Theranostics
13.3
Impact Factor
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Special Issues
Top
INTRODUCTION
MATERIALS AND METHODS
RESULTS
DISCUSSION
Abbreviations
Supplementary Material
Acknowledgements
References
INTRODUCTION
MATERIALS AND METHODS
RESULTS
DISCUSSION
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(5):1399-1410. doi:10.7150/thno.21072 This issue Cite
Research Paper
Mesenchymal stem cells-derived exosomes are more immunosuppressive than microparticles in inflammatory arthritis
Stella Cosenza1,2, Karine Toupet1,2, Marie Maumus 1,2, Patricia Luz-Crawford3, Olivier Blanc-Brude4, Christian Jorgensen1,2,5*, Danièle Noël1,2,5 ![]() *
*
1. Inserm, U1183, Saint-Eloi Hospital, Montpellier, France;
2. Montpellier University, UFR de Médecine, Montpellier, France;
3. Laboratorio de Inmunología Celular y Molecular, Centro de Investigación Biomédica, Facultad de Medicina, Universidad de Los Andes, Santiago, Chile,
4. Inserm, UMRs-970, Centre de Recherche Cardiovasculaire de Paris, PRES Sorbonne-Paris-Cité, Université Paris-Descartes, France;
5. Clinical immunology and osteoarticular diseases Therapeutic Unit, Hôpital Lapeyronie, Montpellier, France
*: equally contributing authors
Received 2017-5-18; Accepted 2017-11-9; Published 2018-2-3
Citation:
Cosenza S, Toupet K, Maumus M, Luz-Crawford P, Blanc-Brude O, Jorgensen C, Noël D. Mesenchymal stem cells-derived exosomes are more immunosuppressive than microparticles in inflammatory arthritis. Theranostics 2018; 8(5):1399-1410. doi:10.7150/thno.21072. https://www.thno.org/v08p1399.htm
Other stylesAbstract

Objectives: Mesenchymal stem cells (MSCs) release extracellular vesicles (EVs) that display a therapeutic effect in inflammatory disease models. Although MSCs can prevent arthritis, the role of MSCs-derived EVs has never been reported in rheumatoid arthritis. This prompted us to compare the function of exosomes (Exos) and microparticles (MPs) isolated from MSCs and investigate their immunomodulatory function in arthritis.
Methods: MSCs-derived Exos and MPs were isolated by differential ultracentrifugation. Immunosuppressive effects of MPs or Exos were investigated on T and B lymphocytes in vitro and in the Delayed-Type Hypersensitivity (DTH) and Collagen-Induced Arthritis (CIA) models.
Results: Exos and MPs from MSCs inhibited T lymphocyte proliferation in a dose-dependent manner and decreased the percentage of CD4+ and CD8+ T cell subsets. Interestingly, Exos increased Treg cell populations while parental MSCs did not. Conversely, plasmablast differentiation was reduced to a similar extent by MSCs, Exos or MPs. IFN-γ priming of MSCs before vesicles isolation did not influence the immunomodulatory function of isolated Exos or MPs. In DTH, we observed a dose-dependent anti-inflammatory effect of MPs and Exos, while in the CIA model, Exos efficiently decreased clinical signs of inflammation. The beneficial effect of Exos was associated with fewer plasmablasts and more Breg-like cells in lymph nodes.
Conclusions: Both MSCs-derived MPs and Exos exerted an anti-inflammatory role on T and B lymphocytes independently of MSCs priming. However, Exos were more efficient in suppressing inflammation in vivo. Our work is the first demonstration of the therapeutic potential of MSCs-derived EVs in inflammatory arthritis.
Keywords: mesenchymal stem cells, extracellular vesicles, trophic factors, cell therapy, rheumatoid arthritis
INTRODUCTION
Mesenchymal stem or stromal cells (MSCs) are multipotent progenitor cells, which can be isolated from many tissues, such as bone marrow, adipose tissue, synovium or Wharton's jelly [1]. MSCs are characterized by a multilineage differentiation potential and paracrine function through the release of multiple growth factors, chemokines and cytokines. One major role of MSCs is to suppress proliferation and function of cells of both innate and adaptive immunity [2]. We and others have demonstrated that MSCs priming by the inflammatory environment (interferon (IFN)-γ with interleukin (IL)-1α, IL-1β or TNF-α) is required for their immunomodulatory effect [3-6]. Thanks to this property, they are widely investigated for their therapeutic properties in a variety of inflammatory and autoimmune diseases, including type 2 diabetes, experimental autoimmune encephalomyelitis or rheumatoid arthritis (RA). Among inflammatory models, the collagen-induced arthritis (CIA) murine model is of particular interest since it reproduces the main symptoms of RA both at clinical and biological levels [7]. It is a widely used model of arthritis, easily reproducible and useful for therapies evaluation. However, the model is time consuming, requiring 45 days for full arthritis development, and incidence is not 100%. Another model of inflammation is delayed-type hypersensitivity (DTH) model, which is highly reproducible and induced in one week. This is therefore a rapid, easy to manipulate and useful model for anti-inflammatory treatment evaluation [6]. A number of molecules secreted by MSCs have been shown to mediate their immunoregulatory function, including indoleamine-2,3-dioxygenase (IDO), inducible nitric oxide synthase (iNOS), prostaglandin E2 (PGE2), TNF stimulated gene (TSG)-6, and human leukocyte antigen (HLA)-G. Indeed, MSCs that were deficient for IL6, IL1 receptor antagonist (IL1RA) or glucocorticoid-induced leucine zipper (GILZ) had partly lost their immunosuppressive capacity in the murine collagen-induced arthritis (CIA) model [8-11]. In addition to being released in the extracellular milieu, a number of factors are proposed to be packaged into extracellular vesicles (EVs) and migrate throughout the body via the bloodstream. EVs are small vesicles produced by virtually all cell types, characterized by a phospholipid bilayer and containing a large variety of proteins, mRNAs, and miRNAs [12]. Two main types of EVs are exosomes, or small vesicles (diameter below 150 nm), produced in the endosomal compartment in so-called multivesicular bodies, and microvesicles, or microparticles (ranging from 150 to 1000 nm in diameter), released by budding of the plasma membrane. In addition to size, the International Society of Extracellular Vesicles recently proposed minimal criteria to define EVs, including morphology, mechanism of cellular release and biochemical parameters [13]. Therapeutic efficacy of MSCs-derived EVs (MSCs-EVs) has been reported in different disease models, such as myocardial infarction and reperfusion injury, liver and kidney injury, hind limb ischemia and inflammatory diseases (for review, see [14]). While much interest in MSCs-EVs for the treatment of many diseases has been shown, little is known on their exact function. Moreover, no study has evaluated the role of MSCs-derived EVs in pre-clinical models relevant for rheumatoid arthritis [15]. In inflammatory conditions, an inhibitory function of MSCs-EVs on immune cell activation has been claimed in some studies while others reported absence of immunomodulatory effect [16]. Such discrepancies between studies might be due to differences in isolation protocols, but also to the activation state of MSCs during EVs production. Moreover, the respective roles of different subtypes of EVs are poorly investigated. Indeed, the first objective of this study was to compare the immunosuppressive function of small vesicles/exosomes (Exos) versus larger microparticles (MPs), both in vitro and in vivo in a model of inflammatory arthritis. The second objective was to determine whether pre-activation of MSCs during preparation of conditioned media might impact the immunomodulatory effect of EVs.
MATERIALS AND METHODS
Mesenchymal stem cell culture
Bone marrow MSCs were isolated from C57BL/6 mice and characterized by phenotyping and trilineage differentiation potential [17]. They were maintained in proliferative medium consisting in DMEM, 100 µg/mL penicillin/streptomycin, 2 mM glutamine and supplemented with 10% foetal calf serum (FCS).
Mesenchymal stem cell-derived extracellular vesicle production and characterization
MSCs were seeded at 6x104 cells/cm² in proliferative medium for 24 h and then with production medium for 48 h. Production medium consisted of proliferative medium supplemented with 3% EVs-free FCS, obtained by overnight ultracentrifugation of DMEM plus 20% FCS at 100,000 × g. When indicated, MSCs were activated with 20 ng/mL IFN-γ. Using an anti-viral functional assay, activity of the recombinant protein was measured to be 0.3-0.9 ng/mL (R&D systems, France). MSCs-conditioned medium (CM) was recovered from 150 mm diameter culture dishes containing 3-5x106 MSCs in 12 mL. The number of apoptotic MSCs was checked by Annexin 5 labelling and flow cytometry quantification and was always below 5% before ultracentrifugation. MSCs-CM (distributed in 6 tubes containing 38.5 mL each) was centrifuged at 300 × g for 10 min and 2,500 × g for 25 min to remove detached cells and debris/apoptotic bodies, respectively. CM was used pure in some experiments or further centrifuged for EVs isolation. For total EVs, CM was centrifuged at 100,000 × g for 2 h in polyallomer tubes using a SW28 Ti Swinging-Bucket rotor (k factor 246; Beckman Coulter, Villepinte). Total EVs pellets were rinsed in phosphate-buffered saline (PBS), centrifuged at 100,000 × g for 2 h and suspended in 100 µL of PBS. For MPs, CM was centrifuged at 18,000 × g for 1 h; the pellet was washed and finally suspended in PBS. For Exos, supernatant from the MPs fraction was filtered through a 0.22 µm porous membrane and centrifuged at 100,000 × g for 2 h. The pellet was washed and suspended in PBS. EVs preparations were normalized to total protein content as quantified by BCA assay (Sigma, Saint-Quentin Fallavier, France).
Size distribution of EVs was determined by Nanoparticle Tracking Analysis (NTA) using a NanoSight LM10-12 instrument as advised by the manufacturer (NTA 3.1 build 3.1.54; Malvern Instruments, Orsay) using the following parameters: camera level 13; threshold 5; 22.4°C; 3 videos per analyzed sample. Visualization of EVs was assessed by transmission electron microscopy. EVs suspensions were loaded on Formvar-coated grids and negatively stained with uranyl acetate for 15 min. Grids were observed using a Tecnai F20 FEI 200KV microscope.
Flow cytometry analysis
For apoptosis, MSCs were labelled using the Annexin V-PE apoptosis detection kit following the manufacturer's instructions (eBioscience). For EVs, 1 µg equivalent protein was incubated with 4 µm aldehyde/sulfate latex beads (ThermoFisher Scientific) at 4°C overnight and free reactive sites on the beads were filled by adding 100 mM glycine. EVs-coated beads were centrifuged at 3000 × g for 20 min and washed 3 times in PBS. EVs-coated beads were stained using 1 µL of fluorophore-conjugated specific antibodies (0.2 mg/mL) for CD9, CD29, CD44, CD81, SCA-1 (BD Biosciences, Le Pont de Claix) for 30 min and data acquisition was performed using FACS Canto II cytometer (BD Biosciences, Le Pont de Claix). For cell analysis, total splenocytes or isolated cell subsets were incubated in PBS containing 0.2% bovine serum albumin (BSA) and 1 µL of fluorophore-conjugated antibodies (0.2 mg/mL) specific for CD4, CD8, CD19, CD25, CD138, B220 or respective isotype controls (BD-Bioscience) at 4 °C for 20 min. For intracellular staining, cells were stimulated with PMA (50 ng/mL), ionomycin (1 µg/mL) and brefeldin A (10 µg/mL) for 4 h. Then, cells were stained with specific Abs against mouse CD4 and CD25 before being fixed and permeabilized with Perm/Fix solution (eBioscience). Finally, 1 µL of fluorophore-conjugated antibodies (0.2 mg/mL) specific for IL10, IL17, IFN-γ, isotypic controls (BD-Bioscience) or FOXP3 (eBioscience) was added for 30 min in the dark. Flow cytometry analyses were done with Diva software (BD Pharmingen, Le Pont-de-Claix, France).
T lymphocyte proliferation assay and immune cell subset isolation and differentiation
Murine splenocytes were isolated as described [10]. CD4+ or CD8+ T lymphocytes were isolated from spleen using the Dynabeads Untouched Mouse CD4 or CD8 cells kit (ThermoFisher Scientific). After magnetic separation, supernatants containing untouched CD4+ or CD8+ T cells were recovered. T cells were cultured in activation medium consisting of IMDM (Life Technologies) containing 10% heat-inactivated FCS, 2 mM L-glutamine, 100 U/mL penicillin, 100 μg/mL streptomycin (Lonza), 0.1 mM non-essential amino acids, 1 mM sodium pyruvate, 20 mM HEPES and 50 μM of beta-mercaptoethanol (Life Technologies). A proliferation assay was performed with 2x105 immune cells stimulated with 5 μg/mL of concanavalin A (ConA) and CM from MSCs (100 µL), MSCs or EVs was added for 3 days. When indicated, IFN-γ (20 ng/mL)-primed MSCs were added to the proliferation assay. Cell proliferation was measured using CellTiter-Glo® Assay (Promega).
B lymphocytes were isolated from spleen using the CD43 Microbeads kit (Miltenyi Biotech) and plated (105 cells/well) for activation as described [10]. Briefly, B cells were labeled with 2 µM of Cell Trace Violet™ (CTV; Molecular Probes, Eugene, OR, USA) at 37°C for 10 min. B cells were cultured in activation medium (see above) without beta-mercaptoethanol. B cells were activated with 2.5 µg/mL of CpG-containing oligodeoxynucleotide (CpG-ODN) 1826 (5'TCC-ATG-ACG-TTC-CTG-ACG-TT-3´) (Enzo Life Science, Villeurbanne, France), 2.5 µg/mL unconjugated goat F(ab´)2 anti-mouse IgM (Jackson Immunoresearch, Suffolk, UK), 25 ng/mL CD40L and 1000 U of IL2 (R&D Systems). When indicated, MSCs (ratio: 1 MSC/5 cells) or different MPs or Exos amounts were added for 3 days.
ELISA assays
Cytokines were quantified in culture supernatants or sera by ELISA (R&D Systems, Lille). For cytokine production, 2x106 splenocytes were stimulated with 25 μg/mL of bovine collagen II (bCII) and supernatants were collected after 48 h. Total Ig from B lymphocyte supernatants or mice sera was quantified as described [10]. MPs and Exos preparations were resuspended in PBS and submitted to 4 cycles of 5 s of sonication (XL2020 Sonicator Ultrasonic liquid processor, Misonix, Delta Labo, Avignon) before protein quantification.
Delayed-T hypersensitivity model
Experiments were conducted in accordance with the Ethical Committee for animal experimentation of the Languedoc-Roussillon (Approval CEEA-LR-2016050918509993). BALB/c mice were immunized by injection of chick ovalbumin (cOVA) emulsified in 100 µL complete Freund's adjuvant at the base of the tail. The recall was done at day 5 with 30 µg cOVA in 30 µL saline solution or 30 µL of increasing quantities of EVs mixed with the cOVA solution in fat pad as described [18]. The left footpad was injected with 30 µL saline solution as a negative control. At day 6, the thickness of the footpads was measured using a caliper before euthanizing the mice. Thickness increment was calculated as the difference between the immunized footpads at day 6 (right-left) and the unimmunized footpads at day 0 (right-left).
Collagen-induced arthritis model
Arthritis was induced in 9-week-old DBA/1 mice in accordance with the Ethical Committee for animal experimentation of the Languedoc-Roussillon (Approval APAFIS#5347-2016050917427820). Briefly, bCII (2 mg/mL) was diluted in acetic acid (0.05 M) and emulsified in Freund´s complete adjuvant (Thermoscientific, Rockford, IL, USA) as described [10]. The suspension (100 µL) was injected intradermally at the base of the tail at day 0. At day 21, a boost with bCII in Freund´s incomplete adjuvant was administered. EVs were injected intravenously at day 18 and 24. Clinical signs of arthritis were scored as reported [19]. At euthanasia, blood, draining lymph nodes and spleens were collected for immune cell analysis. Hind limbs were fixed in 4% formaldehyde for X-ray micro-computed tomography (µCT).
Bone parameter analyses
After fixation of hind limbs in 4% formaldehyde, paws were scanned in a µCT scanner SkyScan 1176 (Bruker, Belgium) using the following parameters: 50 kV, 500 µA, 0.5 mm aluminium filter, 180°. Scans were reconstructed using NRecon software (Bruker) and bone parameters were quantified with CTAn software (Bruker) on the cuneiform bone of ankles.
Statistical analyses
Statistical analysis was performed with GraphPad 6 Prism Software. In experiments where number of replicates was under 7, we assumed that data distribution was not Gaussian and we used a nonparametric Mann-Whitney test for comparing data between two groups (control versus treated). For paired multiple groups, we used non-parametric Kruskal-Wallis test followed by Dunn's multiple comparison test. For comparing data between 3 groups and two variables where replicates were ≥ 15, normality was checked and we used a two-way ANOVA followed by Tukey's multiple comparison test. The statistical test used is indicated in the figure legend. A p value < 0.05 was considered significant.
RESULTS
Cryopreserved EVs lose their immunosuppressive function
In the present study, we aimed to investigate the immunosuppressive function of total EVs recovered after serial ultracentrifugation (Fig. 1A). We first compared the effect of IFN-γ-primed MSCs versus naïve MSCs on T cell proliferation and observed a tendency to higher immunosuppressive effect of primed MSCs in the proliferation assay (Fig. 1B). We then tested the CM of naïve or IFN-γ-primed MSCs before and after centrifugation. Like MSCs, pre-centrifugation supernatants exerted an immunosuppressive function on splenocytes. Supernatants from primed MSCs tended to be more suppressive (Fig. 1C). Post-centrifugation supernatants lost their immunomodulatory effects. This indicated that the immunosuppressive components were retained in the EVs-containing pellets. Indeed, EVs incubated on activated splenocytes displayed a dose-dependent immunosuppressive effect when EVs were isolated from primed MSCs (Fig. 1D). Importantly, we found that the immunomodulatory activity of EVs was lost after freeze-thawing (Fig. 1E). We therefore characterized EVs preparations kept at 4°C overnight or after freeze-thawing at -80°C. By TEM analysis, the major difference observed was aggregation or fusion of a fraction of EVs after freezing (Fig. 1F). The immunophenotype of EVs was positive and similar for Sca-1, CD44, CD29 (Fig. 1G). By contrast, both EVs preparations were positive for CD81 but a second CD81bright EVs population was detected at 4°C. By NTA analysis, we observed an overall similar distribution of EVs and a tendency for a lower total number of vesicles after thawing, which was however not significantly different from EVs kept at 4°C (Fig. 1H). By contrast, the median size of EVs was significantly lower when submitted to a freeze-thaw cycle. Therefore, due to the loss of immunosuppressive function after EVs freezing, all subsequent analyses were performed using freshly prepared EVs kept at 4°C less than 24 h.
Figure 1
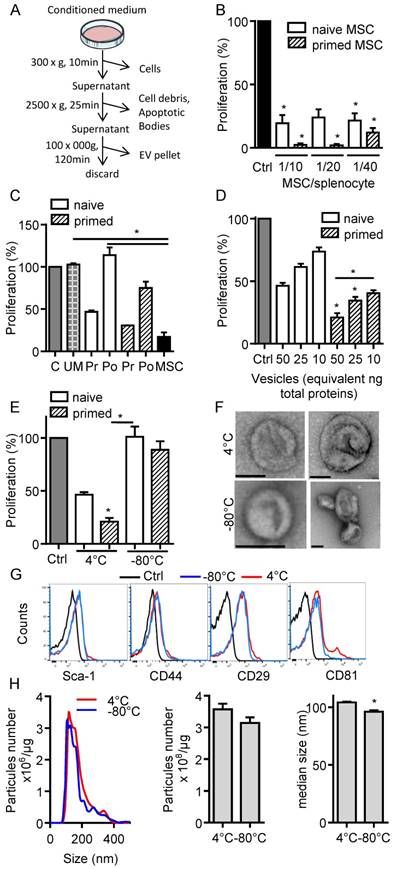
Freshly isolated extracellular vesicles from murine MSCs exert immunosuppressive functions. (A) Experimental protocol for isolation of total extracellular vesicles (EVs) using differential ultracentrifugation. (B) Proliferation of Concanavalin A-activated murine splenocytes cultured alone for 3 days (Ctrl) or incubated with naïve or IFN-γ (20 ng/mL)-primed MSCs (n=3 biological replicates). (C) Proliferation of Concanavalin A-activated murine splenocytes cultured alone for 3 days (C) or incubated with ultracentrifuged production medium (UM) or naïve MSCs or MSCs-conditioned medium (CM) pre (Pr)- or post (Po)-100,000 × g centrifugation according to (A). CM was depleted in cells and debris (by 300 × g and 2500 × g centrifugation steps). MSCs were naïve or primed with 20 ng/mL IFN-γ (n=3 biological replicates). (D) Proliferation of Concanavalin A-activated murine splenocytes cultured alone for 3 days (Ctrl) or incubated with increasing amounts of EVs (n=4 biological replicates). (E) Proliferation of Concanavalin A-activated murine splenocytes cultured alone for 3 days (Ctrl) or incubated with 50 ng of freshly isolated or freeze-thawed EVs (n=4 biological replicates). (F) TEM analysis of freshly isolated (4°C) or freeze-thawed EVs (-80°C). Bar is 100 nm. (G) Expression of MSCs membrane markers (Sca-1, CD44, CD29) and of CD81 exosomal marker on freshly isolated (4°C) or freeze-thawed EVs (-80°C) analyzed by flow cytometry. (H) Number and median size of freshly isolated (4°C) or freeze-thawed EVs (-80°C) by Nano Tracking Analysis. Statistical analysis used a non-parametric Kruskal-Wallis test with Dunn's multiple comparison post-test (B, C, D, E) or a Mann-Whitney test (H). *: p<0.05.
Characterization of exosomes and microparticles
To better characterize MSCs-derived EVs, we used serial ultracentrifugation steps as described [20] and different techniques to analyse the isolated vesicles as were recently recommended by the International Society for Extracellular Vesicles [13]. We recovered MPs and small size EVs (Exos) at 18,000 × g and 100,000 × g, respectively (Fig. 2A). EVs preparations were normalized to the quantity of total proteins: 0.7±0.2 µg and 1.08±0.1 µg proteins/106 MSCs in Exos and MPs fractions respectively, with no difference between EVs from naïve and primed MSCs (Fig. 2B). TEM analysis of MPs and Exos confirmed the presence of round shaped vesicles surrounded by a bilayer membrane with heterogeneous sizes of MPs (Fig. 2C). By NTA analysis, we observed heterogeneity of MPs ranging from 152±1.5 nm to 572±40 nm and from 163±5.9 nm to 524±20 nm in diameter for naïve and primed MSCs, respectively (Fig. 2D, F). EVs concentration was ~1-2x109 EVs/µg of proteins for MPs and Exos (Fig. 2E). No difference in size was noticed between naïve and primed MSCs. Importantly, small size EVs (fractions A/B in Fig. 2D, right) accounted for 75% of MPs (Fig. 1G). In Exos, a homogeneous population of EVs measuring 121±2 nm (naïve MSCs) and 111±4.5 nm (primed MSCs) was detected. Flow cytometry analysis detected expression of CD9 and CD81 in Exos, while expression of MSCs-derived membrane markers Sca-1, CD44, CD29 was detected in MPs, independently of priming (Fig. 2H). Using Western blotting analysis, preferential expression of the exosome-associated proteins Hsp70, Tsg101 and Alix was detected in Exos (not shown). The enrichment of Exos and MPs preparations was illustrated by the presence of the vast majority of Exos (~120 nm) in the 100,000 × g fraction, while MPs (>150 nm) were in the 18,000 × g fraction.
Exos and MPs exhibited similar immunosuppressive effects on T lymphocytes
We next investigated the immunosuppressive properties of MPs and Exos in a proliferation assay. Both fractions decreased the proliferation of ConA-activated splenocytes to a similar extent and in a dose-dependent manner, independently of MSCs priming (Fig. 3A). Among affected lymphocyte subpopulations, MSCs, MPs and Exos tended to reduce the percentage of CD8+IFN-γ+ but MSCs seemed more potent in reducing the number of CD4+IFN-γ+ T lymphocytes (Fig. 3B). Increase of the CD4+IL10+ Tr1 regulatory cell population was observed with MSCs, MPs and Exos, although this was not statistically significant. Interestingly, the CD4+CD25+Foxp3+ Treg population tended to increase only when Exos and MPs were added. We then isolated CD4+ or CD8+ T cells and measured their proliferation rate in the presence of EVs. Both MPs and Exos were unable to reduce the proliferation of CD8+ or CD4+ T lymphocytes, indicating that the inhibitory effect of MPs and Exos on T lymphocytes was indirect (Fig. 3C). Altogether, our data indicated that MPs and Exos exerted an indirect inhibitory effect on T lymphocyte proliferation through Tr1 and Treg induction, respectively.
Figure 2
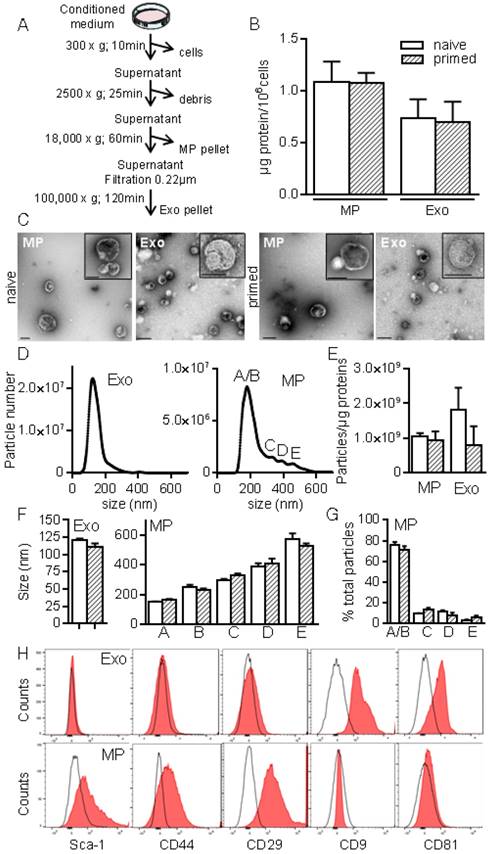
Isolation and characterization of exosomes and microparticles isolated from murine MSCs. (A) Experimental protocol for isolation of microparticles (MPs) and exosomes (Exos) from MSCs-conditioned medium using differential ultracentrifugation. (B) Quantification of EVs produced by 106 MSCs and expressed as equivalent protein. MSCs were naïve or primed with 20 ng/mL IFN-γ (n=5 biological replicates). (C) Representative pictures of MPs (18K) and Exos (100 k) by transmission electron microscopy. Bars represent 200 nm in large pictures and inserts for MPs; for Exos, bars are 200 nm for large pictures and 100 nm for inserts. (D) Number and size of Exos (left) and MPs (right) detected in 1 mL (corresponding to 1 µg EVs equivalent protein) by Nano Tracking Analysis. Letters (A to E) indicate various population peaks (n=3 biological replicates). (E) Quantification of Exos and MPs particle numbers related to the quantity of protein (n=3 biological replicates). (F) Mean size of Exos (left) and MPs (right) in the fractions represented in (D) (n=3 biological replicates). (G) Percentage of MPs in each fraction (A to E) related to total MPs (n=3 biological replicates). (H) Expression of MSCs membrane markers (Sca-1, CD44, CD29) and of exosomal markers (CD9, CD81) on Exos (top) and MPs (bottom) isolated from naïve MSCs analyzed by flow cytometry.
Exos and MPs exhibited similar immunosuppressive effects on B lymphocytes
We then investigated the role of EVs on B lymphocyte differentiation as in [10]. Plasmablast generation tended to be inhibited in the presence of MSCs, MPs and Exos (Fig. 4A). Inhibition of plasmablast differentiation was associated with lower levels of total IgG produced in coculture supernatants (Fig. 4B). However, addition of Exos and MPs on differentiating plasmablasts did not change cytokine production.
We next looked for the presence of factors known to be involved in the immunosuppressive effect of MSCs, in particular those known to impact T and B lymphocytes differentiation and function. While Exos or MPs did not convey IL-6, both conveyed TGF-β1 and PGE2 with no difference between naïve and primed MSCs (Fig. 4C). IL1RA was the most abundant factor in both types of EVs.
Prevention of CIA and DTH by Exos and MPs
First, we wanted to evaluate in vivo the anti-inflammatory effects of EVs in a relatively simple model of inflammation, the DTH model. This experiment was designed to determine the most efficient dose of Exos or MPs in vivo. As with MSCs administration, infusion of MPs tended to reduce inflammation, as measured by paw swelling, and a dose of 250 ng Exos or MPs was the most efficient (Fig. 5A). The dose of 250 ng (corresponding to the production of ~2.5x105 MSCs) was defined as our standard condition. We then compared the effects of total EVs, MPs and Exos injected IV at day 18 and 24 after immunization in the CIA model. We previously demonstrated efficacy of MSCs injection in this model at these time points [8-11, 21]. The rationale of evaluating EVs was that MPs and Exos might exert different anti-inflammatory potencies in vivo and EVs preparations might display intermediate effects. Indeed, complete protection from arthritis was observed with total EVs. 5% incidence and very low clinical scores were obtained with Exos (Fig. 5B-C). In contrast, MPs did not significantly reduce arthritis symptoms but still tended to decrease both score and incidence. Analysis by µCT imaging revealed less bone degradation, as indicated by lower bone surface erosion (area/volume) and higher bone thickness, in mice treated with Exos or MPs (Fig. 5D-F). These findings with DTH and CIA studies supported an anti-inflammatory role of both Exos and MPs, with Exos being more potent than MPs at equal quantity, to suppress arthritis signs.
Figure 3
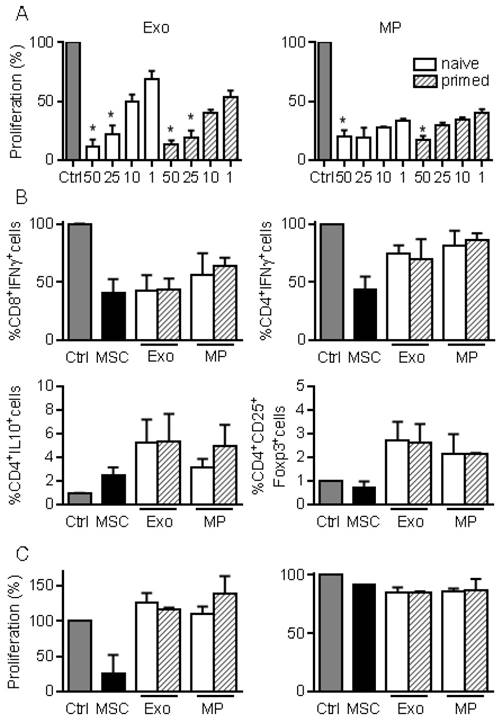
MPs and Exos exert immunosuppressive functions on T lymphocyte subsets. (A) Proliferation of Concanavalin A-activated murine splenocytes cultured alone for 3 days (Ctrl) or incubated with increasing amounts (ng) of Exos (left) or MPs (right) from naïve or IFN-γ primed MSCs (n=5 biological replicates). (B) Percentage of CD8+IFN-γ+ cytotoxic T cells, CD4+IFN-γ+ Th1 cells, CD4+IL-10+ Tr1 cells and CD4+CD25+Foxp3+ Treg cells in splenocytes (n=4) when incubated alone or with naïve MSCs and 25 ng of Exos or MPs. (C) Proliferation of Concanavalin A-activated sorted CD4+ (left) and CD8+ (right) T lymphocytes cultured alone for 3 days (Ctrl) or incubated with naïve MSCs and 50 ng of Exos or MPs from naïve or IFN-γ primed MSCs (n=3 biological replicates). Statistical analysis used a non-parametric Kruskal-Wallis test with Dunn's multiple comparison post-test. *: p<0.05 compared to Ctrl.
Exos were more efficient than MPs in preventing mice from developing CIA
To further investigate whether Exos populations display higher immunomodulatory effects than MPs, we performed a second study injecting 250 ng of Exos and 600 ng of MPs. Because MPs did not significantly decrease arthritic signs in the previous experiment, we decided to inject 2.4-fold more MPs than Exos in order to firmly conclude the absence of MPs effects. Exos infusion significantly reduced paw swelling and global clinical score compared to CIA control or MPs (Fig. 6A). Although MPs decreased the fore paw swelling score, they did not significantly protect from arthritis, as shown by the global arthritis clinical score. Looking at immune cell populations in lymph nodes, we observed no difference in the percentages of Th1, Th17, CD4+Foxp3+ Treg cell subsets. However, a significant decrease in CD4+IL-10+ Tr1 cells was noticed in mice receiving Exos or MPs (Fig. 6B). Interestingly, we detected a significant decrease in plasmablast percentage and increase in CD19+IL-10+ Breg-like populations. We also measured cytokines in culture supernatants of lymph node cells. Upon ConA activation, levels of IL-6 and IL-1β were significantly down-regulated in lymph nodes from Exos-treated mice, but only IL-6 was after MPs infusion (Fig. 6C). Of interest, IL-10 did not change when lymph node cells were stimulated with ConA, whereas IL-10 was significantly up-regulated when cells from Exos-treated mice were stimulated with type II collagen. Finally, we did not detect IL-6 in mouse sera but type II collagen-specific IgG2a/IgG1 antibodies ratios were significantly decreased at all time points in Exos-treated mice (Fig. 6D). These data indicated a shift from a Th1-type toward a Th2-type immune response in Exos-treated mice as compared to control arthritic mice. These results indicated that in vivo, MSCs-derived Exos exerted an antigen-specific anti-inflammatory effect on arthritic mice, which was likely due to decreased plasmablast differentiation and generation of Breg cells.
Figure 4
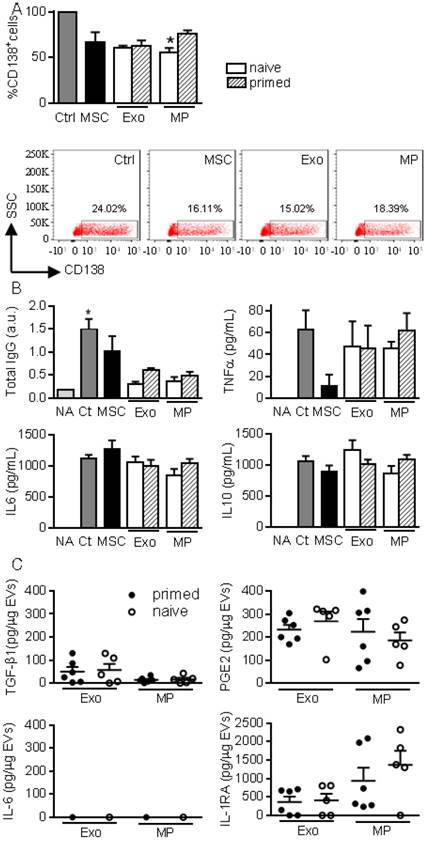
MPs and Exos exert immunosuppressive functions on B lymphocytes. (A) Percentage of CD138+ plasmablasts obtained after activation (Ctrl) or culturing with naïve MSCs or 50 ng Exos or MPs from naïve or IFN-γ primed MSCs (n=5 biological replicates). Representative flow cytometry pictures are shown below. (B) Concentration of total IgG, TNFα, IL6, IL10 in supernatants from plasmablasts in (A) as expressed in arbitrary unit (a.u.) or pg/mL (n=5 biological replicates). (C) Amounts of TGF-β1, PGE2, IL-6, IL1-RA in 1 µg of Exos or MPs as evaluated by ELISA (n=5 biological replicates). Statistical analysis used a non-parametric Kruskal-Wallis test with Dunn's multiple comparison post-test. *: p<0.05compared to Ctrl.
Figure 5
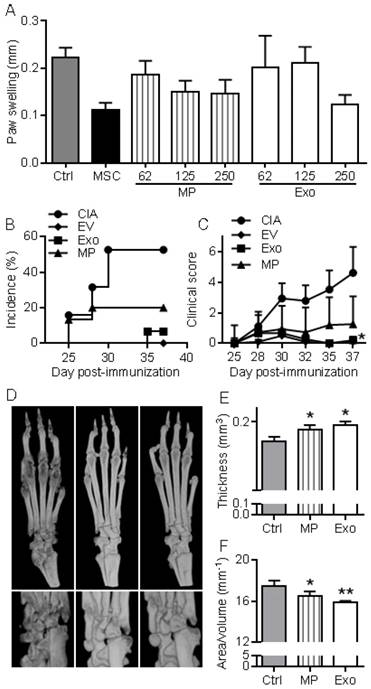
MPs and Exos suppress inflammation in the DTH and CIA models. (A) Inhibition of inflammation in the Delayed-Type Hypersensitivity (DTH) model with increasing doses of MPs or Exos from IFN-γ primed MSCs, as measured by swelling of hind paws at day 6 (n=7 biological replicates). (B) Incidence of mice with inflammation in the collagen-induced arthritis (CIA) model until day 37 at euthanasia (n=15 biological replicates). (C) Inhibition of inflammation as measured by clinical score in the same mice as in (B). (D) Representative 3D reconstruction images of hind paws by µCT analysis showing bone degradation in feet (top) or ankles (bottom) from a Ctrl mouse (left), MPs-treated mouse (middle) and Exos-treated mouse (right). (E) Mean thickness of cuneiform bone from Ctrl mice or mice treated with MPs or Exos as evaluated by histomorphometric µCT analysis. (F) Mean bone degradation of cuneiform bone as evaluated by area/volume parameter by µCT. Statistical analysis used a nonparametric Kruskal-Wallis test followed by Dunn's multiple comparison test (A, E, F) or a two-way ANOVA followed by Tukey's multiple comparison test (C). *: p<0.05 or **: p<0.01 compared to Ctrl.
Figure 6
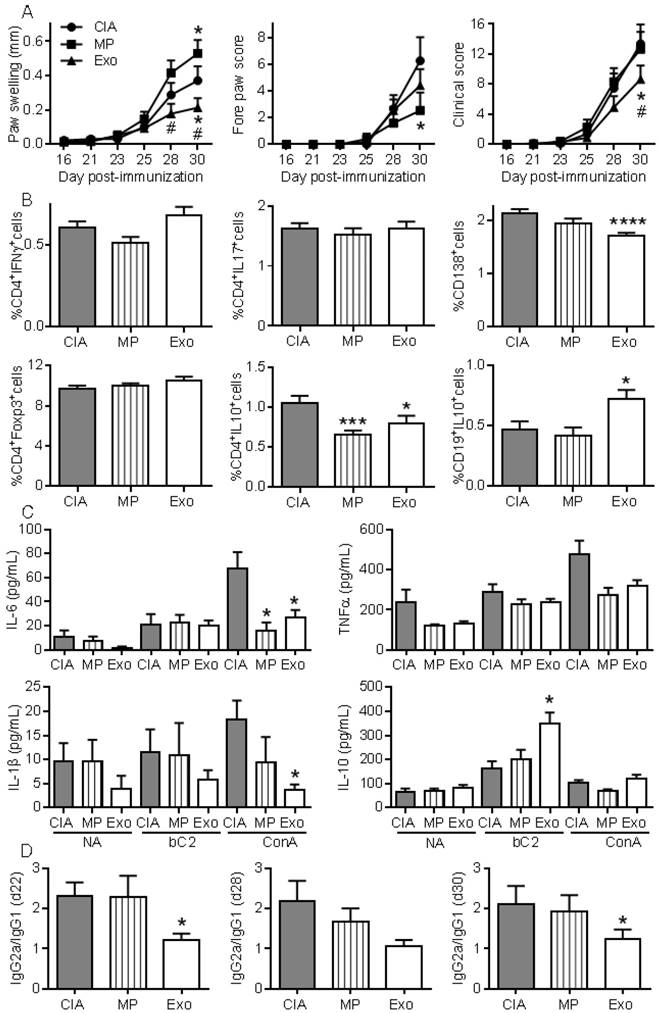
Exos are efficient immunosuppressive agents in CIA by decreasing plasmablast differentiation and generating Breg cells. (A) Effect of 250 ng Exos or 600 ng MPs on inflammation in the CIA model as measured by hind paw swelling, fore paw score and global clinical score until day 30 at euthanasia. (B) Determination of the percentage of CD4+IFNγ+ Th1 cells, CD4+IL17+ Th17 cells, CD4+Foxp3+ Treg cells, CD4+IL10+ Tr1 cells, CD138+ plasmablasts and CD19+IL10+ Breg cells in lymph nodes from control CIA mice and mice treated with Exo or MPs. (C) Quantification of IL-6, TNF-α, IL-1β and IL-10 in supernatants from resting lymph nodes (NA), activated with specific type II collagen (bC2) or ConA. (D) Type II collagen-specific IgG2a/IgG1 antibody ratios in sera of CIA Ctrl mice or mice treated with Exos or MPs at the indicated time points during CIA. Statistical analysis used a two-way ANOVA followed by Tukey's multiple comparison test (A) or a nonparametric Kruskal-Wallis test followed by Dunn's multiple comparison test (B, C, D) with n=15 biological replicates and *: p<0.05; ***: p<0.001 or ****: p<0.00001 compared to CIA Ctrl.
DISCUSSION
This study provides the first evidence that MSCs-derived EVs exert an immunomodulatory effect in an inflammatory arthritis model. Moreover, this is one of the few studies reporting a differential functional role in vivo for MPs and Exos isolated from the same MSCs-conditioned medium.
Here we showed for the first time that in vitro, MSCs-derived MPs and Exos exert similar immunosuppressive functions by decreasing T and B lymphocyte proliferation and inducing Treg cell populations. However, the inhibitory effects of both Exos and MPs on T lymphocyte proliferation were not direct, indicating that other immune cell subsets were responsible for this inhibition. We have shown that Exos and MPs can impact maturation of macrophages, expressing lower levels of TNFα and higher levels of IL10 [38]. Whether Exos and MPs-dependent macrophage polarization might be involved in T lymphocyte proliferation is currently under investigation. Importantly, Exos and MPs were efficient at inducing Treg cell subsets. These results were obtained with EVs isolated from murine MSCs, while a number of studies previously reported immunosuppressive effects for EVs isolated from human MSCs [22-25]. Most studies used either total EVs or Exos. Only one compared MPs and Exos using murine and human MSCs-derived EVs in allogeneic or xenogeneic settings but it failed to show any anti-inflammatory effect [26]. Other studies reported no or low functional effects of EVs on T cell proliferation but reported an indirect inhibition of B and NK cell proliferation [22, 24, 27]. These discrepancies may be attributed to isolation protocols, MSCs sources, culture conditions during MSCs expansion and/or the use of freeze-thawed EVs. Of interest, we clearly demonstrated here that freeze-thawing of EVs at -80°C was detrimental for their functional role. By TEM analysis, we observed images of aggregation or fusion of EVs and a fraction of vesicles with disrupted membranes after freeze-thawing of EVs, suggesting that release of active biomolecules likely occured. The number of vesicles also tended to decrease after freezing. This may be related to loss of vesicles after membrane rupture, or aggregation or fusion as has already been reported [28]. Finally, we noticed a decrease in the median size of EVs, which may reflect preferential loss of large EVs. Whether these differences play a role on the function of EVs is however unclear. Another possibility could be the alteration of proteins at the EVs membrane surface, preventing binding to a specific receptor on target cells. Further investigation is needed to understand how freeze-thawing impacts the immunosuppressive effect of EVs. This is of importance for clinical translation where the use of EVs would rely on preconditioned standardized batches and it highlights the need for evaluating other means of EVs preservation, such as lyophilisation. Other authors reported no alteration of immunomodulatory effects of thawed MSCs-derived MPs or Exos [26]. This suggested that other parameters influence the functionality of EVs.
Priming human MSCs before EVs isolation has been shown to increase the immunomodulatory functions of EVs [24], while another report indicated that MSCs priming did not enhance the immunomodulatory effects of MSCs-derived EVs [25]. Our results are in line with the latter study. We did not observe any increased function of MPs or Exos from primed murine MSCs although CM from primed MSCs exerted a slightly higher anti-inflammatory effect on T cell proliferation, suggesting that soluble factors not packaged inside EVs might be involved. One factor that has been shown to participate to the immunomodulatory function of murine MSCs is inducible nitric oxide synthase (iNOS) [8]. iNOS is involved in the production of nitric oxide (NO) that suppresses T cell responsiveness [6]. Whether iNOS protein or mRNA can be packaged inside EVs from naïve or primed MSCs and released inside target cells to exert a functional role is unlikely but warrants investigation. Conversely, release of NO by MSCs in the extracellular milieu could explain the higher immunomodulatory effect of primed MSCs-CM.
Both types of EVs behaved differently from parental MSCs, even though they exerted a similar role on immune cells. Exos and MPs were less efficient in suppressing Th1 cell proliferation but we found, intriguingly, that they were more potent in inducing CD4+CD25+Foxp3+ Treg and CD4+IL-10+ Tr1 cell populations in vitro. The reason for this differential activity is not known but preliminary analyses detected TGF-β1 within EVs cargo. Although the quantities detected are rather low and may not be relevant for a functional effect of MSCs-derived EVs, the presence of TGF-β1 as mRNA and protein in MSCs-derived EVs has already been reported [29]. In this study, depletion of EVs from culture supernatants reduced the number of TGF-β transcripts in co-incubated peripheral blood mononuclear cells and TGF-β1 mRNA transfer was proposed to be responsible for Treg cell generation. Also detected in MPs and Exos were PGE2 and IL1RA, which have been shown to be important mediators in the in vitro anti-inflammatory effects of MSCs and in inflammatory arthritis [8, 10]. Further analysis of EVs cargo is needed to help answer whether these factors or others mediate the therapeutic efficacy of EVs.
The second key finding is the immunomodulatory functions of MSCs-derived EVs in inflammatory diseases. We have previously reported that MSCs can exert anti-inflammatory effects at a dose of 2.5x105 in the DTH model, and they protect mice from arthritis when injected at a dose of 106 cells at day 18 and 24 after immunization [8-11, 21, 30]. MSCs-mediated protection from inflammation was estimated to be ~50% in terms of paw swelling increase in both models. The quantity of MPs and Exos produced by 106 MSCs is ~1 µg. In the DTH study, we used the same quantity of Exos or MPs as that produced by 2.5x105 MSCs in 48 h and observed similar efficacy. In the CIA study, we used 3-fold or 2-fold less Exos or MPs, respectively, than the quantity produced by 106 MSCs in 48 h. Paw swelling and clinical score were decreased by 35% as compared to CIA control. We may therefore assume that the quantity of MPs or Exos produced by a defined number of MSCs in 48 h reproduce most of the anti-inflammatory functions of the same number of injected cells.
We demonstrated in vivo that Exos, but not MPs, were able to significantly decrease signs of arthritis. To firmly conclude that MPs are less immunosuppressive, we increased the quantity of MPs injected by 2.4-fold as compared to Exos, but did not observe a therapeutic effect. This confirmed that MPs were less efficient than Exos at suppressing inflammation in this model. One possible explanation is that the in vivo biodistribution of Exos and MPs may differ. While EVs from MSCs were shown to preferentially home to spleen, lungs and liver where they were taken up by patrolling macrophages [31], the respective homing of MPs and Exos has not been investigated. However, it has been reported that small nanoparticles (under 100 nm) are less prone to opsonisation and preferentially accumulate in liver and in spleen [32]. Indeed, differential homing of the two types of vesicles to spleen and liver could help elucidate their differential functional effects on inflammation.
The therapeutic efficacy of Exos was not related to a significant decrease of Th1 and Th17 lymphocytes or increase of CD4+CD25+Foxp3+ cells although there was a trend. We even observed a significant decrease of Tr1 cells. The reason for this is not known but we can hypothesize that in vitro conditions do not reflect the in vivo environment, which is much more complex, or that timing of analysis was not appropriate. Indeed, it has been reported that Tr1 expansion in vivo can be transient and correlates with the intensity of inflammation [33]. Analysis of the immune response at euthanasia might have missed Tr1 cell expansion. Importantly, the main role of Exos was to inhibit plasmablast differentiation while inducing IL-10-expressing Breg cells. This result is supported by previous reports that EVs bind preferentially to B lymphocytes and less effectively to other immune cells in vitro [24, 34]. Although our results are the first demonstration of the therapeutic efficacy of MSCs-derived Exos in experimental arthritis, a recent report has shown that MSCs-derived Exos can decrease osteoarthritic symptoms in an inflammatory model of osteoarthritis [35]. Indeed, EVs isolated from dendritic cells, neutrophils or artificial vesicles have shown therapeutic potential in osteoarthritis or RA [12, 36, 37], the present study uncovers new therapeutic opportunities for MSCs-derived EVs in the treatment of rheumatic diseases.
Abbreviations
MSCs: mesenchymal stem/stromal cells; EVs: extracellular vesicles; Exos: exosomes; MPs: microparticles; RA: rheumatoid arthritis; CIA: collagen-induced arthritis; DTH: delayed hypersensitivity; CM: conditioned medium; NTA: nano tracking analysis; FACS: fluorescence-activated cell sorting; PBS: phosphate buffered saline; BSA: bovine serum albumin; PMA: phytohaemaglutinin; conA: concanavalin A; FCS: fetal calf serum; PE: phycoerythrin; bCII: bovine collagen II; cOVA: chicken ovalbumin; IFN: interferon; IL: interleukin; TNF: tumor necrosis factor; IDO: indoleamine-2,3-dioxygenase; iNOS: inducible nitric oxide synthase; PGE2: prostaglandin E2; TSG-6: TNF stimulated gene; HLA-G: human leukocyte antigen; IL1RA: IL1 receptor antagonist; GILZ: glucocorticoid-induced leucine zipper.
Supplementary Material
Supplementary figure S1.
Acknowledgements
We thank Géraldine Pénarier (SANOFI, Montpellier) and Marie Morille (ICGM, Montpellier) for providing access and useful advice for Nano Tracking Analysis of Evs, Marc Mathieu for Western blot analysis and Marc Piechaczyck for providing access to the ultracentrifuge from IGMM (Montpellier, France). Thanks to the “Réseau des Animaleries de Montpellier” animal facility and the “Réseau d'Histologie Expérimentale de Montpellier” histology facility for processing our animal tissues.
Contributors
DN, CJ designed the experiments. Experimental work was performed by SC, KT, MM with contributions from PLC, OBB. SC, KT, MM, DN analyzed the data and prepared the manuscript. SC, KT, MM, PLC, OBB, CJ, DN contributed to writing of the manuscript and final approval.
Funding
Work in the laboratory Inserm U1183 was supported by the Inserm Institute and the University of Montpellier. This project has received funding from the European Union's Horizon 2020 Programme (project ADIPOA2, grant agreement no: 643809). The materials presented and views expressed here are the responsibility of the authors only. The EU Commission takes no responsibility for any use made of the information set out. Study was also supported by the Fondation de l'Avenir, Paris, France (AP-RMA-2015-013). We thank the Agence Nationale pour la Recherche for support of the national infrastructure: "ECELLFRANCE: Development of a national adult mesenchymal stem cell based therapy platform" (ANR-11-INSB-005).
Competing Interest
The authors have no financial or personal conflict of interest to disclose.
References
1. Jorgensen C, Noel D. Mesenchymal stem cells in osteoarticular diseases. Regen Med. 2011;6:44-51
2. Castro-Manrreza ME, Montesinos JJ. Immunoregulation by mesenchymal stem cells: biological aspects and clinical applications. J Immunol Res. 2015;2015:394917
3. Kavanagh DP, Robinson J, Kalia N. Mesenchymal Stem Cell Priming: Fine-tuning Adhesion and Function. Stem Cell Rev. 2014;10:587-99
4. Maumus M, Roussignol G, Toupet K, Penarier G, Bentz I, Teixeira S. et al. Utility of a Mouse Model of Osteoarthritis to Demonstrate Cartilage Protection by IFNgamma-Primed Equine Mesenchymal Stem Cells. Front Immunol. 2016;7:392
5. Djouad F, Plence P, Bony C, Tropel P, Apparailly F, Sany J. et al. Immunosuppressive effect of mesenchymal stem cells favors tumor growth in allogeneic animals. Blood. 2003;102:3837-44
6. Ren G, Zhang L, Zhao X, Xu G, Zhang Y, Roberts AI. et al. Mesenchymal stem cell-mediated immunosuppression occurs via concerted action of chemokines and nitric oxide. Cell Stem Cell. 2008;2:141-50
7. Kannan K, Ortmann RA, Kimpel D. Animal models of rheumatoid arthritis and their relevance to human disease. Pathophysiology. 2005;12:167-81
8. Bouffi C, Bony C, Courties G, Jorgensen C, Noel D. IL-6-dependent PGE2 secretion by mesenchymal stem cells inhibits local inflammation in experimental arthritis. PLoS One. 2010;5:e14247
9. Luz-Crawford P, Tejedor G, Mausset-Bonnefont AL, Beaulieu E, Morand EF, Jorgensen C. et al. Gilz governs the therapeutic potential of mesenchymal stem cells by inducing a switch from pathogenic to regulatory Th17 cells. Arthritis Rheumatol. 2015;67:1514-24
10. Luz-Crawford P, Djouad F, Toupet K, Bony C, Franquesa M, Hoogduijn MJ. et al. Mesenchymal Stem Cell-Derived Interleukin 1 Receptor Antagonist Promotes Macrophage Polarization and Inhibits B Cell Differentiation. Stem Cells. 2016;34:483-92
11. Luz-Crawford P, Ipseiz N, Espinosa-Carrasco G, Caicedo A, Tejedor G, Toupet K. et al. PPARbeta/delta directs the therapeutic potential of mesenchymal stem cells in arthritis. Ann Rheum Dis. 2016;75:2166-74
12. Maumus M, Jorgensen C, Noel D. Mesenchymal stem cells in regenerative medicine applied to rheumatic diseases: role of secretome and exosomes. Biochimie. 2013;95:2229-34
13. Lotvall J, Hill AF, Hochberg F, Buzas EI, Di Vizio D, Gardiner C. et al. Minimal experimental requirements for definition of extracellular vesicles and their functions: a position statement from the International Society for Extracellular Vesicles. J Extracell Vesicles. 2014;3:26913
14. Heldring N, Mager I, Wood MJ, Le Blanc K, Andaloussi SE. Therapeutic Potential of Multipotent Mesenchymal Stromal Cells and Their Extracellular Vesicles. Hum Gene Ther. 2015;26:506-17
15. Cosenza S, Ruiz M, Maumus M, Jorgensen C, Noel D. Pathogenic or Therapeutic Extracellular Vesicles in Rheumatic Diseases: Role of Mesenchymal Stem Cell-Derived Vesicles. Int J Mol Sci. 2017;18:889
16. Burrello J, Monticone S, Gai C, Gomez Y, Kholia S, Camussi G. Stem Cell-Derived Extracellular Vesicles and Immune-Modulation. Front Cell Dev Biol. 2016;4:83
17. Maria AT, Toupet K, Maumus M, Fonteneau G, Le Quellec A, Jorgensen C. et al. Human adipose mesenchymal stem cells as potent anti-fibrosis therapy for systemic sclerosis. J Autoimmun. 2016;70:31-9
18. Domergue S, Bony C, Maumus M, Toupet K, Frouin E, Rigau V. et al. Comparison between Stromal Vascular Fraction and Adipose Mesenchymal Stem Cells in Remodeling Hypertrophic Scars. PLoS One. 2016;11:e0156161
19. Toupet K, Maumus M, Luz-Crawford P, Lombardo E, Lopez-Belmonte J, van Lent P. et al. Survival and biodistribution of xenogenic adipose mesenchymal stem cells is not affected by the degree of inflammation in arthritis. PLoS One. 2015;10:e0114962
20. Thery C, Amigorena S, Raposo G, Clayton A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol. 2006 Chapter 3: Unit 3 22
21. Bouffi C, Bony C, Jorgensen C, Noel D. Skin fibroblasts are potent suppressors of inflammation in experimental arthritis. Ann Rheum Dis. 2011;70:1671-6
22. Del Fattore A, Luciano R, Pascucci L, Goffredo BM, Giorda E, Scapaticci M. et al. Immunoregulatory Effects of Mesenchymal Stem Cell-Derived Extracellular Vesicles on T Lymphocytes. Cell Transplant. 2015;24:2615-27
23. Blazquez R, Sanchez-Margallo FM, de la Rosa O, Dalemans W, Alvarez V, Tarazona R. et al. Immunomodulatory Potential of Human Adipose Mesenchymal Stem Cells Derived Exosomes on in vitro Stimulated T Cells. Front Immunol. 2014;5:556
24. Di Trapani M, Bassi G, Midolo M, Gatti A, Kamga PT, Cassaro A. et al. Differential and transferable modulatory effects of mesenchymal stromal cell-derived extracellular vesicles on T, B and NK cell functions. Sci Rep. 2016;6:24120
25. Monguio-Tortajada M, Roura S, Galvez-Monton C, Pujal JM, Aran G, Sanjurjo L. et al. Nanosized UCMSC-derived extracellular vesicles but not conditioned medium exclusively inhibit the inflammatory response of stimulated T cells: implications for nanomedicine. Theranostics. 2017;7:270-84
26. Matula Z, Nemeth A, Lorincz P, Szepesi A, Brozik A, Buzas EI. et al. The Role of Extracellular Vesicle and Tunneling Nanotube-Mediated Intercellular Cross-Talk Between Mesenchymal Stem Cells and Human Peripheral T Cells. Stem Cells Dev. 2016;25:1818-32
27. Conforti A, Scarsella M, Starc N, Giorda E, Biagini S, Proia A. et al. Microvescicles derived from mesenchymal stromal cells are not as effective as their cellular counterpart in the ability to modulate immune responses in vitro. Stem Cells Dev. 2014;23:2591-9
28. Brisson AR, Tan S, Linares R, Gounou C, Arraud N. Extracellular vesicles from activated platelets: a semiquantitative cryo-electron microscopy and immuno-gold labeling study. Platelets. 2017;28:263-71
29. Favaro E, Carpanetto A, Lamorte S, Fusco A, Caorsi C, Deregibus MC. et al. Human mesenchymal stem cell-derived microvesicles modulate T cell response to islet antigen glutamic acid decarboxylase in patients with type 1 diabetes. Diabetologia. 2014;57:1664-73
30. Bony C, Cren M, Domergue S, Toupet K, Jorgensen C, Noel D. Adipose Mesenchymal Stem Cells Isolated after Manual or Water-jet-Assisted Liposuction Display Similar Properties. Front Immunol. 2016;6:655
31. Grange C, Tapparo M, Bruno S, Chatterjee D, Quesenberry PJ, Tetta C. et al. Biodistribution of mesenchymal stem cell-derived extracellular vesicles in a model of acute kidney injury monitored by optical imaging. Int J Mol Med. 2014;33:1055-63
32. Wiklander OP, Nordin JZ, O'Loughlin A, Gustafsson Y, Corso G, Mager I. et al. Extracellular vesicle in vivo biodistribution is determined by cell source, route of administration and targeting. J Extracell Vesicles. 2015;4:26316
33. Foussat A, Cottrez F, Brun V, Fournier N, Breittmayer JP, Groux H. A comparative study between T regulatory type 1 and CD4+CD25+ T cells in the control of inflammation. J Immunol. 2003;171:5018-26
34. Budoni M, Fierabracci A, Luciano R, Petrini S, Di Ciommo V, Muraca M. The immunosuppressive effect of mesenchymal stromal cells on B lymphocytes is mediated by membrane vesicles. Cell Transplant. 2013;22:369-79
35. Zhu Y, Wang Y, Zhao B, Niu X, Hu B, Li Q. et al. Comparison of exosomes secreted by induced pluripotent stem cell-derived mesenchymal stem cells and synovial membrane-derived mesenchymal stem cells for the treatment of osteoarthritis. Stem Cell Res Ther. 2017;8:64
36. Withrow J, Murphy C, Liu Y, Hunter M, Fulzele S, Hamrick MW. Extracellular vesicles in the pathogenesis of rheumatoid arthritis and osteoarthritis. Arthritis Res Ther. 2016;18:286
37. Headland SE, Jones HR, Norling LV, Kim A, Souza PR, Corsiero E. et al. Neutrophil-derived microvesicles enter cartilage and protect the joint in inflammatory arthritis. Sci Transl Med. 2015;7:315ra190
38. Cosenza S, Ruiz M, Toupet K, Jorgensen C, Noël D. Mesenchymal stem cells derived exosomes and microparticles protect cartilage and bone from degradation in osteoarthritis. Scientific reports. 2017;7:16214
Author contact
![]() Corresponding author: D. Noël, Inserm U1183, IRMB, Hôpital Saint-Eloi, 80 avenue Augustin Fliche, 34295 Montpellier cedex 5, France. Tel: +33 4 67 33 04 73 - Fax: +33 4 67 33 01 13 - E-mail: daniele.noelfr
Corresponding author: D. Noël, Inserm U1183, IRMB, Hôpital Saint-Eloi, 80 avenue Augustin Fliche, 34295 Montpellier cedex 5, France. Tel: +33 4 67 33 04 73 - Fax: +33 4 67 33 01 13 - E-mail: daniele.noelfr
Citation styles
APA
Cosenza, S., Toupet, K., Maumus, M., Luz-Crawford, P., Blanc-Brude, O., Jorgensen, C., Noël, D. (2018). Mesenchymal stem cells-derived exosomes are more immunosuppressive than microparticles in inflammatory arthritis. Theranostics, 8(5), 1399-1410. https://doi.org/10.7150/thno.21072.
ACS
Cosenza, S.; Toupet, K.; Maumus, M.; Luz-Crawford, P.; Blanc-Brude, O.; Jorgensen, C.; Noël, D. Mesenchymal stem cells-derived exosomes are more immunosuppressive than microparticles in inflammatory arthritis. Theranostics 2018, 8 (5), 1399-1410. DOI: 10.7150/thno.21072.
NLM
Cosenza S, Toupet K, Maumus M, Luz-Crawford P, Blanc-Brude O, Jorgensen C, Noël D. Mesenchymal stem cells-derived exosomes are more immunosuppressive than microparticles in inflammatory arthritis. Theranostics 2018; 8(5):1399-1410. doi:10.7150/thno.21072. https://www.thno.org/v08p1399.htm
CSE
Cosenza S, Toupet K, Maumus M, Luz-Crawford P, Blanc-Brude O, Jorgensen C, Noël D. 2018. Mesenchymal stem cells-derived exosomes are more immunosuppressive than microparticles in inflammatory arthritis. Theranostics. 8(5):1399-1410.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.