induced apoptosis of pancreatic cancer in vitro and the tumorigenicity of pancreatic cancer cells in vivo by activating the NF-κB signaling pathway [31]. Importantly, pancreatic cancer with highly activated NF-κB displays aggressive pathological features and has poor treatment outcomes. Although small molecule inhibitors of NF-κB have not yet progressed to clinical trials, several researchers have studied the inhibition of NF-κB using in vitro and in vivo models of pancreatic cancer. Blockade of the NF-κB pathway not only inhibits pancreatic cancer proliferation, epithelial-mesenchymal transition properties, and invasion capacity, but also increases the sensitivity of pancreatic cancer cells to chemotherapeutic drugs [30, 32]. These studies demonstrated that NF-κB activation plays an important role in pancreatic cancer progression. Accordingly, further understanding of the pathways that regulate the NF-κB pathway may provide novel therapeutic targets for pancreatic cancer. In the present study, we found that TRIM31 promoted K63-linked polyubiquitination of TRAF2 and sustained the activation of NF-κB. Consistently, overexpressing TRIM31 dramatically enhanced, whereas silencing TRIM31 inhibited, pancreatic cancer sensitivity to gemcitabine, both in vitro and in vivo. Therefore, our results represent a novel mechanism of NF-κB activation in pancreatic cancer, and indicated that targeting TRIM31 might lead to the development of a novel therapeutic strategy for patients with pancreatic cancer.
The family of tripartite motif (TRIM)-containing proteins is defined as a subfamily of the RING type E3 ubiquitin ligase family and contains more than 70 members in humans. TRIM family proteins are involved in a wide range of biological processes, and their alterations are associated with diverse pathological conditions, such as developmental disorders, viral infections, and tumorigenesis [33, 34]. For example, TRIM19 is localized in promyelocytic leukemia (PML)-nuclear bodies (PML-NBs) in the nucleus and regulates the response to various cellular stresses, DNA repair, and viral infection [35, 36]. TRIM25 is abundantly expressed in multiple female tumors, including osteoblasts, breast, and ovarian cancers, and its overexpression not only promotes cancer development and progression, but also correlates with poor prognosis in patients with cancer [37, 38]. Urano et al. reported that overexpression of TRIM25 causes G2 arrest by mediating ubiquitylation and degradation of 14-3-3σ in breast cancer [39]. Additionally, Horn et al. showed that TRIM32 overexpression was coupled to the inhibition of TNF and ultraviolet (UV) irradiation-induced apoptosis, and that the upregulation of TRIM32 promoted UVB-induced squamous cell carcinomas proliferation, motility, and transformation [40]. Consistently, we found that TRIM31 was upregulated in pancreatic cancer and that TRIM31 overexpression promoted pancreatic cancer cells drug tolerance, both in vitro and in vivo, which is in agreement with oncogenic-effect of TRIM family members. However, the mechanism of TRIM31 upregulation in pancreatic cancer remains unknown. Interestingly, TCGA analysis showed that TRIM31 is highly amplified (33.2%) in pancreatic cancer. At the same time, according to the ECR Browser (https://ecrbrowser.dcode.org/), high levels of NF-κB could be recruited to the promoter region of TRIM31. These findings suggested that overexpression of TRIM31 in pancreatic cancer might be associated with genomic amplification or NF-κB-mediated transcriptional regulation. Thus, it will be of great interest to further study the mechanism of TRIM31 upregulation in pancreatic cancer.
With respect to the multitude of anti-apoptotic pathways in pancreatic cancer, many molecular targets might have high potential as novel therapy strategies. Huang et al. showed that obatoclax potentiates TRAIL-mediated apoptosis by unsequestering Bak and Bim from Bcl-2/Bcl-x(L) proteins, subsequently enhancing Bid-mediated cross-talk between the mitochondrial and death receptor-mediated apoptotic pathways [41]. Vogler and colleagues provide evidence that XIAP inhibitors prime pancreatic carcinoma cells for TRAIL-induced apoptosis and potentiate the antitumor activity of TRAIL against established pancreatic carcinoma. Using the chicken chorioallantoic membrane model, Vogler also demonstrated that XIAP inhibition significantly enhances TRAIL-mediated apoptosis and suppression of pancreatic cancer growth, both in vitro and in vivo [42, 43]. Collectively, these findings establish a strong rationale for therapeutic targeting of anti-apoptotic signaling in pancreatic cancer. Therefore, uncovering a novel regulatory molecule that can simultaneously regulate these anti-apoptotic genes might be a promising strategy to overcome anti-apoptotic resistance in the treatment of pancreatic cancer. Our data demonstrated that TRIM31 overexpression in pancreatic cancer activates the NF-κB signaling pathway, resulting in the upregulation of multiple anti-apoptosis downstream genes. Meanwhile, inhibition of TRIM31 sensitized pancreatic cancer cell lines to gemcitabine cytotoxicity by inactivating the NF-κB signaling pathway, which in turn resulted in downregulation of the anti-apoptosis downstream genes. These data suggested that TRIM31 could contribute to NF-κB activation and thereby represents a novel target for anti-apoptosis resistance in pancreatic cancer.
Studies have demonstrated that TRIM family proteins may have distinct functions in different cell types. For instance, overexpression of TRIM25 could promote proliferation and invasion of colorectal cancer cells through TGF-β signaling. However, upregulation of TRIM25 inhibits hepatocellular carcinoma progression by targeting metastasis associated 1 protein [44-45]. Similarly, TRIM31 was also found to be downregulated in non-small cell lung cancer and ovarian cancer, in which it serves as a potential tumor suppressor, but upregulated in gastric cancer and hepatocellular carcinoma, in which it functions as an onco-protein [46-49]. Collectively, these findings suggest that whether TRIM31 functions as a tumor suppressor or as onco-protein depends on the tumor type. In the current study, we found that higher TRIM31 expression was associated with a shorter survival and earlier relapse survival in patients with pancreatic cancer. TRIM31 overexpression in tumor cells conferred gemcitabine resistance on pancreatic cancer, whereas, inhibition of TRIM31 sensitized pancreatic cancer cell lines to gemcitabine both in vitro and in vivo. TRIM31 promoted K63-linked polyubiquitination of tumor necrosis factor receptor-associated factor 2 (TRAF2) and sustained the activation of nuclear transcription factor kappa B (NF-κB) in pancreatic cancer cells. Thus, these findings reveal a novel mechanism for constitutive NF-κB activation in pancreatic cancer, and also highlight an important oncogenic role for TRIM31 in pancreatic cancer progression and chemoresistance.
In summary, our results provide evidence that overexpression of TRIM31 in human pancreatic cancer might be important in the acquisition of a drug resistance/poor prognosis phenotype. This suggested that TRIM31 functions as an oncoprotein in pancreatic cancer progression and may serve as a novel potential therapeutic biomarker. Furthermore, the functional and mechanistic studies of TRIM31 presented in this study indicated that TRIM31 plays a critical role in controlling pancreatic cancer gemcitabine resistance by activating the NF-κB signaling pathway. Therefore, understanding the biological function and molecular mechanisms of TRIM31 in pancreatic cancer progression and chemoresistance may establish TRIM31 as a potential therapeutic target for the treatment of pancreatic cancer.
Abbreviations
EGF: epidermal growth factor; GSEA: gene set enrichment analysis; HPDECs: human pancreatic duct epithelial cells; IHC: immunohistochemistry; IKK: I kappa B kinase; IL-1β: interleukin 1β; MAVS: mitochondrial antiviral-signaling protein; NEMO: NF-kappa-B essential modulator; NF-κB: nuclear transcription factor kappa B; NLRP3: NLR family pyrin domain containing 3; shRNA: short hairpin RNA; siRNA: interfering RNA; TABs: TAK1-binding proteins; TAK1: TGF-β-activated kinase 1; TNF-α: tumor necrosis factor-α; TRAFs: TNF receptor associated factors; TRAF2: tumor necrosis factor receptor-associated factor 2; TRIM31: tripartite motif containing 31; RIP: receptor interacting protein.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work was supported by the National Natural Science Foundation of China [grant numbers 81672906, 81660483, 81560477].
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M. et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359-86
2. Weisbeck A, Jansen RJ. Nutrients and the pancreas: an epigenetic perspective. Nutrients. 2017;15(3):9
3. Zhang Q, Chen S, Zeng L, Chen Y, Lian G, Qian C. et al. New developments in the early diagnosis of pancreatic cancer. Expert Rev Gastroenterol Hepatol. 2017;11:149-56
4. Conroy T, Bachet JB, Ayav A, Huguet F, Lambert A, Caramella C. et al. Current standards and new innovative approaches for treatment of pancreatic cancer. Eur J Cancer. 2016;57:10-22
5. Boeck S, Ankerst DP, Heinemann V. The role of adjuvant chemotherapy for patients with resected pancreatic cancer: systematic review of randomized controlled trials and meta-analysis. Oncology. 2007;72:314-21
6. Deplanque G, Demartines N. Pancreatic cancer: are more chemotherapy and surgery needed? Lancet. 2017;389:985-6
7. Habermehl D, Brecht IC, Bergmann F, Welzel T, Rieken S, Werner J. et al. Chemoradiation in patients with isolated recurrent pancreatic cancer - therapeutical efficacy and probability of re-resection. Radiat Oncol. 2013;8:27
8. DiDonato JA, Mercurio F, Karin M. NF-kappaB and the link between inflammation and cancer. Immunol Rev. 2012;246:379-400
9. Karin M. NF-kappaB as a critical link between inflammation and cancer. Cold Spring Harb Perspect Biol. 2009;1:a000141
10. Garg A, Aggarwal BB. Nuclear transcription factor-kappaB as a target for cancer drug development. Leukemia. 2002;16:1053-68
11. Chen ZJ, Fuchs SY. Ubiquitin-dependent activation of NF-kappaB: K63-linked ubiquitin chains: a link to cancer? Cancer Biol Ther. 2004;3:286-8
12. Lang V, Rodriguez MS. Innate link between NF-kappaB activity and ubiquitin-like modifiers. Biochem Soc Trans. 2008;36:853-7
13. Haas AL. Linear polyubiquitylation: the missing link in NF-kappaB signalling. Nat Cell Biol. 2009;11:116-8
14. Ea CK, Deng L, Xia ZP, Pineda G, Chen ZJ. Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol Cell. 2006;22:245-57
15. Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature. 2001;412:346-51
16. Deng L, Wang C, Spencer E, Yang L, Braun A, You J. et al. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell. 2000;103:351-61
17. Ghosh S, Baltimore D. Activation in vitro of NF-kappa B by phosphorylation of its inhibitor I kappa B. Nature. 1990;344:678-82
18. Chen ZJ, Parent L, Maniatis T. Site-specific phosphorylation of IkappaBalpha by a novel ubiquitination-dependent protein kinase activity. Cell. 1996;84:853-62
19. Chandler NM, Canete JJ, Callery MP. Increased expression of NF-kappa B subunits in human pancreatic cancer cells. J Surg Res. 2004;118:9-14
20. Arlt A, Gehrz A, Muerkoster S, Vorndamm J, Kruse ML, Folsch UR. et al. Role of NF-kappaB and Akt/PI3K in the resistance of pancreatic carcinoma cell lines against gemcitabine-induced cell death. Oncogene. 2003;22:3243-51
21. Zhang Y, Banerjee S, Wang ZW, Marciniak DJ, Majumdar AP, Sarkar FH. Epidermal growth factor receptor-related protein inhibits cell growth and induces apoptosis of BxPC3 pancreatic cancer cells. Cancer Res. 2005;65:3877-82
22. Arlt A, Rosenstiel P, Kruse ML, Grohmann F, Minkenberg J, Perkins ND. et al. IEX-1 directly interferes with RelA/p65 dependent transactivation and regulation of apoptosis. Biochim Biophys Acta. 2008;1783:941-52
23. Liu B, Zhang M, Chu H, Zhang H, Wu H, Song G. et al. The ubiquitin E3 ligase TRIM31 promotes aggregation and activation of the signaling adaptor MAVS through Lys63-linked polyubiquitination. Nat Immunol. 2017;18:214-24
24. Song H, Liu B, Huai W, Yu Z, Wang W, Zhao J. et al. The E3 ubiquitin ligase TRIM31 attenuates NLRP3 inflammasome activation by promoting proteasomal degradation of NLRP3. Nat Commun. 2016;7:13727
25. Sugiura T, Miyamoto K. Characterization of TRIM31, upregulated in gastric adenocarcinoma, as a novel RBCC protein. J Cell Biochem. 2008;105:1081-91
26. Dokmanovic M, Chang BD, Fang J, Roninson IB. Retinoid-induced growth arrest of breast carcinoma cells involves co-activation of multiple growth-inhibitory genes. Cancer Biol Ther. 2002;1:24-7
27. Uwagawa T, Yanaga K. Effect of NF-kappaB inhibition on chemoresistance in biliary-pancreatic cancer. Surg Today. 2015;45:1481-8
28. Wang W, Abbruzzese JL, Evans DB, Larry L, Cleary KR, Chiao PJ. The nuclear factor-kappa B RelA transcription factor is constitutively activated in human pancreatic adenocarcinoma cells. Clin Cancer Res. 1999;5:119-27
29. Sclabas GM, Fujioka S, Schmidt C, Evans DB, Chiao PJ. NF-kappaB in pancreatic cancer. J Gastrointest Cancer. 2003;33:15-26
30. Nomura A, Majumder K, Giri B, Dauer P, Dudeja V, Roy S. et al. Inhibition of NF-kappa B pathway leads to deregulation of epithelial-mesenchymal transition and neural invasion in pancreatic cancer. Lab Invest. 2016;96:1268-78
31. Wang L, Zhou W, Zhong Y, Huo Y, Fan P, Zhan S. et al. Overexpression of G protein-coupled receptor GPR87 promotes pancreatic cancer aggressiveness and activates NF-kappaB signaling pathway. Mol Cancer. 2017;16:61
32. Horiuchi T, Uwagawa T, Shirai Y, Saito N, Iwase R, Haruki K. et al. New treatment strategy with nuclear factor-kappaB inhibitor for pancreatic cancer. J Surg Res. 2016;206:1-8
33. Hatakeyama S. TRIM proteins and cancer. Nat Rev Cancer. 2011;11:792-804
34. Cambiaghi V, Giuliani V, Lombardi S, Marinelli C, Toffalorio F, Pelicci PG. TRIM proteins in cancer. Adv Exp Med Biol. 2012;770:77-91
35. Ferbeyre G, de Stanchina E, Querido E, Baptiste N, Prives C, Lowe SW. PML is induced by oncogenic ras and promotes premature senescence. Genes Dev. 2000;14:2015-27
36. Masroori N, Merindol N, Berthoux L. The interferon-induced antiviral protein PML (TRIM19) promotes the restriction and transcriptional silencing of lentiviruses in a context-specific, isoform-specific fashion. Retrovirology. 2016;13:19
37. Suzuki T, Urano T, Tsukui T, Horie-Inoue K, Moriya T, Ishida T. et al. Estrogen-responsive finger protein as a new potential biomarker for breast cancer. Clin Cancer Res. 2005;11:6148-54
38. Sakuma M, Akahira J, Suzuki T, Inoue S, Ito K, Moriya T. et al. Expression of estrogen-responsive finger protein (Efp) is associated with advanced disease in human epithelial ovarian cancer. Gynecol Oncol. 2005;99:664-70
39. Urano T, Saito T, Tsukui T, Fujita M, Hosoi T, Muramatsu M. et al. Efp targets 14-3-3 sigma for proteolysis and promotes breast tumour growth. Nature. 2002;417:871-5
40. Horn EJ, Albor A, Liu Y, El-Hizawi S, Vanderbeek GE, Babcock M. et al. RING protein Trim32 associated with skin carcinogenesis has anti-apoptotic and E3-ubiquitin ligase properties. Carcinog. 2004;25:157-67
41. Huang S, Okumura K, Sinicrope FA. BH3 mimetic obatoclax enhances TRAIL-mediated apoptosis in human pancreatic cancer cells. Clin Cancer Res. 2009;15:150-9
42. Vogler M, Walczak H, Stadel D, Haas TL, Genze F, Jovanovic M. et al. Small molecule XIAP inhibitors enhance TRAIL-induced apoptosis and antitumor activity in preclinical models of pancreatic carcinoma. Cancer Res. 2009;69:2425-34
43. Vogler M, Walczak H, Stadel D, Haas TL, Genze F, Jovanovic M. et al. Targeting XIAP bypasses Bcl-2-mediated resistance to TRAIL and cooperates with TRAIL to suppress pancreatic cancer growth in vitro and in vivo. Cancer Res. 2008;68:7956-65
44. Zang HL, Ren SN, Cao H, Tian XF. The ubiquitin ligase TRIM25 inhibits hepatocellular carcinoma progression by targeting metastasis associated 1 protein. IUBMB Life. 2017;69:795-801
45. Sun N, Xue Y, Dai T, Li X, Zheng N. Tripartite motif containing 25 promotes proliferation and invasion of colorectal cancer cells through TGF-β signaling. Biosci Rep. 2017;12(4):37-46
46. Li H, Zhang Y, Zhang Y, Bai X, Peng Y, He P. TRIM31 is downregulated in non-small cell lung cancer and serves as a potential tumor suppressor. Tumour Biol. 2014;35:5747-52
47. Wei Z, Liu Y, Wang Y, Zhang Y, Luo Q, Man X, Wei F, Yu X. Downregulation of Foxo3 and TRIM31 by miR-551b in side population promotes cell proliferation, invasion, and drug resistance of ovarian cancer. Med Oncol. 2016;33:126
48. Sugiura T. The cellular level of TRIM31, an RBCC protein overexpressed in gastric cancer, is regulated by multiple mechanisms including the ubiquitin-proteasome system. Cell Biol Int. 2011;35:657-61
49. Guo P, Ma X, Zhao W, Huai W, Li T, Qiu Y, Zhang Y, Han L. TRIM31 is upregulated in hepatocellular carcinoma and promotes disease progression by inducing ubiquitination of TSC1-TSC2 complex. Oncogene. 2018;37:478-88
Author contact
![]() Corresponding author: Chengyi Sun, E-mail: sunchengyi2014com; Tel/Fax: +86-851-6771326.
Corresponding author: Chengyi Sun, E-mail: sunchengyi2014com; Tel/Fax: +86-851-6771326.
Citation styles
APA
Yu, C., Chen, S., Guo, Y., Sun, C. (2018). Oncogenic TRIM31 confers gemcitabine resistance in pancreatic cancer via activating the NF-κB signaling pathway. Theranostics, 8(12), 3224-3236. https://doi.org/10.7150/thno.23259.
ACS
Yu, C.; Chen, S.; Guo, Y.; Sun, C. Oncogenic TRIM31 confers gemcitabine resistance in pancreatic cancer via activating the NF-κB signaling pathway. Theranostics 2018, 8 (12), 3224-3236. DOI: 10.7150/thno.23259.
NLM
Yu C, Chen S, Guo Y, Sun C. Oncogenic TRIM31 confers gemcitabine resistance in pancreatic cancer via activating the NF-κB signaling pathway. Theranostics 2018; 8(12):3224-3236. doi:10.7150/thno.23259. https://www.thno.org/v08p3224.htm
CSE
Yu C, Chen S, Guo Y, Sun C. 2018. Oncogenic TRIM31 confers gemcitabine resistance in pancreatic cancer via activating the NF-κB signaling pathway. Theranostics. 8(12):3224-3236.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.