Impact Factor
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and methods
Results
Discussion
Conclusion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(6):1678-1689. doi:10.7150/thno.22502 This issue Cite
Research Paper
Surface Enhanced Raman Spectroscopy (SERS) for the Multiplex Detection of Braf, Kras, and Pik3ca Mutations in Plasma of Colorectal Cancer Patients
Xiaozhou Li1,2 ![]() , Tianyue Yang1, Caesar Siqi Li3, Youtao Song4, Hong Lou2, Dagang Guan5, Lili Jin2
, Tianyue Yang1, Caesar Siqi Li3, Youtao Song4, Hong Lou2, Dagang Guan5, Lili Jin2
1. School of Science, Shenyang Ligong University, 110159, Shenyang, China
2. School of Life Science, Liaoning University, 110036, Shenyang, China
3. College of Medicine, Northeast Ohio Medical University, 44272, Rootstown, USA
4. College of Environmental Sciences, Liaoning University, 110036, Shenyang, China
5. Oncology Department, Shengjing Hospital of China Medical University, 110022, Shenyang, China
Received 2017-8-23; Accepted 2017-11-24; Published 2018-2-12
Abstract

In this paper, we discuss the use of a procedure based on polymerase chain reaction (PCR) and surface enhanced Raman spectroscopy (SERS) (PCR-SERS) to detect DNA mutations.
Methods: This method was implemented by first amplifying DNA-containing target mutations, then by annealing probes, and finally by applying SERS detection. The obtained SERS spectra were from a mixture of fluorescence tags labeled to complementary sequences on the mutant DNA. Then, the SERS spectra of multiple tags were decomposed to component tag spectra by multiple linear regression (MLR).
Results: The detection limit was 10-11 M with a coefficient of determination (R2) of 0.88. To demonstrate the applicability of this process on real samples, the PCR-SERS method was applied on blood plasma taken from 49 colorectal cancer patients to detect six mutations located at the BRAF, KRAS, and PIK3CA genes. The mutation rates obtained by the PCR-SERS method were in concordance with previous research. Fisher's exact test showed that only two detected mutations at BRAF (V600E) and PIK3CA (E542K) were significantly positively correlated with right-sided colon cancer. No other clinical feature such as gender, age, cancer stage, or differentiation was correlated with mutation (V600E at BRAF, G12C, G12D, G12V, G13D at KRAS, and E542K at PIK3CA). Visually, a dendrogram drawn through hierarchical clustering analysis (HCA) supported the results of Fisher's exact test. The clusters drawn by all six mutations did not conform to the distributions of cancer stages, differentiation or cancer positions. However, the cluster drawn by the two mutations of V600E and E542K showed that all samples with those mutations belonged to the right-sided colon cancer group.
Conclusion: The suggested PCR-SERS method is multiplexed, flexible in probe design, easy to incorporate into existing PCR conditions, and was sensitive enough to detect mutations in blood plasma.
Keywords: colorectal cancer, gene, surface enhanced Raman scattering, PCR, mutation
Introduction
Colorectal cancer is the third and second most common cancer in men and women respectively, responsible for about 10% of the global cancer incidence[1]. The incidence of colorectal cancer is higher in developed countries such as Europe and the Americas due to higher intake of calories and animal fat. In China, colorectal cancer has also become a leading cause of death[2]. Although influenced by many external conditions, cancers including colorectal cancer are diseases that may be directly caused by harmful gene mutations[3]. Both point mutations and deletions have an important role in tumorigenesis, promotion, invasion, and metastasis of cancer, as well as chemotherapy resistance[4]. The detection of these mutated genes is potentially useful for diagnosis[5], therapy selection[6] and prognosis[7] for colorectal cancer. The key "driver genes" for tumorigenesis regulate the three cellular processes of cell fate, cell survival and genome maintenance, which can all be classified into 12 signaling pathways[8]. Among these genes, BRAF, KRAS, and PIK3CA are the three participants in the cell survival process that are the most commonly mutated in colorectal cancer patients. As mutations exist at the molecular level, detection is a rather complicated process at present. Conventional methods for mutation detection include real-time PCR (RT-PCR), amplification refractory mutation system (ARMS), and DNA sequencing, among others[9]. However, those methods can be complicated, time-consuming, and may require sophisticated equipment. Compared to these detection methods, Raman spectroscopy is rapid, easy to operate, and non-invasive. As an inelastic scattering spectroscopy method, Raman spectroscopy can examine solutions in glass containers without any special preparation and provide 'fingerprint' spectral patterns for those chemicals[10]. However, in the biomedical field, analytes are usually of very low concentration and Raman spectroscopy use is less common. This is due to the challenges of sample degradation, background fluorescence, and low scattering cross section[10]. Surface enhanced Raman spectroscopy (SERS) can conquer the above-mentioned shortcomings while maintaining the advantages of normal Raman and enhancing the signals of Raman scattering by up to 14 orders of magnitude[11]. This is accomplished through the surface plasmonics effect, which works by adsorbing target molecules on noble metal surfaces. The enhancement mechanism can be further classified into physical (electromagnetic) and chemical mechanisms, with the physical mechanism serving as the major contributor[12]. Due to its nano-metal based nature, SERS is flexible enough to be incorporated with optical labels or other biomedical techniques[13, 14]. As such, SERS has been widely investigated for biofluid and DNA analysis and has shown promising results.
The detection of DNA by SERS can be categorized into direct and indirect (through DNA tags) methods[15]. The direct detection method allows SERS to detect single base mismatches and methylations in DNA duplexes[16]. In indirect detection, DNA sequences can be detected using SERS tags by SERS spectra or electrochemically driven melting curve [17, 18], in combination with chromategraphic separations such as lateral flow assay[19, 20] or enrichment methods such as magnetic beads[21]. However, the small scale of base modifications only permits modifications on short DNA sequences to be detected without amplification. For the detection of mutations in long sequence genes that exist in blood or tissue, certain amplification methods are essential. The amplification process not only increases the concentration of target sequences, but can also limit the gene sequences to short lengths that include the target mutations. Examples of amplification methods that have been used before to detect gene mutations include polymerase chain reaction (PCR)[22, 23], exponential strand displacement amplification (SDA) [24], and ARMS[25]. The combination of such amplification methods and SERS can be accomplished either by melting the target PCR product onto immobilized probes on SERS substrates [22], by using amplification products as linking sequences to bind capture probes and reporter probes[24], or by directly detecting fluorescence-labeled primers that have participated in the amplification[25]. These papers ingeniously utilized biological amplification methods in combination with SERS.
In this paper, we attempted to combine PCR and SERS by adding mutation-specific probes in the PCR product and then subsequently detecting the incorporated probes with SERS. After removing unincorporated probes, the remaining probes bound to the target mutations were extracted for SERS detection. This process converted the problem of detecting mutations into the problem of detecting probes. The advantage of this method is the exploitable multiplex detection ability and easy utilization of existing PCR conditions without any modification of primers. The suggested PCR-SERS method was first tested via application on three mixtures with known mutation types. Then, the plasma of 49 colorectal cancer patients was screened for six different mutations (distributed across BRAF, KRAS, PIK3CA). Finally, the relationship between the six mutation types and clinical characteristics of the 49 patients were investigated by Fisher's exact test and hierarchical clustering analysis (HCA). The results indicated that the PCR-SERS method has a detection limit efficient for detecting plasma mutations (10-11 M) and that it can be used for the detection of up to three mutation types simultaneously.
Materials and methods
Materials and cells
All chemicals used in this experiment were purchased at the highest grade from Sigma Aldrich (St. Louis, USA). Primers and fluorescence tagged probes were purchased from Sangon Biotech (Shanghai, China). The OVCAR cell line was bought from Bioleaf Corporation (Shanghai, China).
DNA solutions
Three DNA mixtures with known mutation types were prepared for testing the multiplex ability of the suggested PCR-SERS method. As most patients harbor zero to three mutations in the six mutation types at BRAF, KRAS, and PIK3CA[26], we prepared each of the three DNA mixtures to have one, two, and three mutations. The mutation compositions and concentrations of the three DNA mixtures are displayed in Table 1.
Mutation compositions and concentrations of the three DNA mixtures
| BRAF-V600E | KRAS-G12C | KRAS-G12D | |
|---|---|---|---|
| Mixture #1 | 10-10 M | NA | NA |
| Mixture #2 | 10-10 M | 10-10 M | NA |
| Mixture #3 | 10-10 M | 10-10 M | 10-10 M |
To determine the detection limit, wild-type KRAS solutions with six increasing concentrations ranging from 10-12 to 10-10 M (10-12, 10-11, 20-11, 50-11, 80-11, and 10-10 M) were prepared. As the size of DNA fragments in blood plasma was most frequently 70-200 bp[27], wild-type KRAS of 180 bp in length was used as the detection target to simulate real blood DNA conditions. Wild-type KRAS genes were obtained via PCR on an OVCAR cell line containing wild-type KRAS genes. Primers and PCR conditions were identical to the ones used on the blood plasma samples in the sample DNA section. OVCAR were cultured at 37oC in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum. DNA in the OVCAR cell line was extracted using the QIAmp DNA mini kit (Qiagen, Hilden, Germany).
Sample plasma
Blood samples were collected from 49 colorectal cancer patients from the Shengjing Hospital of China Medical University. Plasma was obtained by adding EDTA anticoagulant into the blood and centrifuging the blood at 3900 rcf for 10 min at 4oC. The resulting plasma was collected in a 1.5 mL Eppendorf tube and stored at -80oC. Plasma DNA was extracted with a QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer's recommendations. The obtained DNA was stored at -20oC. Clinicopathological information (gender, age, TNM stages, differentiation, and cancer position) was obtained through medical records (Table 2). This study was conducted under the approval of the Ethics Committee in accordance with the principles of the Declaration of Helsinki.
Clinicopathological information of the 49 colorectal cancer patients
| Clinical characteristics | Number (percentage) |
|---|---|
| Gender | |
| Female | 23 (47%) |
| Male | 26 (53%) |
| Age | |
| ≤39 | 6 (12%) |
| 40-59 | 13 (27%) |
| ≥60 | 30 (61%) |
| TNM stages | |
| I | 6 (12%) |
| II | 7 (14%) |
| III | 18 (37%) |
| IV | 18 (37%) |
| Differentiation | |
| Well | 4 (8%) |
| Moderate | 36 (73%) |
| Poor | 9 (18%) |
| Position | |
| Left-sided colon | 12 (24%) |
| Right-sided colon | 10 (20%) |
| Rectum | 27 (55%) |
PCR and probes
For each DNA sample taken from the 49 colorectal cancer patients, six mutations located at the BRAF, KRAS, and PIK3CA genes were amplified using PCR. The six mutations were V600E at BRAF exon 15, G12C, G12D, G12V, G13D at KRAS exon 2, and E542K at PIK3CA exon 9. One multiplex PCR was designed to amplify all six mutations with primers and PCR conditions adopted and modified from the paper by Lurkin et al[28]. As the PCR was not allele specific, three pairs of primers targeting BRAF, KRAS, and PIK3CA were used (Table 3). PCR reactions were performed in a total volume of 25 μL containing 1x PCR buffer, 2 μL extracted DNA, 0.15 μM of each primer pair, 2.5 mM MgCl2, 0.3 mM dNTP, and 1.25 units DNA polymerase. Thermal cycling conditions involved a 5 min initial denaturation at 95oC, a 45-cycle amplification step (95oC for 30 s, 55oC for 50 s, and 72oC for 45 s), and a final extension step at 72oC for 10 min.
Primers in the multiplex PCR targeting BRAF, KRAS, and PIK3CA
| Genes | Primers (5'⟶3') | Product size (bp) |
|---|---|---|
| BRAF | F: TCTTCATGAAGACCTCACAGT R: CCAGACAACTGTTCAAACTGA | 96 |
| KRAS | F: GGCCTGCTGAAAATGACTG R: GGTCCTGCACCAGTAATATG | 163 |
| PIK3CA | F: AGTAACAGACTAGCTAGAGA R: ATTTTAGCACTTACCTGTGAC | 139 |
Six probe sequences labeled with the different fluorescence tags of R6G, Cy3, Cy5, TAMRA, ROX, and FAM were used for targeting the six mutation types, and were purified for the following SERS detection. 1 μL probe mix was first added and annealed to the PCR product by heating to 95oC for 5 min, and subsequently the mixture was cooled. The resulting mixtures were then purified using the PCR purification kit (Qiagen, USA) to remove unincorporated probes. Then, the purified DNA/probes were suspended in a formamide elution buffer and were heated to 65oC for 5 min to remove sDNA as stated by Graham et al[25]. Table 4 lists the probe sequences for the six mutation types.
Probe sequences targeting six mutation types
| Genes | Mutations | Probe sequences (5'⟶3') |
|---|---|---|
| BRAF exon 15 | 1799T>A | R6G-CTAGCTACAGAGAAATCTCG |
| KRAS exon 2 | 34G>T | Cy3-TTGGAGCTTGTGGCGTAGGCA |
| 35G>A | Cy5-TTGGAGCTGATGGCGTAGGCA | |
| 35G>T | TAMRA-TTGGAGCTGTTGGCGTAGGCA | |
| 38G>A | ROX-TTGGAGCTGGTGACGTAGGCA | |
| PIK3CA exon 9 | 1624G>A | FAM-ATCCTCTCTCTAAAATCACTGAGC |
SERS
Ag colloids were synthesized according to a modified Lee and Meisel procedure[29]. Briefly, 90 mg silver nitrate was suspended in 500 mL distilled water at 45oC. The mixtures were heated to boiling under stirring. 10 mL of 1% sodium citrate solution were added to the mixture when the boiling commenced. The boiling was continued for 90 min with continuous stirring.
SERS spectra were recorded on a Renishaw Raman microspectrometer system (Gloucestershire, UK) equipped with a He-Ne laser (λ = 632 nm, beam diameter: 1.5 μm) and an electrically cooled CCD detector. The laser power at the sample was 4 mW. Exposure time of the CCD was 10 s. The SERS measurements were done by mixing target probes, Ag colloid and spermine (0.1 M) in a 1:1:1 volume ratio. The spermine acted as a colloid aggregating agent and allowed the probe to adsorb the Ag colloid[30]. The positively charged spermine and the negatively charged probe sequences can form stable electrostatic attraction. Probe sequences will be positioned in the interparticle hot spots, which can ensure high enhancement and reproducibility[31].
Statistical analysis
Statistical analyses were all performed in the R language environment (https://www.r-project.org). To eliminate noise, all spectra were pre-processed with smoothing and baseline correction. Normalization was not performed to avoid any removal of information from the spectra. All samples were measured three times and the averaged spectra were used.
Multiple linear regression (MLR) was utilized on the obtained SERS spectra for deconvolution into component spectra[32, 33]. For each SERS spectrum obtained from the PCR methods, the spectrum was deconvoluted by the combination of weighted SERS spectra of each reference tag. The analysis was done by inputting the SERS spectra of the mixtures and the SERS spectra of the six tags. The resultant data was a data matrix with one line composing the six coefficients of one sample.
SERS of mixture = coefficient 1 × SERS of tag 1 + coefficient 2 × SERS of tag 2 + ... + coefficient 6 × SERS of tag 6 + Constant
The obtained coefficients represent the weights of each component tag, indicating corresponding mutations. In the MLR, the coefficients of each tag were obtained by comparison with the SERS spectrum of the corresponding tag itself; the differences in enhancement factors between different tags does not influence the MLR results.
The correlation between mutation types and the five clinical characteristics (gender, age, TNM stages, differentiation and cancer position) were analyzed using Fisher's exact test. Finally, dendrograms were drawn by hierarchical clustering analysis (HCA) to visualize the grouping of samples based on binary mutation status.
Results
PCR-SERS
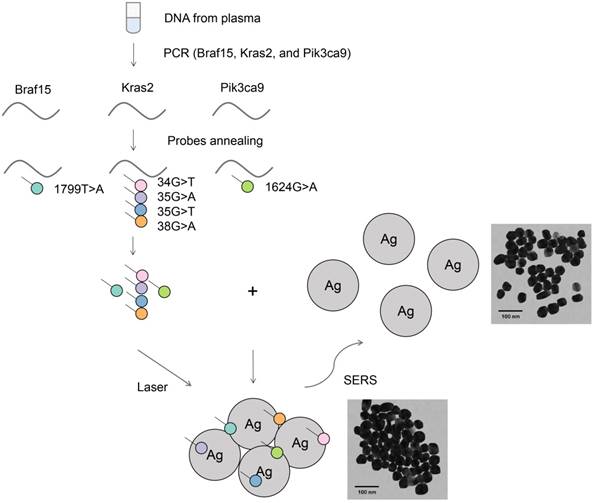
Figure 1 illustrates the suggested PCR-SERS method for the detection of plasma gene mutations. Primers were designed to amplify selected gene regions that enclose the target mutation points. This results in the amplification of all wild-type and all six types of targeted mutant DNA within this region. After PCR, the PCR product was added to and annealed with florescence-labeled probe sequences that were complementary to the target mutations. This annealing process resulted in gene sequences with target mutations labeled with dyes, while wild-type or non-targeted mutations were unlabeled. Then, purification was performed on the probe-labeled PCR products to remove unbound probes. This process ensured that only probes bound to PCR products (probes that harbored existing mutations) remained. The above annealing and removal process transformed the problem of detecting mutations in genes into the problem of detecting fluorescence tags on probes.
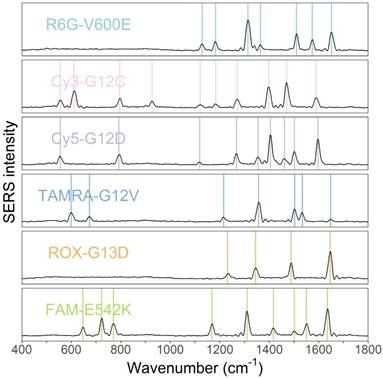
The obtained SERS spectra of the probe mixtures were then deconvoluted into SERS spectra of individual tags by MLR. The SERS spectra of all tags that were labeled to probe sequences were measured and used as reference spectra (independent variables) in the MLR (Figure 2). The weights of each tag were obtained through the coefficients of MLR. Peak numbers for the six fluorescence tags ranged from 3 to 10. Despite some overlap, the six spectra of the tags have differing peak profiles. This difference makes the use of MLR possible.
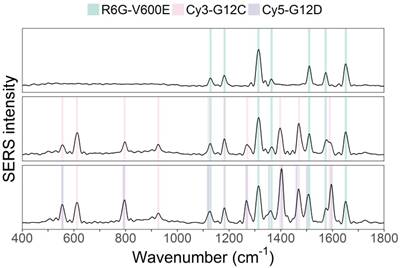
As a test, three mixtures with known DNA mutation content were created and then measured by the suggested PCR-SERS method. One solution contained BRAF-V600E), another V600E and KRAS-G12C, and the last V600E, G12C, and KRAS-G12D. The three mutations (V600E, G12C, and G12D) were mixed at the ratios 1:0:0, 1:1:0, and 1:1:1. This combination of mutations was selected since most colon cancer patients harbored fewer than three of the six targeted mutations studied in this experiment. Figure 3 shows the SERS spectra of the three mutation mixtures. From the spectra, we can see that the PCR-SERS method successfully retained the feature peaks of each fluorescence tag in the output spectra, even when the tags were mixed.
MLR analysis was utilized on the resulting three SERS spectra to identify the composition of each spectrum. The inputs of MLR were seven SERS spectra, each with two dimensions (wavenumber and intensity). One of the seven inputs was the SERS spectrum obtained from PCR-SERS (dependent variable), and the other six were reference SERS spectra of the fluorescence tags used in this study (independent variables). The outputs of MLR were the coefficients (weights) of each reference spectrum. Table 5 shows the resultant MLR coefficients for the three mixtures. The mixing ratio of each mixture is also listed in the right columns of Table 5 for better comparison. The table indicates that the obtained coefficient values were less than 0.078 away from the actual mixing ratios (and expected values) of 1.0.
Schematic of the PCR-SERS method for the detection of DNA mutations in blood. Genes at Braf15, Kras2, and Pik3ca9 were amplified using mutation non-specific primers. Six mutation types in the three genes were detected using specific probes by probe annealing and subsequent separation. The final SERS measurements were conducted by mixing tags and Ag colloids together.
MLR coefficients for the three DNA mixtures
| Mixtures | MLR coefficients | Actual mixing ratios | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Ref 1 (V600E) | Ref 2 (G12C) | Ref 3 (G12D) | Ref 4 (G12V) | Ref 5 (G13D) | Ref 6 (E542K) | Ref 1 (V600E) | Ref 2 (G12C) | Ref 3 (G12D) | |
| #1 | 0.975 | 0.027 | -0.005 | -0.006 | 0.017 | 0.007 | 1 | 0 | 0 |
| #2 | 0.925 | 0.975 | 0.019 | -0.0003 | -0.001 | 0.045 | 1 | 1 | 0 |
| #3 | 0.926 | 0.961 | 0.992 | 0.078 | -0.074 | 0.038 | 1 | 1 | 1 |
SERS spectra of the six fluorescence tags of R6G, Cy3, Cy5, TAMRA, ROX, and FAM. The six tags probing mutations V600E, G12C, G12D, G12V, G13D, and E542K, respectively.
SERS spectra results of PCR-SERS applied on the three mixtures with mixing ratios of 1:0:0, 1:1:0, and 1:1:1, respectively.
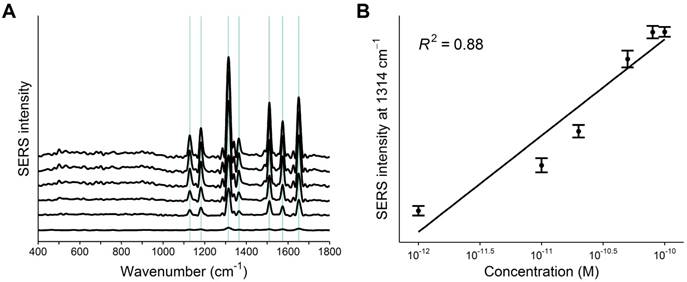
Next, the detection limit of the PCR-SERS method was found by trialing the method on six wild-type KRAS gene solutions of varying concentrations (10-12, 10-11, 20-11, 50-11, 80-11, and 10-10 M). Figure 4A shows the obtained SERS spectra. The concentration of KRAS in solution correlated with spectral feature peak intensity. A linear regression line was drawn using the highest spectral peak of R6G located at 1314 cm-1 (Figure 4B). The determination coefficient (R2) of the regression line was 0.88. The detection limit was calculated by tripling the standard deviation of the highest peak height and was found to be 5.15×10-11 M with a root mean square error (RMSE) of 1.25×10-11 M.
Application on sample plasma
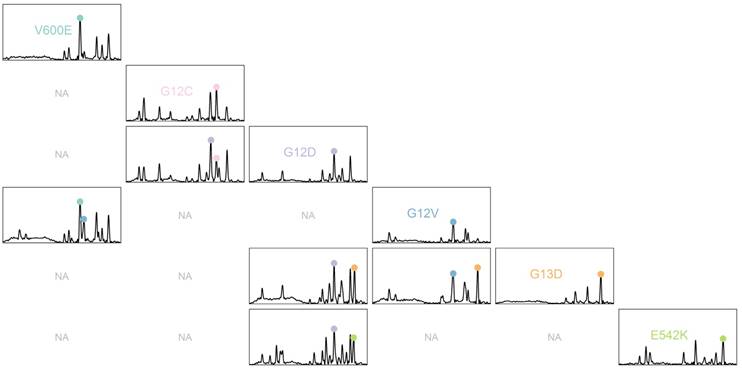
The PCR-SERS method was applied on real plasma samples taken from 49 colorectal cancer patients. Six mutations of interest at BRAF, KRAS, and PIK3CA were detected as mentioned in the method above. Figure 5 shows the obtained SERS spectra. For better differentiation, a pairs plot was drawn according to mutation type. Sub-plots on the diagonal line represented samples with only one mutation. Apparent feature peaks for corresponding tags can be identified in the averaged spectra in each of the sub-plots.
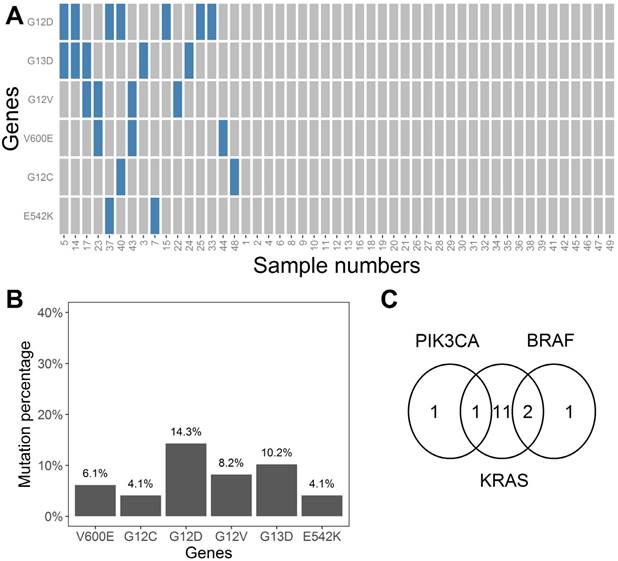
The MLR coefficients that represent whether each mutation exists are shown in Table S1. These coefficients are numbers ranging between 0 and 1. We considered coefficients more than 0.5 as indicative of a positive result for mutation state, and vice versa. The value of 0.5 was selected to minimize the interference of noise. Based on the binary converted coefficients, a heatmap was drawn (Figure 6A) to show the distribution of the six mutation types across all 49 samples. The heatmap indicated that 67% (33 samples) of the samples did not have any of the specified six mutations. The incidence of the BRAF-V600E, KRAS-G12C, KRAS-G12D, KRAS-G12V, KRAS-G13C, and PIK3CA-E542K mutations in our samples was 6.1%, 4.1%, 14.3%, 8.2%, 10.2%, and 4.1%, respectively (Figure 6B). Seven samples harbored two mutations, and nine samples harbored one mutation (Figure 6A). In general, two samples harbored PIK3CA mutations, 14 samples harbored KRAS mutations, and three samples harbored BRAF mutations. One sample had both PIK3CA and KRAS mutations, and two samples had both KRAS and BRAF mutations (Figure 6C).
Relationship between clinical characteristics and mutation profile
Finally, the correlations between the clinical characteristics of our participants and their mutation states were explored by Fisher's exact test. P values obtained through Fisher's exact test that represent the correlations are listed in Table 6. Included in this test were five factors: gender, age, and the clinical characteristics of TNM stage, differentiation, and cancer position. The results indicated that among the observed factors, cancer position had a statistically significant correlation with the presence of the BRAF-V600E (p=0.007) and PIK3CA-E542K (p=0.038) mutations.
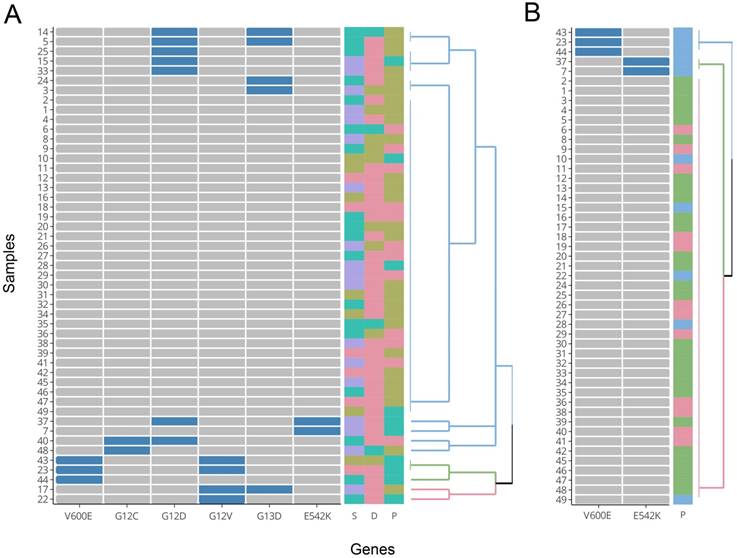
For a better visualization of the relationship between mutations and the three clinical features of TNM stage, differentiation, and cancer position, HCA was performed. The HCA results were displayed using a dendrogram (Figure 7). In the figure, each of the 49 rows represents one sample, and each of the columns represents one mutation type. Cells that are blue indicate positive expression of the mutation and grey colour indicates negative expression. In Figure 7A, all six mutation types were included in the construction of the cluster. The colored squares on the right indicate states of the three clinical features of TNM stage (S), differentiation (D) and cancer position (P), respectively. The grouping of the three clinical features (colour distribution of each column) does not show apparent correlations with the branches of the HCA clustering. Due to the fact that right-sided colorectal cancer was associated with higher mutation percentages at BRAF-V600E and PIK3CA-E542K versus left-colon cancer in our samples, the mutation states of BRAF-V600E and PIK3CA-E542K alone were used to construct a new hierarchical clustering. From Figure 7B, we can see that all samples with a V600E or E542K mutation presented with right-sided colon cancer (blue squares on top of Figure 7B). However, there were still 5 samples with right-sided colon cancer that did not possess a corresponding V600E or E542K mutation. These results mirrored Fisher's exact test and indicated that the two aforementioned mutations were significantly correlated with right-sided colon cancer.
P values obtained through Fisher's exact test
| Clinical features | Ref 1 (V600E) | Ref 2 (G12C) | Ref 3 (G12D) | Ref 4 (G12V) | Ref 5 (G13D) | Ref 6 (E542K) |
|---|---|---|---|---|---|---|
| Gender | 0.237 | 0.215 | 1.000 | 0.612 | 0.018 | 1.000 |
| Age | 0.693 | 0.145 | 0.411 | 1.000 | 0.813 | 0.630 |
| TNM stages | 0.168 | 1.000 | 0.540 | 0.413 | 0.840 | 0.724 |
| Differentiation | 0.612 | 0.189 | 0.284 | 1.000 | 0.524 | 1.000 |
| Cancer position | 0.007 (<0.05) | 0.702 | 0.752 | 0.050 | 0.222 | 0.038 (<0.05) |
A) PCR-SERS spectra of the six KRAS wild-type solutions with increasing concentrations (10-12, 10-11, 20-11, 50-11, 80-11, and 10-10 M). B) Linear regression line drawn by the peak intensities of the highest peak at 1314 cm-1.
SERS spectra of PCR products of plasma samples illustrated using pairs plots. Sub-plots with non-existent mutation combinations are marked as "NA".
A) Heatmap indicating states of six mutation types for all 49 plasma samples. Blue and gray rectangles represent the existence or non-existence of certain mutations, respectively. B) Mutation percentages for the six mutations (V600E, G12C, G12D, G12V, G13D, and E542K) in the 49 plasma samples. C) Venn diagram showing the occurrence and overlap of the mutations at BRAF, PIK3CA, and KRAS
Discussion
The development of colorectal cancer is a multi-step and complex process that involves a number of genes[1]. Both the clinical behavior and response to therapy of a tumor are associated with genetic composition. Therefore, the clinical value of detecting DNA mutations continues to increase. Blood is a good candidate for mutation detection due to its ease of sampling relative to other tissues. Critically, blood also contains cell-free DNA (cfDNA), which is partially released by tumor cells undergoing apoptosis and necrosis[34]. Although the tumor origin of cfDNA makes it a valuable biomarker, the low frequency of mutations and interference from wild-type sequences are both challenges for cancer gene detection[34]. These drawbacks demand a detection method with high sensitivity. Fortunately, SERS serves as a highly sensitive spectroscopy method that has been largely investigated for the detection of genes and DNA[15]. The combination of SERS and biological amplification methods can further increase detection sensitivity. For example, a Taqman assay-based SERS detection method reached a detection limit of 7×10-15 mol/m3 for DNA sequences[35], an ion-mediated cascade amplification SERS method had a detection limit of 30 fM for target DNA[36], and a single base extension-based SERS method detected methylation down to 3 pM[37]. These combinations either utilize dyes to probe target sequences, Ag nano-particles tagged by complementary sequences with target mutations, or they use dye-labeled single bases in the amplification process.
A simple multiplex genotyping method based on SERS was put forward by Graham et al[25]. This research used an amplification refractory mutation system (ARMS) to amplify different mutations with different primers. By labeling different primers with different dyes, the attached dyes could predict the existence of certain mutations. In this research, we used the mutation non-specific primer pairs to amplify target genes, and used dye-labeled probes to tag the existence of mutations. This method is easily applied to existing PCR conditions and only requires additional probe sequences. Due to the narrow bands of SERS, this method is especially convenient for detecting multiple mutations simultaneously. One drawback of this method however is that the mutation sequences should be known beforehand in order to design the probes. MLR was first used for spectral deconvolution by Lutz et al[32] and then Yuan et al[33]. In Lutz's research, the spectral fitting error of MLR was 1-2% of the total signal intensity. Such a low fitting error demonstrated the efficiency of MLR deconvolution on spectral data. In this experiment, by using MLR for the determination of mutation states, six mutation types were discriminated successfully in a single measurement. The detection limit of this method reached a level of 10-11 M, which in our opinion made the suggested PCR-SERS method competent for cfDNA detection. This value is comparable with reports from previous research that examined the combination of PCR and SERS for cancer cell mutation detection (0.2 fM)[38]. This value is also competitive with traditional mutation detection methods (with sensitivities ranging from 0.01% to 10%)[39].
The genes examined in this paper (BRAF, KRAS, and PIK3CA) were all frequently mutated and have been widely studied in colorectal cancer patients. These three genes were found to be indicative of clinicopathological features and therapy selection. BRAF mutations appear to be associated with unfavourable clinicopathological features and often resulted in a poorer prognosis[40, 41]. The frequency of BRAF mutations is 2-5 times higher in high-grade tumors than in low-grade tumors[42]. Wild-type KRAS was found to be a necessary condition for panitumumab efficacy in patients with colorectal cancer[3]. Mutant KRAS on the other hand was found to be associated with the mucinous subtype of cancer and was associated with greater differentiation, high-grade dysplasis, and metastases[43-45]. Research has indicated that PIK3CA may predict resistance to anti-EGFR (epidermal growth factor receptor) therapy in late stage colorectal cancer[46]. The mutation percentages obtained by PCR-SERS of the six mutation types (BRAF-V600E, KRAS-G12C, KRAS-G12D, KRAS-G12V, KRAS-G13C, and PIK3CA-E542K) all had good concordance with previous research. In this paper, the mutation percentages obtained from the 49 samples for BRAF, KRAS, and PIK3CA were 6.1%, 28.6%, and 4.1%, respectively. In previous research, mutation percentages for BRAF, KRAS, and PIK3CA were found to be 5-22%[42], 45%[45], and 15-20%[47], respectively. As only one to four mutation types were detected for each of the three genes in this experiment, the mutation percentages are lower than published values. For correlation between clinical features and mutation profiles, our results indicated that BRAF-V600E (p=0.007) and PIK3CA-E542K mutations (p=0.038) occurred more often in patients with right-sided colon cancer than left-sided colon and rectal cancer. These results are consistent with the reports by Sclafani et al[42] and are reasonable given that cancer in the right colon is biologically distinct versus cancer in the left colon[1].
Compared to conventional mutation detection methods, the PCR-SERS protocol has the advantages of rapidity, multiplex ability, high sensitivity, and simple data analysis (Table 7)[9, 48, 49]. In comparison with fluorescence, which is the main means of detection in comparable methods, SERS are fingerprint spectra of secondary structures of molecules. This feature of SERS makes SERS more capable of multiplex detection without the need of separation process such as electrophoresis. As an extension to multiplex PCR, PCR-SERS eases the detection step by measuring the tagged probes using SERS, and improves the sensitivity of multiplex PCR. By using subsequent analysis such as MLR, the existence of certain mutations can be determined. New mutation detection methods can be created by varying or combining certain steps. For example, multiplex ligation-dependent probe amplification (MLPA) can be used to extend the ligation assay for the quantitative assessment of mutations[49]. Further research can be conducted on the combination of SERS with other PCR-like methods.
Conclusion
In conclusion, a PCR-SERS method for multiplex mutation detection was proposed, verified and applied on real plasma samples taken from colorectal cancer patients. This PCR-SERS method utilized dye-labeled probes, which attached to targeted mutations and were extracted for detection using SERS. MLR was used on the obtained SERS spectra to deconvolute the mixture spectra into the composing tag spectra. The effectiveness of this technique was verified on three prepared mixtures with known gene mutations. The determined detection limit was 5.15×10-11 M, which is competent for cfDNA detection. For the 49 patient samples, the PCR-SERS method obtained mutation frequencies of 6.1%, 28.6%, and 4.1% for the BRAF, KRAS, and PIK3CA genes, respectively. Among the six mutation types studied, the BRAF-V600E and PIK3CA-E542K mutations occurred more often in samples from right-sided colon cancer patients than those from left-sided colon and rectal cancer patients. Overall, this PCR-SERS method is a multiplex mutation detection method that is highly sensitive for potential use in clinical mutation screening.
Results of HCA. A) Cluster drawn by all six mutation types (with groupings of TNM stages, differentiation, and cancer position shown on the right). B) Cluster drawn by mutations of V600E and E542K (with grouping of cancer position shown on the right). D: differentiation (poor: brown; moderate: red; well: cyan). S: TNM stages (I: red; II: brown; III: cyan; IV: purple). P: cancer position (left colon: red; right colon: cyan; rectum: brown).
Comparison of the main steps and features of conventional molecular mutation detection methods
| Method | Unknown mutation | Quantitative | Amplification | Differentiation | Detection | Advantages | Disadvantages |
|---|---|---|---|---|---|---|---|
| PCR-SERS in this paper | - | + | multiplex PCR with mutation-specific probes | tags on probes | SERS | rapid multiplex detection, high sensitivity, simple data analysis | uniform SERS substrate |
| Multiplex PCR | - | - | PCR with multiple pairs of primers | size differences | electrophoresis | reduces time and labor requirements | low sensitivity and specificity |
| Real-time PCR | - | + | PCR amplicon is monitored as the PCR progresses | tags on probes | fluorescence | high sensitivity, can be multiplexed, no post-PCR analysis, simple data analysis | expensive in equipment and staff training |
| ARMS | - | - | allele-specific amplification | size differences, tags on primers | electrophoresis, fluorescence | high sensitivity, quick, inexpensive, simple to devise | high cost |
| Ligation assay | - | - | primers annealed to adjacent sites | tags on primers | fluorescence | can be multiplexed, can be used on a single nucleotide or a few adjacent nucleotides | linear amplification rather than exponential one |
| Sequencing | + | + | PCR amplicon used as the template | tags on the dideoxynucleotide | fluorescence, mass spectroscopy | a “gold standard” mutation screening technique, high-throughput, high-accuracy | expensive |
| Single strand conformational polymorphism (SSCP) | + | - | optional PCR | different DNA conformation | fluorescence, electrophoresis | high sensitivity, detects close to 100% of point mutations | complex |
| Denaturing gradient gel electrophoresis (DGGE) | + | - | optional PCR | different melting behavior | fluorescence, electrophoresis | high sensitivity, detects ~80-90% of point mutations | low throughput, complex primer design |
Abbreviations
ARM: amplification refractory mutation system; cfDNA: cell-free DNA; DMEM: Dulbecco's modified Eagle's medium; HCA: hierarchical clustering analysis; MLR: multiple linear regression; PCR: polymerase chain reaction; RMSE: root mean square error; SDA: strand displacement amplification; SERS: surface enhanced Raman spectroscopy.
Supplementary Material
Table S1.
Acknowledgements
We thank Shenyang Shengjing Hospital of the China Medical University for providing blood samples of colorectal cancer patients and all the histophalogical information. This work was sponsored by the Natural Science Foundation of China (NSFC) (31570154) and the Liaoning Science & Technology Project (F14-231-1-34).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Stewart BW, Wild C. World cancer report 2014. Lyon, France: International Agency for Research on Cancer. 2014
2. Chen W, Zheng R, Baade PD. et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115-32
3. Amado RG, Wolf M, Peeters M. et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26:1626-34
4. Fleischhacker M, Dietrich D, Liebenberg V. et al. The role of DNA methylation as biomarkers in the clinical management of lung cancer. Expert Rev Respir Med. 2013;7:363-83
5. Minamoto T, Ronai Z. Gene mutation as a target for early detection in cancer diagnosis. Crit Rev Oncol Hematol. 2001;40:195-213
6. Roock W, Vriendt V, Normanno N. et al. KRAS, BRAF, PIK3CA, and PTEN mutations: implications for targeted therapies in metastatic colorectal cancer. Lancet Oncol. 2011;12:594-603
7. Lievre A, Blons H, Laurent-Puig P. Oncogenic mutations as predictive factors in colorectal cancer. Oncogene. 2010;29:3033-43
8. Vogelstein B, Papadopoulos N, Velculescu VE. et al. Cancer genome landscapes. Science. 2013;339:1546-58
9. Mahdieh N, Rabbani B. An overview of mutation detection methods in genetic disorders. Iran J Pediatr. 2013;23:375-88
10. Ewen Smith W. Modern Raman spectroscopy - a practical approach. Coord Chem Rev. 1981;35:253-7
11. Peng HI, Miller BL. Recent advancements in optical DNA biosensors: exploiting the plasmonic effects of metal nanoparticles. Analyst. 2011;136:436-47
12. Moskovits M. Surface-enhanced Raman spectroscopy: A brief retrospective. J Raman Spectrosc. 2005;36:485-96
13. Hudson SD, Chumanov G. Bioanalytical applications of SERS (surface-enhanced Raman spectroscopy). Anal Bioanal Chem. 2009;394:679-86
14. Wang Y, Yan B, Chen L. SERS tags: novel optical nanoprobes for bioanalysis. Chem Rev. 2013;113:1391-428
15. De Silva Indrasekara AS, Fabris L. SERS-based approaches toward genetic profiling. Bioanalysis. 2015;7:263-78
16. Guerrini L, Krpetić Ž, van Lierop D, Alvarez-Puebla RA, Graham D. Direct Surface-Enhanced Raman Scattering Analysis of DNA Duplexes. Angew Chem Int Ed Engl. 2015;127:1160-4
17. Johnson RP, Richardson JA, Brown T. et al. A label-free, electrochemical SERS-based assay for detection of DNA hybridization and discrimination of mutations. J Am Chem Soc. 2012;134:14099-107
18. Papadopoulou E, Goodchild SA, Cleary DW. et al. Using surface-enhanced Raman spectroscopy and electrochemically driven melting to discriminate Yersinia pestis from Y. pseudotuberculosis based on single nucleotide polymorphisms within unpurified polymerase chain reaction amplicons. Anal Chem. 2015;87:1605-12
19. Fu X, Cheng Z, Yu J. et al. A SERS-based lateral flow assay biosensor for highly sensitive detection of HIV-1 DNA. Biosens Bioelectron. 2016;78:530-7
20. Wang X, Choi N, Cheng Z. et al. Simultaneous Detection of Dual Nucleic Acids Using a SERS-Based Lateral Flow Assay Biosensor. Anal Chem. 2017;89:1163-9
21. Cheng Z, Choi N, Wang R. et al. Simultaneous Detection of Dual Prostate Specific Antigens Using Surface-Enhanced Raman Scattering-Based Immunoassay for Accurate Diagnosis of Prostate Cancer. ACS Nano. 2017;11:4926-33
22. Mahajan S, Richardson J, Brown T. et al. SERS-melting: a new method for discriminating mutations in DNA sequences. J Am Chem Soc. 2008;130:15589-601
23. Wee EJH, Wang Y, Tsao SC-H. et al. Simple, Sensitive and Accurate Multiplex Detection of Clinically Important Melanoma DNA Mutations in Circulating Tumour DNA with SERS Nanotags. Theranostics. 2016;6:1506-13
24. Huang SQ, Hu J, Zhu G. et al. Sensitive detection of point mutation using exponential strand displacement amplification-based surface enhanced Raman spectroscopy. Biosens Bioelectron. 2015;65:191-7
25. Graham D, Mallinder BJ, Whitcombe D. et al. Simple multiplex genotyping by surface-enhanced resonance Raman scattering. Anal Chem. 2002;74:1069-74
26. Baldus SE, Schaefer KL, Engers R. et al. Prevalence and heterogeneity of KRAS, BRAF, and PIK3CA mutations in primary colorectal adenocarcinomas and their corresponding metastases. Clin Cancer Res. 2010;16:790-9
27. Jahr S, Hentze H, Englisch S. et al. DNA fragments in the blood plasma of cancer patients: quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001;61:1659-65
28. Lurkin I, Stoehr R, Hurst CD. et al. Two multiplex assays that simultaneously identify 22 possible mutation sites in the KRAS, BRAF, NRAS and PIK3CA genes. PLoS One. 2010;5:e8802
29. Munro CH, Smith WE, Garner M. et al. Characterization of the Surface of a Citrate-Reduced Colloid Optimized for Use as a Substrate for Surface-Enhanced Resonance Raman Scattering. Langmuir. 1995;11:3712-20
30. Faulds K, Jarvis R, Smith WE. et al. Multiplexed detection of six labelled oligonucleotides using surface enhanced resonance Raman scattering (SERRS). Analyst. 2008;133:1505-12
31. van Lierop D, Krpetic Z, Guerrini L. et al. Positively charged silver nanoparticles and their effect on surface-enhanced Raman scattering of dye-labelled oligonucleotides. Chem Commun (Camb). 2012;48:8192-4
32. Lutz BR, Dentinger CE, Nguyen LN. et al. Spectral analysis of multiplex Raman probe signatures. ACS Nano. 2008;2:2306-14
33. Yuan H, Liu Y, Fales AM. et al. Quantitative surface-enhanced resonant Raman scattering multiplexing of biocompatible gold nanostars for in vitro and ex vivo detection. Anal Chem. 2013;85:208-12
34. Schwarzenbach H, Hoon DSB, Pantel K. Cell-free nucleic acids as biomarkers in cancer patients. Nat Rev Cancer. 2011;11:426-37
35. Harper MM, Robertson B, Ricketts A. et al. Specific detection of DNA through coupling of a TaqMan assay with surface enhanced Raman scattering (SERS). Chem Commun (Camb). 2012;48:9412-4
36. Shi M, Zheng J, Tan Y. et al. Ultrasensitive detection of single nucleotide polymorphism in human mitochondrial DNA utilizing ion-mediated cascade surface-enhanced Raman spectroscopy amplification. Anal Chem. 2015;87:2734-40
37. Hu J, Zhang CY. Single base extension reaction-based surface enhanced Raman spectroscopy for DNA methylation assay. Biosens Bioelectron. 2012;31:451-7
38. Ye S, Yang Y, Xiao J. et al. Surface-enhanced Raman scattering assay combined with autonomous DNA machine for detection of specific DNA and cancer cells. Chem Commun (Camb). 2012;48:8535-7
39. Diaz LA, Diaz JR, Bardelli A. Liquid biopsies: genotyping circulating tumor DNA. J Clin Oncol. 2014;32:579-86
40. Clancy C, Burke JP, Kalady MF. et al. BRAF mutation is associated with distinct clinicopathological characteristics in colorectal cancer: a systematic review and meta-analysis. Colorectal Dis. 2013;15:E711-E718
41. National comprehensive cancer network: Fort Washington, USA. Clinical practice guidelines in oncology - colon cancer. Version 3.2015. https://www.nccn.org/professionals/physician_gls/f_guidelines.asp#site
42. Sclafani F, Gullo G, Sheahan K. et al. BRAF mutations in melanoma and colorectal cancer: A single oncogenic mutation with different tumour phenotypes and clinical implications. Crit Rev Oncol Hematol. 2013;87:55-68
43. Zhang J, Zheng J, Yang Y. et al. Molecular spectrum of KRAS, NRAS, BRAF and PIK3CA mutations in Chinese colorectal cancer patients: analysis of 1,110 cases. Sci Rep. 2015;5:18678
44. Yi C, Huang Y, Yu X. et al. Clinicopathologic distribution of KRAS and BRAF mutations in a Chinese population with colorectal cancer precursor lesions. Oncotarget. 2016;7:17265-74
45. Cicenas J, Tamosaitis L, Kvederaviciute K. et al. KRAS, NRAS and BRAF mutations in colorectal cancer and melanoma. Med Oncol. 2017;34:26
46. Sartore-Bianchi A, Martini M, Molinari F. et al. PIK3CA mutations in colorectal cancer are associated with clinical resistance to EGFR-targeted monoclonal antibodies. Cancer Res. 2009;69:1851-7
47. Ogino S, Lochhead P, Giovannucci E. et al. Discovery of colorectal cancer PIK3CA mutation as potential predictive biomarker: power and promise of molecular pathological epidemiology. Oncogene. 2014;33:2949-55
48. Taylor CF, Taylor GR. Current and emerging techniques for diagnostic mutation detection: an overview of methods for mutation detection. Methods Mol Med. 2004;92:9-44
49. Coleman WB, Tsongalis GJ. Molecular Diagnostics. Totowa, USA: Humana Press. 2005
Author contact
![]() Corresponding author: lixiaozhouedu.cn
Corresponding author: lixiaozhouedu.cn